
Anti-Inflammatory Activity of N-Docosahexaenoylethanolamine and N-Eicosapentaenoylethanolamine in a Mouse Model of Lipopolysaccharide-Induced Neuroinflammation


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
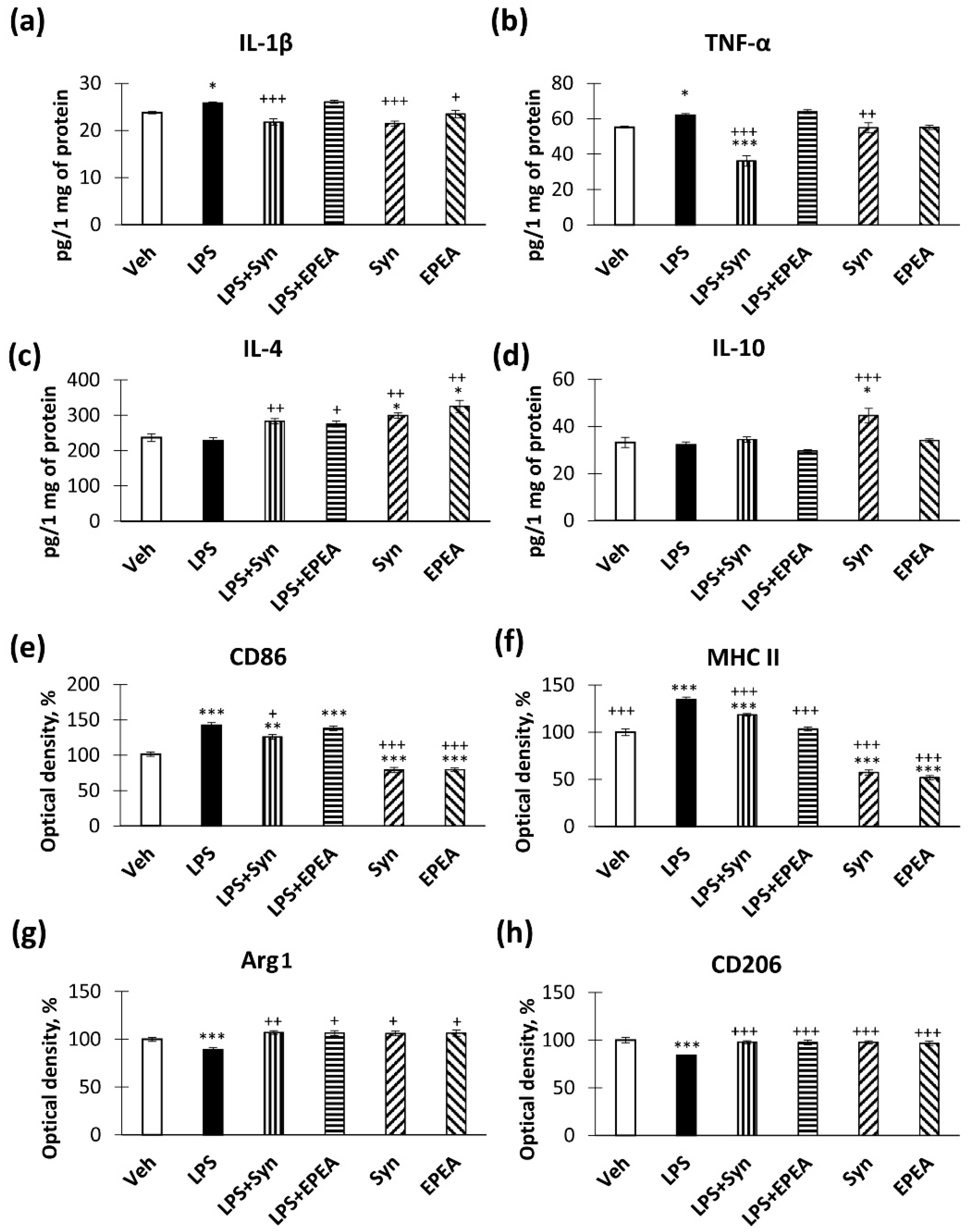
2.1. In Vitro Studies of Cytokines Production
2.2. In Vivo Studies of Pro- and Anti-Inflammatory Factors Production
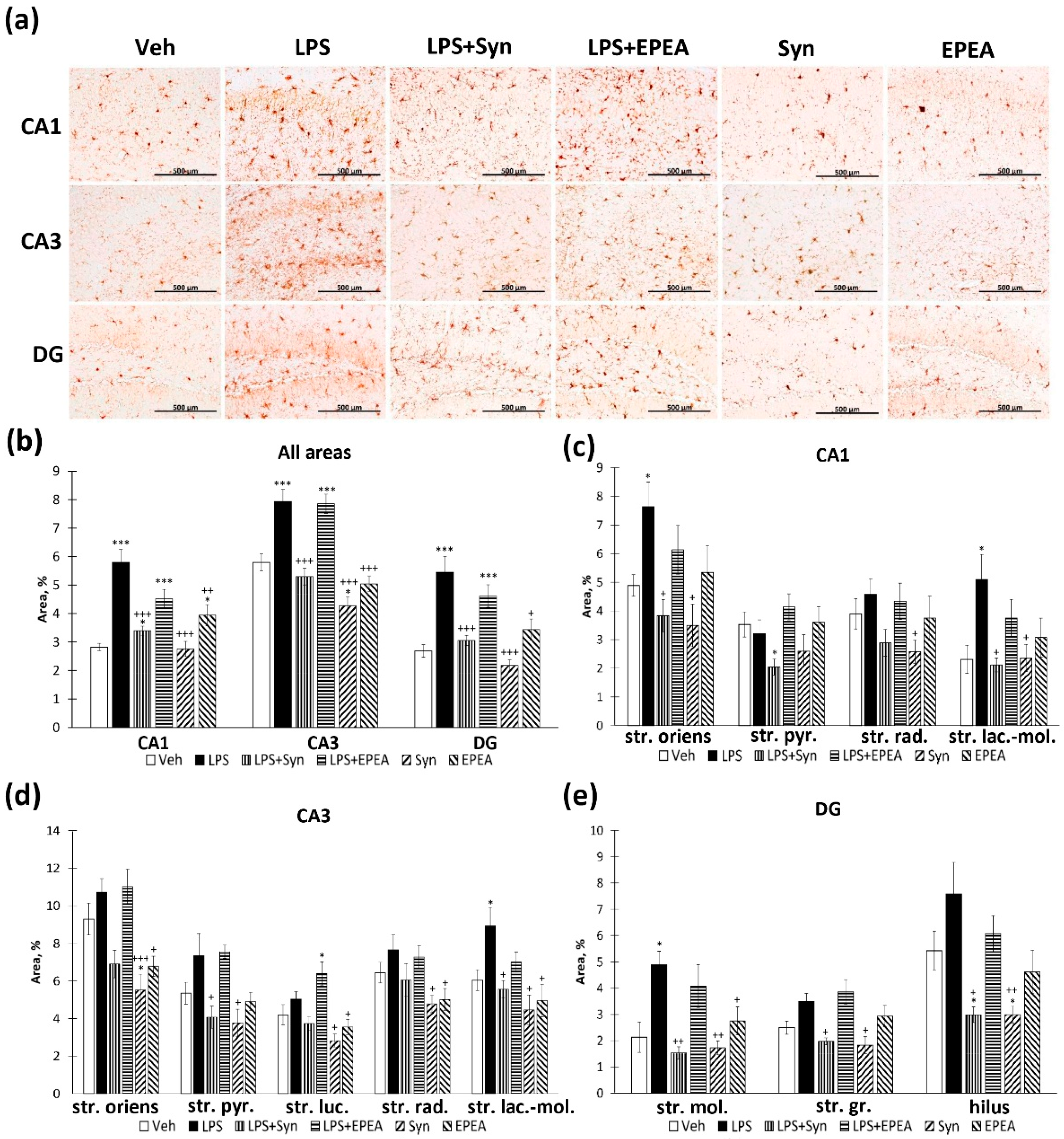
2.3. Microglial Activity in LPS, Synaptamide and EPEA Treatment
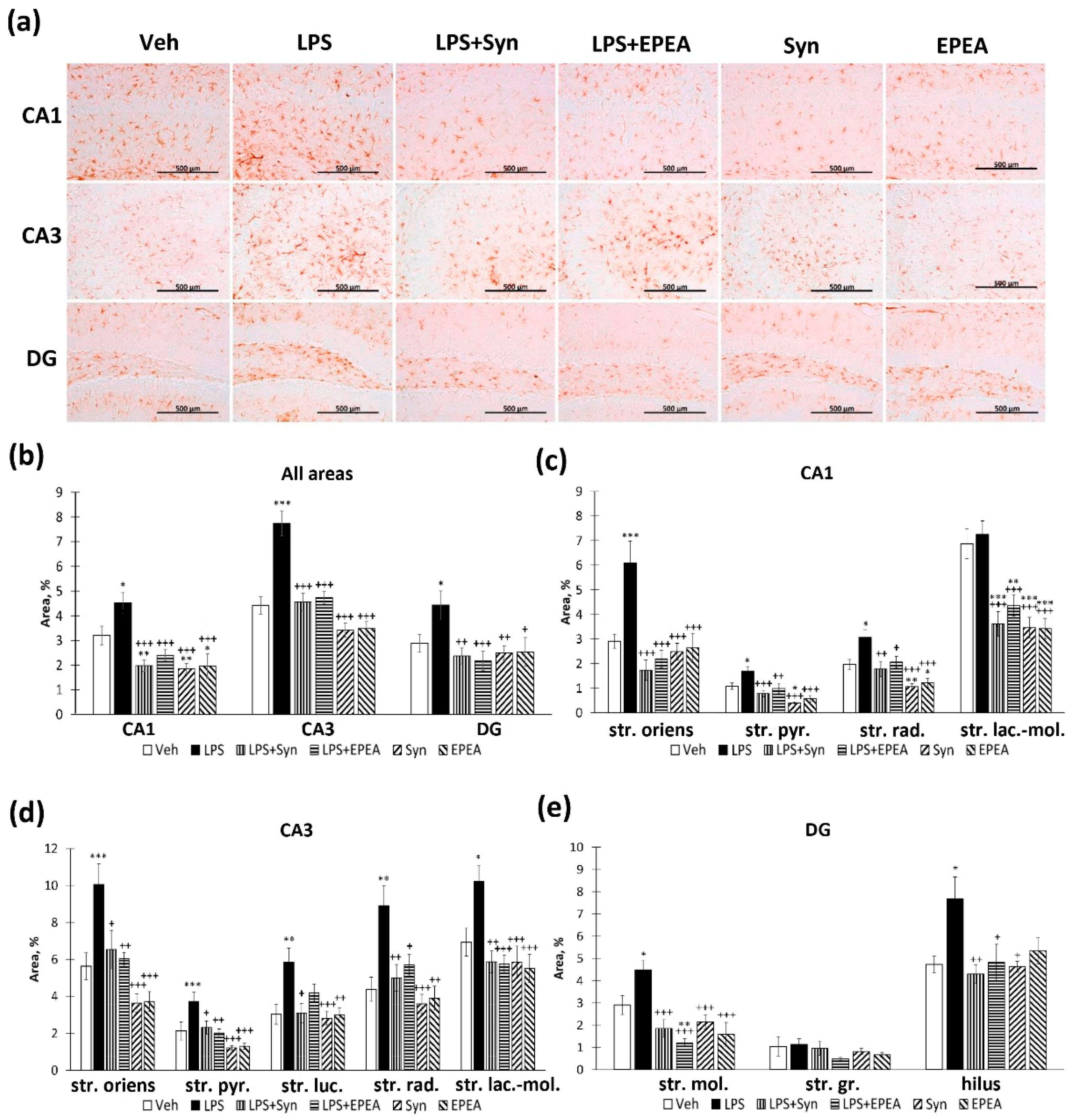
2.4. Astroglial Activity in LPS, Synaptamide and EPEA Treatment
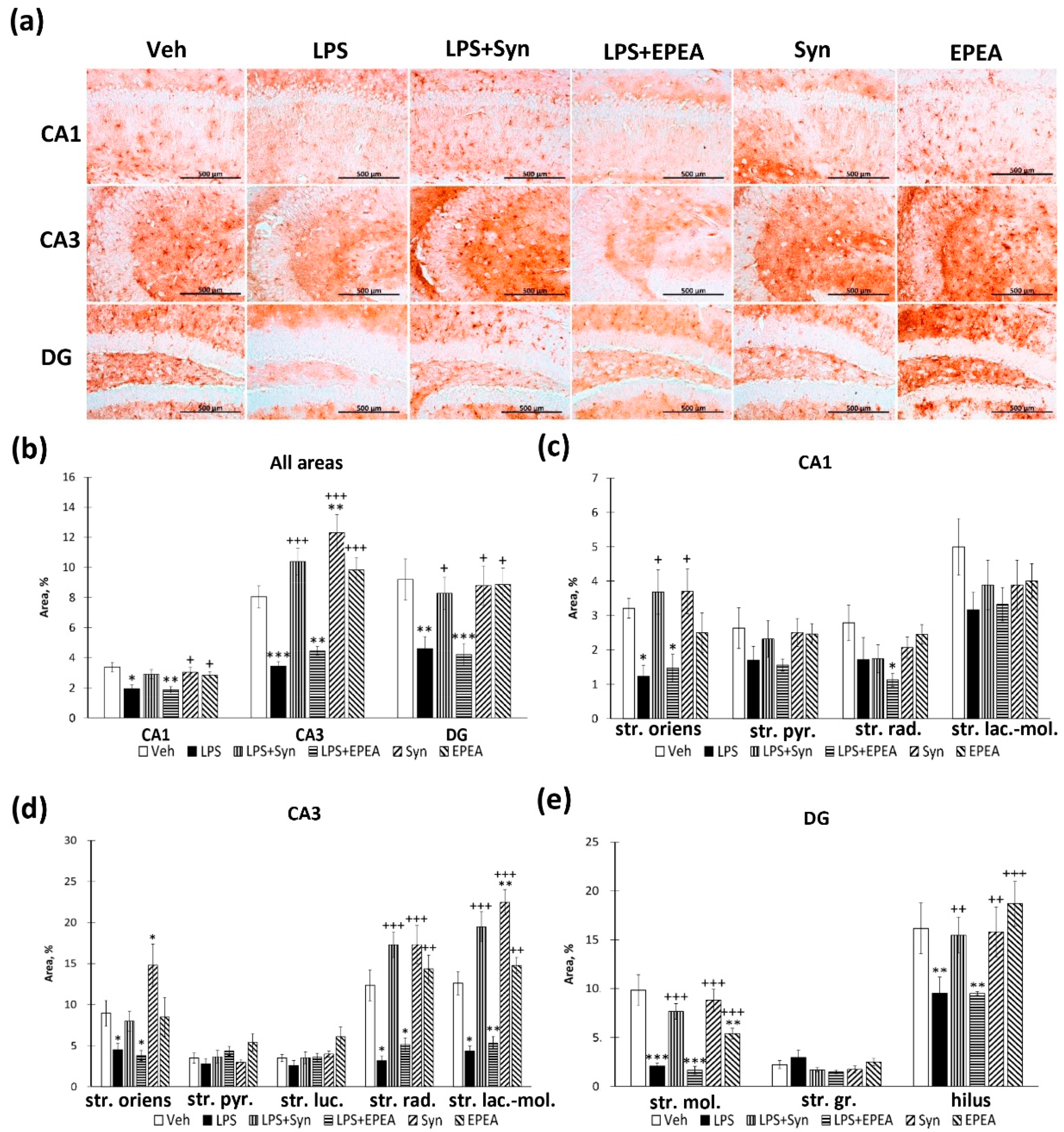
2.5. BDNF Immunoreactivity in LPS, Synaptamide and EPEA Treatment
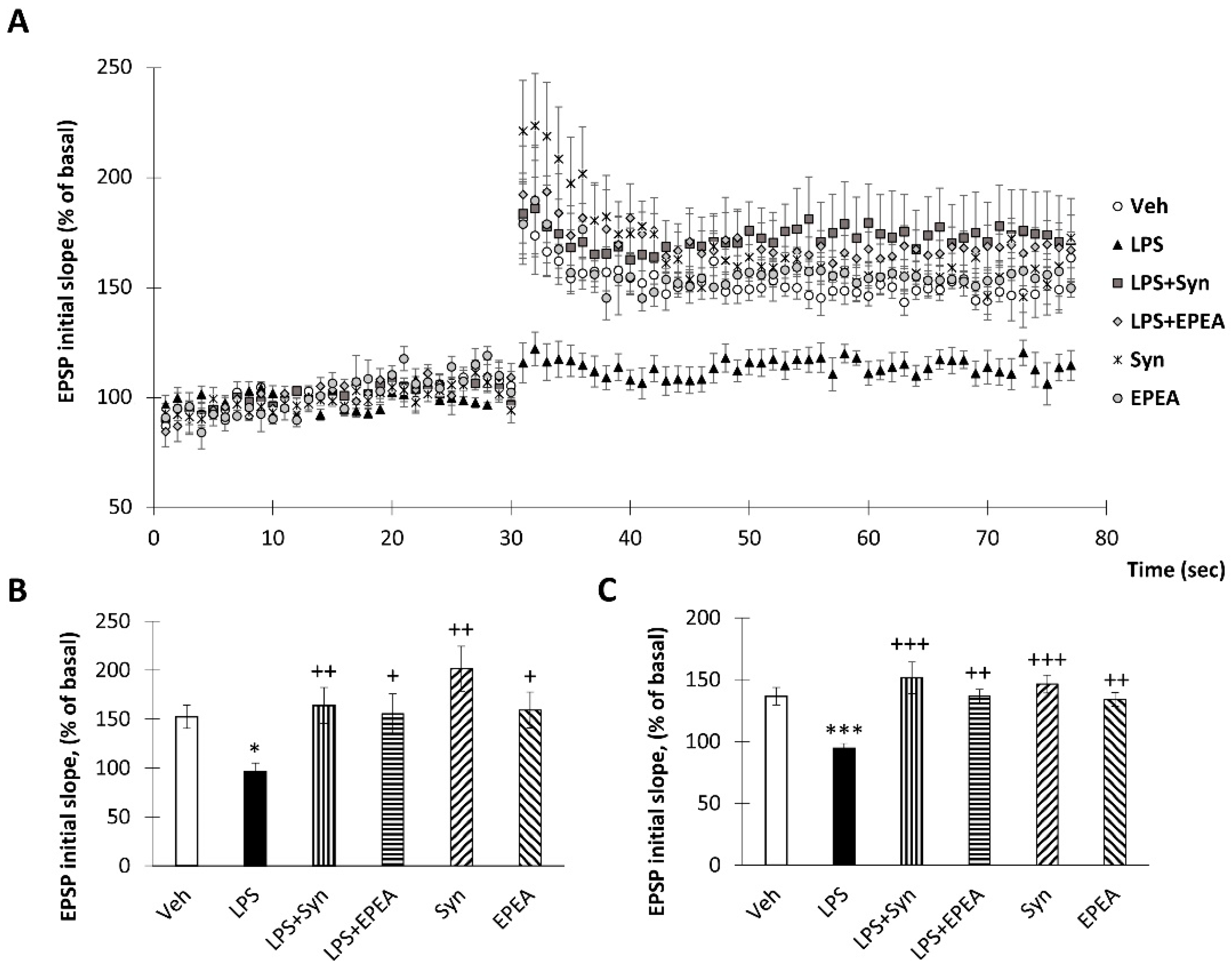
2.6. Synaptamide and EPEA Prevents Synaptic Plasticity Impairment
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Animals and Treatments
4.3. N-docosahexaenoylethanolamine and N-eicosapentanoylethanolamine Preparation
4.4. ELISA
4.5. Immunohistochemical Studies
4.6. Electrophysiological Recordings
4.7. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Overton, M.; Pihlsgård, M.; Elmståhl, S. Prevalence and incidence of mild cognitive impairment across subtypes, age, and sex. Dement. Geriatr. Cogn. Disord. 2019, 47, 219–232. [Google Scholar] [CrossRef] [PubMed]
- Fourrier, C.; Remus-Borel, J.; Greenhalgh, A.D.; Guichardant, M.; Bernoud-Hubac, N.; Lagarde, M.; Joffre, C.; Layé, S. Docosahexaenoic acid-containing choline phospholipid modulates LPS-induced neuroinflammation in vivo and in microglia in vitro. J. Neuroinflammation 2017, 14, 170. [Google Scholar] [CrossRef] [PubMed]
- Minogue, A.M. Role of infiltrating monocytes/macrophages in acute and chronic neuroinflammation: Effects on cognition, learning and affective behaviour. Prog. Neuropsychopharmacol. Biol. Psychiatry 2017, 79, 15–23. [Google Scholar] [CrossRef]
- Belarbi, K.; Jopson, T.; Tweedie, D.; Arellano, C.; Luo, W.; Greig, N.H.; Rosi, S. TNF-α protein synthesis inhibitor restores neuronal function and reverses cognitive deficits induced by chronic neuroinflammation. J. Neuroinflammation 2012, 9, 1–3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, C.M.; Mulcahey, T.A.; Filipek, N.C.; Wise, P.M. Production of proinflammatory cytokines and chemokines during neuroinflammation: Novel roles for estrogen receptors α and β. Endocrinology 2010, 151, 4916–4925. [Google Scholar] [CrossRef] [Green Version]
- Khan, M.S.; Ali, T.; Kim, M.W.; Jo, M.H.; Jo, M.G.; Badshah, H.; Kim, M.O. Anthocyanins protect against LPS-induced oxidative stress-mediated neuroinflammation and neurodegeneration in the adult mouse cortex. Neurochem. Int. 2016, 100, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Wu, Z.; Hayashi, Y.; Nakanishi, H. Age-dependent neuroinflammatory responses and deficits in long-term potentiation in the hippocampus during systemic inflammation. Neuroscience 2012, 216, 133–142. [Google Scholar] [CrossRef]
- Garden, G.A. Epigenetics and the modulation of neuroinflammation. Neurotherapeutics 2013, 10, 782–788. [Google Scholar] [CrossRef] [Green Version]
- Sanderson, D.J.; Bannerman, D.M. The role of habituation in hippocampus-dependent spatial working memory tasks: Evidence from GluA1 AMPA receptor subunit knockout mice. Hippocampus 2012, 22, 981–994. [Google Scholar] [CrossRef] [Green Version]
- Giansante, G.; Marte, A.; Romei, A.; Prestigio, C.; Onofri, F.; Benfenati, F.; Baldelli, P.; Valente, P. Presynaptic L-type Ca2+ channels increase glutamate release probability and excitatory strength in the hippocampus during chronic neuroinflammation. J. Neurosci. 2020, 40, 6825–6841. [Google Scholar] [CrossRef]
- Lai, A.Y.; Swayze, R.D.; El-Husseini, A.; Song, C. Interleukin-1 beta modulates AMPA receptor expression and phosphorylation in hippocampal neurons. J. Neuroimmunol. 2006, 175, 97–106. [Google Scholar] [CrossRef] [PubMed]
- Rosi, S.; Ramirez-Amaya, V.; Hauss-Wegrzyniak, B.; Wenk, G.L. Chronic brain inflammation leads to a decline in hippocampal NMDA-R1 receptors. J. Neuroinflammation 2004, 1, 12. [Google Scholar] [CrossRef] [Green Version]
- Francija, E.; Petrovic, Z.; Brkic, Z.; Mitic, M.; Radulovic, J.; Adzic, M. Disruption of the NMDA receptor GluN2A subunit abolishes inflammation-induced depression. Behav. Brain Res. 2019, 359, 550–559. [Google Scholar] [CrossRef] [PubMed]
- Golia, M.T.; Poggini, S.; Alboni, S.; Garofalo, S.; Ciano Albanese, N.; Viglione, A.; Ajmone-Cat, M.A.; St-Pierre, A.; Brunello, N.; Limatola, C.; et al. Interplay between inflammation and neural plasticity: Both immune activation and suppression impair LTP and BDNF expression. Brain Behav. Immun. 2019, 81, 484–494. [Google Scholar] [CrossRef] [PubMed]
- Yirmiya, R.; Goshen, I. Immune modulation of learning, memory, neural plasticity and neurogenesis. Brain Behav. Immun. 2011, 25, 181–213. [Google Scholar] [CrossRef] [PubMed]
- Asatryan, A.; Bazan, N.G. Molecular mechanisms of signaling via the docosanoid neuroprotectin D1 for cellular homeostasis and neuroprotection. J. Biol. Chem. 2017, 292, 12390–12397. [Google Scholar] [CrossRef] [Green Version]
- Schwab, J.M.; Chiang, N.; Arita, M.; Serhan, C.N. Resolvin E1 and protectin D1 activate inflammation-resolution programmes. Nature 2007, 447, 869–874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xian, W.; Li, T.; Li, L.; Hu, L.; Cao, J. Maresin 1 attenuates the inflammatory response and mitochondrial damage in mice with cerebral ischemia/reperfusion in a SIRT1-dependent manner. Brain Res. 2019, 1711, 83–90. [Google Scholar] [CrossRef]
- Kim, H.Y.; Spector, A.A.; Xiong, Z.M. A synaptogenic amide N-docosahexaenoylethanolamide promotes hippocampal development. Prostaglandins Other Lipid Mediat. 2011, 96, 114–120. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.Y.; Spector, A.A. N-Docosahexaenoylethanolamine: A neurotrophic and neuroprotective metabolite of docosahexaenoic acid. Mol. Aspects Med. 2018, 64, 34–44. [Google Scholar] [CrossRef]
- Rashid, M.A.; Katakura, M.; Kharebava, G.; Kevala, K.; Kim, H.Y. N-Docosahexaenoylethanolamine is a potent neurogenic factor for neural stem cell differentiation. J. Neurochem. 2013, 125, 869–884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, T.; Chen, H.; Kevala, K.; Lee, J.W.; Kim, H.Y. N-Docosahexaenoylethanolamine ameliorates LPS-induced neuroinflammation via cAMP/PKA-dependent signaling. J. Neuroinflammation 2016, 13, 284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, T.; Chen, H.; Kim, H.Y. GPR110 (ADGRF1) mediates anti-inflammatory effects of N-docosahexaenoylethanolamine. J. Neuroinflammation 2019, 16, 225. [Google Scholar] [CrossRef] [PubMed]
- Tyrtyshnaia, A.A.; Egorova, E.L.; Starinets, A.A.; Ponomarenko, A.I.; Ermolenko, E.V.; Manzhulo, I.V. N-Docosahexaenoylethanolamine attenuates neuroinflammation and improves hippocampal neurogenesis in rats with sciatic nerve chronic constriction injury. Mar. Drugs 2020, 18, 516. [Google Scholar] [CrossRef]
- Huang, B.X.; Hu, X.; Kwon, H.S.; Fu, C.; Lee, J.W.; Southall, N.; Marugan, J.; Kim, H.Y. Synaptamide activates the adhesion GPCR GPR110 (ADGRF1) through GAIN domain binding. Commun. Biol. 2020, 3, 109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berger, A.; Crozier, G.; Bisogno, T.; Cavaliere, P.; Innis, S.; Di Marzo, V. Anandamide and diet: Inclusion of dietary arachidonate and docosahexaenoate leads to increased brain levels of the corresponding N-acylethanolamines in piglets. Proc. Natl. Acad. Sci. USA 2001, 98, 6402–6406. [Google Scholar] [CrossRef] [Green Version]
- Balvers, M.G.; Verhoeckx, K.C.; Plastina, P.; Wortelboer, H.M.; Meijerink, J.; Witkamp, R.F. Docosahexaenoic acid and eicosapentaenoic acid are converted by 3T3-L1 adipocytes to N-acyl ethanolamines with anti-inflammatory properties. Biochim. Biophys. Acta 2010, 1801, 1107–1114. [Google Scholar] [CrossRef]
- Meijerink, J.; Plastina, P.; Vincken, J.P.; Poland, M.; Attya, M.; Balvers, M.; Gruppen, H.; Gabriele, B.; Witkamp, R.F. The ethanolamide metabolite of DHA, docosahexaenoylethanolamine, shows immunomodulating effects in mouse peritoneal and RAW264.7 macrophages: Evidence for a new link between fish oil and inflammation. Br. J. Nutr. 2011, 105, 1798–1807. [Google Scholar] [CrossRef] [Green Version]
- McDougle, D.R.; Watson, J.E.; Abdeen, A.A.; Adili, R.; Caputo, M.P.; Krapf, J.E.; Johnson, R.W.; Kilian, K.A.; Holinstat, M.; Das, A. Anti-inflammatory ω-3 endocannabinoid epoxides. Proc. Natl. Acad. Sci. USA 2017, 114, E6034–E6043. [Google Scholar] [CrossRef] [Green Version]
- Cherry, J.D.; Olschowka, J.A.; O’Banion, M.K. Neuroinflammation and M2 microglia: The good, the bad, and the inflamed. J. Neuroinflammation 2014, 11, 1–5. [Google Scholar] [CrossRef] [Green Version]
- Ohgidani, M.; Kato, T.A.; Haraguchi, Y.; Matsushima, T.; Mizoguch, Y.; Murakawa-Hirachi, T.; Sagata, N.; Monji, A.; Kanba, S. Microglial CD206 gene has potential as a state marker of bipolar disorder. Front. Immunol. 2017, 7, 676. [Google Scholar] [CrossRef] [Green Version]
- Savchenko, V.L.; McKanna, J.A.; Nikonenko, I.R.; Skibo, G.G. Microglia and astrocytes in the adult rat brain: Comparative immunocytochemical analysis demonstrates the efficacy of lipocortin 1 immunoreactivity. Neuroscience 2000, 96, 195–203. [Google Scholar] [CrossRef]
- Jinno, S.; Fleischer, F.; Eckel, S.; Schmidt, V.; Kosaka, T. Spatial arrangement of microglia in the mouse hippocampus: A stereological study in comparison with astrocytes. Glia 2007, 55, 1334–1347. [Google Scholar] [CrossRef] [PubMed]
- Lana, D.; Ugolini, F.; Melani, A.; Nosi, D.; Pedata, F.; Giovannini, M.G. The neuron-astrocyte-microglia triad in CA3 after chronic cerebral hypoperfusion in the rat: Protective effect of dipyridamole. Exp. Gerontol. 2017, 96, 46–62. [Google Scholar] [CrossRef] [PubMed]
- Kowiański, P.; Lietzau, G.; Czuba, E.; Waśkow, M.; Steliga, A.; Moryś, J. BDNF: A key factor with multipotent impact on brain signaling and synaptic plasticity. Cell. Mol. Neurobiol. 2018, 38, 579–593. [Google Scholar] [CrossRef]
- Lima Giacobbo, B.; Doorduin, J.; Klein, H.C.; Dierckx, R.A.J.O.; Bromberg, E.; de Vries, E.F.J. Brain-derived neurotrophic factor in brain disorders: Focus on neuroinflammation. Mol. Neurobiol. 2019, 56, 3295–3312. [Google Scholar] [CrossRef] [Green Version]
- Brigadski, T.; Leßmann, V. The physiology of regulated BDNF release. Cell Tissue Res. 2020, 382, 15–45. [Google Scholar] [CrossRef]
- Rodgers, K.R.; Lin, Y.; Langan, T.J.; Iwakura, Y.; Chou, R.C. Innate immune functions of astrocytes are dependent upon tumor necrosis factor-alpha. Sci. Rep. 2020, 10, 7047. [Google Scholar] [CrossRef]
- Ben Haim, L.; Carrillo-de Sauvage, M.A.; Ceyzériat, K.; Escartin, C. Elusive roles for reactive astrocytes in neurodegenerative diseases. Front. Cell. Neurosci. 2015, 9, 278. [Google Scholar] [CrossRef] [Green Version]
- Hirsch, E.C.; Hunot, S. Neuroinflammation in Parkinson’s disease: A target for neuroprotection? Lancet Neurol. 2009, 8, 382–397. [Google Scholar] [CrossRef]
- Acaz-Fonseca, E.; Ortiz-Rodriguez, A.; Azcoitia, I.; Garcia-Segura, L.M.; Arevalo, M.A. Notch signaling in astrocytes mediates their morphological response to an inflammatory challenge. Cell Death Discov. 2019, 5, 85. [Google Scholar] [CrossRef]
- Ali, M.; Heyob, K.; Rogers, L.K. DHA suppresses primary macrophage inflammatory responses via Notch 1/Jagged 1 signaling. Sci. Rep. 2016, 6, 22276. [Google Scholar] [CrossRef] [Green Version]
- Kellner, Y.; Gödecke, N.; Dierkes, T.; Thieme, N.; Zagrebelsky, M.; Korte, M. The BDNF effects on dendritic spines of mature hippocampal neurons depend on neuronal activity. Front. Synaptic Neurosci. 2014, 6, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lipsky, R.H.; Marini, A.M. Brain-derived neurotrophic factor in neuronal survival and behavior-related plasticity. Ann. N. Y. Acad. Sci. 2007, 1122, 130–143. [Google Scholar] [CrossRef]
- Mondelli, V.; Cattaneo, A.; Murri, M.B.; Di Forti, M.; Handley, R.; Hepgul, N.; Miorelli, A.; Navari, S.; Papadopoulos, A.S.; Aitchison, K.J. Stress and inflammation reduce brain-derived neurotrophic factor expression in first-episode psychosis: A pathway to smaller hippocampal volume. J. Clin. Psychiatry 2011, 72, 1677–1684. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Li, J.; Zheng, J.; Qin, S. Reactive astrocytes in neurodegenerative diseases. Aging Dis. 2019, 10, 664–675. [Google Scholar] [CrossRef] [Green Version]
- Liddelow, S.A.; Barres, B.A. Reactive astrocytes: Production, function, and therapeutic potential. Immunity 2017, 46, 957–967. [Google Scholar] [CrossRef] [Green Version]
- Maher, F.O.; Nolan, Y.; Lynch, M.A. Downregulation of IL-4-induced signalling in hippocampus contributes to deficits in LTP in the aged rat. Neurobiol. Aging 2005, 26, 717–745. [Google Scholar] [CrossRef] [PubMed]
- Lyons, A.; Griffin, R.J.; Costelloe, C.E.; Clarke, R.M.; Lynch, M.A. IL-4 attenuates the neuroinflammation induced by amyloid-β in vivo and in vitro. J. Neurochem. 2007, 101, 771–852. [Google Scholar] [CrossRef]
- Batista, C.R.A.; Gomes, G.F.; Candelario-Jalil, E.; Fiebich, B.L.; de Oliveira, A.C.P. Lipopolysaccharide-Induced Neuroinflammation as a Bridge to Understand Neurodegeneration. Int. J. Mol. Sci. 2019, 20, 2293. [Google Scholar] [CrossRef] [Green Version]
- Muhammad, T.; Ikram, M.; Ullah, R.; Rehman, S.U.; Kim, M.O. Hesperetin, a Citrus Flavonoid, Attenuates LPS-Induced Neuroinflammation, Apoptosis and Memory Impairments by Modulating TLR4/NF-κB Signaling. Nutrients 2019, 11, 648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghanbari, M.M.; Loron, A.G.; Sayyah, M. The ω-3 endocannabinoid docosahexaenoyl ethanolamide reduces seizure susceptibility in mice by activating cannabinoid type 1 receptors. Brain Res. Bull. 2021, 170, 74–80. [Google Scholar] [CrossRef] [PubMed]
- Paton, K.F.; Shirazi, R.; Vyssotski, M.; Kivell, B.M. N-docosahexaenoyl ethanolamine (synaptamide) has antinociceptive effects in male mice. Eur. J. Pain 2020, 24, 1990–1998. [Google Scholar] [CrossRef]
- Alharthi, N.; Christensen, P.; Hourani, W.; Ortori, C.; Barrett, D.A.; Bennett, A.J.; Chapman, V.; Alexander, S.P.H. n-3 polyunsaturated N-acylethanolamines are CB2 cannabinoid receptor-preferring endocannabinoids. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2018, 1863, 1433–1440. [Google Scholar] [CrossRef]
- Artmann, A.; Petersen, G.; Hellgren, L.I.; Boberg, J.; Skonberg, C.; Nellemann, C.; Hansen, S.H.; Hansen, H.S. Influence of dietary fatty acids on endocannabinoid and N-acylethanolamine levels in rat brain, liver and small intestine. Biochim. Biophys. Acta 2008, 1781, 200–212. [Google Scholar] [CrossRef] [PubMed]
- Wood, J.T.; Williams, J.S.; Pandarinathan, L.; Janero, D.R.; Lammi-Keefe, C.J.; Makriyannis, A. Dietary docosahexaenoic acid supplementation alters select physiological endocannabinoid-system metabolites in brain and plasma. J. Lipid Res. 2010, 51, 1416–1423. [Google Scholar] [CrossRef] [Green Version]
- Latyshev, N.A.; Ermolenko, E.V.; Kasyanov, S.P. Concentration and purification of polyunsaturated fatty acids from squid liver processing wastes. Eur. J. Lipid Sci. Technol. 2014, 116, 1608–1613. [Google Scholar] [CrossRef]
- Svetashev, V.I. Mild method for preparation of 4, 4-dimethyloxazoline derivatives of polyunsaturated fatty acids for GC–MS. Lipids 2011, 46, 463–470. [Google Scholar] [CrossRef] [PubMed]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tyrtyshnaia, A.; Konovalova, S.; Bondar, A.; Ermolenko, E.; Sultanov, R.; Manzhulo, I. Anti-Inflammatory Activity of N-Docosahexaenoylethanolamine and N-Eicosapentaenoylethanolamine in a Mouse Model of Lipopolysaccharide-Induced Neuroinflammation. Int. J. Mol. Sci. 2021, 22, 10728. https://doi.org/10.3390/ijms221910728
Tyrtyshnaia A, Konovalova S, Bondar A, Ermolenko E, Sultanov R, Manzhulo I. Anti-Inflammatory Activity of N-Docosahexaenoylethanolamine and N-Eicosapentaenoylethanolamine in a Mouse Model of Lipopolysaccharide-Induced Neuroinflammation. International Journal of Molecular Sciences. 2021; 22(19):10728. https://doi.org/10.3390/ijms221910728
Chicago/Turabian StyleTyrtyshnaia, Anna, Sophia Konovalova, Anatoly Bondar, Ekaterina Ermolenko, Ruslan Sultanov, and Igor Manzhulo. 2021. "Anti-Inflammatory Activity of N-Docosahexaenoylethanolamine and N-Eicosapentaenoylethanolamine in a Mouse Model of Lipopolysaccharide-Induced Neuroinflammation" International Journal of Molecular Sciences 22, no. 19: 10728. https://doi.org/10.3390/ijms221910728