
New Insights in the Involvement of the Endocannabinoid System and Natural Cannabinoids in Nicotine Dependence

Abstract
:
1. Introduction
2. Neurobiological Mechanisms of Nicotine Dependence
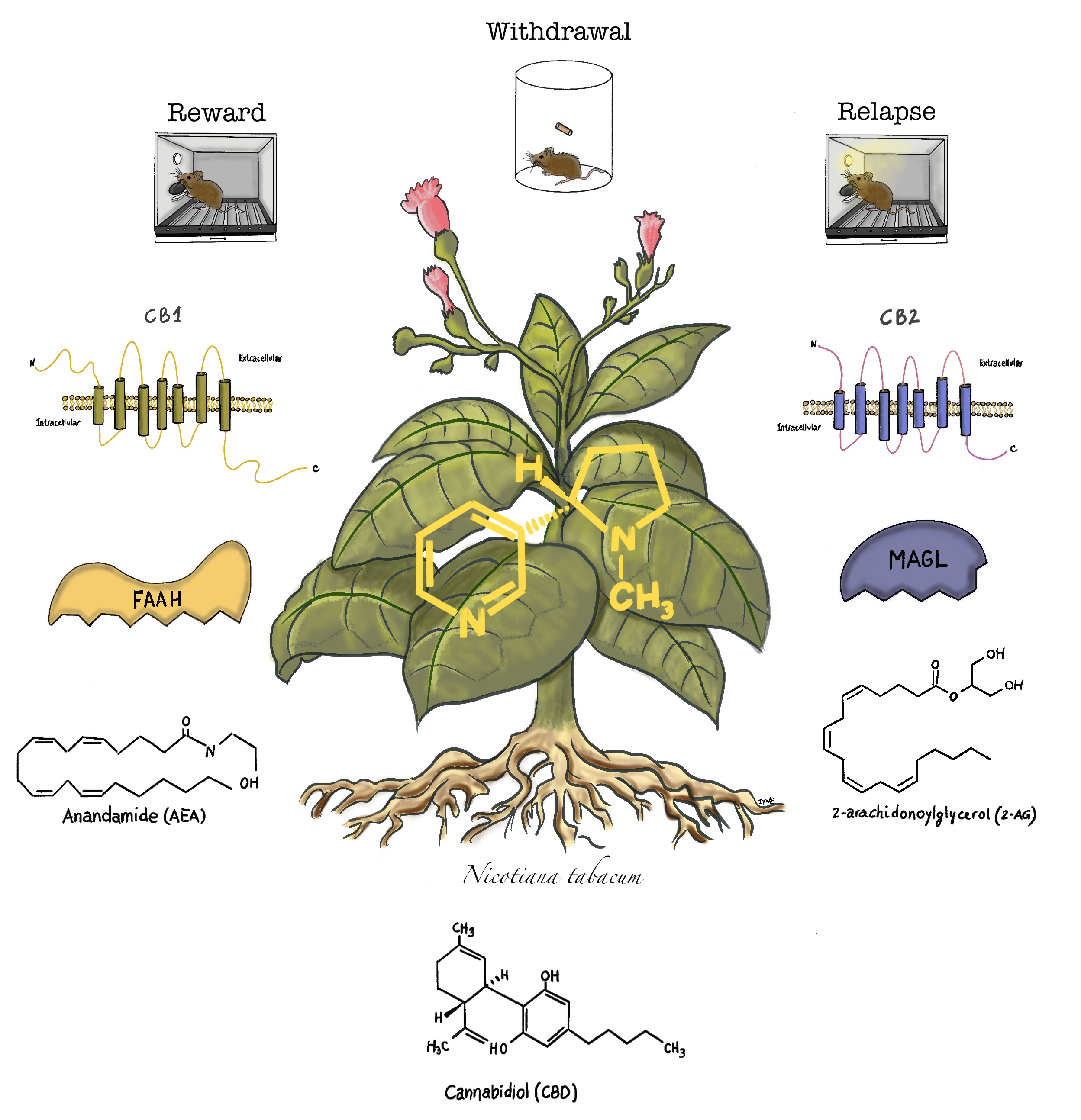
3. The Endogenous Cannabinoid System and Cannabinoid Compounds
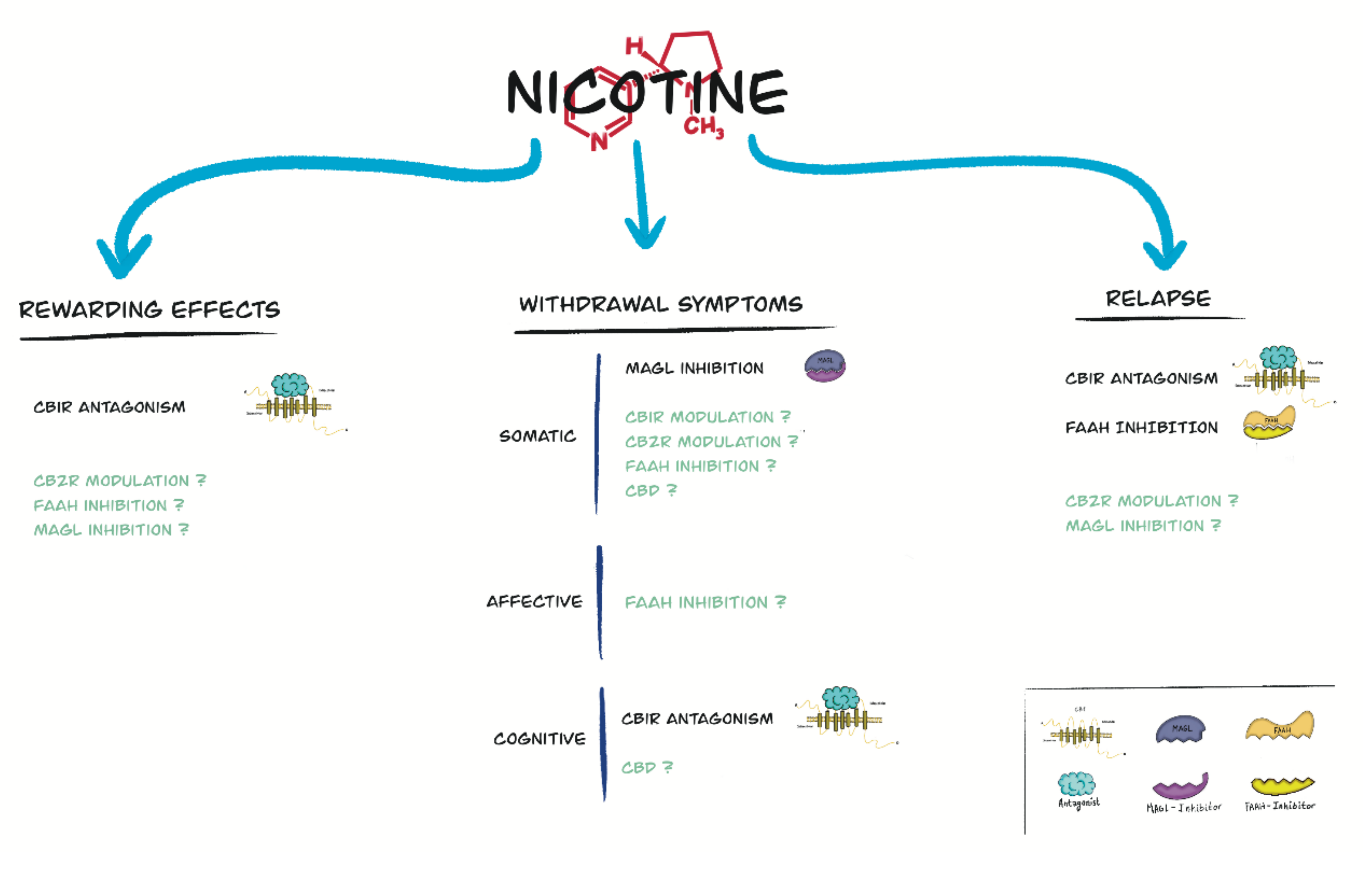
4. Role of the Endocannabinoid System and Cannabinoid Compounds in Nicotine Reward
5. Role of the Endocannabinoid System and Cannabinoid Compounds in Nicotine Withdrawal
6. Role of the Endocannabinoid System and Cannabinoid Compounds in Relapse to Nicotine-Seeking Behavior
7. Effects of Cannabinoid Exposure on Nicotine Addictive Properties
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- National Institute on Drug Abuse (NIDA). Cigarettes and Other Tobacco Products DrugFacts. Available online: https://www.drugabuse.gov/publications/drugfacts/cigarettes-other-tobacco-products (accessed on 12 November 2021).
- Gowing, L.R.; Ali, R.L.; Allsop, S.; Marsden, J.; Turf, E.E.; West, R.; Witton, J. Global statistics on addictive behaviours: 2014 status report. Addiction 2015, 110, 904–919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- WHO. Report on the Global Tobacco Epidemic 2019: Offer Help to Quit Tobacco Use. Available online: https://www.who.int/publications/i/item/9789241516204 (accessed on 12 November 2021).
- Jha, P.; Peto, R. Global Effects of Smoking, of Quitting, and of Taxing Tobacco. N. Engl. J. Med. 2014, 370, 60–68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jha, P. Avoidable global cancer deaths and total deaths from smoking. Nat. Rev. Cancer 2009, 9, 655–664. [Google Scholar] [CrossRef] [PubMed]
- Goodchild, M.; Nargis, N.; D’Espaignet, E.T. Global economic cost of smoking-attributable diseases. Tob. Control 2017, 27, 58–64. [Google Scholar] [CrossRef]
- Tobacco Packaging Market. Global Industry Trends, Share, Size, Growth, Opportunity and Forecast 2021–2026. Available online: https://www.researchandmarkets.com/reports/5263963/tobacco-packaging-market-global-industry-trends (accessed on 12 November 2021).
- Digiacomo, S.I.; Jazayeri, M.-A.; Barua, R.S.; Ambrose, J.A. Environmental Tobacco Smoke and Cardiovascular Disease. Int. J. Environ. Res. Public Health 2018, 16, 96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mainali, P.; Pant, S.; Rodriguez, A.P.; Deshmukh, A.; Mehta, J.L. Tobacco and Cardiovascular Health. Cardiovasc. Toxicol. 2014, 15, 107–116. [Google Scholar] [CrossRef]
- Szyfter, K.; Napierała, M.; Florek, E.; Braakhuis, B.J.M.; Takes, R.P.; Rodrigo, J.P.; Rinaldo, A.; Silver, C.E.; Ferlito, A. Molecular and health effects in the upper respiratory tract associated with tobacco smoking other than cigarettes. Int. J. Cancer 2018, 144, 2635–2643. [Google Scholar] [CrossRef] [PubMed]
- Vanker, A.; Gie, R.; Zar, H. The association between environmental tobacco smoke exposure and childhood respiratory disease: A review. Expert Rev. Respir. Med. 2017, 11, 661–673. [Google Scholar] [CrossRef] [PubMed]
- Snodgrass, A.M.; Tan, P.T.; E Soh, S.; Goh, A.; Shek, L.P.; van Bever, H.P.; Gluckman, P.D.; Godfrey, K.M.; Chong, Y.S.; Saw, S.M.; et al. Tobacco smoke exposure and respiratory morbidity in young children. Tob. Control 2015, 25, e75–e82. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, K.; Gowers, K.H.C.; Lee-Six, H.; Chandrasekharan, D.P.; Coorens, T.; Maughan, E.F.; Beal, K.; Menzies, A.; Millar, F.; Anderson, E.; et al. Tobacco smoking and somatic mutations in human bronchial epithelium. Nature 2020, 578, 266–272. [Google Scholar] [CrossRef] [PubMed]
- Alexandrov, L.B.; Ju, Y.S.; Haase, K.; Van Loo, P.; Martincorena, I.; Nik-Zainal, S.; Totoki, Y.; Fujimoto, A.; Nakagawa, H.; Shibata, T.; et al. Mutational signatures associated with tobacco smoking in human cancer. Science 2016, 354, 618–622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Islami, F.; Torre, L.A.; Jemal, A. Global trends of lung cancer mortality and smoking prevalence. Transl. Lung Cancer Res. 2015, 4, 327–338. [Google Scholar] [CrossRef] [PubMed]
- Grando, S.A. Connections of nicotine to cancer. Nat. Rev. Cancer 2014, 14, 419–429. [Google Scholar] [CrossRef] [PubMed]
- Bialous, S.A.; Sarna, L. Lung Cancer and Tobacco. Nurs. Clin. N. Am. 2017, 52, 53–63. [Google Scholar] [CrossRef]
- World Health Organization. WHO Global Report: Mortality Attributable to Tobacco; WHO: Geneva, Switzerland, 2012; ISBN 9789241564434. [Google Scholar]
- Ashare, R.L.; Wetherill, R. The Intersection of Sex Differences, Tobacco Use, and Inflammation: Implications for Psychiatric Disorders. Curr. Psychiatry Rep. 2018, 20, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Donny, E.C.; White, C.M. A review of the evidence on cigarettes with reduced addictiveness potential. Int. J. Drug Policy 2021, 103436. [Google Scholar] [CrossRef] [PubMed]
- Konstantinou, E.; Fotopoulou, F.; Drosos, A.; Dimakopoulou, N.; Zagoriti, Z.; Niarchos, A.; Makrynioti, D.; Kouretas, D.; Farsalinos, K.; Lagoumintzis, G.; et al. Tobacco-specific nitrosamines: A literature review. Food Chem. Toxicol. 2018, 118, 198–203. [Google Scholar] [CrossRef]
- Niu, J.; Zhao, X.; Jin, Y.; Yang, G.; Li, Z.; Wang, J.; Zhao, R.; Li, Z. Determination of aromatic amines in the urine of smokers using a porous organic framework (JUC-Z2)-coated solid-phase microextraction fiber. J. Chromatogr. A 2018, 1555, 37–44. [Google Scholar] [CrossRef] [PubMed]
- Mohammadi, S.; Domeno, C.; Nerin, I.; Aznar, M.; Samper, P.; Khayatian, G.; Nerin, C. Toxic compounds from tobacco in placenta samples analyzed by UPLC-QTOF-MS. J. Pharm. Biomed. Anal. 2017, 145, 331–338. [Google Scholar] [CrossRef]
- Eldridge, A.; Betson, T.; Gama, M.V.; McAdam, K. Variation in tobacco and mainstream smoke toxicant yields from selected commercial cigarette products. Regul. Toxicol. Pharmacol. 2015, 71, 409–427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Auer, R.; Concha-Lozano, N.; Jacot-Sadowski, I.; Cornuz, J.; Berthet, A. Heat-Not-Burn Tobacco Cigarettes. JAMA Intern. Med. 2017, 177, 1050–1052. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benowitz, N.L.; Fraiman, J.B. Cardiovascular effects of electronic cigarettes. Nat. Rev. Cardiol. 2017, 14, 447–456. [Google Scholar] [CrossRef]
- Fetterman, J.L.; Weisbrod, R.M.; Feng, B.; Bastin, R.; Tuttle, S.T.; Holbrook, M.; Baker, G.; Robertson, R.M.; Conklin, D.J.; Bhatnagar, A.; et al. Flavorings in Tobacco Products Induce Endothelial Cell Dysfunction. Arter. Thromb. Vasc. Biol. 2018, 38, 1607–1615. [Google Scholar] [CrossRef]
- Uchiyama, S.; Noguchi, M.; Takagi, N.; Hayashida, H.; Inaba, Y.; Ogura, H.; Kunugita, N. Simple Determination of Gaseous and Particulate Compounds Generated from Heated Tobacco Products. Chem. Res. Toxicol. 2018, 31, 585–593. [Google Scholar] [CrossRef] [PubMed]
- Walley, S.C.; Wilson, K.M.; Winickoff, J.P.; Groner, J. A Public Health Crisis: Electronic Cigarettes, Vape, and JUUL. Pediatrics 2019, 143, e20182741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giulietti, F.; Filipponi, A.; Rosettani, G.; Giordano, P.; Iacoacci, C.; Spannella, F.; Sarzani, R. Pharmacological Approach to Smoking Cessation: An Updated Review for Daily Clinical Practice. High Blood Press. Cardiovasc. Prev. 2020, 27, 349–362. [Google Scholar] [CrossRef] [PubMed]
- Paterson, D.; Nordberg, A. Neuronal nicotinic receptors in the human brain. Prog. Neurobiol. 2000, 61, 75–111. [Google Scholar] [CrossRef]
- Maurer, S.V.; Williams, C.L. The Cholinergic System Modulates Memory and Hippocampal Plasticity via Its Interactions with Non-Neuronal Cells. Front. Immunol. 2017, 8, 1489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Changeux, J.-P.; Bertrand, D.; Corringer, P.-J.; Dehaene, S.; Edelstein, S.; Léna, C.; Le Novère, N.; Marubio, L.; Picciotto, M.; Zoli, M. Brain nicotinic receptors: Structure and regulation, role in learning and reinforcement. Brain Res. Rev. 1998, 26, 198–216. [Google Scholar] [CrossRef]
- Le Novère, N.; Grutter, T.; Changeux, J.-P. Models of the extracellular domain of the nicotinic receptors and of agonist- and Ca2+-binding sites. Proc. Natl. Acad. Sci. USA 2002, 99, 3210–3215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dani, J.A. Neuronal Nicotinic Acetylcholine Receptor Structure and Function and Response to Nicotine. Int. Rev. Neurobiol. 2015, 124, 3–19. [Google Scholar] [CrossRef] [Green Version]
- McLaughlin, I.; Dani, J.A.; De Biasi, M. Nicotine Withdrawal; Springer: Cham, Switzerland, 2015; pp. 99–123. [Google Scholar]
- Stoker, A.K.; Markou, A. Unraveling the neurobiology of nicotine dependence using genetically engineered mice. Curr. Opin. Neurobiol. 2013, 23, 493–499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walters, C.L.; Brown, S.; Changeux, J.-P.; Martin, B.; Damaj, M.I. The β2 but not α7 subunit of the nicotinic acetylcholine receptor is required for nicotine-conditioned place preference in mice. Psychopharmacology 2006, 184, 339–344. [Google Scholar] [CrossRef] [PubMed]
- Brunzell, D.H.; McIntosh, J.M. Alpha7 Nicotinic Acetylcholine Receptors Modulate Motivation to Self-Administer Nicotine: Implications for Smoking and Schizophrenia. Neuropsychopharmacology 2011, 37, 1134–1143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bierut, L.J.; Stitzel, J.; Wang, J.C.; Hinrichs, A.L.; Grucza, R.; Xuei, X.; Saccone, N.L.; Saccone, S.F.; Bertelsen, S.; Fox, L.; et al. Variants in Nicotinic Receptors and Risk for Nicotine Dependence. Am. J. Psychiatry 2008, 165, 1163–1171. [Google Scholar] [CrossRef] [PubMed]
- Prochaska, J.J.; Benowitz, N.L. Current advances in research in treatment and recovery: Nicotine addiction. Sci. Adv. 2019, 5, eaay9763. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.; Zhao-Shea, R.; McIntosh, J.M.; Gardner, P.D.; Tapper, A.R. Nicotine Persistently Activates Ventral Tegmental Area Dopaminergic Neurons via Nicotinic Acetylcholine Receptors Containing α4 and α6 Subunits. Mol. Pharmacol. 2012, 81, 541–548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Antolin-Fontes, B.; Ables, J.L.; Görlich, A.; Ibañez-Tallon, I. The habenulo-interpeduncular pathway in nicotine aversion and withdrawal. Neuropharmacology 2014, 96, 213–222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frahm, S.; Ślimak, M.A.; Ferrarese, L.; Santos-Torres, J.; Antolin-Fontes, B.; Auer, S.; Filkin, S.; Pons, S.; Fontaine, J.-F.; Tsetlin, V.; et al. Aversion to Nicotine Is Regulated by the Balanced Activity of β4 and α5 Nicotinic Receptor Subunits in the Medial Habenula. Neuron 2011, 70, 522–535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benowitz, N.L. Nicotine Addiction. N. Engl. J. Med. 2010, 362, 2295–2303. [Google Scholar] [CrossRef] [PubMed]
- Heishman, S.J.; Taylor, R.C.; Henningfield, J.E. Nicotine and Smoking: A Review of Effects on Human Performance. Exp. Clin. Psychopharmacol. 1994, 2, 345–395. [Google Scholar] [CrossRef]
- Pulvers, K.; Scheuermann, T.S.; Emami, A.S.; Basora, B.; Luo, X.; Khariwala, S.S.; Ahluwalia, J.S. Reasons for Smoking Among Tri-Ethnic Daily and Nondaily Smokers. Nicotine Tob. Res. 2014, 16, 1567–1576. [Google Scholar] [CrossRef] [Green Version]
- Hall, F.S.; Der-Avakian, A.; Gould, T.J.; Markou, A.; Shoaib, M.; Young, J.W. Negative affective states and cognitive impairments in nicotine dependence. Neurosci. Biobehav. Rev. 2015, 58, 168–185. [Google Scholar] [CrossRef] [Green Version]
- Zoli, M.; Pistillo, F.; Gotti, C. Diversity of native nicotinic receptor subtypes in mammalian brain. Neuropharmacology 2015, 96, 302–311. [Google Scholar] [CrossRef] [PubMed]
- Markou, A. Neurobiology of nicotine dependence. Philos. Trans. R. Soc. B Biol. Sci. 2008, 363, 3159–3168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, Y.; Peng, C.; Arvin, M.C.; Jin, X.-T.; Kim, V.; Ramsey, M.D.; Wang, Y.; Banala, S.; Wokosin, D.L.; McIntosh, J.M.; et al. Nicotinic Cholinergic Receptors in VTA Glutamate Neurons Modulate Excitatory Transmission. Cell Rep. 2018, 23, 2236–2244. [Google Scholar] [CrossRef] [Green Version]
- Koob, G.F.; Volkow, N.D. Neurocircuitry of Addiction. Neuropsychopharmacology 2009, 35, 217–238. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Sun, X. Desensitized nicotinic receptors in brain. Brain Res. Rev. 2005, 48, 420–437. [Google Scholar] [CrossRef]
- De Biasi, M.; Dani, J.A. Reward, Addiction, Withdrawal to Nicotine. Annu. Rev. Neurosci. 2011, 34, 105–130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fasoli, F.; Moretti, M.; Zoli, M.; Pistillo, F.; Crespi, A.; Clementi, F.; Mc Clure-Begley, T.; Marks, M.; Gotti, C.; Fasoli, F.; et al. In vivo chronic nicotine exposure differentially and reversibly affects upregulation and stoichiometry of α4β2 nicotinic receptors in cortex and thalamus. Neuropharmacology 2016, 108, 324–331. [Google Scholar] [CrossRef] [PubMed]
- Brody, A.L.; Mandelkern, M.A.; London, E.D.; Olmstead, R.E.; Farahi, J.; Scheibal, D.; Jou, J.; Allen, V.; Tiongson, E.; Chefer, S.I.; et al. Cigarette Smoking Saturates Brain α4β2 Nicotinic Acetylcholine Receptors. Arch. Gen. Psychiatry 2006, 63, 907–915. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brody, A.L.; Mukhin, A.G.; Mamoun, M.S.; Luu, T.; Neary, M.; Liang, L.; Shieh, J.; Sugar, C.A.; Rose, J.E.; Mandelkern, M.A. Brain Nicotinic Acetylcholine Receptor Availability and Response to Smoking Cessation Treatment. JAMA Psychiatry 2014, 71, 797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cosgrove, K.P.; Esterlis, I.; Sandiego, C.; Petrulli, R.; Morris, E.D. Imaging Tobacco Smoking with PET and SPECT. Neuropharmacol. Nicotine Depend. 2015, 24, 1–17. [Google Scholar] [CrossRef]
- Koob, G.F. Addiction is a Reward Deficit and Stress Surfeit Disorder. Front. Psychiatry 2013, 4, 72. [Google Scholar] [CrossRef] [Green Version]
- Le Foll, B.; Goldberg, S.R. Effects of Nicotine in Experimental Animals and Humans: An Update on Addictive Properties. In Handbook of Experimental Pharmacology; Springer: Berlin/Heidelberg, Germany, 2009; Volume 192, pp. 335–367. [Google Scholar] [CrossRef] [Green Version]
- Allen, S.S.; Bade, T.; Hatsukami, D.; Center, B. Craving, withdrawal, and smoking urges on days immediately prior to smoking relapse. Nicotine Tob. Res. 2008, 10, 35–45. [Google Scholar] [CrossRef]
- Ashare, R.L.; Schmidt, H.D. Optimizing treatments for nicotine dependence by increasing cognitive performance during withdrawal. Expert Opin. Drug Discov. 2014, 9, 579–594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hendricks, P.S.; Delucchi, K.L.; Benowitz, N.L.; Hall, S.M. Clinical Significance of Early Smoking Withdrawal Effects and Their Relationships with Nicotine Metabolism: Preliminary Results from a Pilot Study. Nicotine Tob. Res. 2013, 16, 615–620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hughes, J.R. Effects of abstinence from tobacco: Valid symptoms and time course. Nicotine Tob. Res. 2007, 9, 315–327. [Google Scholar] [CrossRef] [Green Version]
- Wesnes, K.A.; Edgar, C.; Kezic, I.; Salih, H.M.; de Boer, P. Effects of nicotine withdrawal on cognition in a clinical trial setting. Psychopharmacology 2013, 229, 133–140. [Google Scholar] [CrossRef]
- Jackson, A.; Papke, R.L.; Damaj, M.I. Pharmacological modulation of the α7 nicotinic acetylcholine receptor in a mouse model of mecamylamine-precipitated nicotine withdrawal. Psychopharmacology 2018, 235, 1897–1905. [Google Scholar] [CrossRef]
- Stoker, A.K.; Olivier, B.; Markou, A. Role of α7- and β4-Containing Nicotinic Acetylcholine Receptors in the Affective and Somatic Aspects of Nicotine Withdrawal: Studies in Knockout Mice. Behav. Genet. 2011, 42, 423–436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jackson, K.J.; Martin, B.R.; Changeux, J.P.; Damaj, M.I. Differential Role of Nicotinic Acetylcholine Receptor Subunits in Physical and Affective Nicotine Withdrawal Signs. J. Pharmacol. Exp. Ther. 2008, 325, 302–312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kutlu, M.G.; Gould, T.J. Effects of drugs of abuse on hippocampal plasticity and hippocampus-dependent learning and memory: Contributions to development and maintenance of addiction. Learn. Mem. 2016, 23, 515–533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yildirim, E.; Connor, D.A.; Gould, T.J. ABT-089, but not ABT-107, ameliorates nicotine withdrawal-induced cognitive deficits in C57BL6/J mice. Behav. Pharmacol. 2015, 26, 241–248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X. Characterization of Cue-Induced Reinstatement of Nicotine-Seeking Behavior in Smoking Relapse. In Neuropathology of Drug Addictions and Substance Misuse; Academic Press: Cambridge, MA, USA, 2016; pp. 237–245. [Google Scholar] [CrossRef]
- Garcia-Rivas, V.; Deroche-Gamonet, V. Not all smokers appear to seek nicotine for the same reasons: Implications for preclinical research in nicotine dependence. Addict. Biol. 2018, 24, 317–334. [Google Scholar] [CrossRef] [PubMed]
- Wardle, M.C.; Munafò, M.R.; De Wit, H. Effect of social stress during acute nicotine abstinence. Psychopharmacology 2011, 218, 39–48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feltenstein, M.W.; Ghee, S.M.; See, R.E. Nicotine self-administration and reinstatement of nicotine-seeking in male and female rats. Drug Alcohol Depend. 2012, 121, 240–246. [Google Scholar] [CrossRef] [Green Version]
- Nygard, S.K.; Hourguettes, N.J.; Sobczak, G.G.; Carlezon, W.A.; Bruchas, M.R. Stress-Induced Reinstatement of Nicotine Preference Requires Dynorphin/Kappa Opioid Activity in the Basolateral Amygdala. J. Neurosci. 2016, 36, 9937–9948. [Google Scholar] [CrossRef] [Green Version]
- Plaza-Zabala, A.; Flores, A.; Martin-García, E.; Saravia, R.; Maldonado, R.; Berrendero, F. A Role for Hypocretin/Orexin Receptor-1 in Cue-Induced Reinstatement of Nicotine-Seeking Behavior. Neuropsychopharmacology 2013, 38, 1724–1736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Plaza-Zabala, A.; Martín-García, E.; de Lecea, L.; Maldonado, R.; Berrendero, F. Hypocretins Regulate the Anxiogenic-Like Effects of Nicotine and Induce Reinstatement of Nicotine-Seeking Behavior. J. Neurosci. 2010, 30, 2300–2310. [Google Scholar] [CrossRef] [PubMed]
- Bruijnzeel, A.W. Neuropeptide systems and new treatments for nicotine addiction. Psychopharmacology 2016, 234, 1419–1437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berrendero, F.; Plaza-Zabala, A.; Galeote, L.; Flores, A.; Bura, S.A.; Kieffer, B.L.; Maldonado, R. Influence of δ-Opioid Receptors in the Behavioral Effects of Nicotine. Neuropsychopharmacology 2012, 37, 2332–2344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Butler, K.; Le Foll, B. Novel therapeutic and drug development strategies for tobacco use disorder: Endocannabinoid modulation. Expert Opin. Drug Discov. 2020, 15, 1065–1080. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.-C.; Mackie, K. An Introduction to the Endogenous Cannabinoid System. Biol. Psychiatry 2015, 79, 516–525. [Google Scholar] [CrossRef] [Green Version]
- Van Eenige, R.; van der Stelt, M.; Rensen, P.C.N.; Kooijman, S. Regulation of Adipose Tissue Metabolism by the Endocannabinoid System. Trends Endocrinol. Metab. 2018, 29, 326–337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Acharya, N.; Penukonda, S.; Shcheglova, T.; Hagymasi, A.T.; Basu, S.; Srivastava, P.K. Endocannabinoid system acts as a regulator of immune homeostasis in the gut. Proc. Natl. Acad. Sci. USA 2017, 114, 5005–5010. [Google Scholar] [CrossRef] [Green Version]
- Maccarrone, M.; Bab, I.; Bíró, T.; Cabral, G.A.; Dey, S.K.; Di Marzo, V.; Konje, J.C.; Kunos, G.; Mechoulam, R.; Pacher, P.; et al. Endocannabinoid signaling at the periphery: 50 years after THC. Trends Pharmacol. Sci. 2015, 36, 277–296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ibarra-Lecue, I.; Pilar-Cuéllar, F.; Muguruza, C.; Florensa-Zanuy, E.; Díaz, Á.; Urigüen, L.; Castro, E.; Pazos, A.; Callado, L.F. The endocannabinoid system in mental disorders: Evidence from human brain studies. Biochem. Pharmacol. 2018, 157, 97–107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mato, S.; Pilar-Cuéllar, F.; Valdizán, E.M.; González-Maeso, J.; Rodríguez-Puertas, R.; Meana, J.; Sallés, J.; Crespo-Facorro, B.; Pazos, Á. Selective up-regulation of cannabinoid CB1 receptor coupling to Go-proteins in suicide victims with mood disorders. Biochem. Pharmacol. 2018, 157, 258–265. [Google Scholar] [CrossRef] [PubMed]
- Ranganathan, M.; Cortes-Briones, J.; Radhakrishnan, R.; Thurnauer, H.; Planeta, B.; Skosnik, P.; Gao, H.; Labaree, D.; Neumeister, A.; Pittman, B.; et al. Reduced Brain Cannabinoid Receptor Availability in Schizophrenia. Biol. Psychiatry 2015, 79, 997–1005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manzanares, J.; Cabañero, D.; Puente, N.; García-Gutiérrez, M.S.; Grandes, P.; Maldonado, R. Role of the endocannabinoid system in drug addiction. Biochem. Pharmacol. 2018, 157, 108–121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, S.S.-J.; Mackie, K. Distribution of the Endocannabinoid System in the Central Nervous System. Endocannabinoids 2015, 231, 59–93. [Google Scholar] [CrossRef]
- Atwood, B.; Mackie, K. CB2: A cannabinoid receptor with an identity crisis. Br. J. Pharmacol. 2010, 160, 467–479. [Google Scholar] [CrossRef] [Green Version]
- Di Marzo, V.; Bisogno, T.; De Petrocellis, L. The Biosynthesis, Fate and Pharmacological Properties of Endocannabinoids. In Handbook of Experimental Pharmacology; Springer: Berlin/Heidelberg, Germany, 2005; pp. 147–185. [Google Scholar] [CrossRef]
- Wilson, R.I.; Nicoll, R.A. Endocannabinoid Signaling in the Brain. Science 2002, 296, 678–682. [Google Scholar] [CrossRef] [Green Version]
- Okamoto, Y.; Tsuboi, K.; Ueda, N. Chapter 1 Enzymatic Formation of Anandamide. Vitam. Horm. 2009, 81, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Dinh, T.P.; Freund, T.F.; Piomelli, D. A role for monoglyceride lipase in 2-arachidonoylglycerol inactivation. Chem. Phys. Lipids 2002, 121, 149–158. [Google Scholar] [CrossRef] [Green Version]
- Cravatt, B.F.; Giang, D.K.; Mayfield, S.P.; Boger, D.L.; Lerner, R.A.; Gilula, N.B. Molecular characterization of an enzyme that degrades neuromodulatory fatty-acid amides. Nature 1996, 384, 83–87. [Google Scholar] [CrossRef]
- Aymerich, M.S.; Aso, E.; Abellanas, M.; Tolon, R.M.; Ramos, J.A.; Ferrer, I.; Romero, J.; Fernández-Ruiz, J. Cannabinoid pharmacology/therapeutics in chronic degenerative disorders affecting the central nervous system. Biochem. Pharmacol. 2018, 157, 67–84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muldoon, P.P.; Lichtman, A.H.; Parsons, L.H.; Damaj, M.I. The role of fatty acid amide hydrolase inhibition in nicotine reward and dependence. Life Sci. 2012, 92, 458–462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muldoon, P.P.; Chen, J.; Harenza, J.L.; Abdullah, R.A.; Sim-Selley, L.J.; Cravatt, B.F.; Miles, M.F.; Chen, X.; Lichtman, A.H.; I Damaj, M. Inhibition of monoacylglycerol lipase reduces nicotine withdrawal. Br. J. Pharmacol. 2014, 172, 869–882. [Google Scholar] [CrossRef] [Green Version]
- Morales, P.; Hurst, D.P.; Reggio, P.H. Molecular Targets of the Phytocannabinoids: A Complex Picture. Phytocannabinoids 2017, 103, 103–131. [Google Scholar] [CrossRef] [Green Version]
- Gaoni, Y.; Mechoulam, R. Isolation, Structure, and Partial Synthesis of an Active Constituent of Hashish. J. Am. Chem. Soc. 1964, 86, 1646–1647. [Google Scholar] [CrossRef]
- Mechoulam, R.; Shvo, Y. Hashish—I: The Structure of Cannabidiol. Tetrahedron 1963, 19, 2073–2078. [Google Scholar] [CrossRef]
- Huestis, M.A.; Gorelick, D.A.; Heishman, S.J.; Preston, K.; Nelson, R.A.; Moolchan, E.T.; Frank, R.A. Blockade of Effects of Smoked Marijuana by the CB1-Selective Cannabinoid Receptor Antagonist SR141716. Arch. Gen. Psychiatry 2001, 58, 322–328. [Google Scholar] [CrossRef]
- Fernández-Ruiz, J.; Galve-Roperh, I.; Sagredo, O.; Guzmán, M. Possible therapeutic applications of cannabis in the neuropsychopharmacology field. Eur. Neuropsychopharmacol. 2020, 36, 217–234. [Google Scholar] [CrossRef]
- Schoedel, K.A.; Szeto, I.; Setnik, B.; Sellers, E.M.; Levy-Cooperman, N.; Mills, C.; Etges, T.; Sommerville, K. Abuse potential assessment of cannabidiol (CBD) in recreational polydrug users: A randomized, double-blind, controlled trial. Epilepsy Behav. 2018, 88, 162–171. [Google Scholar] [CrossRef] [Green Version]
- Chye, Y.; Christensen, E.; Solowij, N.; Yücel, M. The Endocannabinoid System and Cannabidiol’s Promise for the Treatment of Substance Use Disorder. Front. Psychiatry 2019, 10, 63. [Google Scholar] [CrossRef]
- Navarrete, F.; García-Gutiérrez, M.S.; Gasparyan, A.; Austrich-Olivares, A.; Manzanares, J. Role of Cannabidiol in the Therapeutic Intervention for Substance Use Disorders. Front. Pharmacol. 2021, 12, 626010. [Google Scholar] [CrossRef]
- Cohen, C.; Perrault, G.; Voltz, C.; Steinberg, R.; Soubrié, P. SR141716, a central cannabinoid (CB1) receptor antagonist, blocks the motivational and dopamine-releasing effects of nicotine in rats. Behav. Pharmacol. 2002, 13, 451–463. [Google Scholar] [CrossRef]
- Schindler, C.W.; Redhi, G.H.; Vemuri, K.; Makriyannis, A.; Le Foll, B.; Bergman, J.; Goldberg, S.R.; Justinova, Z. Blockade of Nicotine and Cannabinoid Reinforcement and Relapse by a Cannabinoid CB1-Receptor Neutral Antagonist AM4113 and Inverse Agonist Rimonabant in Squirrel Monkeys. Neuropsychopharmacology 2016, 41, 2283–2293. [Google Scholar] [CrossRef] [Green Version]
- Shoaib, M. The cannabinoid antagonist AM251 attenuates nicotine self-administration and nicotine-seeking behaviour in rats. Neuropharmacology 2008, 54, 438–444. [Google Scholar] [CrossRef]
- Simonnet, A.; Cador, M.; Caille, S. Nicotine reinforcement is reduced by cannabinoid CB1 receptor blockade in the ventral tegmental area. Addict. Biol. 2012, 18, 930–936. [Google Scholar] [CrossRef] [PubMed]
- Forget, B.; Coen, K.M.; Le Foll, B. Inhibition of fatty acid amide hydrolase reduces reinstatement of nicotine seeking but not break point for nicotine self-administration—comparison with CB1 receptor blockade. Psychopharmacology 2009, 205, 613–624. [Google Scholar] [CrossRef] [PubMed]
- Gueye, A.B.; Pryslawsky, Y.; Trigo, J.M.; Poulia, N.; Delis, F.; Antoniou, K.; Loureiro, M.; LaViolette, S.R.; Vemuri, K.; Makriyannis, A.; et al. The CB1Neutral Antagonist AM4113 Retains the Therapeutic Efficacy of the Inverse Agonist Rimonabant for Nicotine Dependence and Weight Loss with Better Psychiatric Tolerability. Int. J. Neuropsychopharmacol. 2016, 19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castañé, A.; Valjent, E.; Ledent, C.; Parmentier, M.; Maldonado, R.; Valverde, O. Lack of CB1 cannabinoid receptors modifies nicotine behavioural responses, but not nicotine abstinence. Neuropharmacology 2002, 43, 857–867. [Google Scholar] [CrossRef]
- Le Foll, B.; Goldberg, S.R. Rimonabant, a CB1 Antagonist, Blocks Nicotine-Conditioned Place Preferences. Neuroreport 2004, 15, 2139–2143. [Google Scholar] [CrossRef] [PubMed]
- Merritt, L.L.; Martin, B.R.; Walters, C.; Lichtman, A.H.; Damaj, M.I. The Endogenous Cannabinoid System Modulates Nicotine Reward and Dependence. J. Pharmacol. Exp. Ther. 2008, 326, 483–492. [Google Scholar] [CrossRef] [Green Version]
- Forget, B.; Hamon, M.; Thiébot, M.-H. Cannabinoid CB1 receptors are involved in motivational effects of nicotine in rats. Psychopharmacology 2005, 181, 722–734. [Google Scholar] [CrossRef]
- Forget, B.; Barthélémy, S.; Saurini, F.; Hamon, M.; Thiébot, M.-H. Differential involvement of the endocannabinoid system in short- and long-term expression of incentive learning supported by nicotine in rats. Psychopharmacology 2006, 189, 59–69. [Google Scholar] [CrossRef]
- Azizi, F.; Fartootzadeh, R.; Alaei, H.; Reisi, P. Effects of concurrent blockade of OX2 and CB1 receptors in the ventral tegmental area on nicotine-induced place preference in rats. Neurosci. Lett. 2018, 684, 121–126. [Google Scholar] [CrossRef]
- Fartootzadeh, R.; Azizi, F.; Alaei, H.; Reisi, P. Functional crosstalk of nucleus accumbens CB1 and OX2 receptors in response to nicotine-induced place preference. Neurosci. Lett. 2019, 698, 160–164. [Google Scholar] [CrossRef]
- Hashemizadeh, S.; Sardari, M.; Rezayof, A. Basolateral amygdala CB1 cannabinoid receptors mediate nicotine-induced place preference. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2014, 51, 65–71. [Google Scholar] [CrossRef] [PubMed]
- Cheer, J.; Wassum, K.M.; Sombers, L.A.; Heien, M.L.A.V.; Ariansen, J.L.; Aragona, B.J.; Phillips, P.; Wightman, R.M. Phasic Dopamine Release Evoked by Abused Substances Requires Cannabinoid Receptor Activation. J. Neurosci. 2007, 27, 791–795. [Google Scholar] [CrossRef] [PubMed]
- Robinson, J.D.; Cinciripini, P.M.; Karam-Hage, M.; Aubin, H.-J.; Dale, L.C.; Niaura, R.; Anthenelli, R.M.; the STRATUS Group. Pooled analysis of three randomized, double-blind, placebo controlled trials with rimonabant for smoking cessation. Addict. Biol. 2017, 23, 291–303. [Google Scholar] [CrossRef] [PubMed]
- Gamaleddin, I.; Zvonok, A.; Makriyannis, A.; Goldberg, S.R.; Le Foll, B. Effects of a Selective Cannabinoid CB2 Agonist and Antagonist on Intravenous Nicotine Self Administration and Reinstatement of Nicotine Seeking. PLoS ONE 2012, 7, e29900. [Google Scholar] [CrossRef] [Green Version]
- Navarrete, F.; Rodriguez-Arias, M.; Martin-García, E.; Navarro, D.; García-Gutiérrez, M.S.; Aguilar, M.A.; Aracil-Fernández, A.; Berbel, P.; Miñarro, J.; Maldonado, R.; et al. Role of CB2 Cannabinoid Receptors in the Rewarding, Reinforcing, and Physical Effects of Nicotine. Neuropsychopharmacology 2013, 38, 2515–2524. [Google Scholar] [CrossRef] [Green Version]
- Ignatowska-Jankowska, B.M.; Muldoon, P.P.; Lichtman, A.H.; Damaj, M.I. The cannabinoid CB2 receptor is necessary for nicotine-conditioned place preference, but not other behavioral effects of nicotine in mice. Psychopharmacology 2013, 229, 591–601. [Google Scholar] [CrossRef] [Green Version]
- Canseco-Alba, A.; Schanz, N.; Sanabria, B.; Zhao, J.; Lin, Z.; Liu, Q.-R.; Onaivi, E.S. Behavioral effects of psychostimulants in mutant mice with cell-type specific deletion of CB2 cannabinoid receptors in dopamine neurons. Behav. Brain Res. 2018, 360, 286–297. [Google Scholar] [CrossRef]
- He, Y.; Galaj, E.; Bi, G.; Wang, X.; Gardner, E.; Xi, Z. β-Caryophyllene, a dietary terpenoid, inhibits nicotine taking and nicotine seeking in rodents. Br. J. Pharmacol. 2019, 177, 2058–2072. [Google Scholar] [CrossRef]
- He, X.-H.; Galaj, E.; Bi, G.-H.; He, Y.; Hempel, B.; Wang, Y.-L.; Gardner, E.L.; Xi, Z.-X. β-caryophyllene, an FDA-Approved Food Additive, Inhibits Methamphetamine-Taking and Methamphetamine-Seeking Behaviors Possibly via CB2 and Non-CB2 Receptor Mechanisms. Front. Pharmacol. 2021, 12, 722476. [Google Scholar] [CrossRef]
- Liu, Q.; Yu, J.; Li, X.; Guo, Y.; Sun, T.; Luo, L.; Ren, J.; Jiang, W.; Zhang, R.; Yang, P.; et al. Cannabinoid receptor GPR55 activation blocks nicotine use disorder by regulation of AMPAR phosphorylation. Psychopharmacology 2021, 238, 3335–3346. [Google Scholar] [CrossRef] [PubMed]
- Gamaleddin, I.; Guranda, M.; Goldberg, S.R.; Le Foll, B. The selective anandamide transport inhibitor VDM11 attenuates reinstatement of nicotine seeking behaviour, but does not affect nicotine intake. Br. J. Pharmacol. 2011, 164, 1652–1660. [Google Scholar] [CrossRef] [Green Version]
- Gamaleddin, I.; Guranda, M.; Scherma, M.; Fratta, W.; Makriyannis, A.; Vadivel, S.K.; Goldberg, S.R.; Le Foll, B. AM404 attenuates reinstatement of nicotine seeking induced by nicotine-associated cues and nicotine priming but does not affect nicotine- and food-taking. J. Psychopharmacol. 2013, 27, 564–571. [Google Scholar] [CrossRef] [Green Version]
- Scherma, M.; Panlilio, L.V.; Fadda, P.; Fattore, L.; Gamaleddin, I.; Le Foll, B.; Justinová, Z.; Mikics, E.; Haller, J.; Medalie, J.; et al. Inhibition of Anandamide Hydrolysis by Cyclohexyl Carbamic Acid 3′-Carbamoyl-3-yl Ester (URB597) Reverses Abuse-Related Behavioral and Neurochemical Effects of Nicotine in Rats. J. Pharmacol. Exp. Ther. 2008, 327, 482–490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Justinova, Z.; Panlilio, L.V.; Moreno-Sanz, G.; Redhi, G.H.; Auber, A.; E Secci, M.; Mascia, P.; Bandiera, T.; Armirotti, A.; Bertorelli, R.; et al. Effects of Fatty Acid Amide Hydrolase (FAAH) Inhibitors in Non-Human Primate Models of Nicotine Reward and Relapse. Neuropsychopharmacology 2015, 40, 2185–2197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mascia, P.; Pistis, M.; Justinova, Z.; Panlilio, L.V.; Luchicchi, A.; Lecca, S.; Scherma, M.; Fratta, W.; Fadda, P.; Barnes, C.; et al. Blockade of Nicotine Reward and Reinstatement by Activation of Alpha-Type Peroxisome Proliferator-Activated Receptors. Biol. Psychiatry 2011, 69, 633–641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pavon, F.J.; Serrano, A.; Sidhpura, N.; Polis, I.; Stouffer, D.; De Fonseca, F.R.; Cravatt, B.F.; Martin-Fardon, R.; Parsons, L.H. Fatty acid amide hydrolase (FAAH) inactivation confers enhanced sensitivity to nicotine-induced dopamine release in the mouse nucleus accumbens. Addict. Biol. 2017, 23, 723–734. [Google Scholar] [CrossRef] [PubMed]
- Scherma, M.; Justinová, Z.; Zanettini, C.; Panlilio, L.V.; Mascia, P.; Fadda, P.; Fratta, W.; Makriyannis, A.; Vadivel, S.K.; Gamaleddin, I.; et al. The anandamide transport inhibitor AM404 reduces the rewarding effects of nicotine and nicotine-induced dopamine elevations in the nucleus accumbens shell in rats. Br. J. Pharmacol. 2011, 165, 2539–2548. [Google Scholar] [CrossRef] [Green Version]
- Luchicchi, A.; Lecca, S.; Carta, S.; Pillolla, G.; Muntoni, A.L.; Yasar, S.; Goldberg, S.R.; Pistis, M. Effects of fatty acid amide hydrolase inhibition on neuronal responses to nicotine, cocaine and morphine in the nucleus accumbens shell and ventral tegmental area: Involvement of PPAR-α nuclear receptors. Addict. Biol. 2010, 15, 277–288. [Google Scholar] [CrossRef] [Green Version]
- Trigo, J.M.; Le Foll, B. Inhibition of monoacylglycerol lipase (MAGL) enhances cue-induced reinstatement of nicotine-seeking behavior in mice. Psychopharmacology 2016, 233, 1815–1822. [Google Scholar] [CrossRef] [PubMed]
- Muldoon, P.P.; Akinola, L.S.; Schlosburg, J.E.; Lichtman, A.H.; Sim-Selley, L.J.; Mahadevan, A.; Cravatt, B.F.; Damaj, M.I. Inhibition of monoacylglycerol lipase reduces nicotine reward in the conditioned place preference test in male mice. Neuropharmacology 2020, 176, 108170. [Google Scholar] [CrossRef] [PubMed]
- Buczynski, M.W.; Herman, M.A.; Hsu, K.-L.; Natividad, L.A.; Irimia, C.; Polis, I.Y.; Pugh, H.; Chang, J.W.; Niphakis, M.J.; Cravatt, B.F.; et al. Diacylglycerol lipase disinhibits VTA dopamine neurons during chronic nicotine exposure. Proc. Natl. Acad. Sci. USA 2016, 113, 1086–1091. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jackson, K.; Muldoon, P.; De Biasi, M.; Damaj, M. New mechanisms and perspectives in nicotine withdrawal. Neuropharmacology 2014, 96, 223–234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Damaj, M.I.; Kao, W.; Martin, B.R. Characterization of Spontaneous and Precipitated Nicotine Withdrawal in the Mouse. J. Pharmacol. Exp. Ther. 2003, 307, 526–534. [Google Scholar] [CrossRef] [Green Version]
- Berrendero, F.; Mendizabal, V.; Robledo, P.; Galeote, L.; Bilkei-Gorzó, A.; Zimmer, A.; Maldonado, R. Nicotine-Induced Antinociception, Rewarding Effects, and Physical Dependence Are Decreased in Mice Lacking the Preproenkephalin Gene. J. Neurosci. 2005, 25, 1103–1112. [Google Scholar] [CrossRef] [Green Version]
- Balerio, G.N.; Aso, E.; Berrendero, F.; Murtra, P.; Maldonado, R. Delta9-tetrahydrocannabinol decreases somatic and motivational manifestations of nicotine withdrawal in mice. Eur. J. Neurosci. 2004, 20, 2737–2748. [Google Scholar] [CrossRef] [PubMed]
- Saravia, R.; Flores, A.; Plaza-Zabala, A.; Busquets-Garcia, A.; Pastor, A.; de la Torre, R.; Di Marzo, V.; Marsicano, G.; Ozaita, A.; Maldonado, R.; et al. CB 1 Cannabinoid Receptors Mediate Cognitive Deficits and Structural Plasticity Changes During Nicotine Withdrawal. Biol. Psychiatry 2017, 81, 625–634. [Google Scholar] [CrossRef] [Green Version]
- Cippitelli, A.; Astarita, G.; Duranti, A.; Caprioli, G.; Ubaldi, M.; Stopponi, S.; Kallupi, M.; Sagratini, G.; de Fonseca, F.R.; Piomelli, D.; et al. Endocannabinoid Regulation of Acute and Protracted Nicotine Withdrawal: Effect of FAAH Inhibition. PLoS ONE 2011, 6, e28142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hughes, J.R. Effects of abstinence from tobacco: Etiology, animal models, epidemiology, and significance: A subjective review. Nicotine Tob. Res. 2007, 9, 329–339. [Google Scholar] [CrossRef] [PubMed]
- Rosen, R.L.; Borges, A.M.; Kibbey, M.M.; Steinberg, M.L.; Leyro, T.M.; Farris, S.G. Distress intolerance and withdrawal severity among daily smokers: The role of smoking abstinence expectancies. Addict. Behav. 2019, 99, 106048. [Google Scholar] [CrossRef] [PubMed]
- Zhao-Shea, R.; DeGroot, S.; Liu, L.; Vallaster, M.; Pang, X.; Su, Q.; Gao, G.; Rando, O.; Martin, G.; George, O.; et al. Increased CRF signalling in a ventral tegmental area-interpeduncular nucleus-medial habenula circuit induces anxiety during nicotine withdrawal. Nat. Commun. 2015, 6, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simonnet, A.; Zamberletti, E.; Cador, M.; Rubino, T.; Caillé, S. Chronic FAAH inhibition during nicotine abstinence alters habenular CB1 receptor activity and precipitates depressive-like behaviors. Neuropharmacology 2017, 113, 252–259. [Google Scholar] [CrossRef]
- Jacobsen, L.K.; Mencl, W.E.; Constable, R.T.; Westerveld, M.; Pugh, K.R. Impact of smoking abstinence on working memory neurocircuitry in adolescent daily tobacco smokers. Psychopharmacology 2007, 193, 557–566. [Google Scholar] [CrossRef] [PubMed]
- Loughead, J.; Wileyto, E.P.; Ruparel, K.; Falcone, M.; Hopson, R.; Gur, R.; Lerman, C. Working Memory-Related Neural Activity Predicts Future Smoking Relapse. Neuropsychopharmacology 2014, 40, 1311–1320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Merritt, P.S.; Cobb, A.R.; Cook, G.I. Sex differences in the cognitive effects of tobacco abstinence: A pilot study. Exp. Clin. Psychopharmacol. 2012, 20, 258–263. [Google Scholar] [CrossRef]
- Patterson, F.; Jepson, C.; Loughead, J.; Perkins, K.; Strasser, A.A.; Siegel, S.; Frey, J.; Gur, R.; Lerman, C. Working memory deficits predict short-term smoking resumption following brief abstinence. Drug Alcohol Depend. 2010, 106, 61–64. [Google Scholar] [CrossRef] [Green Version]
- Gould, T.J.; Leach, P.T. Cellular, molecular, and genetic substrates underlying the impact of nicotine on learning. Neurobiol. Learn. Mem. 2013, 107, 108–132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leach, P.T.; Cordero, K.A.; Gould, T.J. The effects of acute nicotine, chronic nicotine, and withdrawal from chronic nicotine on performance of a cued appetitive response. Behav. Neurosci. 2013, 127, 303–310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parikh, V.; Cole, R.; Patel, J.; Poole, R.L.; Gould, T.J. Cognitive control deficits during mecamylamine-precipitated withdrawal in mice: Possible links to frontostriatal BDNF imbalance. Neurobiol. Learn. Mem. 2016, 128, 110–116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marsicano, G.; Lafenêtre, P. Roles of the Endocannabinoid System in Learning and Memory. In Behavioral Neurobiology of the Endocannabinoid System; Springer: Berlin/Heidelberg, Germany, 2009; pp. 201–230. [Google Scholar] [CrossRef]
- Mechoulam, R.; Parker, L.A. The Endocannabinoid System and the Brain. Annu. Rev. Psychol. 2013, 64, 21–47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evans, D.E.; Sutton, S.; Jentink, K.G.; Lin, H.-Y.; Park, J.Y.; Drobes, D.J. Cannabinoid receptor 1 (CNR1) gene variant moderates neural index of cognitive disruption during nicotine withdrawal. Genes Brain Behav. 2016, 15, 621–626. [Google Scholar] [CrossRef] [Green Version]
- Morgan, C.J.; Das, R.K.; Joye, A.; Curran, H.V.; Kamboj, S. Cannabidiol reduces cigarette consumption in tobacco smokers: Preliminary findings. Addict. Behav. 2013, 38, 2433–2436. [Google Scholar] [CrossRef] [PubMed]
- Hindocha, C.; Freeman, T.P.; Grabski, M.; Stroud, J.B.; Crudgington, H.; Davies, A.C.; Das, R.K.; Lawn, W.; Morgan, C.J.A.; Curran, H.V. Cannabidiol reverses attentional bias to cigarette cues in a human experimental model of tobacco withdrawal. Addiction 2018, 113, 1696–1705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hindocha, C.; Freeman, T.; Grabski, M.; Crudgington, H.; Davies, A.C.; Stroud, J.B.; Das, R.K.; Lawn, W.; Morgan, C.J.A.; Curran, H.V. The effects of cannabidiol on impulsivity and memory during abstinence in cigarette dependent smokers. Sci. Rep. 2018, 8, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Zuardi, A.W.; Rodrigues, N.P.; Silva, A.L.; Bernardo, S.A.; Hallak, J.E.C.; Guimarães, F.S.; Crippa, J.A.S. Inverted U-Shaped Dose-Response Curve of the Anxiolytic Effect of Cannabidiol during Public Speaking in Real Life. Front. Pharmacol. 2017, 8, 259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peres, F.F.; Levin, R.; Suiama, M.A.; Diana, M.C.; Gouvêa, D.A.; de Almeida, V.; Santos, C.M.; Lungato, L.; Zuardi, A.W.; Hallak, J.E.C.; et al. Cannabidiol Prevents Motor and Cognitive Impairments Induced by Reserpine in Rats. Front. Pharmacol. 2016, 7, 343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saravia, R.; Ten-Blanco, M.; Grande, M.T.; Maldonado, R.; Berrendero, F. Anti-inflammatory agents for smoking cessation? Focus on cognitive deficits associated with nicotine withdrawal in male mice. Brain Behav. Immun. 2019, 75, 228–239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, M.; Adeluyi, A.; Anderson, E.L.; Turner, J.R. Glial cells as therapeutic targets for smoking cessation. Neuropharmacology 2020, 175, 108157. [Google Scholar] [CrossRef] [PubMed]
- Adeluyi, A.; Guerin, L.; Fisher, M.L.; Galloway, A.; Cole, R.D.; Chan, S.S.L.; Wyatt, M.D.; Davis, S.W.; Freeman, L.R.; Ortinski, P.I.; et al. Microglia morphology and proinflammatory signaling in the nucleus accumbens during nicotine withdrawal. Sci. Adv. 2019, 5, eaax7031. [Google Scholar] [CrossRef] [Green Version]
- Smith, L.C.; Tieu, L.; Suhandynata, R.T.; Boomhower, B.; Hoffman, M.; Sepulveda, Y.; Carrette, L.L.G.; Momper, J.D.; Fitzgerald, R.L.; Hanham, K.; et al. Cannabidiol reduces withdrawal symptoms in nicotine-dependent rats. Psychopharmacology 2021, 238, 2201–2211. [Google Scholar] [CrossRef] [PubMed]
- Bolognini, D.; Costa, B.; Maione, S.; Comelli, F.; Marini, P.; Di Marzo, V.; Parolaro, D.; A Ross, R.; A Gauson, L.; Cascio, M.G.; et al. The plant cannabinoid Δ9-tetrahydrocannabivarin can decrease signs of inflammation and inflammatory pain in mice. Br. J. Pharmacol. 2010, 160, 677–687. [Google Scholar] [CrossRef] [Green Version]
- McPartland, J.M.; Duncan, M.; Di Marzo, V.; Pertwee, R. Are cannabidiol and Δ9-tetrahydrocannabivarin negative modulators of the endocannabinoid system? A systematic review. Br. J. Pharmacol. 2015, 172, 737–753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xi, Z.; Muldoon, P.; Wang, X.; Bi, G.; Damaj, M.I.; Lichtman, A.H.; Pertwee, R.G.; Gardner, E.L. Δ 8 -Tetrahydrocannabivarin has potent anti-nicotine effects in several rodent models of nicotine dependence. Br. J. Pharmacol. 2019, 176, 4773–4784. [Google Scholar] [CrossRef] [PubMed]
- Cahill, K.; Ussher, M.H. Cannabinoid type 1 receptor antagonists for smoking cessation. Cochrane Database Syst. Rev. 2011, 2011, CD005353. [Google Scholar] [CrossRef] [PubMed]
- Biała, G.; Budzynska, B. Rimonabant attenuates sensitization, cross-sensitization and cross-reinstatement of place preference induced by nicotine and ethanol. Pharmacol. Rep. 2010, 62, 797–807. [Google Scholar] [CrossRef]
- Cohen, C.; Perrault, G.; Griebel, G.; Soubrié, P. Nicotine-Associated Cues Maintain Nicotine-Seeking Behavior in Rats Several Weeks after Nicotine Withdrawal: Reversal by the Cannabinoid (CB1) Receptor Antagonist, Rimonabant (SR141716). Neuropsychopharmacology 2004, 30, 145–155. [Google Scholar] [CrossRef] [Green Version]
- Biala, G.; Budzynska, B.; Staniak, N. Effects of rimonabant on the reinstatement of nicotine-conditioned place preference by drug priming in rats. Behav. Brain Res. 2009, 202, 260–265. [Google Scholar] [CrossRef]
- De Bruin, N.; Lange, J.; Kruse, C.; Herremans, A.; Schoffelmeer, A.; van Drimmelen, M.; De Vries, T. SLV330, a cannabinoid CB1 receptor antagonist, attenuates ethanol and nicotine seeking and improves inhibitory response control in rats. Behav. Brain Res. 2010, 217, 408–415. [Google Scholar] [CrossRef]
- Agrawal, A.; Madden, P.A.; Bucholz, K.K.; Heath, A.C.; Lynskey, M.T. Transitions to regular smoking and to nicotine dependence in women using cannabis. Drug Alcohol Depend. 2008, 95, 107–114. [Google Scholar] [CrossRef] [Green Version]
- Valjent, E.; Mitchell, J.M.; Besson, M.-J.; Caboche, J.; Maldonado, R. Behavioural and biochemical evidence for interactions between Δ9-tetrahydrocannabinol and nicotine. Br. J. Pharmacol. 2002, 135, 564–578. [Google Scholar] [CrossRef] [Green Version]
- Patton, G.C.; Coffey, C.; Carlin, J.B.; Sawyer, S.M.; Lynskey, M. Reverse gateways? Frequent cannabis use as a predictor of tobacco initiation and nicotine dependence. Addiction 2005, 100, 1518–1525. [Google Scholar] [CrossRef]
- Panlilio, L.V.; Zanettini, C.; Barnes, C.; Solinas, M.; Goldberg, S.R. Prior Exposure to THC Increases the Addictive Effects of Nicotine in Rats. Neuropsychopharmacology 2013, 38, 1198–1208. [Google Scholar] [CrossRef] [Green Version]
- Flores, A.; Maldonado, R.; Berrendero, F. THC exposure during adolescence does not modify nicotine reinforcing effects and relapse in adult male mice. Psychopharmacology 2020, 237, 801–809. [Google Scholar] [CrossRef] [PubMed]
- Dukes, A.J.; Fowler, J.P.; Lallai, V.; Pushkin, A.N.; Fowler, C.D. Adolescent Cannabinoid and Nicotine Exposure Differentially Alters Adult Nicotine Self-Administration in Males and Females. Nicotine Tob. Res. 2020, 22, 1364–1373. [Google Scholar] [CrossRef] [PubMed]
- Hempel, B.J.; Melkumyan, M.; Crissman, M.E.; Winston, C.A.; Madar, J.; Riley, A.L. Pre-conception exposure to THC fails to impact nicotine reward in adult offspring. Pharmacol. Biochem. Behav. 2020, 197, 173001. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
| Compound | Mechanism of Action | Effect on Nicotine Reward | Animal |
|---|---|---|---|
| Rimonabant | CB1R antagonist/inverse agonist | ↓ self-administration | Rats |
| ↓ the break point of a progressive ratio schedule in self-administration | Rats | ||
| ↓ conditioned place preference (short term) | Rats, mice | ||
| AM4113 | CB1R neutral antagonist | ↓ the break point of a progressive ratio schedule in self-administration | Rats |
| AM251 | CB1R antagonist/inverse agonist | ↓ self-administration | Rats |
| ↓ conditioned place preference | Rats | ||
| AM630 | CB2R antagonist/inverse agonist | ≈ self-administration | Rats |
| ↓ self-administration | Mice | ||
| ↓ conditioned place preference | Mice | ||
| SR144528 | CB2R antagonist/inverse agonist | ↓ conditioned place preference | Mice |
| JWH133 | CB2R agonist | ↓ conditioned place preference | Mice |
| AM1241 | CB2R agonist | ≈ self-administration | Rats |
| β-Caryophyllene | CB2R agonist | ↓ dose-dependently self-administration | Rats, mice |
| URB597 | FAAH inhibitor | ≈ self-administration | Rats |
| ↓ conditioned place preference | Rats | ||
| ↓ acquisition of self-administration | Rats | ||
| ↓ nicotine-induced dopamine increase | Rats | ||
| ↓ nicotine reward | Squirrel Monkeys | ||
| URB694 | FAAH inhibitor | ↓ nicotine reward | Squirrel Monkeys |
| VDM11 | AEA transport inhibitor | ≈ self-administration | Rats |
| AM404 | AEA transport inhibitor | ≈ self-administration | Rats |
| JZL184 | MAGL inhibitor | ≈ self-administration | Mice |
| ↓ conditioned place-preference | Mice | ||
| 1,2,3-triazole ureas | DAGL inhibitors | ↓ self-administration | Rats |
| Compound | Mechanism of Action | Effect on Nicotine Wihdrawal | Animal |
|---|---|---|---|
| Rimonabant | CB1R antagonist/inverse agonist | ≈ physical signs of withdrawal | Mice |
| ↓ abstinence-induced cognitive impairments | Mice | ||
| URB597 | FAAH inhibitor | ↑ physical signs of withdrawal | Mice |
| ≈ physical signs of withdrawal | Rats | ||
| ↓ abstinence-induced anxiety | Rats | ||
| ↑ abstinence-induced anhedonia | Rats | ||
| ↑ mild stressor-induced plasmatic corticosterone levels during nicotine withdrawal | Rats | ||
| JZL184 | MAGL inhibitor | ↓ physical signs of withdrawal | Mice |
| ≈ abstinence-induced cognitive impairments | Mice | ||
| O7460 | DAGL inhibitor | ↑ physical signs of withdrawal | Mice |
| ↓ abstinence-induced cognitive impairments | Mice | ||
| Cannabidiol | Multiple targets | ↓ abstinence-induced cognitive impairments | Mice |
| ↓ microglia activation | Mice | ||
| ↓ physical signs of withdrawal | Rats | ||
| ↓ abstinence-induced hyperalgesia | Rats |
| Compound | Mechanism of Action | Effect on Nicotine Relapse | Animal |
|---|---|---|---|
| Rimonabant | CB1R antagonist/inverse agonist | ↓ cue-induced self-administration increased by WIN55,212-2 | Rats |
| ↓ priming- and cue-induced self-administration | Squirrel Monkeys | ||
| SLV330 | CB1R antagonist | ↓ cue-induced self-administration | Rats |
| AM4113 | CB1R neutral antagonist | ↓ priming- and cue-induced self-administration | Squirrel Monkeys |
| ↓ priming-, cue-, and stress-induced self-administration | Rats | ||
| Δ8-THCV | CB1R antagonist + CB2R agonist | ↓ priming- and cue-induced self-administration | Rats |
| WIN55,212-2 | CB1R/CB2R agonist | ↑ cue-induced self-administration | Rats |
| AM630 | CB2R antagonist/inverse agonist | ≈ cue-induced self-administration increased by WIN55,212-2 | Rats |
| ≈ priming- and cue-induced self-administration | Rats | ||
| AM1241 | CB2R agonist | ≈ priming- and cue-induced self-administration | Rats |
| URB597 | FAAH inhibitor | ↓ priming- and cue-induced self-administration | Squirrel Monkeys |
| URB694 | FAAH inhibitor | ↓ priming- and cue-induced self-administration | Squirrel Monkeys |
| VDM11 | AEA transport inhibitor | ↓ priming- and cue-induced self-administration | Rats |
| AM404 | AEA transport inhibitor | ↓ priming- and cue-induced self-administration | Rats |
| JZL184 | MAGL inhibitor | ↑ cue-induced self-administration | Mice |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Saravia, R.; Ten-Blanco, M.; Pereda-Pérez, I.; Berrendero, F. New Insights in the Involvement of the Endocannabinoid System and Natural Cannabinoids in Nicotine Dependence. Int. J. Mol. Sci. 2021, 22, 13316. https://doi.org/10.3390/ijms222413316
Saravia R, Ten-Blanco M, Pereda-Pérez I, Berrendero F. New Insights in the Involvement of the Endocannabinoid System and Natural Cannabinoids in Nicotine Dependence. International Journal of Molecular Sciences. 2021; 22(24):13316. https://doi.org/10.3390/ijms222413316
Chicago/Turabian StyleSaravia, Rocio, Marc Ten-Blanco, Inmaculada Pereda-Pérez, and Fernando Berrendero. 2021. "New Insights in the Involvement of the Endocannabinoid System and Natural Cannabinoids in Nicotine Dependence" International Journal of Molecular Sciences 22, no. 24: 13316. https://doi.org/10.3390/ijms222413316