
Immunotherapy in Advanced Prostate Cancer—Light at the End of the Tunnel?

 ,
,
Abstract
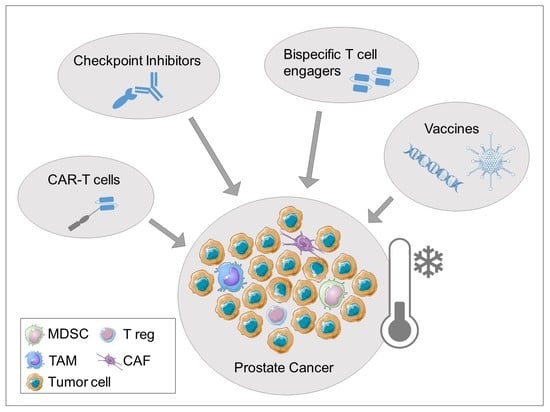
:
1. Background
2. Immune Response and the Role of the Microenvironment in Prostate Cancer
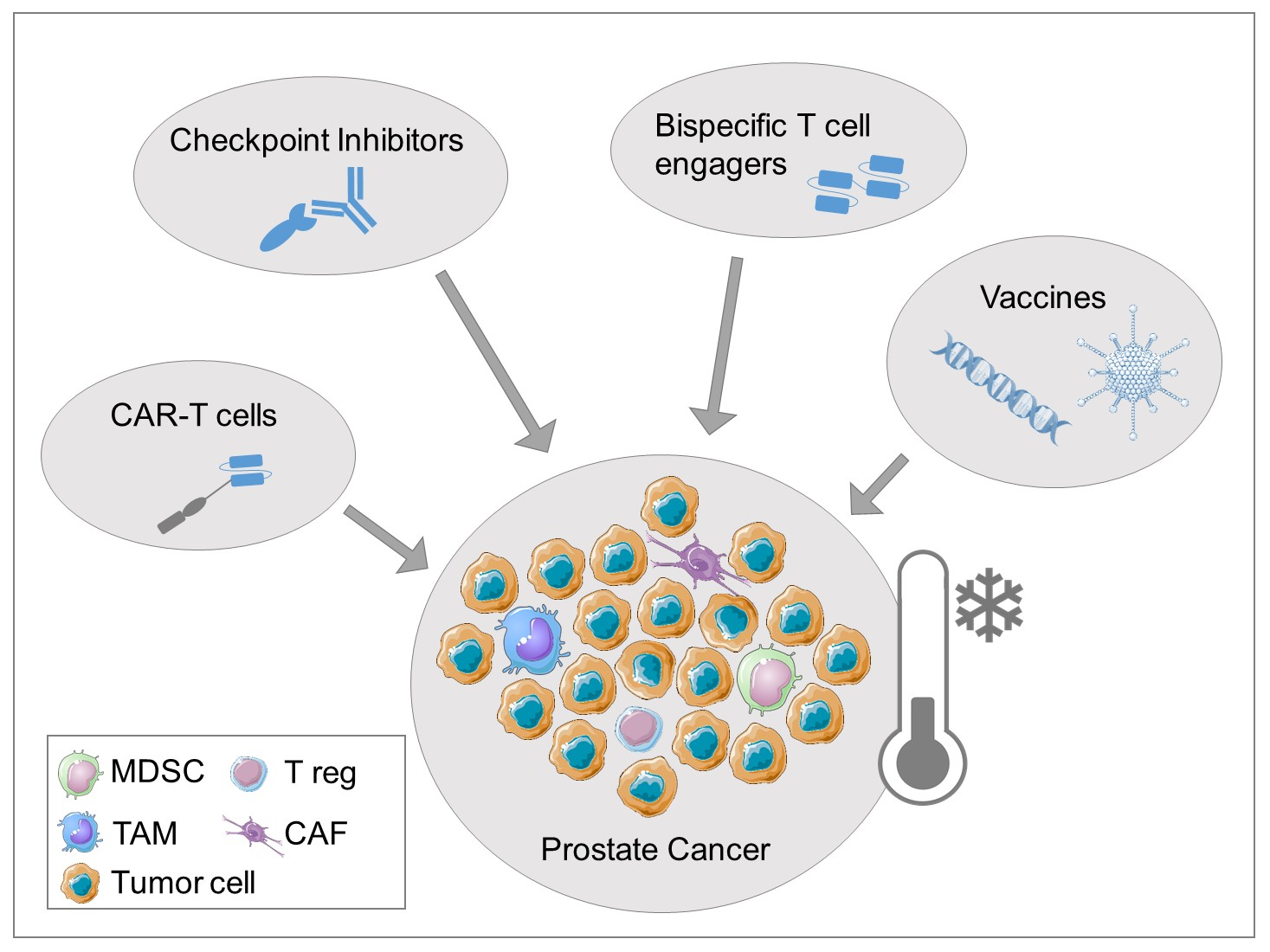
2.1. Intrinsic Factors Influencing Immune Response
2.1.1. Tumor Mutational Burden and Neoantigen Expression
2.1.2. Expression of Programmed Death Ligand-1 (PD-L1)
2.1.3. DNA Repair Defects
2.1.4. Inactivation of PTEN
2.1.5. Androgen Receptor Signaling
2.2. The Role of Tumor Microenvironment in Prostate Cancer
2.2.1. The Tumor Cytokine Milieu
2.2.2. Myeloid-Derived Suppressor Cells (MDSCs)
2.2.3. Tumor-Associated Macrophages (TAMs)
2.2.4. Stromal Cells
2.2.5. Adenosine in PCa
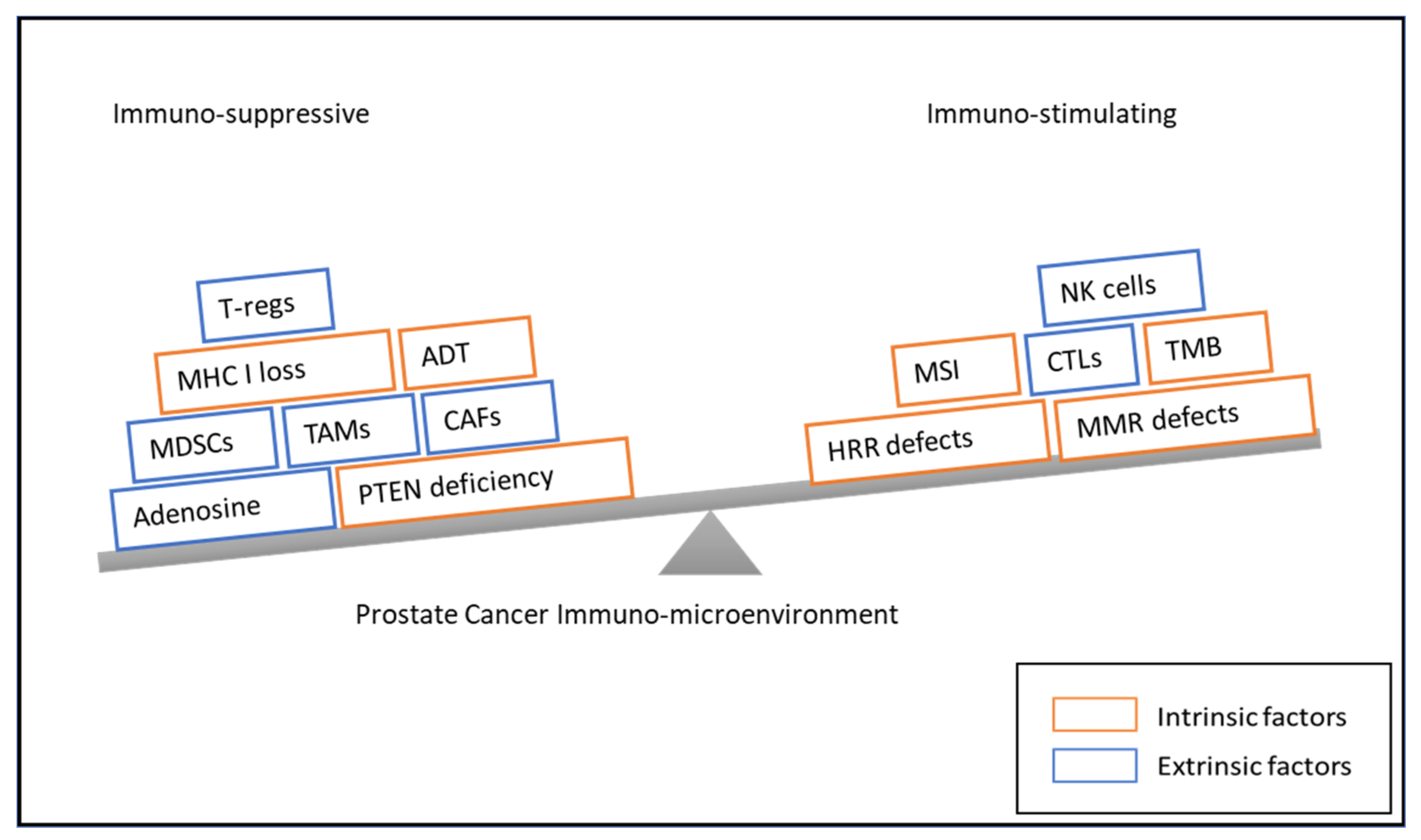
3. Immunotherapeutic Treatment Approaches
3.1. Vaccine-Based Treatment Modalities
3.1.1. Cell-Based Vaccines
3.1.2. Peptide Vaccines
3.1.3. Viral/Bacterial-Based Vaccines
3.1.4. DNA and RNA Vaccines
3.2. Checkpoint Inhibitors
Checkpoint Inhibitor Monotherapy
Anti-CTLA-4 Antibodies
PD-1/PD-L1 Inhibitors
3.3. Checkpoint Inhibitor Combinations
3.3.1. PD-1/PDL-1-Inhibitors and Anti-CTLA-4 Antibodies
3.3.2. PD-1/PD-L1 Antibodies and Androgen Receptor-Targeting Therapies
3.3.3. PD-1/PD-L1 and Chemotherapy
3.3.4. PD-1/PD-L1 and PARP Inhibitors
3.3.5. PD-1/PD-L1-Inhibitors and Tyrosinkinase Inhibitors (TKI)
3.3.6. PD-1/PD-L1 Inhibitors and Radiotherapeutic Approaches
3.4. Bispecific T Cell Engagers
3.5. Chimeric Antigen Receptor T Cells (CAR-T Cells)
4. Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
Abbreviations
| CTLA-4 | cytotoxic T lymphocyte antigen 4 |
| PD-1 | programmed death-1 |
| PD-L1 | PD-1 ligand |
| PCa | prostate cancer |
| TME | tumor microenvironment |
| BiTE | bispecific T cell engager |
| CAR-T cells | chimeric antigen receptor T cells |
| TMИ | tumor mutational burden |
| MHC | major histocompatibility complex |
| CTL | cytotoxic T lymphocytes |
| TILs | tumor-infiltrating lymphocytes |
| DDR | DNA damage repair |
| MMR | mismatch repair |
| MSI | microsatellite instability |
| HRR | homologous recombination repair |
| Treg | regulatory T cells |
| AVPC | aggressive variant of prostate cancer |
| MDSCs | myeloid-derived suppressor cells |
| AK | androgen receptor |
| ADT | androgen-deprivation therapy |
| TAM | tumor-associated macrophages |
| TNF-α | tumor necrosis factor-α |
| MSCs | mesenchymal stromal cells |
| CAFs | cancer-associated fibroblasts |
| EMT | epithelial–mesenchymal transition |
| PAP | prostatic acid phosphatase |
| ATP | adenosine triphosphate |
| DAMP | danger-associated molecular pattern |
| TAA | tumor-associated antigens |
| PSA | prostate-specific antigen |
| PSMA | prostate-specific membrane antigen |
| PSCA | prostate stem cell antigen |
| APC | antigen-presenting cell |
| DC | dendritic cell |
| OS | overall survival |
| mCRPC | metastatic castration-resistant prostate cancer |
| PBMCs | peripheral blood mononuclear cells |
| mHNPC | metastatic hormone-naïve prostate cancer |
| PPV | personalized peptide vaccination |
| PFS | progression-free survival |
| CI | immune checkpoint inhibitor |
| ORR | overall response rate |
| SD | stable disease |
| DCR | disease control rate |
| PR | partial response |
| HRD | homologous recombination deficiency |
| TRAE | Treatment-related adverse events |
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Ferro, M.; Lucarelli, G.; Crocetto, F.; Dolce, P.; Verde, A.; La Civita, E.; Zappavigna, S.; de Cobelli, O.; Di Lorenzo, G.; Facchini, B.A.; et al. First-line systemic therapy for metastatic castration-sensitive prostate cancer: An updated systematic review with novel findings. Crit. Rev. Oncol. Hematol. 2021, 157, 103198. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.R.; Hussain, M.; Saad, F.; Fizazi, K.; Sternberg, C.N.; Crawford, E.D.; Kopyltsov, E.; Park, C.H.; Alekseev, B.; Montesa-Pino, Á.; et al. Darolutamide and Survival in Metastatic, Hormone-Sensitive Prostate Cancer. N. Engl. J. Med. 2022, in press. [Google Scholar] [CrossRef] [PubMed]
- Fizazi, K.; Maldonado, X.; Foulon, S.; Roubaud, G.; McDermott, R.S.; Flechon, A.; Tombal, B.F.; Supiot, S.; Berthold, D.R.; Ronchin, P.; et al. A phase 3 trial with a 2x2 factorial design of abiraterone acetate plus prednisone and/or local radiotherapy in men with de novo metastatic castration-sensitive prostate cancer (mCSPC): First results of PEACE-1. J. Clin. Oncol. 2021, 39, 5000. [Google Scholar] [CrossRef]
- Hofman, M.S.; Emmett, L.; Sandhu, S.; Iravani, A.; Joshua, A.M.; Goh, J.C.; Pattison, D.A.; Tan, T.H.; Kirkwood, I.D.; Ng, S.; et al. [177Lu]Lu-PSMA-617 versus cabazitaxel in patients with metastatic castration-resistant prostate cancer (TheraP): A randomised, open-label, phase 2 trial. Lancet 2021, 397, 797–804. [Google Scholar] [CrossRef]
- De Wit, R.; de Bono, J.; Sternberg, C.N.; Fizazi, K.; Tombal, B.; Wulfing, C.; Kramer, G.; Eymard, J.C.; Bamias, A.; Carles, J.; et al. Cabazitaxel versus Abiraterone or Enzalutamide in Metastatic Prostate Cancer. N. Engl. J. Med. 2019, 381, 2506–2518. [Google Scholar] [CrossRef]
- Sartor, O.; de Bono, J.; Chi, K.N.; Fizazi, K.; Herrmann, K.; Rahbar, K.; Tagawa, S.T.; Nordquist, L.T.; Vaishampayan, N.; El-Haddad, G.; et al. Lutetium-177–PSMA-617 for Metastatic Castration-Resistant Prostate Cancer. N. Engl. J. Med. 2021, 385, 1091–1103. [Google Scholar] [CrossRef]
- De Bono, J.; Mateo, J.; Fizazi, K.; Saad, F.; Shore, N.; Sandhu, S.; Chi, K.N.; Sartor, O.; Agarwal, N.; Olmos, D.; et al. Olaparib for Metastatic Castration-Resistant Prostate Cancer. N. Engl. J. Med. 2020, 382, 2091–2102. [Google Scholar] [CrossRef]
- Alatrash, G.; Jakher, H.; Stafford, P.D.; Mittendorf, E.A. Cancer immunotherapies, their safety and toxicity. Expert Opin. Drug Saf. 2013, 12, 631–645. [Google Scholar] [CrossRef]
- Wolchok, J.D.; Chiarion-Sileni, V.; Gonzalez, R.; Rutkowski, P.; Grob, J.J.; Cowey, C.L.; Lao, C.D.; Wagstaff, J.; Schadendorf, D.; Ferrucci, P.F.; et al. Overall Survival with Combined Nivolumab and Ipilimumab in Advanced Melanoma. N. Engl. J. Med. 2017, 377, 1345–1356. [Google Scholar] [CrossRef]
- Motzer, R.J.; Tannir, N.M.; McDermott, D.F.; Arén Frontera, O.; Melichar, B.; Choueiri, T.K.; Plimack, E.R.; Barthélémy, P.; Porta, C.; George, S.; et al. Nivolumab plus Ipilimumab versus Sunitinib in Advanced Renal-Cell Carcinoma. N. Engl. J. Med. 2018, 378, 1277–1290. [Google Scholar] [CrossRef]
- Shields, M.D.; Marin-Acevedo, J.A.; Pellini, B. Immunotherapy for Advanced Non–Small Cell Lung Cancer: A Decade of Progress. Am. Soc. Clin. Oncol. Educ. Book 2021, 41, e105–e127. [Google Scholar] [CrossRef]
- Kantoff, P.W.; Higano, C.S.; Shore, N.D.; Berger, E.R.; Small, E.J.; Penson, D.F.; Redfern, C.H.; Ferrari, A.C.; Dreicer, R.; Sims, R.B.; et al. Sipuleucel-T immunotherapy for castration-resistant prostate cancer. N. Engl. J. Med. 2010, 363, 411–422. [Google Scholar] [CrossRef] [Green Version]
- Klempner, S.J.; Fabrizio, D.; Bane, S.; Reinhart, M.; Peoples, T.; Ali, S.M.; Sokol, E.S.; Frampton, G.; Schrock, A.B.; Anhorn, R.; et al. Tumor Mutational Burden as a Predictive Biomarker for Response to Immune Checkpoint Inhibitors: A Review of Current Evidence. Oncologist 2020, 25, e147–e159. [Google Scholar] [CrossRef] [Green Version]
- Lawrence, M.S.; Stojanov, P.; Polak, P.; Kryukov, G.V.; Cibulskis, K.; Sivachenko, A.; Carter, S.L.; Stewart, C.; Mermel, C.H.; Roberts, S.A.; et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature 2013, 499, 214–218. [Google Scholar] [CrossRef]
- Ryan, M.J.; Bose, R. Genomic Alteration Burden in Advanced Prostate Cancer and Therapeutic Implications. Front. Oncol. 2019, 9, 1287. [Google Scholar] [CrossRef] [Green Version]
- Wagle, M.C.; Castillo, J.; Srinivasan, S.; Holcomb, T.; Yuen, K.C.; Kadel, E.E.; Mariathasan, S.; Halligan, D.L.; Carr, A.R.; Bylesjo, M.; et al. Tumor Fusion Burden as a Hallmark of Immune Infiltration in Prostate Cancer. Cancer Immunol. Res. 2020, 8, 844–850. [Google Scholar] [CrossRef] [Green Version]
- Haffner, M.C.; Guner, G.; Taheri, D.; Netto, G.J.; Palsgrove, D.N.; Zheng, Q.; Guedes, L.B.; Kim, K.; Tsai, H.; Esopi, D.M.; et al. Comprehensive Evaluation of Programmed Death-Ligand 1 Expression in Primary and Metastatic Prostate Cancer. Am. J. Pathol. 2018, 188, 1478–1485. [Google Scholar] [CrossRef] [Green Version]
- Palicelli, A.; Bonacini, M.; Croci, S.; Magi-Galluzzi, C.; Cañete-Portillo, S.; Chaux, A.; Bisagni, A.; Zanetti, E.; De Biase, D.; Melli, B.; et al. What Do We Have to Know about PD-L1 Expression in Prostate Cancer? A Systematic Literature Review. Part 1: Focus on Immunohistochemical Results with Discussion of Pre-Analytical and Interpretation Variables. Cells 2021, 10, 3166. [Google Scholar] [CrossRef]
- Ebelt, K.; Babaryka, G.; Frankenberger, B.; Stief, C.G.; Eisenmenger, W.; Kirchner, T.; Schendel, D.J.; Noessner, E. Prostate cancer lesions are surrounded by FOXP3+, PD-1+ and B7-H1+ lymphocyte clusters. Eur. J. Cancer 2009, 45, 1664–1672. [Google Scholar] [CrossRef]
- Gevensleben, H.; Dietrich, D.; Golletz, C.; Steiner, S.; Jung, M.; Thiesler, T.; Majores, M.; Stein, J.; Uhl, B.; Müller, S.; et al. The Immune Checkpoint Regulator PD-L1 Is Highly Expressed in Aggressive Primary Prostate Cancer. Clin. Cancer Res. 2016, 22, 1969–1977. [Google Scholar] [CrossRef] [Green Version]
- Baas, W.; Gershburg, S.; Dynda, D.; Delfino, K.; Robinson, K.; Nie, D.; Yearley, J.H.; Alanee, S. Immune Characterization of the Programmed Death Receptor Pathway in High Risk Prostate Cancer. Clin. Genitourin Cancer 2017, 15, 577–581. [Google Scholar] [CrossRef] [PubMed]
- Calagua, C.; Russo, J.; Sun, Y.; Schaefer, R.; Lis, R.; Zhang, Z.; Mahoney, K.; Bubley, G.J.; Loda, M.; Taplin, M.E.; et al. Expression of PD-L1 in Hormone-naïve and Treated Prostate Cancer Patients Receiving Neoadjuvant Abiraterone Acetate plus Prednisone and Leuprolide. Clin. Cancer Res. 2017, 23, 6812–6822. [Google Scholar] [CrossRef] [Green Version]
- Ness, N.; Andersen, S.; Khanehkenari, M.R.; Nordbakken, C.V.; Valkov, A.; Paulsen, E.E.; Nordby, Y.; Bremnes, R.M.; Donnem, T.; Busund, L.T.; et al. The prognostic role of immune checkpoint markers programmed cell death protein 1 (PD-1) and programmed death ligand 1 (PD-L1) in a large, multicenter prostate cancer cohort. Oncotarget 2017, 8, 26789–26801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fankhauser, C.D.; Schüffler, P.J.; Gillessen, S.; Omlin, A.; Rupp, N.J.; Rueschoff, J.H.; Hermanns, T.; Poyet, C.; Sulser, T.; Moch, H.; et al. Comprehensive immunohistochemical analysis of PD-L1 shows scarce expression in castration-resistant prostate cancer. Oncotarget 2018, 9, 10284–10293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palicelli, A.; Croci, S.; Bisagni, A.; Zanetti, E.; De Biase, D.; Melli, B.; Sanguedolce, F.; Ragazzi, M.; Zanelli, M.; Chaux, A.; et al. What Do We Have to Know about PD-L1 Expression in Prostate Cancer? A Systematic Literature Review (Part 6): Correlation of PD-L1 Expression with the Status of Mismatch Repair System, BRCA, PTEN, and Other Genes. Biomedicines 2022, 10, 236. [Google Scholar] [CrossRef]
- Petitprez, F.; Fossati, N.; Vano, Y.; Freschi, M.; Becht, E.; Lucianò, R.; Calderaro, J.; Guédet, T.; Lacroix, L.; Rancoita, P.M.V.; et al. PD-L1 Expression and CD8(+) T-cell Infiltrate are Associated with Clinical Progression in Patients with Node-positive Prostate Cancer. Eur. Urol. Focus 2019, 5, 192–196. [Google Scholar] [CrossRef] [PubMed]
- Sharma, M.; Yang, Z.; Miyamoto, H. Immunohistochemistry of immune checkpoint markers PD-1 and PD-L1 in prostate cancer. Medicine 2019, 98, e17257. [Google Scholar] [CrossRef] [PubMed]
- Abida, W.; Cheng, M.L.; Armenia, J.; Middha, S.; Autio, K.A.; Vargas, H.A.; Rathkopf, D.; Morris, M.J.; Danila, D.C.; Slovin, S.F.; et al. Analysis of the Prevalence of Microsatellite Instability in Prostate Cancer and Response to Immune Checkpoint Blockade. JAMA Oncol. 2019, 5, 471–478. [Google Scholar] [CrossRef]
- Pritchard, C.C.; Morrissey, C.; Kumar, A.; Zhang, X.; Smith, C.; Coleman, I.; Salipante, S.J.; Milbank, J.; Yu, M.; Grady, W.M.; et al. Complex MSH2 and MSH6 mutations in hypermutated microsatellite unstable advanced prostate cancer. Nat. Commun. 2014, 5, 4988. [Google Scholar] [CrossRef] [Green Version]
- Linch, M.; Goh, G.; Hiley, C.; Shanmugabavan, Y.; McGranahan, N.; Rowan, A.; Wong, Y.N.S.; King, H.; Furness, A.; Freeman, A.; et al. Intratumoural evolutionary landscape of high-risk prostate cancer: The PROGENY study of genomic and immune parameters. Ann. Oncol. 2017, 28, 2472–2480. [Google Scholar] [CrossRef]
- Le, D.T.; Durham, J.N.; Smith, K.N.; Wang, H.; Bartlett, B.R.; Aulakh, L.K.; Lu, S.; Kemberling, H.; Wilt, C.; Luber, B.S.; et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science 2017, 357, 409–413. [Google Scholar] [CrossRef] [Green Version]
- Nava Rodrigues, D.; Rescigno, P.; Liu, D.; Yuan, W.; Carreira, S.; Lambros, M.B.; Seed, G.; Mateo, J.; Riisnaes, R.; Mullane, S.; et al. Immunogenomic analyses associate immunological alterations with mismatch repair defects in prostate cancer. J. Clin. Investig. 2018, 128, 4441–4453. [Google Scholar] [CrossRef] [Green Version]
- Abida, W.; Armenia, J.; Gopalan, A.; Brennan, R.; Walsh, M.; Barron, D.; Danila, D.; Rathkopf, D.; Morris, M.; Slovin, S.; et al. Prospective Genomic Profiling of Prostate Cancer Across Disease States Reveals Germline and Somatic Alterations That May Affect Clinical Decision Making. JCO Precis. Oncol. 2017, 2017, 1–16. [Google Scholar] [CrossRef]
- Jenzer, M.; Kess, P.; Nientiedt, C.; Endris, V.; Kippenberger, M.; Leichsenring, J.; Stogbauer, F.; Haimes, J.; Mishkin, S.; Kudlow, B.; et al. The BRCA2 mutation status shapes the immune phenotype of prostate cancer. Cancer Immunol. Immunother. 2019, 68, 1621–1633. [Google Scholar] [CrossRef] [Green Version]
- Heijink, A.M.; Talens, F.; Jae, L.T.; van Gijn, S.E.; Fehrmann, R.S.N.; Brummelkamp, T.R.; van Vugt, M.A.T.M. BRCA2 deficiency instigates cGAS-mediated inflammatory signaling and confers sensitivity to tumor necrosis factor-alpha-mediated cytotoxicity. Nat. Commun. 2019, 10, 100. [Google Scholar] [CrossRef] [Green Version]
- Powles, T.; Loriot, Y.; Ravaud, A.; Vogelzang, N.J.; Duran, I.; Retz, M.; Giorgi, U.D.; Oudard, S.; Bamias, A.; Koeppen, H.; et al. Atezolizumab (atezo) vs. chemotherapy (chemo) in platinum-treated locally advanced or metastatic urothelial carcinoma (mUC): Immune biomarkers, tumor mutational burden (TMB), and clinical outcomes from the phase III IMvigor211 study. J. Clin. Oncol. 2018, 36, 409. [Google Scholar] [CrossRef]
- Powles, T.; Yuen, K.C.; Gillessen, S.; Kadel, E.E.; Rathkopf, D.; Matsubara, N.; Drake, C.G.; Fizazi, K.; Piulats, J.M.; Wysocki, P.J.; et al. Atezolizumab with enzalutamide versus enzalutamide alone in metastatic castration-resistant prostate cancer: A randomized phase 3 trial. Nat. Med. 2022, 28, 144–153. [Google Scholar] [CrossRef]
- Aparicio, A.M.; Shen, L.; Tapia, E.L.; Lu, J.F.; Chen, H.C.; Zhang, J.; Wu, G.; Wang, X.; Troncoso, P.; Corn, P.; et al. Combined Tumor Suppressor Defects Characterize Clinically Defined Aggressive Variant Prostate Cancers. Clin. Cancer Res. 2016, 22, 1520–1530. [Google Scholar] [CrossRef] [Green Version]
- Hamid, A.A.; Gray, K.P.; Shaw, G.; MacConaill, L.E.; Evan, C.; Bernard, B.; Loda, M.; Corcoran, N.M.; Van Allen, E.M.; Choudhury, A.D.; et al. Compound Genomic Alterations of TP53, PTEN, and RB1 Tumor Suppressors in Localized and Metastatic Prostate Cancer. Eur. Urol. 2019, 76, 89–97. [Google Scholar] [CrossRef]
- Robinson, D.; Van Allen, E.M.; Wu, Y.M.; Schultz, N.; Lonigro, R.J.; Mosquera, J.M.; Montgomery, B.; Taplin, M.E.; Pritchard, C.C.; Attard, G.; et al. Integrative clinical genomics of advanced prostate cancer. Cell 2015, 161, 1215–1228. [Google Scholar] [CrossRef] [Green Version]
- Peng, W.; Chen, J.Q.; Liu, C.; Malu, S.; Creasy, C.; Tetzlaff, M.T.; Xu, C.; McKenzie, J.A.; Zhang, C.; Liang, X.; et al. Loss of PTEN Promotes Resistance to T Cell-Mediated Immunotherapy. Cancer Discov. 2016, 6, 202–216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, Y.; Wang, H.; Yang, L.; Zhang, Z.; Li, C.; Yuan, X.; Bu, L.; Chen, L.; Chen, Y.; Li, C.M.; et al. PTEN-L promotes type I interferon responses and antiviral immunity. Cell Mol. Immunol. 2018, 15, 48–57. [Google Scholar] [CrossRef] [PubMed]
- Vidotto, T.; Saggioro, F.P.; Jamaspishvili, T.; Chesca, D.L.; Picanco de Albuquerque, C.G.; Reis, R.B.; Graham, C.H.; Berman, D.M.; Siemens, D.R.; Squire, J.A.; et al. PTEN-deficient prostate cancer is associated with an immunosuppressive tumor microenvironment mediated by increased expression of IDO1 and infiltrating FoxP3+ T regulatory cells. Prostate 2019, 79, 969–979. [Google Scholar] [CrossRef] [PubMed]
- Calcinotto, A.; Spataro, C.; Zagato, E.; Di Mitri, D.; Gil, V.; Crespo, M.; De Bernardis, G.; Losa, M.; Mirenda, M.; Pasquini, E.; et al. IL-23 secreted by myeloid cells drives castration-resistant prostate cancer. Nature 2018, 559, 363–369. [Google Scholar] [CrossRef] [PubMed]
- Zhao, D.; Cai, L.; Lu, X.; Liang, X.; Li, J.; Chen, P.; Ittmann, M.; Shang, X.; Jiang, S.; Li, H.; et al. Chromatin Regulator CHD1 Remodels the Immunosuppressive Tumor Microenvironment in PTEN-Deficient Prostate Cancer. Cancer Discov. 2020, 10, 1374–1387. [Google Scholar] [CrossRef] [PubMed]
- Formaggio, N.; Rubin, M.A.; Theurillat, J.P. Loss and revival of androgen receptor signaling in advanced prostate cancer. Oncogene 2021, 40, 1205–1216. [Google Scholar] [CrossRef] [PubMed]
- Crawford, E.D.; Heidenreich, A.; Lawrentschuk, N.; Tombal, B.; Pompeo, A.C.L.; Mendoza-Valdes, A.; Miller, K.; Debruyne, F.M.J.; Klotz, L. Androgen-targeted therapy in men with prostate cancer: Evolving practice and future considerations. Prostate Cancer Prostatic. Dis. 2019, 22, 24–38. [Google Scholar] [CrossRef] [Green Version]
- Ben-Batalla, I.; Vargas-Delgado, M.E.; von Amsberg, G.; Janning, M.; Loges, S. Influence of Androgens on Immunity to Self and Foreign: Effects on Immunity and Cancer. Front. Immunol. 2020, 11, 1184. [Google Scholar] [CrossRef]
- Gamat, M.; McNeel, D.G. Androgen deprivation and immunotherapy for the treatment of prostate cancer. Endocr. Relat. Cancer 2017, 24, T297–T310. [Google Scholar] [CrossRef]
- Page, S.T.; Plymate, S.R.; Bremner, W.J.; Matsumoto, A.M.; Hess, D.L.; Lin, D.W.; Amory, J.K.; Nelson, P.S.; Wu, J.D. Effect of medical castration on CD4+ CD25+ T cells, CD8+ T cell IFN-gamma expression, and NK cells: A physiological role for testosterone and/or its metabolites. Am. J. Physiol. Endocrinol. Metab. 2006, 290, E856–E863. [Google Scholar] [CrossRef] [Green Version]
- Shen, Y.C.; Ghasemzadeh, A.; Kochel, C.M.; Nirschl, T.R.; Francica, B.J.; Lopez-Bujanda, Z.A.; Carrera Haro, M.A.; Tam, A.; Anders, R.A.; Selby, M.J.; et al. Combining intratumoral Treg depletion with androgen deprivation therapy (ADT): Preclinical activity in the Myc-CaP model. Prostate Cancer Prostatic. Dis. 2018, 21, 113–125. [Google Scholar] [CrossRef]
- Pu, Y.; Xu, M.; Liang, Y.; Yang, K.; Guo, Y.; Yang, X.; Fu, Y.X. Androgen receptor antagonists compromise T cell response against prostate cancer leading to early tumor relapse. Sci. Transl. Med. 2016, 8, 333ra347. [Google Scholar] [CrossRef] [Green Version]
- Obradovic, A.Z.; Dallos, M.C.; Zahurak, M.L.; Partin, A.W.; Schaeffer, E.M.; Ross, A.E.; Allaf, M.E.; Nirschl, T.R.; Liu, D.; Chapman, C.G.; et al. T-Cell Infiltration and Adaptive Treg Resistance in Response to Androgen Deprivation with or without Vaccination in Localized Prostate Cancer. Clin. Cancer Res. 2020, 26, 3182–3192. [Google Scholar] [CrossRef] [Green Version]
- Long, X.; Hou, H.; Wang, X.; Liu, S.; Diao, T.; Lai, S.; Hu, M.; Zhang, S.; Liu, M.; Zhang, H. Immune signature driven by ADT-induced immune microenvironment remodeling in prostate cancer is correlated with recurrence-free survival and immune infiltration. Cell Death Dis. 2020, 11, 779. [Google Scholar] [CrossRef]
- Shiao, S.L.; Chu, G.C.; Chung, L.W. Regulation of prostate cancer progression by the tumor microenvironment. Cancer Lett. 2016, 380, 340–348. [Google Scholar] [CrossRef] [Green Version]
- Stultz, J.; Fong, L. How to turn up the heat on the cold immune microenvironment of metastatic prostate cancer. Prostate Cancer Prostatic. Dis. 2021, 24, 697–717. [Google Scholar] [CrossRef]
- Chulpanova, D.S.; Kitaeva, K.V.; Green, A.R.; Rizvanov, A.A.; Solovyeva, V.V. Molecular Aspects and Future Perspectives of Cytokine-Based Anti-cancer Immunotherapy. Front. Cell Dev. Biol. 2020, 8, 402. [Google Scholar] [CrossRef]
- Mao, C.; Ding, Y.; Xu, N. A Double-Edged Sword Role of Cytokines in Prostate Cancer Immunotherapy. Front. Oncol. 2021, 11, 688489. [Google Scholar] [CrossRef]
- Wang, X.; Yang, L.; Huang, F.; Zhang, Q.; Liu, S.; Ma, L.; You, Z. Inflammatory cytokines IL-17 and TNF-α up-regulate PD-L1 expression in human prostate and colon cancer cells. Immunol. Lett. 2017, 184, 7–14. [Google Scholar] [CrossRef] [Green Version]
- Lundholm, M.; Hägglöf, C.; Wikberg, M.L.; Stattin, P.; Egevad, L.; Bergh, A.; Wikström, P.; Palmqvist, R.; Edin, S. Secreted Factors from Colorectal and Prostate Cancer Cells Skew the Immune Response in Opposite Directions. Sci. Rep. 2015, 5, 15651. [Google Scholar] [CrossRef]
- Wise, G.J.; Marella, V.K.; Talluri, G.; Shirazian, D. Cytokine variations in patients with hormone treated prostate cancer. J. Urol. 2000, 164, 722–725. [Google Scholar] [CrossRef]
- Mariathasan, S.; Turley, S.J.; Nickles, D.; Castiglioni, A.; Yuen, K.; Wang, Y.; Kadel Iii, E.E.; Koeppen, H.; Astarita, J.L.; Cubas, R.; et al. TGFβ attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature 2018, 554, 544–548. [Google Scholar] [CrossRef]
- Kloss, C.C.; Lee, J.; Zhang, A.; Chen, F.; Melenhorst, J.J.; Lacey, S.F.; Maus, M.V.; Fraietta, J.A.; Zhao, Y.; June, C.H. Dominant-Negative TGF-β Receptor Enhances PSMA-Targeted Human CAR T Cell Proliferation And Augments Prostate Cancer Eradication. Mol. Ther. 2018, 26, 1855–1866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tesi, R.J. MDSC; the Most Important Cell You Have Never Heard Of. Trends Pharm. Sci. 2019, 40, 4–7. [Google Scholar] [CrossRef]
- Koinis, F.; Xagara, A.; Chantzara, E.; Leontopoulou, V.; Aidarinis, C.; Kotsakis, A. Myeloid-Derived Suppressor Cells in Prostate Cancer: Present Knowledge and Future Perspectives. Cells 2022, 11, 20. [Google Scholar] [CrossRef] [PubMed]
- De Cicco, P.; Ercolano, G.; Ianaro, A. The New Era of Cancer Immunotherapy: Targeting Myeloid-Derived Suppressor Cells to Overcome Immune Evasion. Front. Immunol. 2020, 11, 1680. [Google Scholar] [CrossRef] [PubMed]
- Kwon, J.T.W.; Bryant, R.J.; Parkes, E.E. The tumor microenvironment and immune responses in prostate cancer patients. Endocr. Relat. Cancer 2021, 28, T95–T107. [Google Scholar] [CrossRef]
- Bezzi, M.; Seitzer, N.; Ishikawa, T.; Reschke, M.; Chen, M.; Wang, G.; Mitchell, C.; Ng, C.; Katon, J.; Lunardi, A.; et al. Diverse genetic-driven immune landscapes dictate tumor progression through distinct mechanisms. Nat. Med. 2018, 24, 165–175. [Google Scholar] [CrossRef]
- Hossain, D.M.; Pal, S.K.; Moreira, D.; Duttagupta, P.; Zhang, Q.; Won, H.; Jones, J.; D’Apuzzo, M.; Forman, S.; Kortylewski, M. TLR9-Targeted STAT3 Silencing Abrogates Immunosuppressive Activity of Myeloid-Derived Suppressor Cells from Prostate Cancer Patients. Clin. Cancer Res. 2015, 21, 3771–3782. [Google Scholar] [CrossRef] [Green Version]
- Idorn, M.; Køllgaard, T.; Kongsted, P.; Sengeløv, L.; Thor Straten, P. Correlation between frequencies of blood monocytic myeloid-derived suppressor cells, regulatory T cells and negative prognostic markers in patients with castration-resistant metastatic prostate cancer. Cancer Immunol. Immunother. 2014, 63, 1177–1187. [Google Scholar] [CrossRef]
- Lopez-Bujanda, Z.; Drake, C.G. Myeloid-derived cells in prostate cancer progression: Phenotype and prospective therapies. J. Leukoc. Biol 2017, 102, 393–406. [Google Scholar] [CrossRef] [Green Version]
- Wen, J.; Huang, G.; Liu, S.; Wan, J.; Wang, X.; Zhu, Y.; Kaliney, W.; Zhang, C.; Cheng, L.; Wen, X.; et al. Polymorphonuclear MDSCs are enriched in the stroma and expanded in metastases of prostate cancer. J. Pathol. Clin. Res. 2020, 6, 171–177. [Google Scholar] [CrossRef]
- Lu, X.; Horner, J.W.; Paul, E.; Shang, X.; Troncoso, P.; Deng, P.; Jiang, S.; Chang, Q.; Spring, D.J.; Sharma, P.; et al. Effective combinatorial immunotherapy for castration-resistant prostate cancer. Nature 2017, 543, 728–732. [Google Scholar] [CrossRef] [Green Version]
- Sternberg, C.; Armstrong, A.; Pili, R.; Ng, S.; Huddart, R.; Agarwal, N.; Khvorostenko, D.; Lyulko, O.; Brize, A.; Vogelzang, N.; et al. Randomized, Double-Blind, Placebo-Controlled Phase III Study of Tasquinimod in Men with Metastatic Castration-Resistant Prostate Cancer. J. Clin. Oncol. 2016, 34, 2636–2643. [Google Scholar] [CrossRef] [PubMed]
- Larionova, I.; Tuguzbaeva, G.; Ponomaryova, A.; Stakheyeva, M.; Cherdyntseva, N.; Pavlov, V.; Choinzonov, E.; Kzhyshkowska, J. Tumor-Associated Macrophages in Human Breast, Colorectal, Lung, Ovarian and Prostate Cancers. Front. Oncol. 2020, 10, 566511. [Google Scholar] [CrossRef]
- Erlandsson, A.; Carlsson, J.; Lundholm, M.; Fält, A.; Andersson, S.-O.; Andrén, O.; Davidsson, S. M2 macrophages and regulatory T cells in lethal prostate cancer. Prostate 2019, 79, 363–369. [Google Scholar] [CrossRef]
- Mantovani, A.; Marchesi, F.; Malesci, A.; Laghi, L.; Allavena, P. Tumour-associated macrophages as treatment targets in oncology. Nat. Rev. Clin. Oncol. 2017, 14, 399–416. [Google Scholar] [CrossRef]
- Lissbrant, I.F.; Stattin, P.; Wikstrom, P.; Damber, J.E.; Egevad, L.; Bergh, A. Tumor associated macrophages in human prostate cancer: Relation to clinicopathological variables and survival. Int. J. Oncol. 2000, 17, 445–496. [Google Scholar] [CrossRef]
- Shimura, S.; Yang, G.; Ebara, S.; Wheeler, T.M.; Frolov, A.; Thompson, T.C. Reduced Infiltration of Tumor-associated Macrophages in Human Prostate Cancer: Association with Cancer Progression1. Cancer Res. 2000, 60, 5857–5861. [Google Scholar]
- Chen, S.; Zhu, G.; Yang, Y.; Wang, F.; Xiao, Y.-T.; Zhang, N.; Bian, X.; Zhu, Y.; Yu, Y.; Liu, F.; et al. Single-cell analysis reveals transcriptomic remodellings in distinct cell types that contribute to human prostate cancer progression. Nat. Cell Biol. 2021, 23, 87–98. [Google Scholar] [CrossRef] [PubMed]
- Thienger, P.; Rubin, M.A. Prostate cancer hijacks the microenvironment. Nat. Cell Biol. 2021, 23, 3–5. [Google Scholar] [CrossRef] [PubMed]
- Seif, F.; Sharifi, L.; Khoshmirsafa, M.; Mojibi, Y.; Mohsenzadegan, M. A Review of Preclinical Experiments toward Targeting M2 Macrophages in Prostate Cancer. Curr. Drug Targets 2019, 20, 789–798. [Google Scholar] [CrossRef] [PubMed]
- Joyce, J.A.; Pollard, J.W. Microenvironmental regulation of metastasis. Nat. Rev. Cancer 2009, 9, 239–252. [Google Scholar] [CrossRef]
- Deryugina, E.I.; Kiosses, W.B. Intratumoral Cancer Cell Intravasation Can Occur Independent of Invasion into the Adjacent Stroma. Cell Rep. 2017, 19, 601–616. [Google Scholar] [CrossRef] [Green Version]
- Bonollo, F.; Thalmann, G.N.; Kruithof-de Julio, M.; Karkampouna, S. The Role of Cancer-Associated Fibroblasts in Prostate Cancer Tumorigenesis. Cancers 2020, 12, 1887. [Google Scholar] [CrossRef]
- Monteran, L.; Erez, N. The Dark Side of Fibroblasts: Cancer-Associated Fibroblasts as Mediators of Immunosuppression in the Tumor Microenvironment. Front. Immunol. 2019, 10, 1835. [Google Scholar] [CrossRef] [Green Version]
- Giannoni, E.; Bianchini, F.; Masieri, L.; Serni, S.; Torre, E.; Calorini, L.; Chiarugi, P. Reciprocal activation of prostate cancer cells and cancer-associated fibroblasts stimulates epithelial-mesenchymal transition and cancer stemness. Cancer Res. 2010, 70, 6945–6956. [Google Scholar] [CrossRef] [Green Version]
- Fukumura, D.; Kloepper, J.; Amoozgar, Z.; Duda, D.G.; Jain, R.K. Enhancing cancer immunotherapy using antiangiogenics: Opportunities and challenges. Nat. Rev. Clin. Oncol. 2018, 15, 325–340. [Google Scholar] [CrossRef]
- Ziani, L.; Safta-Saadoun, T.B.; Gourbeix, J.; Cavalcanti, A.; Robert, C.; Favre, G.; Chouaib, S.; Thiery, J. Melanoma-associated fibroblasts decrease tumor cell susceptibility to NK cell-mediated killing through matrix-metalloproteinases secretion. Oncotarget 2017, 8, 19780–19794. [Google Scholar] [CrossRef] [Green Version]
- Li, T.; Yang, Y.; Hua, X.; Wang, G.; Liu, W.; Jia, C.; Tai, Y.; Zhang, Q.; Chen, G. Hepatocellular carcinoma-associated fibroblasts trigger NK cell dysfunction via PGE2 and IDO. Cancer Lett. 2012, 318, 154–161. [Google Scholar] [CrossRef]
- Ohta, A.; Sitkovsky, M. Role of G-protein-coupled adenosine receptors in downregulation of inflammation and protection from tissue damage. Nature 2001, 414, 916–920. [Google Scholar] [CrossRef] [Green Version]
- Beavis, P.A.; Stagg, J.; Darcy, P.K.; Smyth, M.J. CD73: A potent suppressor of antitumor immune responses. Trends Immunol. 2012, 33, 231–237. [Google Scholar] [CrossRef]
- Stagg, J.; Divisekera, U.; Duret, H.; Sparwasser, T.; Teng, M.W.; Darcy, P.K.; Smyth, M.J. CD73-deficient mice have increased antitumor immunity and are resistant to experimental metastasis. Cancer Res. 2011, 71, 2892–2900. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Wang, L.; Chen, X.; Li, L.; Li, Y.; Ping, Y.; Huang, L.; Yue, D.; Zhang, Z.; Wang, F.; et al. CD39/CD73 upregulation on myeloid-derived suppressor cells via TGF-β-mTOR-HIF-1 signaling in patients with non-small cell lung cancer. Oncoimmunology 2017, 6, e1320011. [Google Scholar] [CrossRef] [Green Version]
- Yu, M.; Guo, G.; Huang, L.; Deng, L.; Chang, C.S.; Achyut, B.R.; Canning, M.; Xu, N.; Arbab, A.S.; Bollag, R.J.; et al. CD73 on cancer-associated fibroblasts enhanced by the A(2B)-mediated feedforward circuit enforces an immune checkpoint. Nat. Commun. 2020, 11, 515. [Google Scholar] [CrossRef]
- Vigano, S.; Alatzoglou, D.; Irving, M.; Ménétrier-Caux, C.; Caux, C.; Romero, P.; Coukos, G. Targeting Adenosine in Cancer Immunotherapy to Enhance T-Cell Function. Front. Immunol. 2019, 10, 925. [Google Scholar] [CrossRef] [Green Version]
- Leone, R.D.; Emens, L.A. Targeting adenosine for cancer immunotherapy. J. Immunother. Cancer 2018, 6, 57. [Google Scholar] [CrossRef] [Green Version]
- Allard, B.; Longhi, M.S.; Robson, S.C.; Stagg, J. The ectonucleotidases CD39 and CD73: Novel checkpoint inhibitor targets. Immunol. Rev. 2017, 276, 121–144. [Google Scholar] [CrossRef] [Green Version]
- Graddis, T.J.; McMahan, C.J.; Tamman, J.; Page, K.J.; Trager, J.B. Prostatic acid phosphatase expression in human tissues. Int. J. Clin. Exp. Pathol. 2011, 4, 295–306. [Google Scholar]
- Bendell, J.; Bauer, T.; Patel, M.; Falchook, G.; Karlix, J.L.; Lim, E.; Mugundu, G.; Mitchell, P.D.; Pouliot, G.P.; Moorthy, G.; et al. Abstract CT026: Evidence of immune activation in the first-in-human Phase Ia dose escalation study of the adenosine 2a receptor antagonist, AZD4635, in patients with advanced solid tumors. Cancer Res. 2019, 79, CT026. [Google Scholar] [CrossRef]
- Wise, D.R.; Gardner, O.; Gilbert, H.N.; Rieger, A.; Paoloni, M.C.; Krishnan, K. A phase Ib/II, open-label, platform study evaluating the efficacy and safety of AB928-based treatment combinations in participants with metastatic castrate-resistant prostate cancer. J. Clin. Oncol. 2020, 38, TPS272. [Google Scholar] [CrossRef]
- Cha, H.R.; Lee, J.H.; Ponnazhagan, S. Revisiting Immunotherapy: A Focus on Prostate Cancer. Cancer Res. 2020, 80, 1615–1623. [Google Scholar] [CrossRef] [Green Version]
- Powers, E.; Karachaliou, G.S.; Kao, C.; Harrison, M.R.; Hoimes, C.J.; George, D.J.; Armstrong, A.J.; Zhang, T. Novel therapies are changing treatment paradigms in metastatic prostate cancer. J. Hematol. Oncol. 2020, 13, 144. [Google Scholar] [CrossRef] [PubMed]
- Maiorano, B.A.; Schinzari, G.; Ciardiello, D.; Rodriquenz, M.G.; Cisternino, A.; Tortora, G.; Maiello, E. Cancer Vaccines for Genitourinary Tumors: Recent Progresses and Future Possibilities. Vaccines 2021, 9, 623. [Google Scholar] [CrossRef] [PubMed]
- Arlen, P.M.; Mohebtash, M.; Madan, R.A.; Gulley, J.L. Promising novel immunotherapies and combinations for prostate cancer. Future Oncol. 2009, 5, 187–196. [Google Scholar] [CrossRef] [Green Version]
- Marshall, C.H.; Park, J.C.; DeWeese, T.L.; King, S.; Afful, M.; Hurrelbrink, J.; Manogue, C.; Cotogno, P.; Moldawer, N.P.; Barata, P.C.; et al. Randomized phase II study of sipuleucel-T (SipT) with or without radium-223 (Ra223) in men with asymptomatic bone-metastatic castrate-resistant prostate cancer (mCRPC). J. Clin. Oncol. 2020, 38, 130. [Google Scholar] [CrossRef]
- Sutherland, S.I.M.; Ju, X.; Horvath, L.G.; Clark, G.J. Moving on From Sipuleucel-T: New Dendritic Cell Vaccine Strategies for Prostate Cancer. Front. Immunol. 2021, 12, 641307. [Google Scholar] [CrossRef]
- Vogelzang, N.J.; Beer, T.M.; Gerritsen, W.; Oudard, S.; Wiechno, P.; Kukielka-Budny, B.; Samal, V.; Hajek, J.; Feyerabend, S.; Khoo, V.; et al. Efficacy and Safety of Autologous Dendritic Cell-Based Immunotherapy, Docetaxel, and Prednisone vs Placebo in Patients with Metastatic Castration-Resistant Prostate Cancer: The VIABLE Phase 3 Randomized Clinical Trial. JAMA Oncol. 2022, e217298, in press. [Google Scholar] [CrossRef]
- Schuhmacher, J.; Heidu, S.; Balchen, T.; Richardson, J.R.; Schmeltz, C.; Sonne, J.; Schweiker, J.; Rammensee, H.G.; Thor Straten, P.; Røder, M.A.; et al. Vaccination against RhoC induces long-lasting immune responses in patients with prostate cancer: Results from a phase I/II clinical trial. J. Immunother Cancer 2020, 8, e001157. [Google Scholar] [CrossRef]
- Lilleby, W.; Gaudernack, G.; Brunsvig, P.F.; Vlatkovic, L.; Schulz, M.; Mills, K.; Hole, K.H.; Inderberg, E.M. Phase I/IIa clinical trial of a novel hTERT peptide vaccine in men with metastatic hormone-naive prostate cancer. Cancer Immunol. Immunother. 2017, 66, 891–901. [Google Scholar] [CrossRef]
- Yoshimura, K.; Minami, T.; Nozawa, M.; Kimura, T.; Egawa, S.; Fujimoto, H.; Yamada, A.; Itoh, K.; Uemura, H. A Phase 2 Randomized Controlled Trial of Personalized Peptide Vaccine Immunotherapy with Low-dose Dexamethasone Versus Dexamethasone Alone in Chemotherapy-naive Castration-resistant Prostate Cancer. Eur. Urol. 2016, 70, 35–41. [Google Scholar] [CrossRef] [Green Version]
- Kantoff, P.W.; Schuetz, T.J.; Blumenstein, B.A.; Glode, L.M.; Bilhartz, D.L.; Wyand, M.; Manson, K.; Panicali, D.L.; Laus, R.; Schlom, J.; et al. Overall survival analysis of a phase II randomized controlled trial of a Poxviral-based PSA-targeted immunotherapy in metastatic castration-resistant prostate cancer. J. Clin. Oncol. 2010, 28, 1099–1105. [Google Scholar] [CrossRef]
- Gulley, J.L.; Borre, M.; Vogelzang, N.J.; Ng, S.; Agarwal, N.; Parker, C.C.; Pook, D.W.; Rathenborg, P.; Flaig, T.W.; Carles, J.; et al. Phase III Trial of PROSTVAC in Asymptomatic or Minimally Symptomatic Metastatic Castration-Resistant Prostate Cancer. J. Clin. Oncol. 2019, 37, 1051–1061. [Google Scholar] [CrossRef]
- Zhou, S.; Gravekamp, C.; Bermudes, D.; Liu, K. Tumour-targeting bacteria engineered to fight cancer. Nat. Rev. Cancer 2018, 18, 727–743. [Google Scholar] [CrossRef]
- Gupta, K.H.; Nowicki, C.; Giurini, E.F.; Marzo, A.L.; Zloza, A. Bacterial-Based Cancer Therapy (BBCT): Recent Advances, Current Challenges, and Future Prospects for Cancer Immunotherapy. Vaccines 2021, 9, 1497. [Google Scholar] [CrossRef]
- Stein, M.N.; Fong, L.; Mega, A.E.; Lam, E.T.; Heyburn, J.W.; GUTIERREZ, A.A.; Parsi, M.; Vangala, S.; Haas, N.B. KEYNOTE-046 (Part B): Effects of ADXS-PSA in combination with pembrolizumab on survival in metastatic, castration-resistant prostate cancer patients with or without prior exposure to docetaxel. J. Clin. Oncol. 2020, 38, 126. [Google Scholar] [CrossRef]
- Gamat-Huber, M.; Jeon, D.; Johnson, L.E.; Moseman, J.E.; Muralidhar, A.; Potluri, H.K.; Rastogi, I.; Wargowski, E.; Zahm, C.D.; McNeel, D.G. Treatment Combinations with DNA Vaccines for the Treatment of Metastatic Castration-Resistant Prostate Cancer (mCRPC). Cancers 2020, 12, 2831. [Google Scholar] [CrossRef]
- McNeel, D.G.; Eickhoff, J.C.; Johnson, L.E.; Roth, A.R.; Perk, T.G.; Fong, L.; Antonarakis, E.S.; Wargowski, E.; Jeraj, R.; Liu, G. Phase II Trial of a DNA Vaccine Encoding Prostatic Acid Phosphatase (pTVG-HP [MVI-816]) in Patients with Progressive, Nonmetastatic, Castration-Sensitive Prostate Cancer. J. Clin. Oncol. 2019, 37, 3507–3517. [Google Scholar] [CrossRef]
- Shore, N.D.; Morrow, M.P.; McMullan, T.; Kraynyak, K.A.; Sylvester, A.; Bhatt, K.; Cheung, J.; Boyer, J.D.; Liu, L.; Sacchetta, B.; et al. CD8(+) T Cells Impact Rising PSA in Biochemically Relapsed Cancer Patients Using Immunotherapy Targeting Tumor-Associated Antigens. Mol. Ther. 2020, 28, 1238–1250. [Google Scholar] [CrossRef]
- Kyriakopoulos, C.E.; Eickhoff, J.C.; Ferrari, A.C.; Schweizer, M.T.; Wargowski, E.; Olson, B.M.; McNeel, D.G. Multicenter Phase I Trial of a DNA Vaccine Encoding the Androgen Receptor Ligand-binding Domain (pTVG-AR, MVI-118) in Patients with Metastatic Prostate Cancer. Clin. Cancer Res. 2020, 26, 5162–5171. [Google Scholar] [CrossRef] [PubMed]
- Stenzl, A.; Feyerabend, S.; Syndikus, I.; Sarosiek, T.; Kübler, H.; Heidenreich, A.; Cathomas, R.; Grüllich, C.; Loriot, Y.; Perez Gracia, S.L.; et al. Results of the randomized, placebo-controlled phase I/IIB trial of CV9104, an mRNA based cancer immunotherapy, in patients with metastatic castration-resistant prostate cancer (mCRPC). Ann. Oncol. 2017, 28, v408–v409. [Google Scholar] [CrossRef]
- Beer, T.M.; Kwon, E.D.; Drake, C.G.; Fizazi, K.; Logothetis, C.; Gravis, G.; Ganju, V.; Polikoff, J.; Saad, F.; Humanski, P.; et al. Randomized, Double-Blind, Phase III Trial of Ipilimumab Versus Placebo in Asymptomatic or Minimally Symptomatic Patients with Metastatic Chemotherapy-Naive Castration-Resistant Prostate Cancer. J. Clin. Oncol. 2017, 35, 40–47. [Google Scholar] [CrossRef] [PubMed]
- Kwon, E.D.; Drake, C.G.; Scher, H.I.; Fizazi, K.; Bossi, A.; van den Eertwegh, A.J.; Krainer, M.; Houede, N.; Santos, R.; Mahammedi, H.; et al. Ipilimumab versus placebo after radiotherapy in patients with metastatic castration-resistant prostate cancer that had progressed after docetaxel chemotherapy (CA184-043): A multicentre, randomised, double-blind, phase 3 trial. Lancet Oncol. 2014, 15, 700–712. [Google Scholar] [CrossRef] [Green Version]
- Cabel, L.; Loir, E.; Gravis, G.; Lavaud, P.; Massard, C.; Albiges, L.; Baciarello, G.; Loriot, Y.; Fizazi, K. Long-term complete remission with Ipilimumab in metastatic castrate-resistant prostate cancer: Case report of two patients. J. Immunother. Cancer 2017, 5, 31. [Google Scholar] [CrossRef] [Green Version]
- Basnet, A.; Khullar, G.; Mehta, R.; Chittoria, N. A Case of Locally Advanced Castration-resistant Prostate Cancer with Remarkable Response to Nivolumab. Clin. Genitourin. Cancer 2017, 15, e881–e884. [Google Scholar] [CrossRef]
- Fizazi, K.; Drake, C.G.; Beer, T.M.; Kwon, E.D.; Scher, H.I.; Gerritsen, W.R.; Bossi, A.; den Eertwegh, A.; Krainer, M.; Houede, N.; et al. Final Analysis of the Ipilimumab Versus Placebo Following Radiotherapy Phase III Trial in Postdocetaxel Metastatic Castration-resistant Prostate Cancer Identifies an Excess of Long-term Survivors. Eur. Urol. 2020, 78, 822–830. [Google Scholar] [CrossRef]
- Subudhi, S.K.; Vence, L.; Zhao, H.; Blando, J.; Yadav, S.S.; Xiong, Q.; Reuben, A.; Aparicio, A.; Corn, P.G.; Chapin, B.F.; et al. Neoantigen responses, immune correlates, and favorable outcomes after ipilimumab treatment of patients with prostate cancer. Sci. Transl. Med. 2020, 12, eaaz3577. [Google Scholar] [CrossRef]
- Antonarakis, E.S.; Piulats, J.M.; Gross-Goupil, M.; Goh, J.; Ojamaa, K.; Hoimes, C.J.; Vaishampayan, U.; Berger, R.; Sezer, A.; Alanko, T.; et al. Pembrolizumab for Treatment-Refractory Metastatic Castration-Resistant Prostate Cancer: Multicohort, Open-Label Phase II KEYNOTE-199 Study. J. Clin. Oncol. 2020, 38, 395–405. [Google Scholar] [CrossRef]
- Topalian, S.L.; Hodi, F.S.; Brahmer, J.R.; Gettinger, S.N.; Smith, D.C.; McDermott, D.F.; Powderly, J.D.; Carvajal, R.D.; Sosman, J.A.; Atkins, M.B.; et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N. Engl. J. Med. 2012, 366, 2443–2454. [Google Scholar] [CrossRef]
- Hansen, A.R.; Massard, C.; Ott, P.A.; Haas, N.B.; Lopez, J.S.; Ejadi, S.; Wallmark, J.M.; Keam, B.; Delord, J.P.; Aggarwal, R.; et al. Pembrolizumab for advanced prostate adenocarcinoma: Findings of the KEYNOTE-028 study. Ann. Oncol. 2018, 29, 1807–1813. [Google Scholar] [CrossRef]
- Petrylak, D.P.; Loriot, Y.; Shaffer, D.R.; Braiteh, F.; Powderly, J.; Harshman, L.C.; Conkling, P.; Delord, J.P.; Gordon, M.; Kim, J.W.; et al. Safety and Clinical Activity of Atezolizumab in Patients with Metastatic Castration-Resistant Prostate Cancer: A Phase I Study. Clin. Cancer Res. 2021, 27, 3360–3369. [Google Scholar] [CrossRef]
- Antonarakis, E.S.; Isaacsson Velho, P.; Fu, W.; Wang, H.; Agarwal, N.; Sacristan Santos, V.; Maughan, B.L.; Pili, R.; Adra, N.; Sternberg, C.N.; et al. CDK12-Altered Prostate Cancer: Clinical Features and Therapeutic Outcomes to Standard Systemic Therapies, Poly (ADP-Ribose) Polymerase Inhibitors, and PD-1 Inhibitors. JCO Precis. Oncol. 2020, 4, 370–381. [Google Scholar] [CrossRef]
- Gao, J.; Ward, J.F.; Pettaway, C.A.; Shi, L.Z.; Subudhi, S.K.; Vence, L.M.; Zhao, H.; Chen, J.; Chen, H.; Efstathiou, E.; et al. VISTA is an inhibitory immune checkpoint that is increased after ipilimumab therapy in patients with prostate cancer. Nat. Med. 2017, 23, 551–555. [Google Scholar] [CrossRef] [Green Version]
- Subudhi, S.K.; Siddiqui, B.A.; Aparicio, A.M.; Yadav, S.S.; Basu, S.; Chen, H.; Jindal, S.; Tidwell, R.S.S.; Varma, A.; Logothetis, C.J.; et al. Combined CTLA-4 and PD-L1 blockade in patients with chemotherapy-naive metastatic castration-resistant prostate cancer is associated with increased myeloid and neutrophil immune subsets in the bone microenvironment. J. Immunother. Cancer 2021, 9, e002919. [Google Scholar] [CrossRef]
- Sharma, P.; Pachynski, R.K.; Narayan, V.; Flechon, A.; Gravis, G.; Galsky, M.D.; Mahammedi, H.; Patnaik, A.; Subudhi, S.K.; Ciprotti, M.; et al. Nivolumab Plus Ipilimumab for Metastatic Castration-Resistant Prostate Cancer: Preliminary Analysis of Patients in the CheckMate 650 Trial. Cancer Cell 2020, 38, 489–499.e483. [Google Scholar] [CrossRef]
- Boudadi, K.; Suzman, D.L.; Anagnostou, V.; Fu, W.; Luber, B.; Wang, H.; Niknafs, N.; White, J.R.; Silberstein, J.L.; Sullivan, R.; et al. Ipilimumab plus nivolumab and DNA-repair defects in AR-V7-expressing metastatic prostate cancer. Oncotarget 2018, 9, 28561–28571. [Google Scholar] [CrossRef] [Green Version]
- Shenderov, E.; Boudadi, K.; Fu, W.; Wang, H.; Sullivan, R.; Jordan, A.; Dowling, D.; Harb, R.; Schonhoft, J.; Jendrisak, A.; et al. Nivolumab plus ipilimumab, with or without enzalutamide, in AR-V7-expressing metastatic castration-resistant prostate cancer: A phase-2 nonrandomized clinical trial. Prostate 2021, 81, 326–338. [Google Scholar] [CrossRef]
- Graff, J.N.; Beer, T.M.; Alumkal, J.J.; Slottke, R.E.; Redmond, W.L.; Thomas, G.V.; Thompson, R.F.; Wood, M.A.; Koguchi, Y.; Chen, Y.; et al. A phase II single-arm study of pembrolizumab with enzalutamide in men with metastatic castration-resistant prostate cancer progressing on enzalutamide alone. J. Immunother. Cancer 2020, 8, e000642. [Google Scholar] [CrossRef]
- Hoimes, C.J.; Graff, J.N.; Tagawa, S.T.; Hwang, C.; Kilari, D.; Tije, A.J.T.; Omlin, A.; McDermott, R.S.; Vaishampayan, U.N.; Elliott, T.; et al. KEYNOTE-199 cohorts (C) 4 and 5: Phase II study of pembrolizumab (pembro) plus enzalutamide (enza) for enza-resistant metastatic castration-resistant prostate cancer (mCRPC). J. Clin. Oncol. 2020, 38, 5543. [Google Scholar] [CrossRef]
- Sweeney, C.J.; Gillessen, S.; Rathkopf, D.; Matsubara, N.; Drake, C.; Fizazi, K.; Piulats, J.M.; Wysocki, P.J.; Buchschacher, G.L.; Doss, J.; et al. Abstract CT014: IMbassador250: A phase III trial comparing atezolizumab with enzalutamide vs enzalutamide alone in patients with metastatic castration-resistant prostate cancer (mCRPC). Cancer Res. 2020, 80, CT014. [Google Scholar] [CrossRef]
- Galluzzi, L.; Buque, A.; Kepp, O.; Zitvogel, L.; Kroemer, G. Immunological Effects of Conventional Chemotherapy and Targeted Anticancer Agents. Cancer Cell 2015, 28, 690–714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fizazi, K.; González Mella, P.; Castellano, D.; Minatta, J.N.; Rezazadeh, A.; Shaffer, D.R.; Vazquez Limon, J.C.; Sánchez López, H.M.; Armstrong, A.J.; Horvath, L.; et al. CheckMate 9KD Arm B final analysis: Efficacy and safety of nivolumab plus docetaxel for chemotherapy-naïve metastatic castration-resistant prostate cancer. Rev. Clin. Oncol. 2021, 39, 12. [Google Scholar] [CrossRef]
- Fizazi, K.; Gonzalez Mella, P.; Castellano, D.; Minatta, J.N.; Rezazadeh Kalebasty, A.; Shaffer, D.; Vazquez Limon, J.C.; Sanchez Lopez, H.M.; Armstrong, A.J.; Horvath, L.; et al. Nivolumab plus docetaxel in patients with chemotherapy-naive metastatic castration-resistant prostate cancer: Results from the phase II CheckMate 9KD trial. Eur. J. Cancer 2022, 160, 61–71. [Google Scholar] [CrossRef]
- Strickland, K.C.; Howitt, B.E.; Shukla, S.A.; Rodig, S.; Ritterhouse, L.L.; Liu, J.F.; Garber, J.E.; Chowdhury, D.; Wu, C.J.; D’Andrea, A.D.; et al. Association and prognostic significance of BRCA1/2-mutation status with neoantigen load, number of tumor-infiltrating lymphocytes and expression of PD-1/PD-L1 in high grade serous ovarian cancer. Oncotarget 2016, 7, 13587–13598. [Google Scholar] [CrossRef] [Green Version]
- Karzai, F.; VanderWeele, D.; Madan, R.A.; Owens, H.; Cordes, L.M.; Hankin, A.; Couvillon, A.; Nichols, E.; Bilusic, M.; Beshiri, M.L.; et al. Activity of durvalumab plus olaparib in metastatic castration-resistant prostate cancer in men with and without DNA damage repair mutations. J. ImmunoTher. Cancer 2018, 6, 141. [Google Scholar] [CrossRef]
- Pachynski, R.K.; Retz, M.; Goh, J.C.; Burotto, M.; Gravis, G.; Castellano, D.; Flechon, A.; Zschaebitz, S.; Shaffer, D.R.; Limon, J.C.V.; et al. CheckMate 9KD cohort A1 final analysis: Nivolumab (NIVO) + rucaparib for post-chemotherapy (CT) metastatic castration-resistant prostate cancer (mCRPC). J. Clin. Oncol. 2021, 39, 5044. [Google Scholar] [CrossRef]
- Petrylak, D.P.; Perez-Gracia, J.L.; Lacombe, L.; Bastos, D.A.; Mahammedi, H.; Kwan, E.M.; Zschäbitz, S.; Armstrong, A.J.; Pachynski, R.K.; Goh, J.C.; et al. 579MO CheckMate 9KD cohort A2 final analysis: Nivolumab (NIVO) + rucaparib for chemotherapy (CT)-naïve metastatic castration-resistant prostate cancer (mCRPC). In Proceedings of the 2021 ESMO Annual Congress, Online, 16–21 September 2021. [Google Scholar]
- Yakes, F.M.; Chen, J.; Tan, J.; Yamaguchi, K.; Shi, Y.; Yu, P.; Qian, F.; Chu, F.; Bentzien, F.; Cancilla, B.; et al. Cabozantinib (XL184), a novel MET and VEGFR2 inhibitor, simultaneously suppresses metastasis, angiogenesis, and tumor growth. Mol. Cancer Ther. 2011, 10, 2298–2308. [Google Scholar] [CrossRef] [Green Version]
- Kwilas, A.R.; Ardiani, A.; Donahue, R.N.; Aftab, D.T.; Hodge, J.W. Dual effects of a targeted small-molecule inhibitor (cabozantinib) on immune-mediated killing of tumor cells and immune tumor microenvironment permissiveness when combined with a cancer vaccine. J. Transl. Med. 2014, 12, 294. [Google Scholar] [CrossRef] [Green Version]
- Tolaney, S.M.; Ziehr, D.R.; Guo, H.; Ng, M.R.; Barry, W.T.; Higgins, M.J.; Isakoff, S.J.; Brock, J.E.; Ivanova, E.V.; Paweletz, C.P.; et al. Phase II and Biomarker Study of Cabozantinib in Metastatic Triple-Negative Breast Cancer Patients. Oncologist 2017, 22, 25–32. [Google Scholar] [CrossRef] [Green Version]
- Agarwal, N.M.B.; Maughan, B.L.; Dorff, T.; Kelly, W.; Fang, B.; McKay, R.; Singh, P.; Pagliaro, L.; Dreicer, R.; Srinivas, S.; et al. Cabozantinib (C) in combination with atezolizumab (A) in patients (pts) with metastatic castration-resistant prostate cancer (mCRPC): Results of expanded cohort 6 of the COSMIC-021 study. Ann. Oncol. 2021, 32, S1283–S1346. [Google Scholar] [CrossRef]
- Postow, M.A.; Callahan, M.K.; Barker, C.A.; Yamada, Y.; Yuan, J.; Kitano, S.; Mu, Z.; Rasalan, T.; Adamow, M.; Ritter, E.; et al. Immunologic correlates of the abscopal effect in a patient with melanoma. N. Engl. J. Med. 2012, 366, 925–931. [Google Scholar] [CrossRef] [Green Version]
- Formenti, S.C.; Rudqvist, N.P.; Golden, E.; Cooper, B.; Wennerberg, E.; Lhuillier, C.; Vanpouille-Box, C.; Friedman, K.; Ferrari de Andrade, L.; Wucherpfennig, K.W.; et al. Radiotherapy induces responses of lung cancer to CTLA-4 blockade. Nat. Med. 2018, 24, 1845–1851. [Google Scholar] [CrossRef]
- Fong, L.; Morris, M.J.; Sartor, O.; Higano, C.S.; Pagliaro, L.; Alva, A.; Appleman, L.J.; Tan, W.; Vaishampayan, U.; Porcu, R.; et al. A Phase Ib Study of Atezolizumab with Radium-223 Dichloride in Men with Metastatic Castration-Resistant Prostate Cancer. Clin. Cancer Res. 2021, 27, 4746–4756. [Google Scholar] [CrossRef]
- Einsele, H.; Borghaei, H.; Orlowski, R.Z.; Subklewe, M.; Roboz, G.J.; Zugmaier, G.; Kufer, P.; Iskander, K.; Kantarjian, H.M. The BiTE (bispecific T-cell engager) platform: Development and future potential of a targeted immuno-oncology therapy across tumor types. Cancer 2020, 126, 3192–3201. [Google Scholar] [CrossRef]
- Offner, S.; Hofmeister, R.; Romaniuk, A.; Kufer, P.; Baeuerle, P.A. Induction of regular cytolytic T cell synapses by bispecific single-chain antibody constructs on MHC class I-negative tumor cells. Mol. Immunol. 2006, 43, 763–771. [Google Scholar] [CrossRef]
- Dang, K.; Castello, G.; Clarke, S.C.; Li, Y.; Balasubramani, A.; Boudreau, A.; Davison, L.; Harris, K.E.; Pham, D.; Sankaran, P.; et al. Attenuating CD3 affinity in a PSMAxCD3 bispecific antibody enables killing of prostate tumor cells with reduced cytokine release. J. Immunother. Cancer 2021, 9, e002488. [Google Scholar] [CrossRef]
- Deegen, P.; Thomas, O.; Nolan-Stevaux, O.; Li, S.; Wahl, J.; Bogner, P.; Aeffner, F.; Friedrich, M.; Liao, M.Z.; Matthes, K.; et al. The PSMA-targeting Half-life Extended BiTE Therapy AMG 160 has Potent Antitumor Activity in Preclinical Models of Metastatic Castration-resistant Prostate Cancer. Clin. Cancer Res. 2021, 27, 2928–2937. [Google Scholar] [CrossRef]
- Tran, B.H.L.; Dorff, T.; Rettig, M.; Lolkema, M.P.; Machiels, J.P.; Rottey, S.; Autio, K.; Greil, R.; Adra, N.; Lemech, C.; et al. #6090 Interim results from a phase 1 study of AMG 160, a half-life extended (HLE), PSMA-targeted, bispecific T-cell engager (BiTE®) immune therapy for metastatic castration-resistant prostate cancer (mCRPC). In Proceedings of the ESMO 2020, Online, 19–21 September 2020. [Google Scholar]
- Lund, M.E.; Howard, C.B.; Thurecht, K.J.; Campbell, D.H.; Mahler, S.M.; Walsh, B.J. A bispecific T cell engager targeting Glypican-1 redirects T cell cytolytic activity to kill prostate cancer cells. BMC Cancer 2020, 20, 1214. [Google Scholar] [CrossRef]
- Yamamoto, K.; Trad, A.; Baumgart, A.; Huske, L.; Lorenzen, I.; Chalaris, A.; Grotzinger, J.; Dechow, T.; Scheller, J.; Rose-John, S. A novel bispecific single-chain antibody for ADAM17 and CD3 induces T-cell-mediated lysis of prostate cancer cells. Biochem. J. 2012, 445, 135–144. [Google Scholar] [CrossRef] [Green Version]
- Giffin, M.J.; Cooke, K.; Lobenhofer, E.K.; Estrada, J.; Zhan, J.; Deegen, P.; Thomas, M.; Murawsky, C.M.; Werner, J.; Liu, S.; et al. AMG 757, a Half-Life Extended, DLL3-Targeted Bispecific T-Cell Engager, Shows High Potency and Sensitivity in Preclinical Models of Small-Cell Lung Cancer. Clin. Cancer Res. 2021, 27, 1526–1537. [Google Scholar] [CrossRef]
- Puca, L.; Gavyert, K.; Sailer, V.; Conteduca, V.; Dardenne, E.; Sigouros, M.; Isse, K.; Kearney, M.; Vosoughi, A.; Fernandez, L.; et al. Delta-like protein 3 expression and therapeutic targeting in neuroendocrine prostate cancer. Sci. Transl. Med. 2019, 11, eaav0891. [Google Scholar] [CrossRef]
- Goebeler, M.E.; Bargou, R.C. T cell-engaging therapies—BiTEs and beyond. Nat. Rev. Clin. Oncol. 2020, 17, 418–434. [Google Scholar] [CrossRef]
- Belmontes, B.; Sawant, D.V.; Zhong, W.; Tan, H.; Kaul, A.; Aeffner, F.; O’Brien, S.A.; Chun, M.; Noubade, R.; Eng, J.; et al. Immunotherapy combinations overcome resistance to bispecific T cell engager treatment in T cell-cold solid tumors. Sci. Transl. Med. 2021, 13, eabd1524. [Google Scholar] [CrossRef] [PubMed]
- Zorko, N.A.; Ryan, C.J. Novel immune engagers and cellular therapies for metastatic castration-resistant prostate cancer: Do we take a BiTe or ride BiKEs, TriKEs, and CARs? Prostate Cancer Prostatic Dis. 2021, 24, 986–996. [Google Scholar] [CrossRef] [PubMed]
- Wolf, P.; Alzubi, J.; Gratzke, C.; Cathomen, T. The potential of CAR T cell therapy for prostate cancer. Nat. Rev. Urol. 2021, 18, 556–571. [Google Scholar] [CrossRef] [PubMed]
- Maude, S.L.; Frey, N.; Shaw, P.A.; Aplenc, R.; Barrett, D.M.; Bunin, N.J.; Chew, A.; Gonzalez, V.E.; Zheng, Z.; Lacey, S.F.; et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N. Engl. J. Med. 2014, 371, 1507–1517. [Google Scholar] [CrossRef] [Green Version]
- Schuster, S.J.; Bishop, M.R.; Tam, C.S.; Waller, E.K.; Borchmann, P.; McGuirk, J.P.; Jäger, U.; Jaglowski, S.; Andreadis, C.; Westin, J.R.; et al. Tisagenlecleucel in Adult Relapsed or Refractory Diffuse Large B-Cell Lymphoma. N. Engl. J. Med. 2019, 380, 45–56. [Google Scholar] [CrossRef]
- Munshi, N.C.; Anderson, L.D., Jr.; Shah, N.; Madduri, D.; Berdeja, J.; Lonial, S.; Raje, N.; Lin, Y.; Siegel, D.; Oriol, A.; et al. Idecabtagene Vicleucel in Relapsed and Refractory Multiple Myeloma. N. Engl. J. Med. 2021, 384, 705–716. [Google Scholar] [CrossRef]
- Porter, D.L.; Hwang, W.T.; Frey, N.V.; Lacey, S.F.; Shaw, P.A.; Loren, A.W.; Bagg, A.; Marcucci, K.T.; Shen, A.; Gonzalez, V.; et al. Chimeric antigen receptor T cells persist and induce sustained remissions in relapsed refractory chronic lymphocytic leukemia. Sci. Transl. Med. 2015, 7, 303ra139. [Google Scholar] [CrossRef] [Green Version]
- Bostwick, D.G.; Pacelli, A.; Blute, M.; Roche, P.; Murphy, G.P. Prostate specific membrane antigen expression in prostatic intraepithelial neoplasia and adenocarcinoma: A study of 184 cases. Cancer 1998, 82, 2256–2261. [Google Scholar] [CrossRef]
- Troyer, J.K.; Beckett, M.L.; Wright, G.L. Detection and characterization of the prostate-specific membrane antigen (PSMA) in tissue extracts and body fluids. Int. J. Cancer 1995, 62, 552–558. [Google Scholar] [CrossRef]
- Ristau, B.T.; O’Keefe, D.S.; Bacich, D.J. The prostate-specific membrane antigen: Lessons and current clinical implications from 20 years of research. Urol. Oncol. 2014, 32, 272–279. [Google Scholar] [CrossRef] [Green Version]
- Reiter, R.E.; Gu, Z.; Watabe, T.; Thomas, G.; Szigeti, K.; Davis, E.; Wahl, M.; Nisitani, S.; Yamashiro, J.; Le Beau, M.M.; et al. Prostate stem cell antigen: A cell surface marker overexpressed in prostate cancer. Proc. Natl. Acad. Sci. USA 1998, 95, 1735–1740. [Google Scholar] [CrossRef] [Green Version]
- Gu, Z.; Thomas, G.; Yamashiro, J.; Shintaku, I.P.; Dorey, F.; Raitano, A.; Witte, O.N.; Said, J.W.; Loda, M.; Reiter, R.E. Prostate stem cell antigen (PSCA) expression increases with high gleason score, advanced stage and bone metastasis in prostate cancer. Oncogene 2000, 19, 1288–1296. [Google Scholar] [CrossRef] [Green Version]
- Lawrence, M.G.; Lai, J.; Clements, J.A. Kallikreins on steroids: Structure, function, and hormonal regulation of prostate-specific antigen and the extended kallikrein locus. Endocr. Rev. 2010, 31, 407–446. [Google Scholar] [CrossRef]
- Lindo, L.; Wilkinson, L.H.; Hay, K.A. Befriending the Hostile Tumor Microenvironment in CAR T-Cell Therapy. Front. Immunol. 2020, 11, 618387. [Google Scholar] [CrossRef]
- Newick, K.; O’Brien, S.; Moon, E.; Albelda, S.M. CAR T Cell Therapy for Solid Tumors. Annu. Rev. Med. 2017, 68, 139–152. [Google Scholar] [CrossRef]
- Junghans, R.P.; Ma, Q.; Rathore, R.; Gomes, E.M.; Bais, A.J.; Lo, A.S.; Abedi, M.; Davies, R.A.; Cabral, H.J.; Al-Homsi, A.S.; et al. Phase I Trial of Anti-PSMA Designer CAR-T Cells in Prostate Cancer: Possible Role for Interacting Interleukin 2-T Cell Pharmacodynamics as a Determinant of Clinical Response. Prostate 2016, 76, 1257–1270. [Google Scholar] [CrossRef]
- Zhang, Q.; Helfand, B.T.; Carneiro, B.A.; Qin, W.; Yang, X.J.; Lee, C.; Zhang, W.; Giles, F.J.; Cristofanilli, M.; Kuzel, T.M. Efficacy against Human Prostate Cancer by Prostate-specific Membrane Antigen-specific, Transforming Growth Factor-β Insensitive Genetically Targeted CD8(+) T-cells Derived from Patients with Metastatic Castrate-resistant Disease. Eur. Urol. 2018, 73, 648–652. [Google Scholar] [CrossRef]
- Carabasi, M.H.; McKean, M.; Stein, M.N.; Schweizer, M.T.; Luke, J.J.; Narayan, V.; Pachynski, R.K.; Parikh, R.A.; Zhang, J.; Fountaine, T.J.; et al. PSMA targeted armored chimeric antigen receptor (CAR) T-cells in patients with advanced mCRPC: A phase I experience. J. Clin. Oncol. 2021, 39, 2534. [Google Scholar] [CrossRef]
- Zheng, Y.; Nandakumar, K.S.; Cheng, K. Optimization of CAR-T Cell-Based Therapies Using Small-Molecule-Based Safety Switches. J. Med. Chem. 2021, 64, 9577–9591. [Google Scholar] [CrossRef]
- Casucci, M.; Falcone, L.; Camisa, B.; Norelli, M.; Porcellini, S.; Stornaiuolo, A.; Ciceri, F.; Traversari, C.; Bordignon, C.; Bonini, C.; et al. Extracellular NGFR Spacers Allow Efficient Tracking and Enrichment of Fully Functional CAR-T Cells Co-Expressing a Suicide Gene. Front. Immunol. 2018, 9, 507. [Google Scholar] [CrossRef] [Green Version]
- Straathof, K.C.; Pulè, M.A.; Yotnda, P.; Dotti, G.; Vanin, E.F.; Brenner, M.K.; Heslop, H.E.; Spencer, D.M.; Rooney, C.M. An inducible caspase 9 safety switch for T-cell therapy. Blood 2005, 105, 4247–4254. [Google Scholar] [CrossRef] [PubMed]
- Brudno, J.N.; Somerville, R.P.; Shi, V.; Rose, J.J.; Halverson, D.C.; Fowler, D.H.; Gea-Banacloche, J.C.; Pavletic, S.Z.; Hickstein, D.D.; Lu, T.L.; et al. Allogeneic T Cells That Express an Anti-CD19 Chimeric Antigen Receptor Induce Remissions of B-Cell Malignancies That Progress after Allogeneic Hematopoietic Stem-Cell Transplantation without Causing Graft-Versus-Host Disease. J. Clin. Oncol. 2016, 34, 1112–1121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Depil, S.; Duchateau, P.; Grupp, S.A.; Mufti, G.; Poirot, L. ’Off-the-shelf’ allogeneic CAR T cells: Development and challenges. Nat. Rev. Drug Discov. 2020, 19, 185–199. [Google Scholar] [CrossRef] [PubMed]
- Martínez Bedoya, D.; Dutoit, V.; Migliorini, D. Allogeneic CAR T Cells: An Alternative to Overcome Challenges of CAR T Cell Therapy in Glioblastoma. Front. Immunol. 2021, 12, 640082. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
| Trial Name | Trial Phase | Estimated Enrolment (pts) | Experimental Therapy | Disease Stage | Required Pre-Treatmet | Primary Endpoint | NCT Number |
|---|---|---|---|---|---|---|---|
| Vaccination (Phase 1/2) | |||||||
| OVM-200-100 | 1 FIH | 36 | OVM-200 | mCRPC or locally advanced | Any first-line therapy | Safety, tolerability | NCT05104515 |
| UR1534 | 1 | 20 | Bcl-xl_42-CAF09b | mHSCP | ADT | Safety | NCT03412786 |
| 17-C-0007 | 1/2 | 29 | PROSTVAC-V/F + Nivo | mCRPC | ADT | Safety | NCT02933255 |
| QuEST1 | 1/2 | 113 | BN-Brachyury + M7824 vs. BN-Brachyury + M7824 + N-803 vs. BN-Brachyury + M7824 + N-803 + Epacadostat | mCRPC | 1 NHA or if MSI high/MMRd: Pembrolizumab or if HRR mutation: Olaparib/Rucaparib | PSA decline of ≥30% (>21 days) and/or OR (RECIST 1.1) | NCT03493945 |
| UW18037 | 2 | 60 | pTVG-HP + Pembrolizumab vs. pTVG-HP + pTVG-AR + Pembrolizumab | mCRPC | ADT | PFS | NCT04090528 |
| PRO-MERIT | 1/2 | 130 | W_pro1 + Cemiplimab | mCRPC | 2-3 lines | DLTs TEAEs ORR (Part 2 Arms 1A and 1B) | NCT04382898 |
| Trial Name | Trial Phase | Estimated Enrolment (pts) | Experimental Therapy | Disease Stage | Required Pretreatment | Primary Endpoint | NCT Number |
| CONTACT-02 | 3 | 580 | Atezolizumab Cabozantinib | mCRPC | 1 NHA docetaxel only in HNPC | Duration of PFS by RECIST1.1 | NCT04446117 |
| EVOLUTION | 2 | 110 | Nivolumab + Ipilimumab + 177 Lu-PSMA | mCRPC | Progression on 1 NHA | PSA-PFS at 1 year | NCT05150236 |
| CheckMate7DX | 3 | 984 | Nivolumab + Docetaxel followed by Nivolumab | mCRPC | 1-2 NHA | rPFS, OS | NCT04100018 |
| KEYNOTE-991 | 3 | 1232 | Pembrolizumab + Enzalutamide | mHNPC | Docetaxel in HNPC allowed | rPFS, OS | NCT04191096 |
| KEYNOTE-641 | 3 | 1200 | Pembrolizumab + Enzalutamide | mCRPC | Chemotherapy-naïve, abiraterone-naïve, or intolerant or progressed on abiraterone | OS, rPFS | NCT03834493 |
| KEYLYNK-010 | 3 | 780 | Pembrolizumab + Olaparib | mCRPC | 1 NHA and Docetaxel | OS, rPFS | NCT03834519 |
| KEYNOTE-921 | 3 | 1000 | Pembrolizumab + Docetaxel | mCRPC | ≤1 NHA or mHSPC or mCRPC | OS, rPFS | NCT03834506 |
| Trial Name | Trial Phase | Estimated Enrolment (pts) | Experimental Therapy | Disease Stage | Required Pretreatment | Primary Endpoint(s) | NCT Number |
| AZD4635 in prostate cancer | 2 | 60 | Module 1: AZD4635 + durvalumab; Module 2: AZD4635 + oleclumab | mCRPC | Progressed on standard of care | ORR, PSA RR (>50%) | NCT04089553 |
| QUEST (combination 1) | 1b/2 | 136 | Cetrelimab + Niraparib | mCRPC | ns | Part 1: incidence of specific toxitities Part 2: ORR | NCT03431350 |
| KRONOS | 1b | 33 | Cetrelimab + Apalutamid | mCRPC | Progression on NHA | Adverse events PSA Response week 12 | NCT03551782 |
| ImmunoProst | 2 | 38 | Nivolumab | +HRD mCRPC 1 | docetaxel | PSA RR (>50%) | NCT03040791 |
| PORTER | 1 | 45 | A: Nivolumab + NKTR-214 B: Nivolumab + SBRT + CDX-301 + Poly-ICLC C: Nivolumab + CDX-301 + INO-5151 | mCRPC | Prior NHA (e.g., abiraterone, enzalutamide, apalautamide) | Incidence and severity of adverse events | NCT03835533 |
| 201808043 CA209-9MW | 1 | 20 | Nivolumab/Ipilimumab/ PROSTVAC/Neoantigen DNA vaccine | mHNPC | Chemohormonal therapy | Safety and Tolerability | NCT03532217 |
| CA209-935 | 2 | 175 | Nivolumab + Ipilimumab (4 times) followed by Nivolumab maintenance | mCRPC with immunogenic signature 2 | 1 line of therapy | Composite response rate 3 | NCT03061539 |
| IMPACT CA209-8JJ (cohort A) | 2 | 40 | Nivolumab + Ipilimumab (4 times) followed by Nivolumab maintenance | mCRPC with CDK12 mutations | ns | PSA RR (>50%) | NCT03570619 |
| INSPIRE CA184-585 | 2 | 75 | Nivolumab + Ipilimumab (4 times) followed by Nivolumab monotherapy | mCRPC with immunogenic phenotype 4 | ns | DCR 5 | NCT04717154 |
| Rad2Nivo CA209-7G6 | 1b/2 | 36 | Nivolumab + Radium223 | Symptomatic mCRPC without visceral Mets | ns | Safety ctDNA reduction after 6 weeks | NCT04109729 |
| PLANE-PC | 2 | 50 | Pembrolizumab + Lenvatinib | Neuroendocrine PCa | ns | rPFS | NCT04848337 |
| Keynote 365 | 1b/2 | 1000 | Cohort A AC: Pembrolizumab + Olaparib Cohort B AC: Pembrolizumab + Docetaxel + Prednisone Cohort C AC: Pembrolizumab + Enzalutamide Cohort D AC: Pembrolizumab + Abiraterone + Prednisone Cohort E AC: Pembrolizumab + Lenvatinib Cohort F t-NE: Pembrolizumab + Lenvatinib Cohort G (AC) Pembrolizumab/Vibostolimab coformulation Cohort H t-NE: Pembrolizumab/Vibostolimab coformulation Cohort I t-NE: Pembrolizumab + Carboplatin + Etoposide | For Cohorts A, B, C, D, E, and G: histologically or cytologically confirmed adenocarcinoma of the prostate without small cell histology Cohorts F, H, and I: neuroendocrine PCa defined by ≥1% neuroendocrine cells in a recent biopsy specimen | Cohort E: Docetaxel + up to 2 NHA Cohort F, G, H, I: Docetaxel + 1 other chemotherapy allowed + up to 2 NHA | 50% PSA RR ORR Number of participants with AEs Number of participants discontinuing study medication due to AEs | NCT02861573 |
| Short trial Title | Trial Phase | Estimated Enrolment (pts) | Experimental Therapy | Disease Stage | Required Pretreatment | Primary Endpoint | NCT Number |
|---|---|---|---|---|---|---|---|
| Safety, Tolerability, Pharmacokinetics, and Efficacy of Acapatamab in Subjects With mCRPC | 1 | 288 | Acapatamab, acapatamab + Pembrolizumab, acapatamab + Etanercept Prophylaxis, acapatamab + Cytochrome P450 Cocktail | mCRPC | ADT, taxane | Safety and tolerability | NCT03792841 |
| A Study of Tarlatamab (AMG 757) in Participants with Neuroendocrine Prostate Cancer | 1b | 60 | Tarlatamab (AMG 757) | Neuroendocrine prostate cancer | 1 line of prior systemic treatment | Safety and tolerability | NCT04702737 |
| Study of AMG 509 in Subjects with Metastatic Castration-Resistant Prostate Cancer | 1 | 110 | AMG 509 | mCRPC | Prior NHA, taxane | Safety and tolerability | NCT04221542 |
| Safety and Efficacy of Therapies for Metastatic Castration-Resistant Prostate Cancer (mCRPC) | 1/2 | 159 | Acapatamab + Enzalutamide, Acapatamab + Abiraterone, Acapatamab + AMG 404 | mCRPC | Safety and tolerability | NCT04631601 | |
| Study with Bispecific Antibody Engaging T cells, in Patients with Progressive Cancer Diseases With Positive PSCA Marker | 1 | 24 | GEM3PSCA | PSCA expressing cancer including prostate carcinoma | Progressive Disease After Standard Systemic Therapy | MTD Incidence and intensity of AEs DLT | NCT03927573 |
| CART-PSMA-TGFβRDN Cells for Castrate-Resistant Prostate Cancer | 1 | 18 | CART-PSMA-TGFβRDN | mCRPC | At least 1 NHA | Safety and tolerability | NCT03089203 |
| P-PSMA-101 CAR-T Cells in the Treatment of Subjects With mCRPC and Advanced Salivary Gland Cancers | 1 | 60 | P-PSMA-101 Rimiducid (safety switch activator) may be administered as indicated | mCRPC | Safety, DLT, efficacy RECIST 1.1 and PCWG3 | NCT04249947 | |
| PSCA-CAR T Cells in Treating Patients with PSCA + mCRPC | 1 | 33 | Autologous Anti-PSCA-CAR-4-1BB/TCRzeta-CD19t-expressing T-lymphocytes | mCRPC | At least 1 NHA | Safety and tolerability Define recommended phase 2 dose | NCT03873805 |
| Safety and Activity Study of PSCA-Targeted CAR-T Cells (BPX-601) in Subjects with Selected Advanced Solid Tumors | 1/2 | 151 | BPX-601: Autologous T cells genetically modified with retrovirus vector containing PSCA-specific CAR and an inducible MyD88/Cluster designation (CD)40 (iMC) co-stimulatory domain Rimiducid: Dimerizer infusion to activate the iMC of the BPX-601 cells for improved proliferation and persistence | mCRPC among others | MTD and/or recommended extension dose of BPX-601 measured by DLT | NCT02744287 | |
| A Study of JNJ-75229414 for Metastatic Castration-Resistant Prostate Cancer Participant | 1 | 60 | KLK2 CAR-T Cells (JNJ-75229414) | mCRPC | At least 1 NHA or one prior chemotherapy | Number and severity of AE, DLT | NCT05022849 |
| Dose-Escalating Trial with UniCAR02-T Cells and PSMA Target Module (TMpPSMA) in Patients with Progressive Disease After Standard Systemic Therapy in Cancers With Positive PSMA Marker | 1 | 35 | UniCAR02-T Cells and PSMA Target Module (TMpPSMA) | mCRPC | Systemic standard therapies | Safety and tolerability, MTD, DLT | NCT04633148 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
von Amsberg, G.; Alsdorf, W.; Karagiannis, P.; Coym, A.; Kaune, M.; Werner, S.; Graefen, M.; Bokemeyer, C.; Merkens, L.; Dyshlovoy, S.A. Immunotherapy in Advanced Prostate Cancer—Light at the End of the Tunnel? Int. J. Mol. Sci. 2022, 23, 2569. https://doi.org/10.3390/ijms23052569
von Amsberg G, Alsdorf W, Karagiannis P, Coym A, Kaune M, Werner S, Graefen M, Bokemeyer C, Merkens L, Dyshlovoy SA. Immunotherapy in Advanced Prostate Cancer—Light at the End of the Tunnel? International Journal of Molecular Sciences. 2022; 23(5):2569. https://doi.org/10.3390/ijms23052569
Chicago/Turabian Stylevon Amsberg, Gunhild, Winfried Alsdorf, Panagiotis Karagiannis, Anja Coym, Moritz Kaune, Stefan Werner, Markus Graefen, Carsten Bokemeyer, Lina Merkens, and Sergey A. Dyshlovoy. 2022. "Immunotherapy in Advanced Prostate Cancer—Light at the End of the Tunnel?" International Journal of Molecular Sciences 23, no. 5: 2569. https://doi.org/10.3390/ijms23052569