
Impairments of Long-Term Synaptic Plasticity in the Hippocampus of Young Rats during the Latent Phase of the Lithium-Pilocarpine Model of Temporal Lobe Epilepsy

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
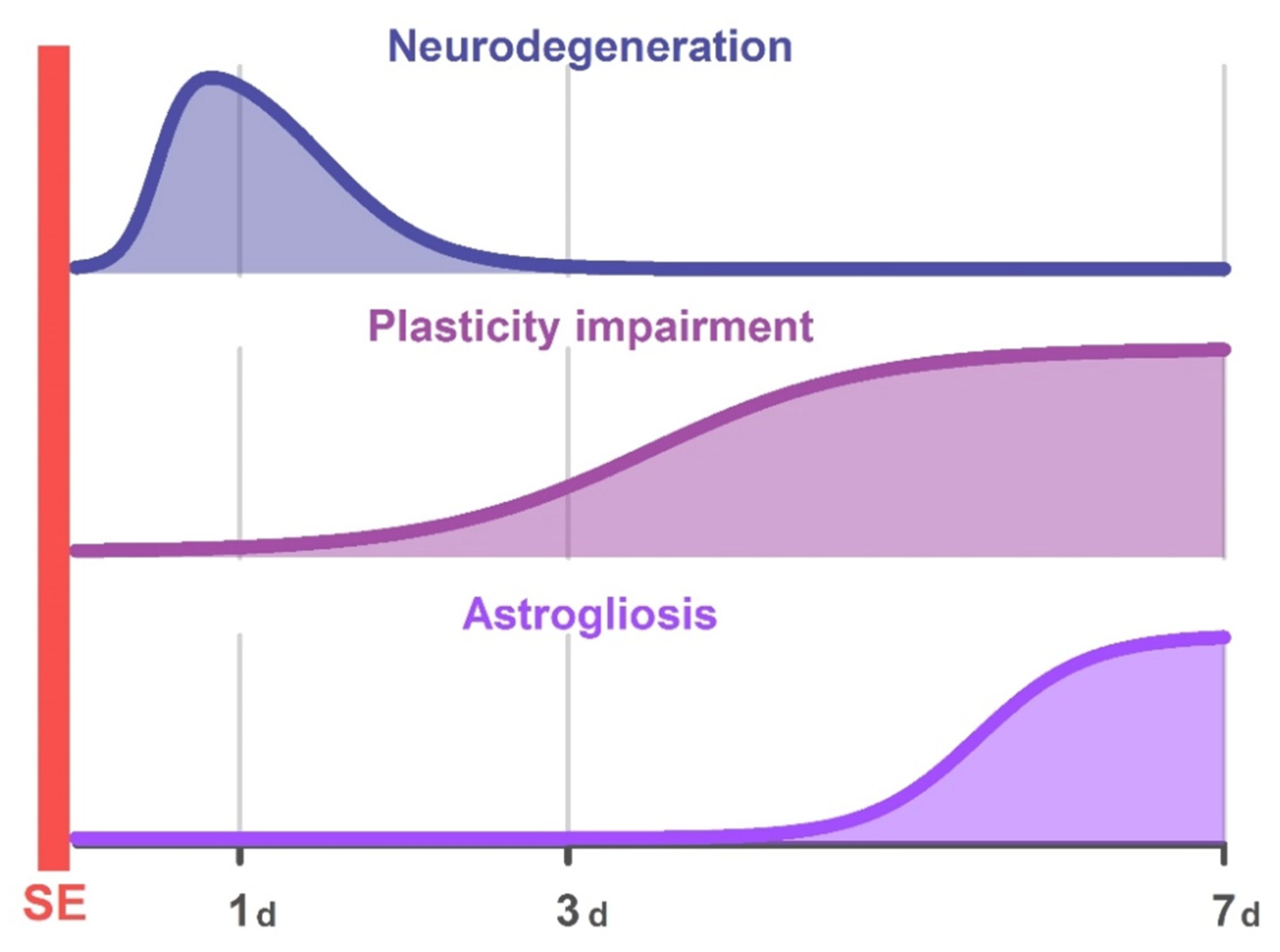
2. Results
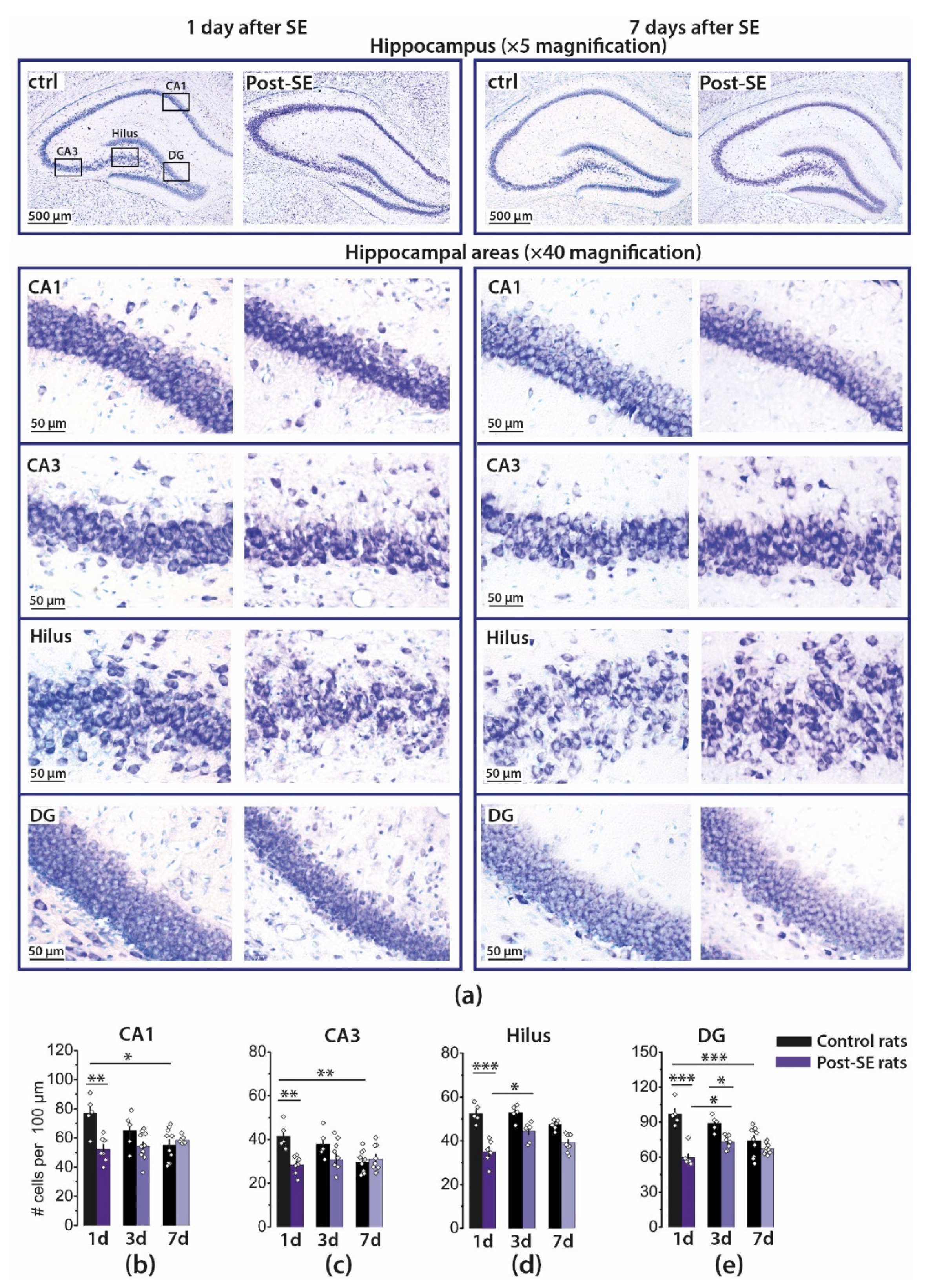
2.1. Pilocarpine-Induced SE Provokes Neuronal Loss in the Hippocampus of Young Rats
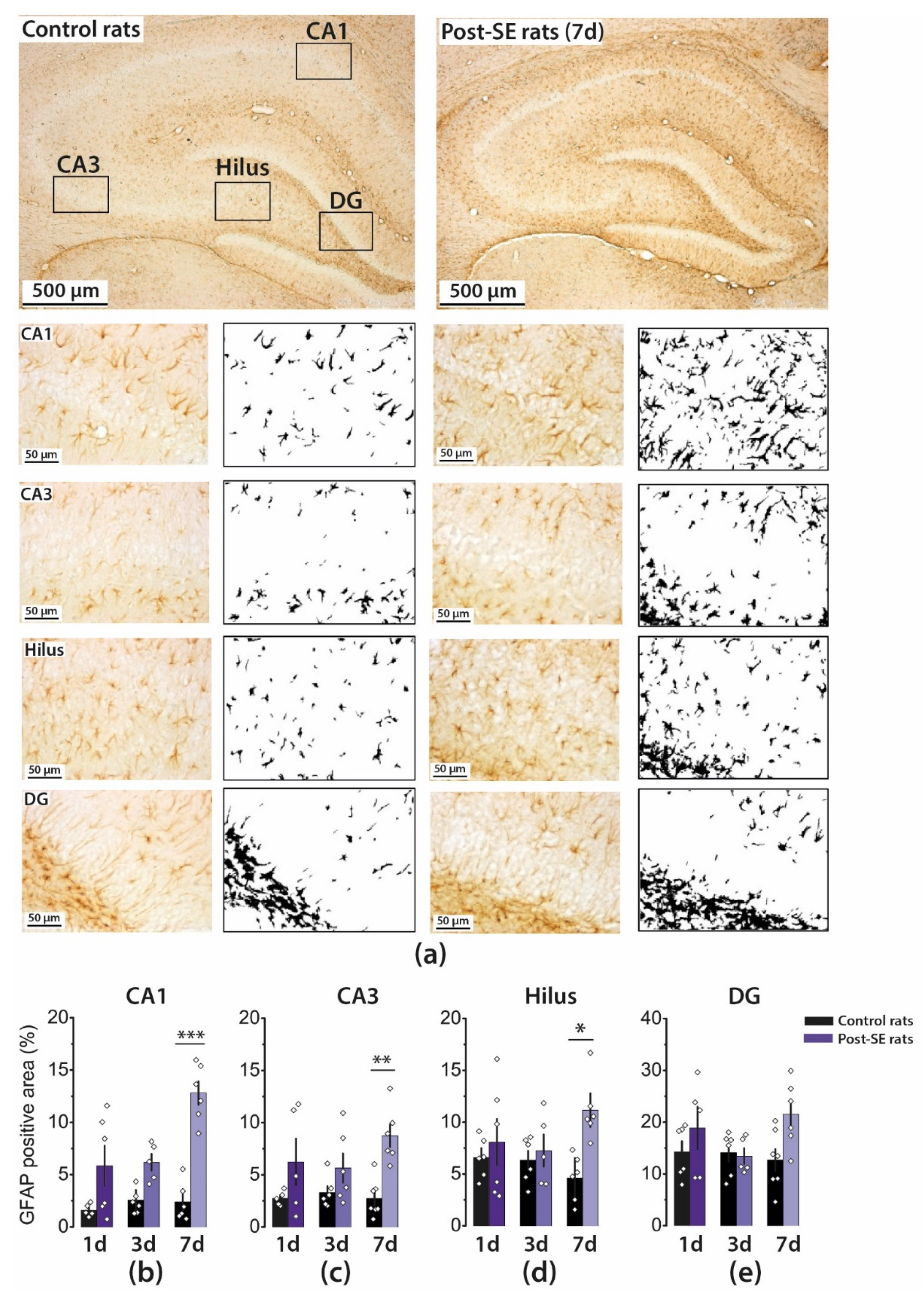
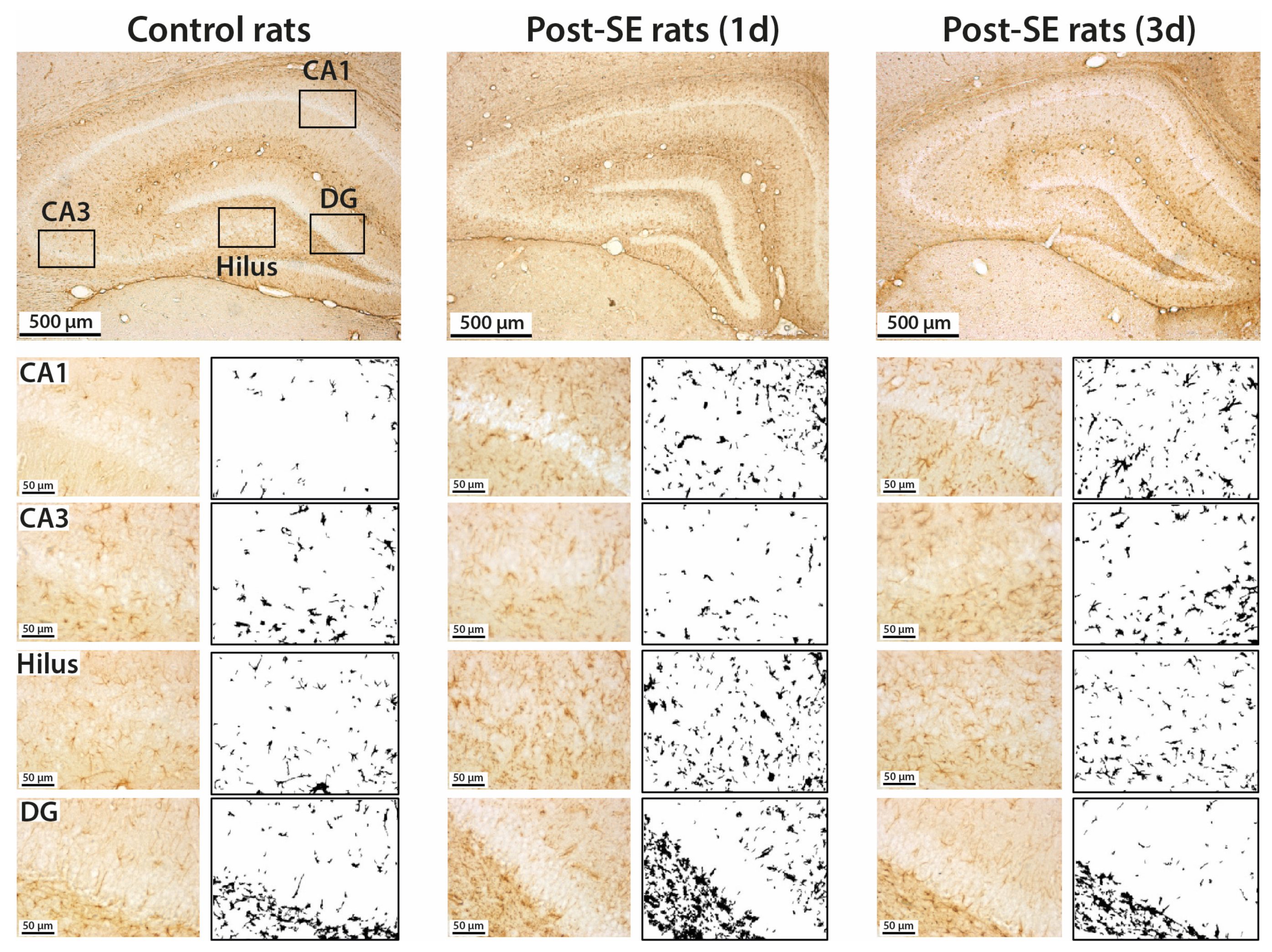
2.2. Astrogliosis in the Rat Hippocampus Develops by the End of the First Week after SE
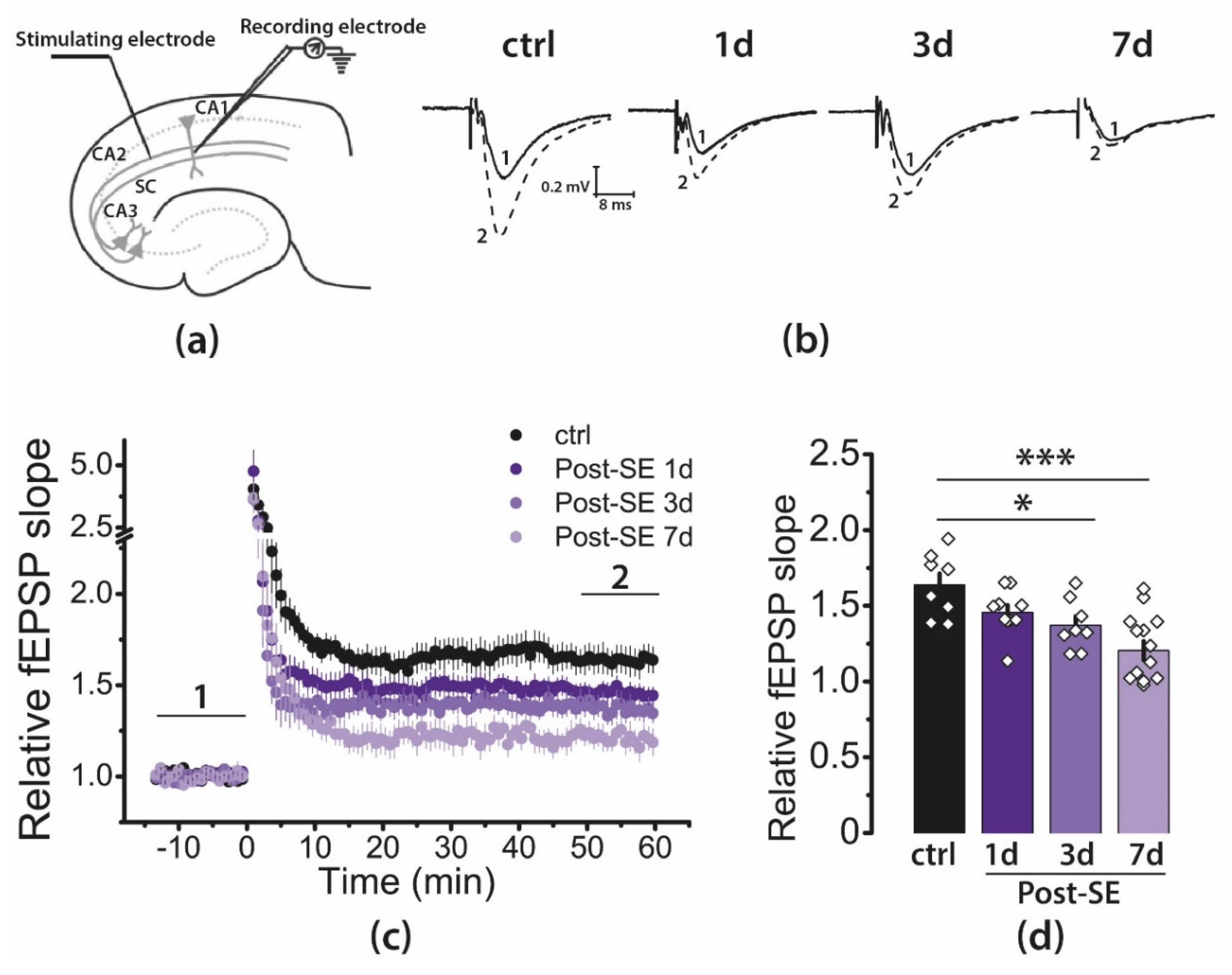
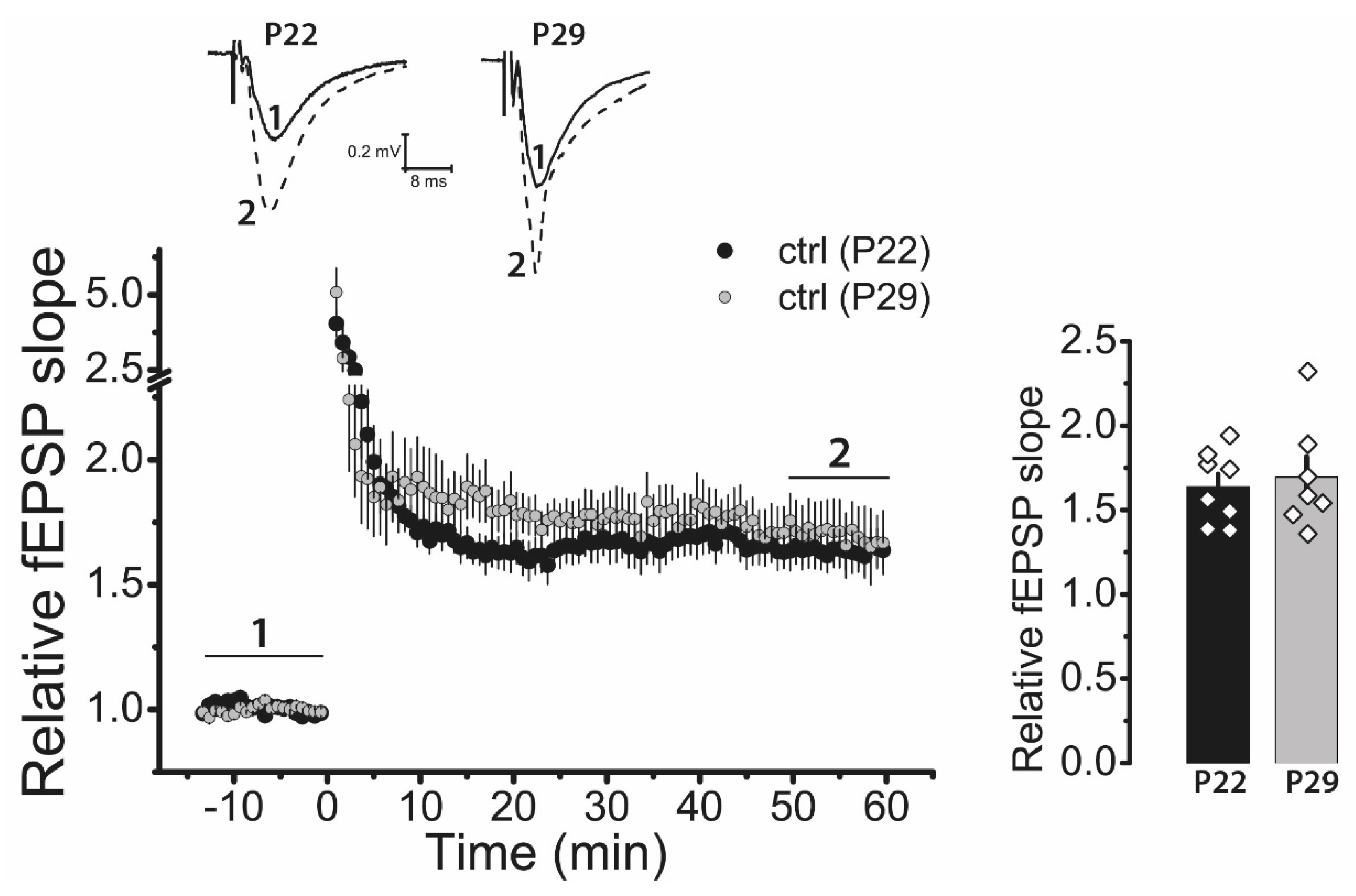
2.3. LTP Is Attenuated in the CA1 Hippocampal Area in Post-SE Rats
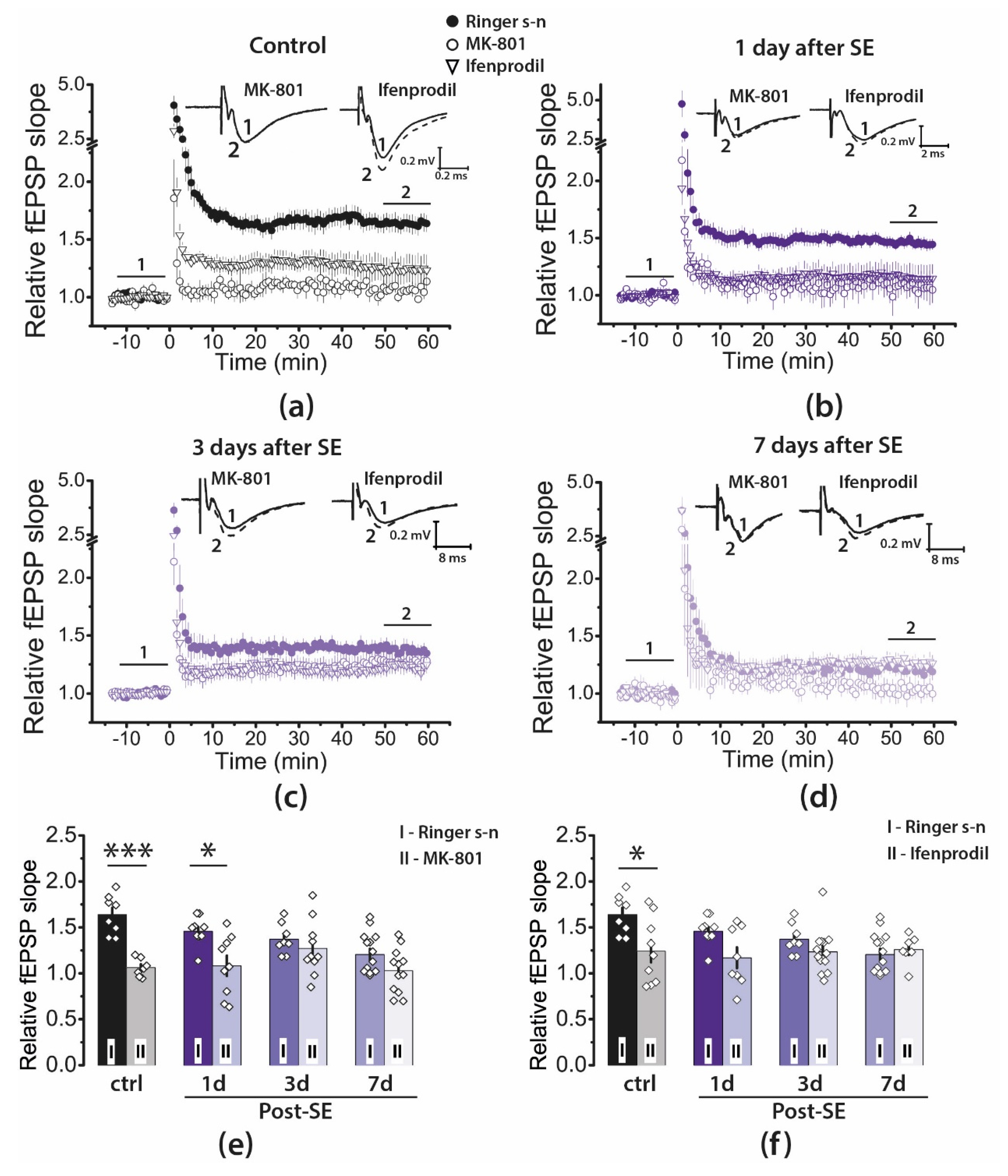
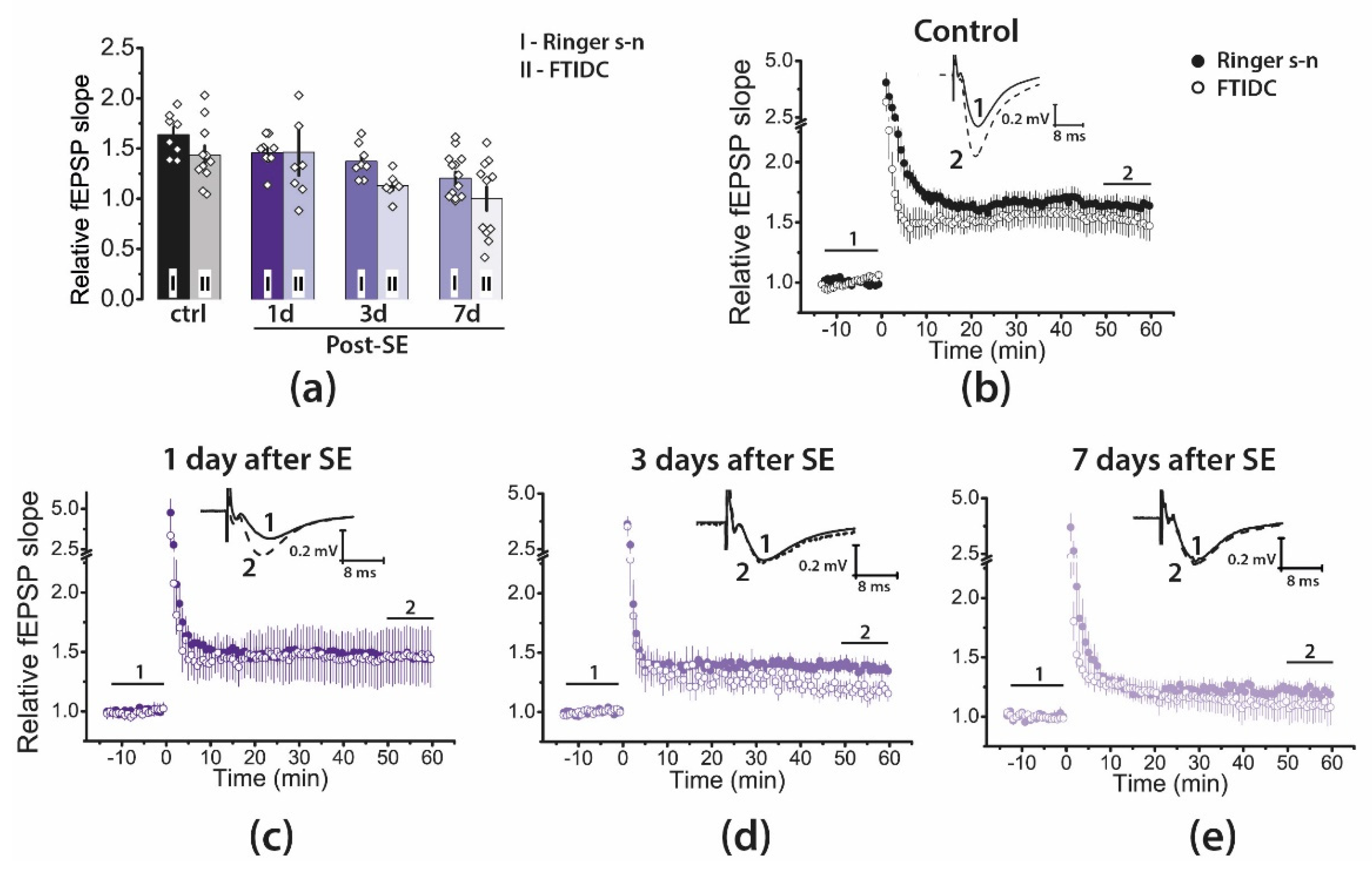
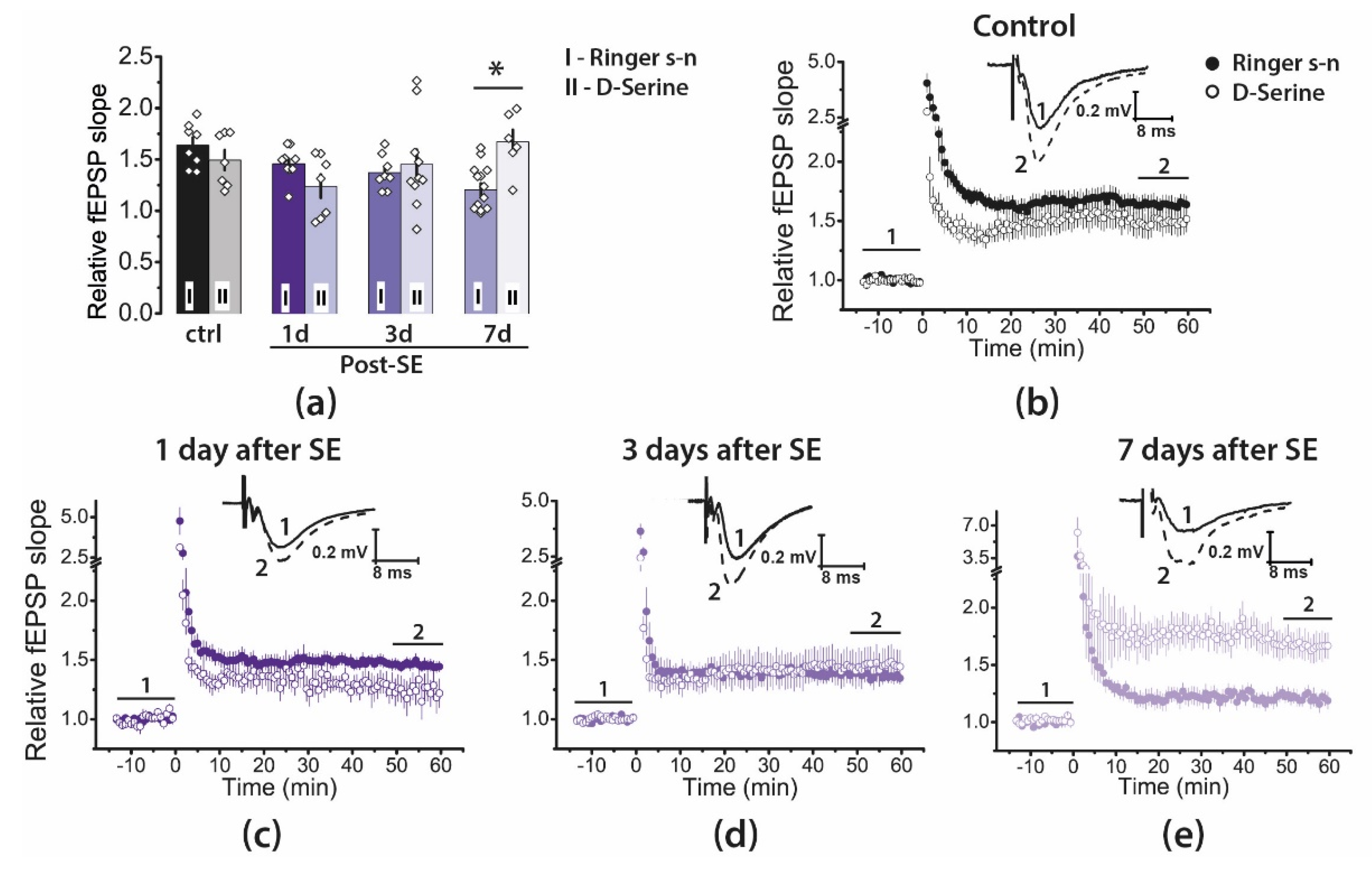
2.4. Pharmacological Properties of LTP Changed during the Latent Period of the Lithium–Pilocarpine Model in Young Rats
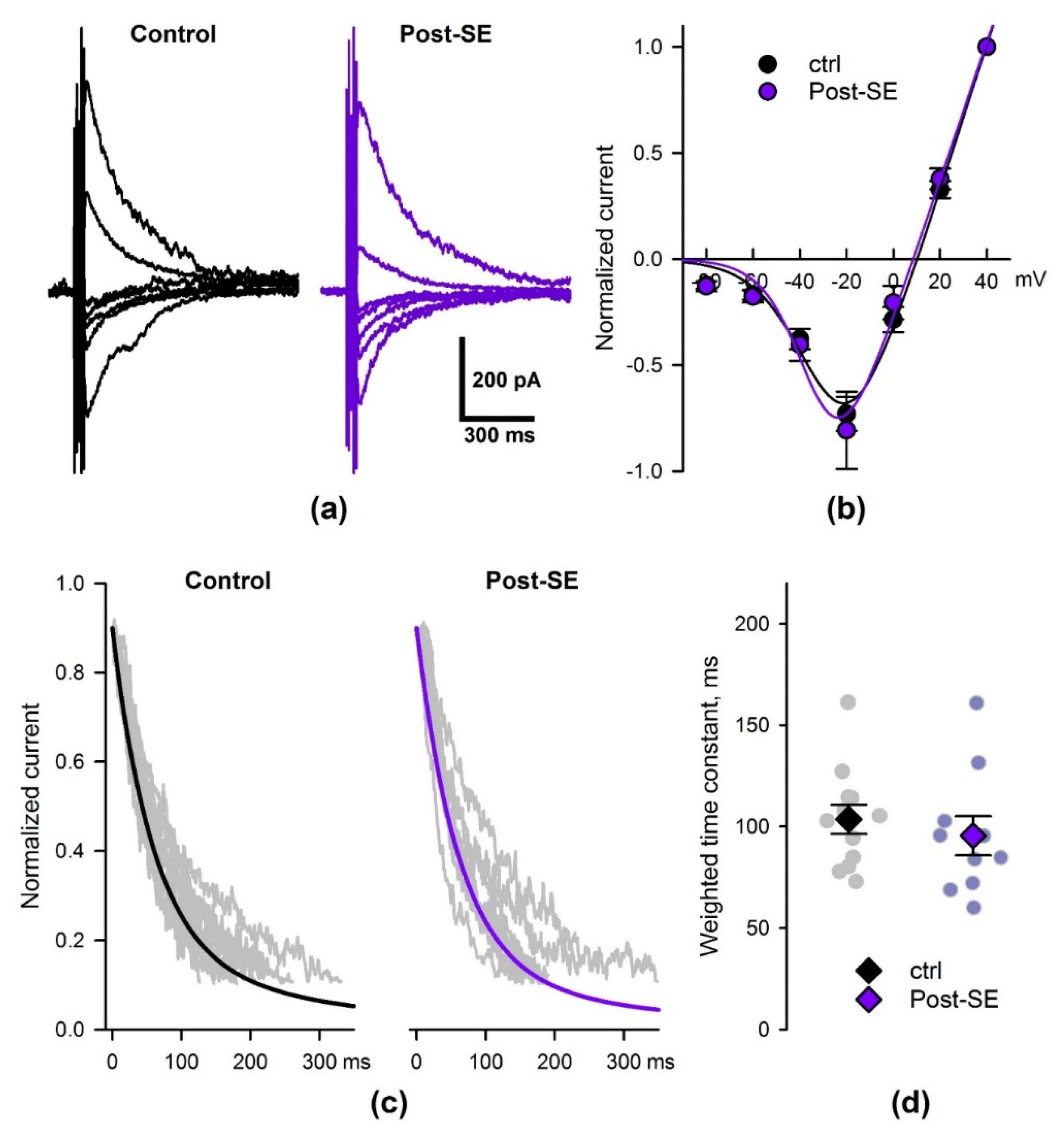
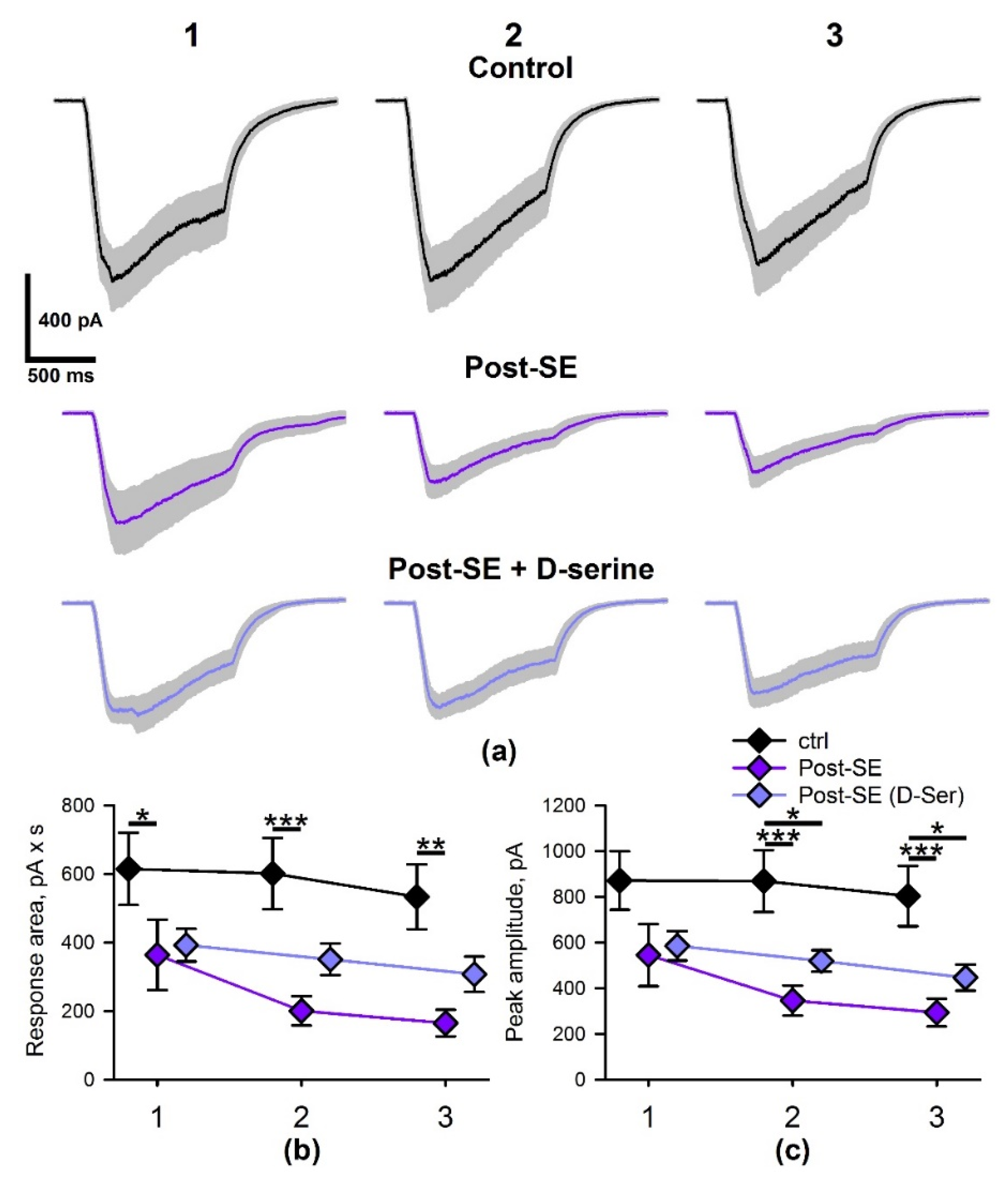
2.5. SE Affects the Properties of NMDAR-Mediated Synaptic Currents
3. Discussion
4. Materials and Methods
4.1. Animals and the Lithium–Pilocarpine Model of Temporal Lobe Epilepsy
4.2. Hippocampal Slice Preparation
4.3. Field Potential Recordings and LTP Induction
4.4. The Whole-Cell Patch-Clamp Recordings
4.5. Histology
4.5.1. Tissue Preparation
4.5.2. Nissl Staining
4.5.3. Immunohistochemistry
4.6. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Appendix A


References
- Chin, J.H.; Vora, N. The global burden of neurologic diseases. Neurology 2014, 83, 349–351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shah, P.; Bassett, D.S.; Wisse, L.E.M.; Detre, J.A.; Stein, J.M.; Yushkevich, P.A.; Shinohara, R.T.; Pluta, J.B.; Valenciano, E.; Daffner, M.; et al. Mapping the structural and functional network architecture of the medial temporal lobe using 7T MRI. Hum. Brain Mapp. 2018, 39, 851–865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frankland, P.W.; Bontempi, B. The organization of recent and remote memories. Nat. Rev. Neurosci. 2005, 6, 119–130. [Google Scholar] [CrossRef] [PubMed]
- Mathern, G.W.; Adelson, P.D.; Cahan, L.D.; Leite, J.P. Hippocampal neuron damage in human epilepsy: Meyer’s hypothesis revisited. Prog. Brain Res. 2002, 135, 237–251. [Google Scholar] [CrossRef]
- Elger, C.E.; Helmstaedter, C.; Kurthen, M. Chronic epilepsy and cognition. Lancet Neurol. 2004, 3, 663–672. [Google Scholar] [CrossRef]
- Pitkänen, A.; Lukasiuk, K. Mechanisms of epileptogenesis and potential treatment targets. Lancet Neurol. 2011, 10, 173–186. [Google Scholar] [CrossRef]
- Smolensky, I.V.; Zubareva, O.E.; Kalemenev, S.V.; Lavrentyeva, V.V.; Dyomina, A.V.; Karepanov, A.A.; Zaitsev, A.V. Impairments in cognitive functions and emotional and social behaviors in a rat lithium-pilocarpine model of temporal lobe epilepsy. Behav. Brain Res. 2019, 372, 112044. [Google Scholar] [CrossRef] [PubMed]
- Reddy, D.; Kuruba, R. Experimental Models of Status Epilepticus and Neuronal Injury for Evaluation of Therapeutic Interventions. Int. J. Mol. Sci. 2013, 14, 18284–18318. [Google Scholar] [CrossRef]
- Avanzini, G.; Depaulis, A.; Tassinari, A.; de Curtis, M.; Curtis, M. de Do seizures and epileptic activity worsen epilepsy and deteriorate cognitive function? Epilepsia 2013, 54, 14–21. [Google Scholar] [CrossRef]
- Bell, B.; Lin, J.J.; Seidenberg, M.; Hermann, B. The neurobiology of cognitive disorders in temporal lobe epilepsy. Nat. Rev. Neurol. 2011, 7, 154–164. [Google Scholar] [CrossRef]
- Martin, S.J.; Grimwood, P.D.; Morris, R.G. Synaptic plasticity and memory: An evaluation of the hypothesis. Annu. Rev. Neurosci. 2000, 23, 649–711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Citri, A.; Malenka, R.C. Synaptic Plasticity: Multiple Forms, Functions, and Mechanisms. Neuropsychopharmacology 2008, 33, 18–41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bliss, T.V.P.; Collingridge, G.L. A synaptic model of memory: Long-term potentiation in the hippocampus. Nature 1993, 361, 31–39. [Google Scholar] [CrossRef] [PubMed]
- Dudek, S.M.; Bear, M.F. Homosynaptic long-term depression in area CA1 of hippocampus and effects of N-methyl-D-aspartate receptor blockade. Proc. Natl. Acad. Sci. USA 1992, 89, 4363–4367. [Google Scholar] [CrossRef] [Green Version]
- Postnikova, T.Y.; Amakhin, D.V.; Trofimova, A.M.; Smolensky, I.V.; Zaitsev, A.V. Changes in Functional Properties of Rat Hippocampal Neurons Following Pentylenetetrazole-induced Status Epilepticus. Neuroscience 2019, 399, 103–116. [Google Scholar] [CrossRef] [PubMed]
- Cruz Del Angel, Y.; Orfila, J.E.; Herson, P.S.; Brooks-Kayal, A.; González, M.I. Down-regulation of AMPA receptors and long-term potentiation during early epileptogenesis. Epilepsy Behav. 2021, 124, 108320. [Google Scholar] [CrossRef] [PubMed]
- Kryukov, K.A.; Kim, K.K.; Magazanik, L.G.; Zaitsev, A.V. Status epilepticus alters hippocampal long-term synaptic potentiation in a rat lithium-pilocarpine model. Neuroreport 2016, 27, 1191–1195. [Google Scholar] [CrossRef] [PubMed]
- Plata, A.; Lebedeva, A.; Denisov, P.; Nosova, O.; Postnikova, T.Y.; Pimashkin, A.; Brazhe, A.; Zaitsev, A.V.; Rusakov, D.A.; Semyanov, A. Astrocytic Atrophy Following Status Epilepticus Parallels Reduced Ca2+ Activity and Impaired Synaptic Plasticity in the Rat Hippocampus. Front. Mol. Neurosci. 2018, 11, 215. [Google Scholar] [CrossRef]
- Zhang, Y.; Cai, G.E.; Yang, Q.; Lu, Q.C.; Li, S.T.; Ju, G. Time-dependent changes in learning ability and induction of long-term potentiation in the lithium–pilocarpine-induced epileptic mouse model. Epilepsy Behav. 2010, 17, 448–454. [Google Scholar] [CrossRef]
- Guli, X.; Tokay, T.; Kirschstein, T.; Köhling, R. Status Epilepticus Enhances Depotentiation after Fully Established LTP in an NMDAR-Dependent but GluN2B-Independent Manner. Neural Plast. 2016, 2016, 6592038. [Google Scholar] [CrossRef] [Green Version]
- Müller, L.; Tokay, T.; Porath, K.; Köhling, R.; Kirschstein, T. Enhanced NMDA receptor-dependent LTP in the epileptic CA1 area via upregulation of NR2B. Neurobiol. Dis. 2013, 54, 183–193. [Google Scholar] [CrossRef]
- Borges, K.; Gearing, M.; McDermott, D.L.; Smith, A.B.; Almonte, A.G.; Wainer, B.H.; Dingledine, R. Neuronal and glial pathological changes during epileptogenesis in the mouse pilocarpine model. Exp. Neurol. 2003, 182, 21–34. [Google Scholar] [CrossRef]
- Holopainen, I.E. Seizures in the developing brain: Cellular and molecular mechanisms of neuronal damage, neurogenesis and cellular reorganization. Neurochem. Int. 2008, 52, 935–947. [Google Scholar] [CrossRef] [PubMed]
- Haas, K.Z.; Sperber, E.F.; Opanashuk, L.A.; Stanton, P.K.; Moshé, S.L. Resistance of immature hippocampus to morphologic and physiologic alterations following status epilepticus or kindling. Hippocampus 2001, 11, 615–625. [Google Scholar] [CrossRef] [PubMed]
- Sankar, R.; Shin, D.H.; Liu, H.; Mazarati, A.; De Vasconcelos, A.P.; Wasterlain, C.G. Patterns of status epilepticus-induced neuronal injury during development and long-term consequences. J. Neurosci. 1998, 18, 8382–8393. [Google Scholar] [CrossRef]
- Curia, G.; Longo, D.; Biagini, G.; Jones, R.S.G.; Avoli, M. The pilocarpine model of temporal lobe epilepsy. J. Neurosci. Methods 2008, 172, 143–157. [Google Scholar] [CrossRef] [PubMed]
- Lévesque, M.; Biagini, G.; de Curtis, M.; Gnatkovsky, V.; Pitsch, J.; Wang, S.; Avoli, M. The pilocarpine model of mesial temporal lobe epilepsy: Over one decade later, with more rodent species and new investigative approaches. Neurosci. Biobehav. Rev. 2021, 130, 274–291. [Google Scholar] [CrossRef] [PubMed]
- Bartolomei, F.; Khalil, M.; Wendling, F.; Sontheimer, A.; Regis, J.; Ranjeva, J.-P.; Guye, M.; Chauvel, P. Entorhinal Cortex Involvement in Human Mesial Temporal Lobe Epilepsy: An Electrophysiologic and Volumetric Study. Epilepsia 2005, 46, 677–687. [Google Scholar] [CrossRef] [Green Version]
- Mathern, G.W.; Kuhlman, P.A.; Mendoza, D.; Pretorius, J.K. Human fascia dentata anatomy and hippocampal neuron densities differ depending on the epileptic syndrome and age at first seizure. J. Neuropathol. Exp. Neurol. 1997, 56, 199–212. [Google Scholar] [CrossRef] [Green Version]
- Nairismagi, J.; Pitkanen, A.; Kettunen, M.I.; Kauppinen, R.A.; Kubova, H. Status Epilepticus in 12-day-old Rats Leads to Temporal Lobe Neurodegeneration and Volume Reduction: A Histologic and MRI Study. Epilepsia 2006, 47, 479–488. [Google Scholar] [CrossRef] [PubMed]
- Ben-Ari, Y.; Holmes, G.L. Effects of seizures on developmental processes in the immature brain. Lancet Neurol. 2006, 5, 1055–1063. [Google Scholar] [CrossRef]
- Verhoog, Q.P.; Holtman, L.; Aronica, E.; van Vliet, E.A. Astrocytes as Guardians of Neuronal Excitability: Mechanisms Underlying Epileptogenesis. Front. Neurol. 2020, 11, 1541. [Google Scholar] [CrossRef]
- Shapiro, L.A.; Wang, L.; Ribak, C.E. Rapid astrocyte and microglial activation following pilocarpine-induced seizures in rats. Epilepsia 2008, 49, 33–41. [Google Scholar] [CrossRef] [Green Version]
- Vizuete, A.F.K.; Hennemann, M.M.; Gonçalves, C.A.; de Oliveira, D.L. Phase-Dependent Astroglial Alterations in Li–Pilocarpine-Induced Status Epilepticus in Young Rats. Neurochem. Res. 2017, 42, 2730–2742. [Google Scholar] [CrossRef] [PubMed]
- Kovalenko, A.A.; Zakharova, M.V.; Zubareva, O.E.; Schwarz, A.P.; Postnikova, T.Y.; Zaitsev, A.V. Alterations in mRNA and protein expression of glutamate receptor subunits following pentylenetetrazole-induced acute seizures in young rats. Neuroscience 2021, 468, 1–15. [Google Scholar] [CrossRef]
- Zubareva, O.E.O.E.; Kovalenko, A.A.A.; Kalemenev, S.V.S.V.; Schwarz, A.P.A.P.; Karyakin, V.B.V.B.; Zaitsev, A.V.A.V. Alterations in mRNA expression of glutamate receptor subunits and excitatory amino acid transporters following pilocarpine-induced seizures in rats. Neurosci. Lett. 2018, 686, 94–100. [Google Scholar] [CrossRef]
- Zaitsev, А.V.; Amakhin, D.V.; Dyomina, A.V.; Zakharova, M.V.; Ergina, J.L.; Postnikova, T.Y.; Diespirov, G.P.; Magazanik, L.G. Synaptic Dysfunction in Epilepsy. J. Evol. Biochem. Physiol. 2021, 57, 542–563. [Google Scholar] [CrossRef]
- Scheefhals, N.; MacGillavry, H.D. Functional organization of postsynaptic glutamate receptors. Mol. Cell. Neurosci. 2018, 91, 82–94. [Google Scholar] [CrossRef]
- Anwyl, R. Metabotropic glutamate receptor-dependent long-term potentiation. Neuropharmacology 2009, 56, 735–740. [Google Scholar] [CrossRef]
- O’neill, N.; McLaughlin, C.; Komiyama, N.; Sylantyev, S. Biphasic modulation of NMDA receptor function by metabotropic glutamate receptors. J. Neurosci. 2018, 38, 9840–9855. [Google Scholar] [CrossRef]
- Lai, T.K.Y.; Zhai, D.; Su, P.; Jiang, A.; Boychuk, J.; Liu, F. The receptor-receptor interaction between mGluR1 receptor and NMDA receptor: A potential therapeutic target for protection against ischemic stroke. FASEB J. 2019, 33, 14423–14439. [Google Scholar] [CrossRef] [Green Version]
- Postnikova, T.Y.; Trofimova, A.M.; Ergina, J.L.; Zubareva, O.E.; Kalemenev, S.V.; Zaitsev, A.V. Transient Switching of NMDA-Dependent Long-Term Synaptic Potentiation in CA3-CA1 Hippocampal Synapses to mGluR1-Dependent Potentiation After Pentylenetetrazole-Induced Acute Seizures in Young Rats. Cell. Mol. Neurobiol. 2019, 39, 287–300. [Google Scholar] [CrossRef] [PubMed]
- Postnikova, T.Y.; Griflyuk, A.V.; Amakhin, D.V.; Kovalenko, A.A.; Soboleva, E.B.; Zubareva, O.E.; Zaitsev, A.V. Early Life Febrile Seizures Impair Hippocampal Synaptic Plasticity in Young Rats. Int. J. Mol. Sci. 2021, 22, 8218. [Google Scholar] [CrossRef]
- Amakhin, D.V.; Malkin, S.L.; Ergina, J.L.; Kryukov, K.A.; Veniaminova, E.A.; Zubareva, O.E.; Zaitsev, A.V. Alterations in Properties of Glutamatergic Transmission in the Temporal Cortex and Hippocampus Following Pilocarpine-Induced Acute Seizures in Wistar Rats. Front. Cell. Neurosci. 2017, 11, 264. [Google Scholar] [CrossRef] [PubMed]
- Iacobucci, G.J.; Popescu, G.K. NMDA receptors: Linking physiological output to biophysical operation. Nat. Rev. Neurosci. 2017, 18, 236–249. [Google Scholar] [CrossRef]
- MacDonald, J.F.; Jackson, M.F.; Beazely, M.A. Hippocampal Long-Term Synaptic Plasticity and Signal Amplification of NMDA Receptors. Crit. Rev. Neurobiol. 2006, 18, 71–84. [Google Scholar] [CrossRef]
- Harney, S.C.; Jane, D.E.; Anwyl, R. Extrasynaptic NR2D-Containing NMDARs Are Recruited to the Synapse during LTP of NMDAR-EPSCs. J. Neurosci. 2008, 28, 11685–11694. [Google Scholar] [CrossRef]
- Debanne, D.; Thompson, S.M.; Gähwiler, B.H. A brief period of epileptiform activity strengthens excitatory synapses in the rat hippocampus in vitro. Epilepsia 2006, 47, 247–256. [Google Scholar] [CrossRef] [PubMed]
- Di Maio, R.; Mastroberardino, P.G.; Hu, X.; Montero, L.; Greenamyre, J.T. Pilocapine alters NMDA receptor expression and function in hippocampal neurons: NADPH oxidase and ERK1/2 mechanisms. Neurobiol. Dis. 2011, 42, 482–495. [Google Scholar] [CrossRef]
- Joshi, S.; Rajasekaran, K.; Sun, H.; Williamson, J.; Kapur, J. Enhanced AMPA receptor-mediated neurotransmission on CA1 pyramidal neurons during status epilepticus. Neurobiol. Dis. 2017, 103, 45–53. [Google Scholar] [CrossRef] [PubMed]
- Abegg, M.H.; Savic, N.; Ehrengruber, M.U.; McKinney, R.A.; Gähwiler, B.H. Epileptiform activity in rat hippocampus strengthens excitatory synapses. J. Physiol. 2004, 554, 439–448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amakhin, D.V.; Soboleva, E.B.; Ergina, J.L.; Malkin, S.L.; Chizhov, A.V.; Zaitsev, A.V. Seizure-Induced Potentiation of AMPA Receptor-Mediated Synaptic Transmission in the Entorhinal Cortex. Front. Cell. Neurosci. 2018, 12, 486. [Google Scholar] [CrossRef] [PubMed]
- Plant, K.; Pelkey, K.A.; Bortolotto, Z.A.; Morita, D.; Terashima, A.; McBain, C.J.; Collingridge, G.L.; Isaac, J.T.R. Transient incorporation of native GluR2-lacking AMPA receptors during hippocampal long-term potentiation. Nat. Neurosci. 2006, 9, 602–604. [Google Scholar] [CrossRef] [PubMed]
- Malkin, S.L.; Amakhin, D.V.; Veniaminova, E.A.; Kim, K.K.; Zubareva, O.E.; Magazanik, L.G.; Zaitsev, A.V. Changes of ampa receptor properties in the neocortex and hippocampus following pilocarpine-induced status epilepticus in rats. Neuroscience 2016, 327, 146–155. [Google Scholar] [CrossRef] [PubMed]
- Lu, W.; Roche, K.W. Posttranslational regulation of AMPA receptor trafficking and function. Curr. Opin. Neurobiol. 2012, 22, 470–479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, H.-K.; Barbarosie, M.; Kameyama, K.; Bear, M.F.; Huganir, R.L. Regulation of distinct AMPA receptor phosphorylation sites during bidirectional synaptic plasticity. Nature 2000, 405, 955–959. [Google Scholar] [CrossRef] [PubMed]
- Paoletti, P.; Bellone, C.; Zhou, Q. NMDA receptor subunit diversity: Impact on receptor properties, synaptic plasticity and disease. Nat. Rev. Neurosci. 2013, 14, 383–400. [Google Scholar] [CrossRef]
- Di Maio, R.; Mastroberardino, P.G.; Hu, X.; Montero, L.M.; Greenamyre, J.T. Thiol oxidation and altered NR2B/NMDA receptor functions in in vitro and in vivo pilocarpine models: Implications for epileptogenesis. Neurobiol. Dis. 2013, 49, 87–98. [Google Scholar] [CrossRef]
- Zhu, X.; Dong, J.; Shen, K.; Bai, Y.; Zhang, Y.; Lv, X.; Chao, J.; Yao, H. NMDA receptor NR2B subunits contribute to PTZ-kindling-induced hippocampal astrocytosis and oxidative stress. Brain Res. Bull. 2015, 114, 70–78. [Google Scholar] [CrossRef]
- Chen, B.; Feng, B.; Tang, Y.; You, Y.; Wang, Y.; Hou, W.; Hu, W.; Chen, Z. Blocking GluN2B subunits reverses the enhanced seizure susceptibility after prolonged febrile seizures with a wide therapeutic time-window. Exp. Neurol. 2016, 283, 29–38. [Google Scholar] [CrossRef]
- Parsons, M.P.; Raymond, L.A. Extrasynaptic NMDA receptor involvement in central nervous system disorders. Neuron 2014, 82, 279–293. [Google Scholar] [CrossRef] [Green Version]
- Franchini, L.; Carrano, N.; Di Luca, M.; Gardoni, F. Synaptic GluN2A-Containing NMDA Receptors: From Physiology to Pathological Synaptic Plasticity. Int. J. Mol. Sci. 2020, 21, 1538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, R.; Huang, F.S.; Abbas, A.K.; Wigström, H. Role of NMDA receptor subtypes in different forms of NMDA-dependent synaptic plasticity. BMC Neurosci. 2007, 8, 55. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.; Wong, T.P.; Pozza, M.F.; Lingenhoehl, K.; Wang, Y.T.; Sheng, M.; Auberson, Y.P.; Wang, Y.T. Role of NMDA Receptor Subtypes in Governing the Direction of Hippocampal Synaptic Plasticity. Science 2004, 304, 1021–1024. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Massey, P.V.; Johnson, B.E.; Moult, P.R.; Auberson, Y.P.; Brown, M.W.; Molnar, E.; Collingridge, G.L.; Bashir, Z.I. Differential roles of NR2A and NR2B-containing NMDA receptors in cortical long-term potentiation and long-term depression. J. Neurosci. 2004, 24, 7821–7828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bartlett, T.E.; Bannister, N.J.; Collett, V.J.; Dargan, S.L.; Massey, P.V.; Bortolotto, Z.A.; Fitzjohn, S.M.; Bashir, Z.I.; Collingridge, G.L.; Lodge, D. Differential roles of NR2A and NR2B-containing NMDA receptors in LTP and LTD in the CA1 region of two-week old rat hippocampus. Neuropharmacology 2007, 52, 60–70. [Google Scholar] [CrossRef]
- Xu, Z.; Chen, R.Q.; Gu, Q.H.; Yan, J.Z.; Wang, S.H.; Liu, S.Y.; Lu, W. Metaplastic regulation of long-term potentiation/long-term depression threshold by activity-dependent changes of NR2A/NR2B ratio. J. Neurosci. 2009, 29, 8764–8773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Traynelis, S.F.; Wollmuth, L.P.; McBain, C.J.; Menniti, F.S.; Vance, K.M.; Ogden, K.K.; Hansen, K.B.; Yuan, H.; Myers, S.J.; Dingledine, R. Glutamate Receptor Ion Channels: Structure, Regulation, and Function. Pharmacol. Rev. 2010, 62, 405–496. [Google Scholar] [CrossRef] [Green Version]
- Diamond, J.S.; Jahr, C.E. Synaptically Released Glutamate Does Not Overwhelm Transporters on Hippocampal Astrocytes During High-Frequency Stimulation. J. Neurophysiol. 2000, 83, 2835–2843. [Google Scholar] [CrossRef] [PubMed]
- Armbruster, M.; Hanson, E.; Dulla, C.G. Glutamate Clearance Is Locally Modulated by Presynaptic Neuronal Activity in the Cerebral Cortex. J. Neurosci. 2016, 36, 10404–10415. [Google Scholar] [CrossRef] [Green Version]
- Bernardinelli, Y.; Muller, D.; Nikonenko, I. Astrocyte-Synapse Structural Plasticity. Neural Plast. 2014, 2014, 232105. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, D.K.; Vargas, J.R.; Wilcox, K.S. Increased coupling and altered glutamate transport currents in astrocytes following kainic-acid-induced status epilepticus. Neurobiol. Dis. 2010, 40, 573–585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zaitsev, A.V.; Smolensky, I.V.; Jorratt, P.; Ovsepian, S.V. Neurobiology, Functions, and Relevance of Excitatory Amino Acid Transporters (EAATs) to Treatment of Refractory Epilepsy. CNS Drugs 2020, 34, 1089–1103. [Google Scholar] [CrossRef] [PubMed]
- Clarkson, C.; Smeal, R.M.; Hasenoehrl, M.G.; White, J.A.; Rubio, M.E.; Wilcox, K.S. Ultrastructural and functional changes at the tripartite synapse during epileptogenesis in a model of temporal lobe epilepsy. Exp. Neurol. 2020, 326, 113196. [Google Scholar] [CrossRef]
- Iacobucci, G.J.; Popescu, G.K. Ca2+-Dependent Inactivation of GluN2A and GluN2B NMDA Receptors Occurs by a Common Kinetic Mechanism. Biophys. J. 2020, 118, 798–812. [Google Scholar] [CrossRef]
- Johnson, J.W.; Ascher, P. Glycine potentiates the NMDA response in cultured mouse brain neurons. Nature 1987, 325, 529–531. [Google Scholar] [CrossRef] [PubMed]
- Benveniste, M.; Clements, J.; Vyklický, L.; Mayer, M.L. A kinetic analysis of the modulation of N-methyl-D-aspartic acid receptors by glycine in mouse cultured hippocampal neurones. J. Physiol. 1990, 428, 333–357. [Google Scholar] [CrossRef] [PubMed]
- Cummings, K.A.; Popescu, G.K. Glycine-dependent activation of NMDA receptors. J. Gen. Physiol. 2015, 145, 513–527. [Google Scholar] [CrossRef] [Green Version]
- Shen, H.-Y.; van Vliet, E.A.; Bright, K.-A.; Hanthorn, M.; Lytle, N.K.; Gorter, J.; Aronica, E.; Boison, D. Glycine transporter 1 is a target for the treatment of epilepsy. Neuropharmacology 2015, 99, 554–565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eichler, S.A.; Kirischuk, S.; Jüttner, R.; Schafermeier, P.K.; Legendre, P.; Lehmann, T.N.; Gloveli, T.; Grantyn, R.; Meier, J.C. Glycinergic tonic inhibition of hippocampal neurons with depolarizing GABAergic transmission elicits histopathological signs of temporal lobe epilepsy. J. Cell. Mol. Med. 2008, 12, 2848–2866. [Google Scholar] [CrossRef] [Green Version]
- Henneberger, C.; Papouin, T.; Oliet, S.H.R.; Rusakov, D.A. Long-term potentiation depends on release of d-serine from astrocytes. Nature 2010, 463, 232–236. [Google Scholar] [CrossRef] [PubMed]
- Racine, R.J.; Milgram, N.W.; Hafner, S. Long-term potentiation phenomena in the rat limbic forebrain. Brain Res. 1983, 260, 217–231. [Google Scholar] [CrossRef]
- Jahr, C.; Stevens, C. Voltage dependence of NMDA-activated macroscopic conductances predicted by single-channel kinetics. J. Neurosci. 1990, 10, 3178–3182. [Google Scholar] [CrossRef] [PubMed]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Postnikova, T.Y.; Diespirov, G.P.; Amakhin, D.V.; Vylekzhanina, E.N.; Soboleva, E.B.; Zaitsev, A.V. Impairments of Long-Term Synaptic Plasticity in the Hippocampus of Young Rats during the Latent Phase of the Lithium-Pilocarpine Model of Temporal Lobe Epilepsy. Int. J. Mol. Sci. 2021, 22, 13355. https://doi.org/10.3390/ijms222413355
Postnikova TY, Diespirov GP, Amakhin DV, Vylekzhanina EN, Soboleva EB, Zaitsev AV. Impairments of Long-Term Synaptic Plasticity in the Hippocampus of Young Rats during the Latent Phase of the Lithium-Pilocarpine Model of Temporal Lobe Epilepsy. International Journal of Molecular Sciences. 2021; 22(24):13355. https://doi.org/10.3390/ijms222413355
Chicago/Turabian StylePostnikova, Tatyana Y., Georgy P. Diespirov, Dmitry V. Amakhin, Elizaveta N. Vylekzhanina, Elena B. Soboleva, and Aleksey V. Zaitsev. 2021. "Impairments of Long-Term Synaptic Plasticity in the Hippocampus of Young Rats during the Latent Phase of the Lithium-Pilocarpine Model of Temporal Lobe Epilepsy" International Journal of Molecular Sciences 22, no. 24: 13355. https://doi.org/10.3390/ijms222413355