
Involvement of the ACE2/Ang-(1–7)/MasR Axis in Pulmonary Fibrosis: Implications for COVID-19

 ,
, {kind=link}
{kind=link}
Abstract
:1. Introduction
2. Pulmonary Fibrosis
2.1. Overview
2.2. Risk Factors
2.3. Pathogenesis
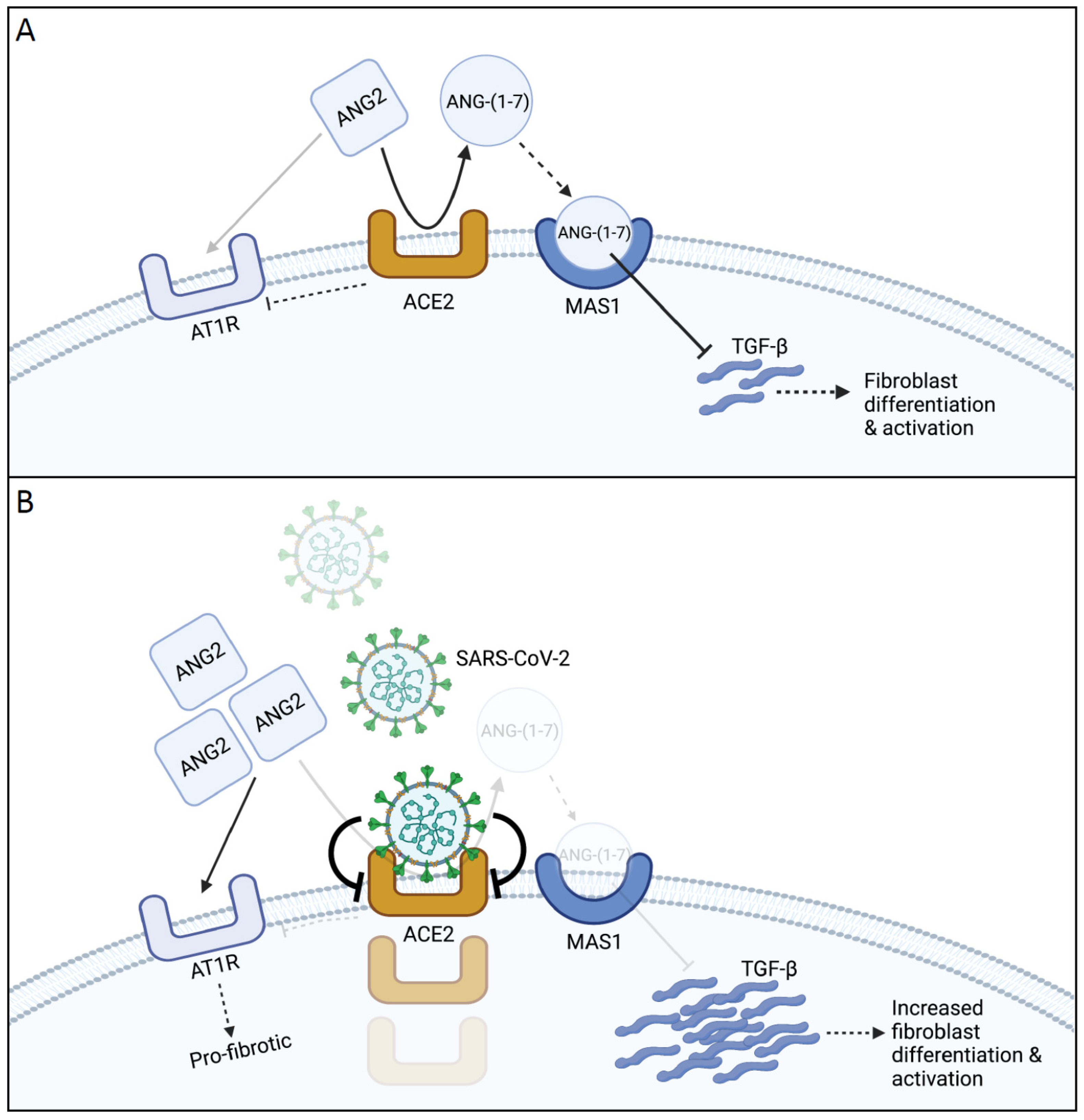
3. The ACE2/Ang-(1–7)/MasR Axis
Fibrosis and the ACE2/Ang-(1–7)/MasR Axis
4. Coronavirus Disease-19 (COVID-19)
Pulmonary Fibrosis in COVID-19
5. Modulation of Pulmonary ACE2 Expression
5.1. Modulation of ACE2 by Cigarette Smoke
5.2. Modulation of ACE2 by Other Inhalation Toxicants
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Antoniou, K.M.; Margaritopoulos, G.A.; Tomassetti, S.; Bonella, F.; Costabel, U.; Poletti, V. Interstitial lung disease. Eur. Respir. Rev. 2014, 23, 40–54. [Google Scholar] [CrossRef] [Green Version]
- Demedts, M.; Wells, A.U.; Anto, J.M.; Costabel, U.; Hubbard, R.; Cullinan, P.; Slabbynck, H.; Rizzato, G.; Poletti, V.; Verbeken, E.K.; et al. Interstitial lung diseases: An epidemiological overview. Eur. Respir. J. Suppl. 2001, 32, 2s–16s. [Google Scholar] [PubMed]
- Demedts, M.; Costabel, U. ATS/ERS international multidisciplinary consensus classification of the idiopathic interstitial pneumonias. Eur. Respir. J. 2002, 19, 794–796. [Google Scholar] [CrossRef] [Green Version]
- Coward, W.R.; Saini, G.; Jenkins, G. The pathogenesis of idiopathic pulmonary fibrosis. Ther. Adv. Respir. Dis. 2010, 4, 367–388. [Google Scholar] [CrossRef] [Green Version]
- Vancheri, C.; Failla, M.; Crimi, N.; Raghu, G. Idiopathic pulmonary fibrosis: A disease with similarities and links to cancer biology. Eur. Respir. J. 2010, 35, 496–504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oh, C.K.; Murray, L.A.; Molfino, N.A. Smoking and idiopathic pulmonary fibrosis. Pulm. Med. 2012, 2012, 808260. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, D.; Brunnemann, K.D.; Gori, G.B.; Wynder, E.L. On the Carcinogenicity of marijuana smoke. In Recent Advances in Phytochemistry; Runeckles, V.C., Ed.; Plenum Press: New York, NY, USA, 1975; pp. 63–81. [Google Scholar]
- Schwartz, R. Legalize marijuana without the smoke. CMAJ 2017, 189, E137–E138. [Google Scholar] [CrossRef] [Green Version]
- Zurier, R.B.; Burstein, S.H. Cannabinoids, inflammation, and fibrosis. FASEB J. 2016, 30, 3682–3689. [Google Scholar] [CrossRef] [Green Version]
- Shenoy, V.; Ferreira, A.J.; Qi, Y.; Fraga-Silva, R.A.; Diez-Freire, C.; Dooies, A.; Jun, J.Y.; Sriramula, S.; Mariappan, N.; Pourang, D.; et al. The angiotensin-converting enzyme 2/angiogenesis-(1-7)/Mas axis confers cardiopulmonary protection against lung fibrosis and pulmonary hypertension. Am. J. Respir. Crit. Care Med. 2010, 182, 1065–1072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chung, M.K.; Karnik, S.; Saef, J.; Bergmann, C.; Barnard, J.; Lederman, M.M.; Tilton, J.; Cheng, F.; Harding, C.V.; Young, J.B.; et al. SARS-CoV-2 and ACE2: The biology and clinical data settling the ARB and ACEI controversy. EBioMedicine 2020, 58, 102907. [Google Scholar] [CrossRef]
- Wang, D.; Hu, B.; Hu, C.; Zhu, F.; Liu, X.; Zhang, J.; Wang, B.; Xiang, H.; Cheng, Z.; Xiong, Y.; et al. Clinical Characteristics of 138 Hospitalized Patients With 2019 Novel Coronavirus-Infected Pneumonia in Wuhan, China. JAMA 2020, 323, 1061–1069. [Google Scholar] [CrossRef]
- Vardavas, C.I.; Nikitara, K. COVID-19 and smoking: A systematic review of the evidence. Tob. Induc. Dis. 2020, 18, 20. [Google Scholar] [CrossRef] [PubMed]
- Dalan, R.; Bornstein, S.R.; El-Armouche, A.; Rodionov, R.N.; Markov, A.; Wielockx, B.; Beuschlein, F.; Boehm, B.O. The ACE-2 in COVID-19: Foe or Friend? Horm. Metab. Res. 2020, 52, 257–263. [Google Scholar] [CrossRef] [PubMed]
- Grillo, F.; Barisione, E.; Ball, L.; Mastracci, L.; Fiocca, R. Lung fibrosis: An undervalued finding in COVID-19 pathological series. Lancet Infect. Dis. 2021, 21, e72. [Google Scholar] [CrossRef]
- Friedman, S.L.; Sheppard, D.; Duffield, J.S.; Violette, S. Therapy for fibrotic diseases: Nearing the starting line. Sci. Transl. Med. 2013, 5, 167sr1. [Google Scholar] [CrossRef] [Green Version]
- Kalchiem-Dekel, O.; Galvin, J.R.; Burke, A.P.; Atamas, S.P.; Todd, N.W. Interstitial Lung Disease and Pulmonary Fibrosis: A Practical Approach for General Medicine Physicians with Focus on the Medical History. J. Clin. Med. 2018, 7, 476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raghu, G.; Remy-Jardin, M.; Myers, J.L.; Richeldi, L.; Ryerson, C.J.; Lederer, D.J.; Behr, J.; Cottin, V.; Danoff, S.K.; Morell, F.; et al. Diagnosis of Idiopathic Pulmonary Fibrosis. An Official ATS/ERS/JRS/ALAT Clinical Practice Guideline. Am. J. Respir. Crit. Care Med. 2018, 198, e44–e68. [Google Scholar] [CrossRef]
- Taskar, V.S.; Coultas, D.B. Is idiopathic pulmonary fibrosis an environmental disease? Proc. Am. Thorac. Soc. 2006, 3, 293–298. [Google Scholar] [CrossRef]
- Sgalla, G.; Iovene, B.; Calvello, M.; Ori, M.; Varone, F.; Richeldi, L. Idiopathic pulmonary fibrosis: Pathogenesis and management. Respir. Res. 2018, 19, 32. [Google Scholar] [CrossRef]
- Chioma, O.S.; Drake, W.P. Role of Microbial Agents in Pulmonary Fibrosis. Yale J. Biol. Med. 2017, 90, 219–227. [Google Scholar]
- Noth, I.; Zhang, Y.; Ma, S.F.; Flores, C.; Barber, M.; Huang, Y.; Broderick, S.M.; Wade, M.S.; Hysi, P.; Scuirba, J.; et al. Genetic variants associated with idiopathic pulmonary fibrosis susceptibility and mortality: A genome-wide association study. Lancet Respir. Med. 2013, 1, 309–317. [Google Scholar] [CrossRef] [Green Version]
- Fingerlin, T.E.; Murphy, E.; Zhang, W.; Peljto, A.L.; Brown, K.K.; Steele, M.P.; Loyd, J.E.; Cosgrove, G.P.; Lynch, D.; Groshong, S.; et al. Genome-wide association study identifies multiple susceptibility loci for pulmonary fibrosis. Nat. Genet. 2013, 45, 613–620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, H.; Peng, G.; Meister, M.; Yao, H.; Yang, J.J.; Zou, M.H.; Liu, Z.R.; Ji, X. Electronic Cigarette Exposure Enhances Lung Inflammatory and Fibrotic Responses in COPD Mice. Front. Pharmacol. 2021, 12, 726586. [Google Scholar] [CrossRef] [PubMed]
- Idiopathic Pulmonary Fibrosis Clinical Research Network; Raghu, G.; Anstrom, K.J.; King, T.E., Jr.; Lasky, J.A.; Martinez, F.J. Prednisone, azathioprine, and N-acetylcysteine for pulmonary fibrosis. N. Engl. J. Med. 2012, 366, 1968–1977. [Google Scholar] [CrossRef]
- Wilson, M.S.; Wynn, T.A. Pulmonary fibrosis: Pathogenesis, etiology and regulation. Mucosal. Immunol. 2009, 2, 103–121. [Google Scholar] [CrossRef] [Green Version]
- Checa, M.; Hagood, J.S.; Velazquez-Cruz, R.; Ruiz, V.; Garcia-De-Alba, C.; Rangel-Escareno, C.; Urrea, F.; Becerril, C.; Montano, M.; Garcia-Trejo, S.; et al. Cigarette Smoke Enhances the Expression of Profibrotic Molecules in Alveolar Epithelial Cells. PLoS ONE 2016, 11, e0150383. [Google Scholar] [CrossRef]
- Spagnolo, P.; Rossi, G.; Cavazza, A. Pathogenesis of idiopathic pulmonary fibrosis and its clinical implications. Expert Rev. Clin. Immunol. 2014, 10, 1005–1017. [Google Scholar] [CrossRef]
- Song, M.; Peng, H.; Guo, W.; Luo, M.; Duan, W.; Chen, P.; Zhou, Y. Cigarette Smoke Extract Promotes Human Lung Myofibroblast Differentiation by the Induction of Endoplasmic Reticulum Stress. Respiration 2019, 98, 347–356. [Google Scholar] [CrossRef]
- Yang, I.V.; Schwartz, D.A. Epigenetics of idiopathic pulmonary fibrosis. Transl. Res. 2015, 165, 48–60. [Google Scholar] [CrossRef] [Green Version]
- Helling, B.A.; Yang, I.V. Epigenetics in lung fibrosis: From pathobiology to treatment perspective. Curr. Opin. Pulm. Med. 2015, 21, 454–462. [Google Scholar] [CrossRef] [Green Version]
- Wuyts, W.A.; Agostini, C.; Antoniou, K.M.; Bouros, D.; Chambers, R.C.; Cottin, V.; Egan, J.J.; Lambrecht, B.N.; Lories, R.; Parfrey, H.; et al. The pathogenesis of pulmonary fibrosis: A moving target. Eur. Respir. J. 2013, 41, 1207–1218. [Google Scholar] [CrossRef]
- Martinez, F.J.; Collard, H.R.; Pardo, A.; Raghu, G.; Richeldi, L.; Selman, M.; Swigris, J.J.; Taniguchi, H.; Wells, A.U. Idiopathic pulmonary fibrosis. Nat. Rev. Dis. Primers 2017, 3, 17074. [Google Scholar] [CrossRef]
- Kim, K.K.; Kugler, M.C.; Wolters, P.J.; Robillard, L.; Galvez, M.G.; Brumwell, A.N.; Sheppard, D.; Chapman, H.A. Alveolar epithelial cell mesenchymal transition develops in vivo during pulmonary fibrosis and is regulated by the extracellular matrix. Proc. Natl. Acad. Sci. USA 2006, 103, 13180–13185. [Google Scholar] [CrossRef] [Green Version]
- Kasai, H.; Allen, J.T.; Mason, R.M.; Kamimura, T.; Zhang, Z. TGF-beta1 induces human alveolar epithelial to mesenchymal cell transition (EMT). Respir. Res. 2005, 6, 56. [Google Scholar] [CrossRef] [Green Version]
- Phillips, R.J.; Burdick, M.D.; Hong, K.; Lutz, M.A.; Murray, L.A.; Xue, Y.Y.; Belperio, J.A.; Keane, M.P.; Strieter, R.M. Circulating fibrocytes traffic to the lungs in response to CXCL12 and mediate fibrosis. J. Clin. Investig. 2004, 114, 438–446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moore, B.B.; Kolodsick, J.E.; Thannickal, V.J.; Cooke, K.; Moore, T.A.; Hogaboam, C.; Wilke, C.A.; Toews, G.B. CCR2-mediated recruitment of fibrocytes to the alveolar space after fibrotic injury. Am. J. Pathol. 2005, 166, 675–684. [Google Scholar] [CrossRef] [Green Version]
- Sava, P.; Ramanathan, A.; Dobronyi, A.; Peng, X.; Sun, H.; Ledesma-Mendoza, A.; Herzog, E.L.; Gonzalez, A.L. Human pericytes adopt myofibroblast properties in the microenvironment of the IPF lung. JCI Insight 2017, 2, e96352. [Google Scholar] [CrossRef] [PubMed]
- Phan, S.H. The myofibroblast in pulmonary fibrosis. Chest 2002, 122, 286S–289S. [Google Scholar] [CrossRef] [PubMed]
- Grimminger, F.; Gunther, A.; Vancheri, C. The role of tyrosine kinases in the pathogenesis of idiopathic pulmonary fibrosis. Eur. Respir. J. 2015, 45, 1426–1433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kapanci, Y.; Desmouliere, A.; Pache, J.C.; Redard, M.; Gabbiani, G. Cytoskeletal protein modulation in pulmonary alveolar myofibroblasts during idiopathic pulmonary fibrosis. Possible role of transforming growth factor beta and tumor necrosis factor alpha. Am. J. Respir. Crit. Care Med. 1995, 152, 2163–2169. [Google Scholar] [CrossRef]
- Zhang, H.Y.; Phan, S.H. Inhibition of myofibroblast apoptosis by transforming growth factor beta(1). Am. J. Respir. Cell Mol. Biol. 1999, 21, 658–665. [Google Scholar] [CrossRef] [PubMed]
- Vaughan, M.B.; Howard, E.W.; Tomasek, J.J. Transforming growth factor-beta1 promotes the morphological and functional differentiation of the myofibroblast. Exp. Cell. Res. 2000, 257, 180–189. [Google Scholar] [CrossRef] [Green Version]
- Willis, B.C.; Liebler, J.M.; Luby-Phelps, K.; Nicholson, A.G.; Crandall, E.D.; du Bois, R.M.; Borok, Z. Induction of epithelial-mesenchymal transition in alveolar epithelial cells by transforming growth factor-beta1: Potential role in idiopathic pulmonary fibrosis. Am. J. Pathol. 2005, 166, 1321–1332. [Google Scholar] [CrossRef]
- Khalil, N.; O’Connor, R.N.; Unruh, H.W.; Warren, P.W.; Flanders, K.C.; Kemp, A.; Bereznay, O.H.; Greenberg, A.H. Increased production and immunohistochemical localization of transforming growth factor-beta in idiopathic pulmonary fibrosis. Am. J. Respir. Cell Mol. Biol. 1991, 5, 155–162. [Google Scholar] [CrossRef]
- Sime, P.J.; Xing, Z.; Graham, F.L.; Csaky, K.G.; Gauldie, J. Adenovector-mediated gene transfer of active transforming growth factor-beta1 induces prolonged severe fibrosis in rat lung. J. Clin. Investig. 1997, 100, 768–776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
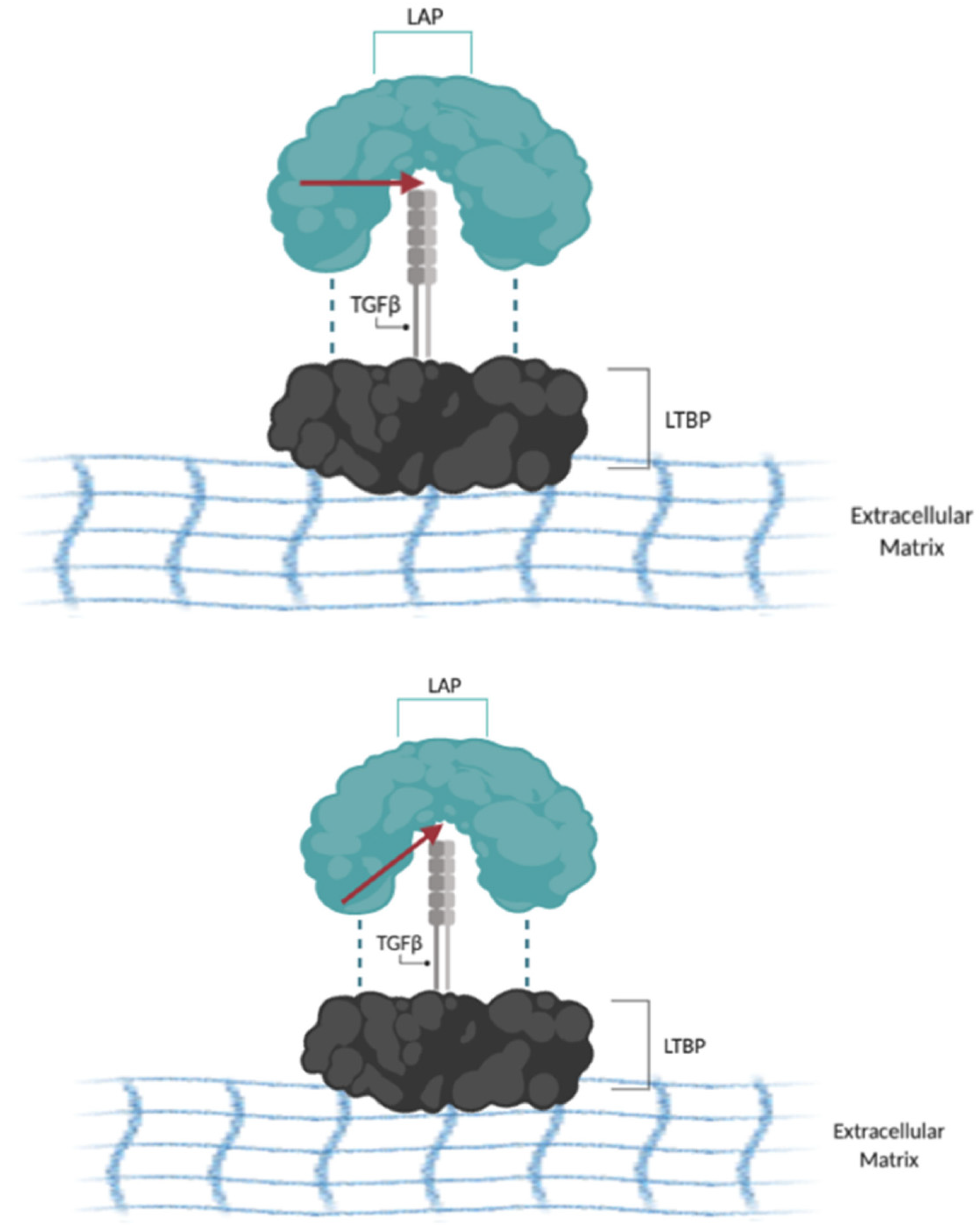
- Wipff, P.J.; Rifkin, D.B.; Meister, J.J.; Hinz, B. Myofibroblast contraction activates latent TGF-beta1 from the extracellular matrix. J. Cell. Biol. 2007, 179, 1311–1323. [Google Scholar] [CrossRef] [Green Version]
- Annes, J.P.; Munger, J.S.; Rifkin, D.B. Making sense of latent TGFbeta activation. J. Cell. Sci. 2003, 116, 217–224. [Google Scholar] [CrossRef] [Green Version]
- Horan, G.S.; Wood, S.; Ona, V.; Li, D.J.; Lukashev, M.E.; Weinreb, P.H.; Simon, K.J.; Hahm, K.; Allaire, N.E.; Rinaldi, N.J.; et al. Partial inhibition of integrin alpha(v)beta6 prevents pulmonary fibrosis without exacerbating inflammation. Am. J. Respir. Crit. Care Med. 2008, 177, 56–65. [Google Scholar] [CrossRef]
- Kubiczkova, L.; Sedlarikova, L.; Hajek, R.; Sevcikova, S. TGF-beta—An excellent servant but a bad master. J. Transl. Med. 2012, 10, 183. [Google Scholar] [CrossRef] [Green Version]
- Kajdaniuk, D.; Marek, B.; Borgiel-Marek, H.; Kos-Kudla, B. Transforming growth factor beta1 (TGFbeta1) in physiology and pathology. Endokrynol. Pol. 2013, 64, 384–396. [Google Scholar] [CrossRef] [Green Version]
- Sakai, N.; Tager, A.M. Fibrosis of two: Epithelial cell-fibroblast interactions in pulmonary fibrosis. Biochim. Biophys. Acta 2013, 1832, 911–921. [Google Scholar] [CrossRef] [Green Version]
- Derynck, R.; Budi, E.H. Specificity, versatility, and control of TGF-beta family signaling. Sci. Signal. 2019, 12. [Google Scholar] [CrossRef] [Green Version]
- Martey, C.A.; Pollock, S.J.; Turner, C.K.; O’Reilly, K.M.; Baglole, C.J.; Phipps, R.P.; Sime, P.J. Cigarette smoke induces cyclooxygenase-2 and microsomal prostaglandin E2 synthase in human lung fibroblasts: Implications for lung inflammation and cancer. Am. J. Physiol. Lung. Cell Mol. Physiol. 2004, 287, L981–L991. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kasembeli, M.M.; Bharadwaj, U.; Robinson, P.; Tweardy, D.J. Contribution of STAT3 to Inflammatory and Fibrotic Diseases and Prospects for its Targeting for Treatment. Int. J. Mol. Sci. 2018, 19, 2299. [Google Scholar] [CrossRef] [Green Version]
- Hamming, I.; Cooper, M.E.; Haagmans, B.L.; Hooper, N.M.; Korstanje, R.; Osterhaus, A.D.; Timens, W.; Turner, A.J.; Navis, G.; van Goor, H. The emerging role of ACE2 in physiology and disease. J. Pathol. 2007, 212, 1–11. [Google Scholar] [CrossRef]
- Gheblawi, M.; Wang, K.; Viveiros, A.; Nguyen, Q.; Zhong, J.C.; Turner, A.J.; Raizada, M.K.; Grant, M.B.; Oudit, G.Y. Angiotensin-Converting Enzyme 2: SARS-CoV-2 Receptor and Regulator of the Renin-Angiotensin System: Celebrating the 20th Anniversary of the Discovery of ACE2. Circ. Res. 2020, 126, 1456–1474. [Google Scholar] [CrossRef] [PubMed]
- Kuba, K.; Imai, Y.; Ohto-Nakanishi, T.; Penninger, J.M. Trilogy of ACE2: A peptidase in the renin-angiotensin system, a SARS receptor, and a partner for amino acid transporters. Pharmacol. Ther. 2010, 128, 119–128. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Zhao, Z.; Wang, Y.; Zhou, Y.; Ma, Y.; Zuo, W. Single-Cell RNA Expression Profiling of ACE2, the Receptor of SARS-CoV-2. Am. J. Respir. Crit. Care Med. 2020, 202, 756–759. [Google Scholar] [CrossRef]
- Aloufi, N.; Traboulsi, H.; Ding, J.; Fonseca, G.J.; Nair, P.; Huang, S.K.; Hussain, S.N.A.; Eidelman, D.H.; Baglole, C.J. Angiotensin-converting enzyme 2 expression in COPD and IPF fibroblasts: The forgotten cell in COVID-19. Am. J. Physiol.-Lung Cell Mol. Physiol. 2021, 320, L152–L157. [Google Scholar] [CrossRef] [PubMed]
- Lambert, D.W.; Hooper, N.M.; Turner, A.J. Angiotensin-converting enzyme 2 and new insights into the renin-angiotensin system. Biochem. Pharmacol. 2008, 75, 781–786. [Google Scholar] [CrossRef]
- Verdecchia, P.; Cavallini, C.; Spanevello, A.; Angeli, F. The pivotal link between ACE2 deficiency and SARS-CoV-2 infection. Eur. J. Intern. Med. 2020, 76, 14–20. [Google Scholar] [CrossRef] [PubMed]
- Flores-Munoz, M.; Work, L.M.; Douglas, K.; Denby, L.; Dominiczak, A.F.; Graham, D.; Nicklin, S.A. Angiotensin-(1-9) attenuates cardiac fibrosis in the stroke-prone spontaneously hypertensive rat via the angiotensin type 2 receptor. Hypertension 2012, 59, 300–307. [Google Scholar] [CrossRef] [Green Version]
- Vickers, C.; Hales, P.; Kaushik, V.; Dick, L.; Gavin, J.; Tang, J.; Godbout, K.; Parsons, T.; Baronas, E.; Hsieh, F.; et al. Hydrolysis of biological peptides by human angiotensin-converting enzyme-related carboxypeptidase. J. Biol. Chem. 2002, 277, 14838–14843. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sparks, M.A.; Crowley, S.D.; Gurley, S.B.; Mirotsou, M.; Coffman, T.M. Classical Renin-Angiotensin system in kidney physiology. Compr. Physiol. 2014, 4, 1201–1228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lelis, D.F.; Freitas, D.F.; Machado, A.S.; Crespo, T.S.; Santos, S.H.S. Angiotensin-(1-7), Adipokines and Inflammation. Metabolism 2019, 95, 36–45. [Google Scholar] [CrossRef] [PubMed]
- Patel, V.B.; Zhong, J.C.; Grant, M.B.; Oudit, G.Y. Role of the ACE2/Angiotensin 1-7 Axis of the Renin-Angiotensin System in Heart Failure. Circ. Res. 2016, 118, 1313–1326. [Google Scholar] [CrossRef] [Green Version]
- Wang, R.; Zagariya, A.; Ibarra-Sunga, O.; Gidea, C.; Ang, E.; Deshmukh, S.; Chaudhary, G.; Baraboutis, J.; Filippatos, G.; Uhal, B.D. Angiotensin II induces apoptosis in human and rat alveolar epithelial cells. Am. J. Physiol. 1999, 276, L885–L889. [Google Scholar] [CrossRef]
- Uhal, B.D.; Dang, M.; Dang, V.; Llatos, R.; Cano, E.; Abdul-Hafez, A.; Markey, J.; Piasecki, C.C.; Molina-Molina, M. Cell cycle dependence of ACE-2 explains downregulation in idiopathic pulmonary fibrosis. Eur. Respir. J. 2013, 42, 198–210. [Google Scholar] [CrossRef] [Green Version]
- Hrenak, J.; Simko, F. Renin-Angiotensin System: An Important Player in the Pathogenesis of Acute Respiratory Distress Syndrome. Int. J. Mol. Sci. 2020, 21, 8038. [Google Scholar] [CrossRef]
- Li, Y.; Cao, Y.; Zeng, Z.; Liang, M.; Xue, Y.; Xi, C.; Zhou, M.; Jiang, W. Angiotensin-converting enzyme 2/angiotensin-(1-7)/Mas axis prevents lipopolysaccharide-induced apoptosis of pulmonary microvascular endothelial cells by inhibiting JNK/NF-kappaB pathways. Sci. Rep. 2015, 5, 8209. [Google Scholar] [CrossRef] [Green Version]
- Meng, Y.; Yu, C.H.; Li, W.; Li, T.; Luo, W.; Huang, S.; Wu, P.S.; Cai, S.X.; Li, X. Angiotensin-converting enzyme 2/angiotensin-(1-7)/Mas axis protects against lung fibrosis by inhibiting the MAPK/NF-kappaB pathway. Am. J. Respir. Cell Mol. Biol. 2014, 50, 723–736. [Google Scholar] [CrossRef]
- Simoes e Silva, A.C.; Silveira, K.D.; Ferreira, A.J.; Teixeira, M.M. ACE2, angiotensin-(1-7) and Mas receptor axis in inflammation and fibrosis. Br. J. Pharmacol. 2013, 169, 477–492. [Google Scholar] [CrossRef] [Green Version]
- Zhang, B.N.; Xu, H.; Gao, X.M.; Zhang, G.Z.; Zhang, X.; Yang, F. Protective Effect of Angiotensin (1-7) on Silicotic Fibrosis in Rats. Biomed. Environ. Sci. 2019, 32, 419–426. [Google Scholar] [CrossRef] [PubMed]
- Delpino, M.V.; Quarleri, J. SARS-CoV-2 Pathogenesis: Imbalance in the Renin-Angiotensin System Favors Lung Fibrosis. Front. Cell Infect. Microbiol. 2020, 10, 340. [Google Scholar] [CrossRef]
- Rauf, A.; Abu-Izneid, T.; Olatunde, A.; Ahmed Khalil, A.; Alhumaydhi, F.A.; Tufail, T.; Shariati, M.A.; Rebezov, M.; Almarhoon, Z.M.; Mabkhot, Y.N.; et al. COVID-19 Pandemic: Epidemiology, Etiology, Conventional and Non-Conventional Therapies. Int. J. Environ. Res. Public Health 2020, 17, 8155. [Google Scholar] [CrossRef] [PubMed]
- Rothan, H.A.; Byrareddy, S.N. The epidemiology and pathogenesis of coronavirus disease (COVID-19) outbreak. J. Autoimmun. 2020, 109, 102433. [Google Scholar] [CrossRef] [PubMed]
- Ciotti, M.; Angeletti, S.; Minieri, M.; Giovannetti, M.; Benvenuto, D.; Pascarella, S.; Sagnelli, C.; Bianchi, M.; Bernardini, S.; Ciccozzi, M. COVID-19 Outbreak: An Overview. Chemotherapy 2019, 64, 215–223. [Google Scholar] [CrossRef]
- Bohn, M.K.; Hall, A.; Sepiashvili, L.; Jung, B.; Steele, S.; Adeli, K. Pathophysiology of COVID-19: Mechanisms Underlying Disease Severity and Progression. Physiology 2020, 35, 288–301. [Google Scholar] [CrossRef]
- Rahman, M.M.; Hasan, M.; Ahmed, A. Potential detrimental role of soluble ACE2 in severe COVID-19 comorbid patients. Rev. Med. Virol. 2021, 31, 1–12. [Google Scholar] [CrossRef]
- Glowacka, I.; Bertram, S.; Herzog, P.; Pfefferle, S.; Steffen, I.; Muench, M.O.; Simmons, G.; Hofmann, H.; Kuri, T.; Weber, F.; et al. Differential downregulation of ACE2 by the spike proteins of severe acute respiratory syndrome coronavirus and human coronavirus NL63. J. Virol. 2010, 84, 1198–1205. [Google Scholar] [CrossRef] [Green Version]
- Lundstrom, A.; Ziegler, L.; Havervall, S.; Rudberg, A.S.; von Meijenfeldt, F.; Lisman, T.; Mackman, N.; Sanden, P.; Thalin, C. Soluble angiotensin-converting enzyme 2 is transiently elevated in COVID-19 and correlates with specific inflammatory and endothelial markers. J. Med. Virol. 2021, 93, 5908–5916. [Google Scholar] [CrossRef]
- Binns, C.; Low, W.Y.; Kyung, L.M. The COVID-19 Pandemic: Public Health and Epidemiology. Asia Pac. J. Public Health 2020, 32, 140–144. [Google Scholar] [CrossRef]
- Bazdyrev, E.; Rusina, P.; Panova, M.; Novikov, F.; Grishagin, I.; Nebolsin, V. Lung Fibrosis after COVID-19: Treatment Prospects. Pharmaceuticals 2021, 14, 807. [Google Scholar] [CrossRef] [PubMed]
- Rumende, C.M.; Susanto, E.C.; Sitorus, T.P. The Management of Pulmonary Fibrosis in COVID-19. Acta Med. Indones. 2021, 53, 233–241. [Google Scholar]
- Letellier, A.; Gibelin, A.; Voiriot, G.; Fartoukh, M.; Djibre, M. Destructive pulmonary fibrosis after severe COVID-19 pneumonia. Int. J. Infect. Dis. 2020, 100, 377–378. [Google Scholar] [CrossRef]
- Li, H.H.; Liu, C.C.; Hsu, T.W.; Lin, J.H.; Hsu, J.W.; Li, A.F.; Yeh, Y.C.; Hung, S.C.; Hsu, H.S. Upregulation of ACE2 and TMPRSS2 by particulate matter and idiopathic pulmonary fibrosis: A potential role in severe COVID-19. Part Fibre Toxicol. 2021, 18, 11. [Google Scholar] [CrossRef]
- Zhang, C.; Wu, Z.; Li, J.W.; Tan, K.; Yang, W.; Zhao, H.; Wang, G.Q. Discharge may not be the end of treatment: Pay attention to pulmonary fibrosis caused by severe COVID-19. J. Med. Virol. 2021, 93, 1378–1386. [Google Scholar] [CrossRef] [PubMed]
- Ojo, A.S.; Balogun, S.A.; Williams, O.T.; Ojo, O.S. Pulmonary Fibrosis in COVID-19 Survivors: Predictive Factors and Risk Reduction Strategies. Pulm. Med. 2020, 2020, 6175964. [Google Scholar] [CrossRef]
- Huang, W.; Wu, Q.; Chen, Z.; Xiong, Z.; Wang, K.; Tian, J.; Zhang, S. The potential indicators for pulmonary fibrosis in survivors of severe COVID-19. J. Infect. 2021, 82, e5–e7. [Google Scholar] [CrossRef] [PubMed]
- Kayhan, S.; Kocakoc, E. Pulmonary Fibrosis Due to COVID-19 Pneumonia. Korean J. Radiol. 2020, 21, 1273–1275. [Google Scholar] [CrossRef] [PubMed]
- Zou, J.N.; Sun, L.; Wang, B.R.; Zou, Y.; Xu, S.; Ding, Y.J.; Shen, L.J.; Huang, W.C.; Jiang, X.J.; Chen, S.M. The characteristics and evolution of pulmonary fibrosis in COVID-19 patients as assessed by AI-assisted chest HRCT. PLoS ONE 2021, 16, e0248957. [Google Scholar] [CrossRef]
- Gentile, F.; Aimo, A.; Forfori, F.; Catapano, G.; Clemente, A.; Cademartiri, F.; Emdin, M.; Giannoni, A. COVID-19 and risk of pulmonary fibrosis: The importance of planning ahead. Eur. J. Prev. Cardiol. 2020, 27, 1442–1446. [Google Scholar] [CrossRef]
- Yu, M.; Liu, Y.; Xu, D.; Zhang, R.; Lan, L.; Xu, H. Prediction of the Development of Pulmonary Fibrosis Using Serial Thin-Section CT and Clinical Features in Patients Discharged after Treatment for COVID-19 Pneumonia. Korean J. Radiol. 2020, 21, 746–755. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.J.; Xu, J.; Yin, J.M.; Li, L.; Hou, W.; Zhang, L.L.; Zhou, Z.; Yu, Y.Z.; Li, H.J.; Feng, Y.M.; et al. Lower Circulating Interferon-Gamma Is a Risk Factor for Lung Fibrosis in COVID-19 Patients. Front. Immunol. 2020, 11, 585647. [Google Scholar] [CrossRef] [PubMed]
- de Lang, A.; Osterhaus, A.D.; Haagmans, B.L. Interferon-gamma and interleukin-4 downregulate expression of the SARS coronavirus receptor ACE2 in Vero E6 cells. Virology 2006, 353, 474–481. [Google Scholar] [CrossRef] [Green Version]
- Salka, K.; Abutaleb, K.; Chorvinsky, E.; Thiruvengadam, G.; Arroyo, M.; Gomez, J.L.; Gutierrez, M.J.; Pillai, D.K.; Jaiswal, J.K.; Nino, G. IFN Stimulates ACE2 Expression in Pediatric Airway Epithelial Cells. Am. J. Respir. Cell Mol. Biol. 2021, 64, 515–518. [Google Scholar] [CrossRef] [PubMed]
- Mahmudpour, M.; Roozbeh, J.; Keshavarz, M.; Farrokhi, S.; Nabipour, I. COVID-19 cytokine storm: The anger of inflammation. Cytokine 2020, 133, 155151. [Google Scholar] [CrossRef]
- Smith, J.C.; Sausville, E.L.; Girish, V.; Yuan, M.L.; Vasudevan, A.; John, K.M.; Sheltzer, J.M. Cigarette smoke exposure and inflammatory signaling increase the expression of the SARS-CoV-2 receptor ACE2 in the respiratory tract. Dev. Cell 2020, 53, 514–529. [Google Scholar] [CrossRef]
- Liu, A.; Zhang, X.; Li, R.; Zheng, M.; Yang, S.; Dai, L.; Wu, A.; Hu, C.; Huang, Y.; Xie, M.; et al. Overexpression of the SARS-CoV-2 receptor ACE2 is induced by cigarette smoke in bronchial and alveolar epithelia. J. Pathol. 2021, 253, 17–30. [Google Scholar] [CrossRef]
- Russo, P.; Bonassi, S.; Giacconi, R.; Malavolta, M.; Tomino, C.; Maggi, F. COVID-19 and Smoking. Is Nicotine the Hidden Link? Eur. Respir. J. 2020. [Google Scholar] [CrossRef]
- Tomchaney, M.; Contoli, M.; Mayo, J.; Baraldo, S.; Li, S.; Cabel, C.R.; Bull, D.A.; Lick, S.; Malo, J.; Knoper, S.; et al. Paradoxical effects of cigarette smoke and COPD on SARS-CoV-2 infection and disease. BMC Pulm. Med. 2021, 21, 275. [Google Scholar] [CrossRef] [PubMed]
- Civiletto, C.W.; Aslam, S.; Hutchison, J. Electronic Delivery (Vaping) Of Cannabis and Nicotine; StatPearls: Treasure Island, FL, USA, 2019. [Google Scholar]
- Lee, A.C.; Chakladar, J.; Li, W.T.; Chen, C.; Chang, E.Y.; Wang-Rodriguez, J.; Ongkeko, W.M. Tobacco, but Not Nicotine and Flavor-Less Electronic Cigarettes, Induces ACE2 and Immune Dysregulation. Int. J. Mol. Sci. 2020, 21, 5513. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Management of Substance Abuse. Available online: https://www.who.int/substance_abuse/facts/cannabis/en/ (accessed on 22 November 2021).
- Ware, M.A. Cannabis and the lung: No more smoking gun? Ann. Am. Thorac. Soc. 2013, 10, 248. [Google Scholar] [CrossRef]
- Morris, R.R. Human pulmonary histopathological changes from marijuana smoking. J. Forensic. Sci. 1985, 30, 345–349. [Google Scholar] [CrossRef]
- Phan, T.D.; Lau, K.K.; Li, X. Lung bullae and pulmonary fibrosis associated with marijuana smoking. Australas. Radiol. 2005, 49, 411–414. [Google Scholar] [CrossRef]
- Fligiel, S.E.; Beals, T.F.; Tashkin, D.P.; Paule, M.G.; Scallet, A.C.; Ali, S.F.; Bailey, J.R.; Slikker, W., Jr. Marijuana exposure and pulmonary alterations in primates. Pharmacol. Biochem. Behav. 1991, 40, 637–642. [Google Scholar] [CrossRef]
- Zhang, S.; Gu, H.; Hu, N. Role of Peroxisome Proliferator-Activated Receptor gamma in Ocular Diseases. J. Ophthalmol. 2015, 2015, 275435. [Google Scholar] [CrossRef] [Green Version]
- Kokeny, G.; Calvier, L.; Hansmann, G. PPARgamma and TGFbeta-Major Regulators of Metabolism, Inflammation, and Fibrosis in the Lungs and Kidneys. Int. J. Mol. Sci. 2021, 22, 431. [Google Scholar] [CrossRef] [PubMed]
- Carmona-Hidalgo, B.; Gonzalez-Mariscal, I.; Garcia-Martin, A.; Prados, M.E.; Ruiz-Pino, F.; Appendino, G.; Tena-Sempere, M.; Munoz, E. Delta9-Tetrahydrocannabinolic Acid markedly alleviates liver fibrosis and inflammation in mice. Phytomedicine 2021, 81, 153426. [Google Scholar] [CrossRef]
- Parlar, A.; Arslan, S.O.; Yumrutas, O.; Elibol, E.; Yalcin, A.; Uckardes, F.; Aydin, H.; Dogan, M.F.; Kayhan Kustepe, E.; Ozer, M.K. Effects of cannabinoid receptor 2 synthetic agonist, AM1241, on bleomycin induced pulmonary fibrosis. Biotech Histochem. 2021, 96, 48–59. [Google Scholar] [CrossRef]
- Wang, B.; Kovalchuk, A.; Li, D.; Rodriguez-Juarez, R.; Ilnytskyy, Y.; Kovalchuk, I.; Kovalchuk, O. In search of preventive strategies: Novel high-CBD Cannabis sativa extracts modulate ACE2 expression in COVID-19 gateway tissues. Aging 2020, 12, 22425–22444. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.C.; Yang, D.; Nicolaescu, V.; Best, T.J.; Ohtsuki, T.; Chen, S.N.; Friesen, J.B.; Drayman, N.; Mohamed, A.; Dann, C.; et al. Cannabidiol Inhibits SARS-CoV-2 Replication and Promotes the Host Innate Immune Response. bioRxiv 2021. [Google Scholar] [CrossRef]
- Anil, S.M.; Shalev, N.; Vinayaka, A.C.; Nadarajan, S.; Namdar, D.; Belausov, E.; Shoval, I.; Mani, K.A.; Mechrez, G.; Koltai, H. Cannabis compounds exhibit anti-inflammatory activity in vitro in COVID-19-related inflammation in lung epithelial cells and pro-inflammatory activity in macrophages. Sci. Rep. 2021, 11, 1462. [Google Scholar] [CrossRef]
- Paland, N.; Pechkovsky, A.; Aswad, M.; Hamza, H.; Popov, T.; Shahar, E.; Louria-Hayon, I. The Immunopathology of COVID-19 and the Cannabis Paradigm. Front. Immunol. 2021, 12, 631233. [Google Scholar] [CrossRef]
- Esposito, G.; Pesce, M.; Seguella, L.; Sanseverino, W.; Lu, J.; Corpetti, C.; Sarnelli, G. The potential of cannabidiol in the COVID-19 pandemic. Br. J. Pharmacol. 2020, 177, 4967–4970. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Morganstein, T.; Haidar, Z.; Trivlidis, J.; Azuelos, I.; Huang, M.J.; Eidelman, D.H.; Baglole, C.J. Involvement of the ACE2/Ang-(1–7)/MasR Axis in Pulmonary Fibrosis: Implications for COVID-19. Int. J. Mol. Sci. 2021, 22, 12955. https://doi.org/10.3390/ijms222312955
Morganstein T, Haidar Z, Trivlidis J, Azuelos I, Huang MJ, Eidelman DH, Baglole CJ. Involvement of the ACE2/Ang-(1–7)/MasR Axis in Pulmonary Fibrosis: Implications for COVID-19. International Journal of Molecular Sciences. 2021; 22(23):12955. https://doi.org/10.3390/ijms222312955
Chicago/Turabian StyleMorganstein, Taylor, Zahraa Haidar, Joshua Trivlidis, Ilan Azuelos, Megan Jiaxin Huang, David H. Eidelman, and Carolyn J. Baglole. 2021. "Involvement of the ACE2/Ang-(1–7)/MasR Axis in Pulmonary Fibrosis: Implications for COVID-19" International Journal of Molecular Sciences 22, no. 23: 12955. https://doi.org/10.3390/ijms222312955