
Properties and Mechanisms of Flavin-Dependent Monooxygenases and Their Applications in Natural Product Synthesis



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
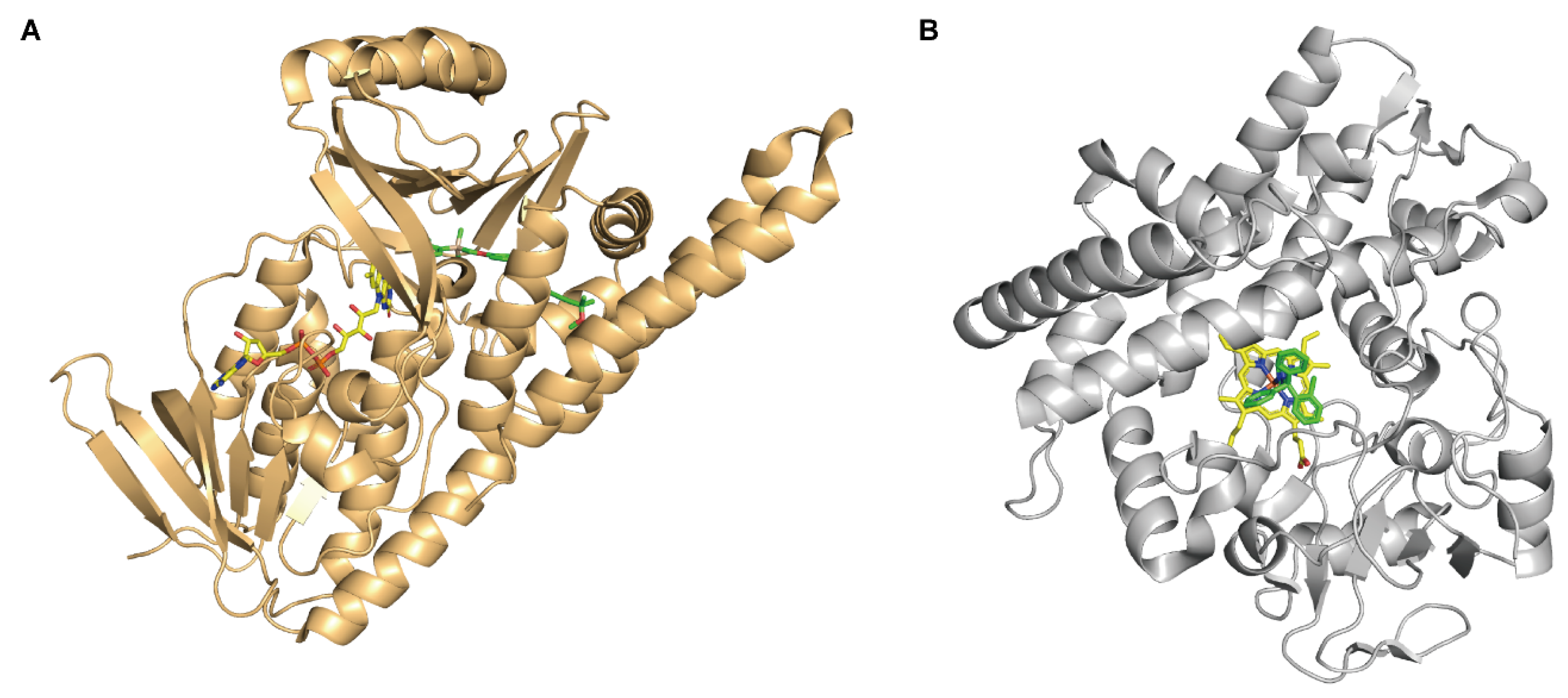
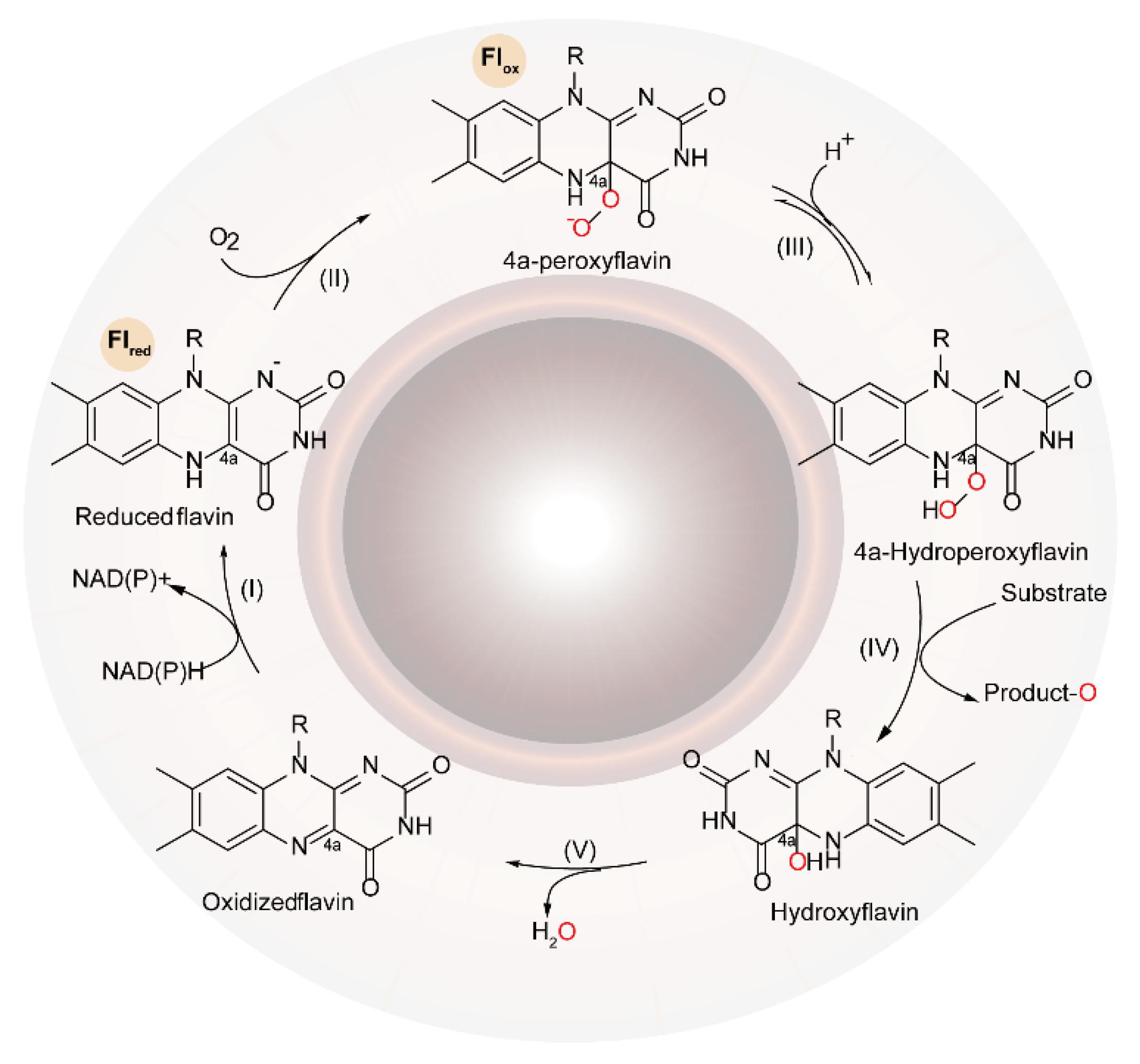
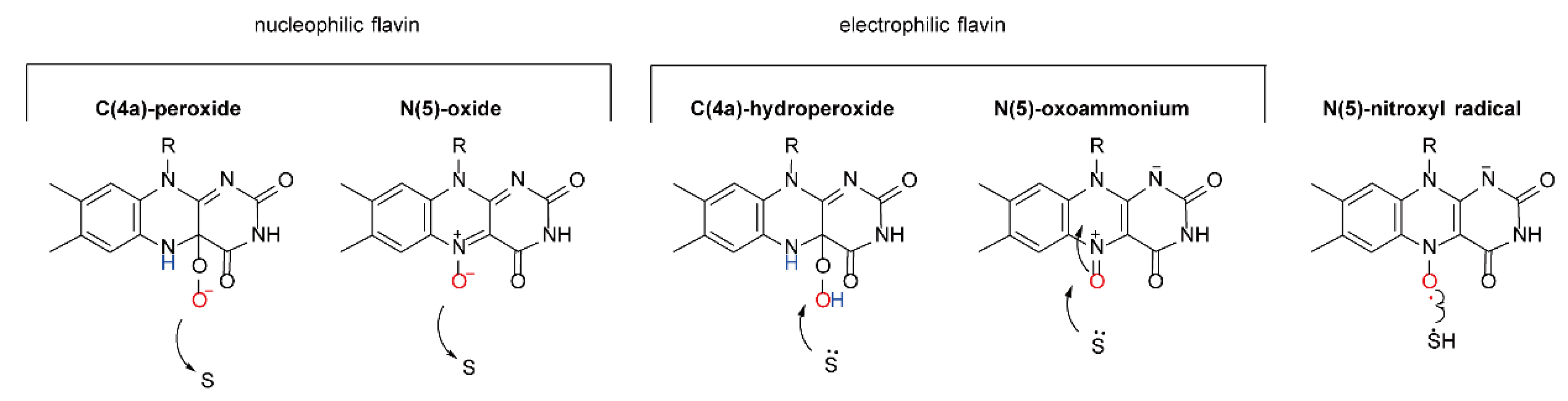
2. The Characteristics and Functional Mechanisms of FMOs
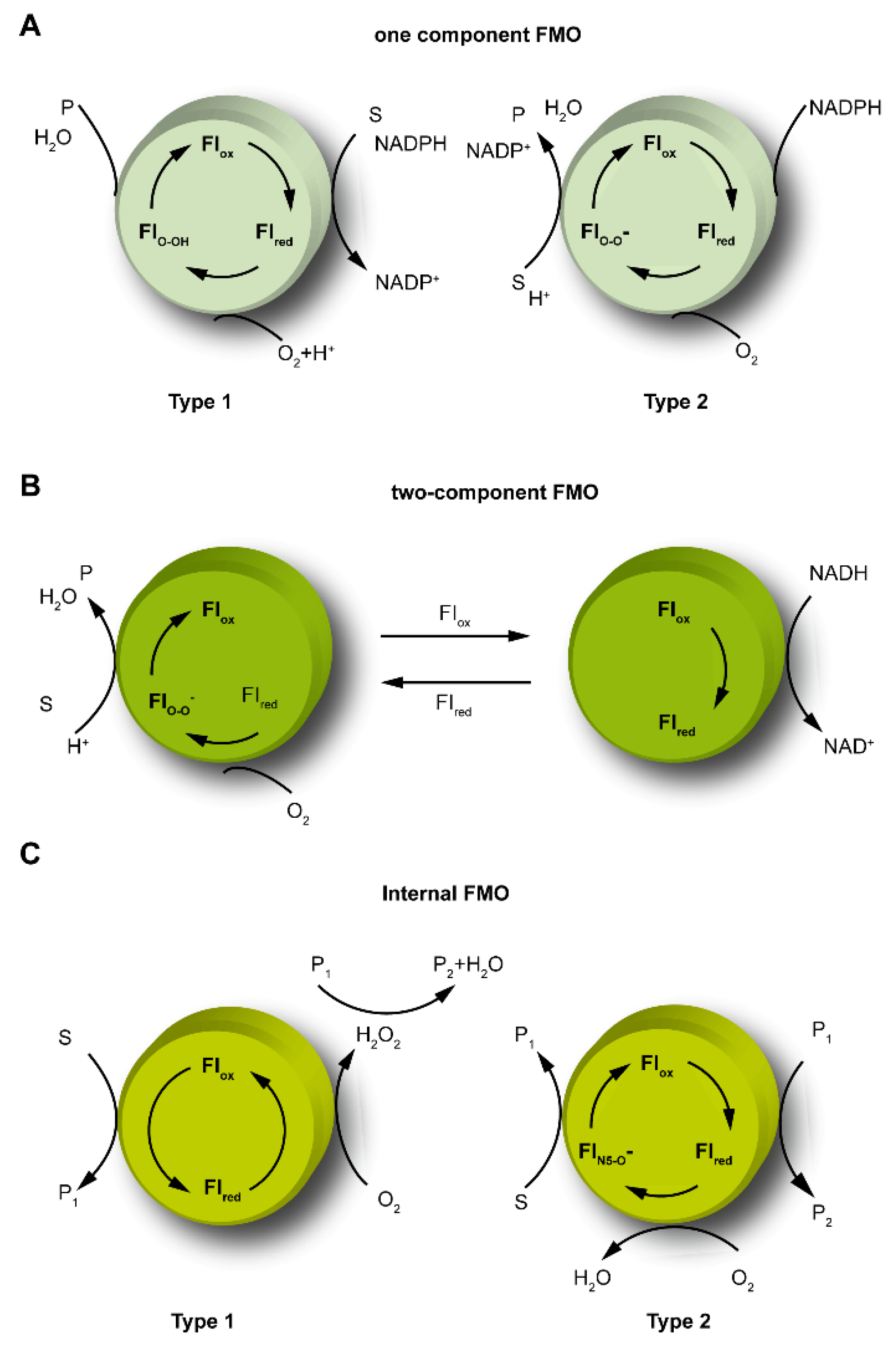
2.1. The Catalytic Mechanism for One-Component FMOs
2.2. The Catalytic Mechanism for Two-Component FMOs
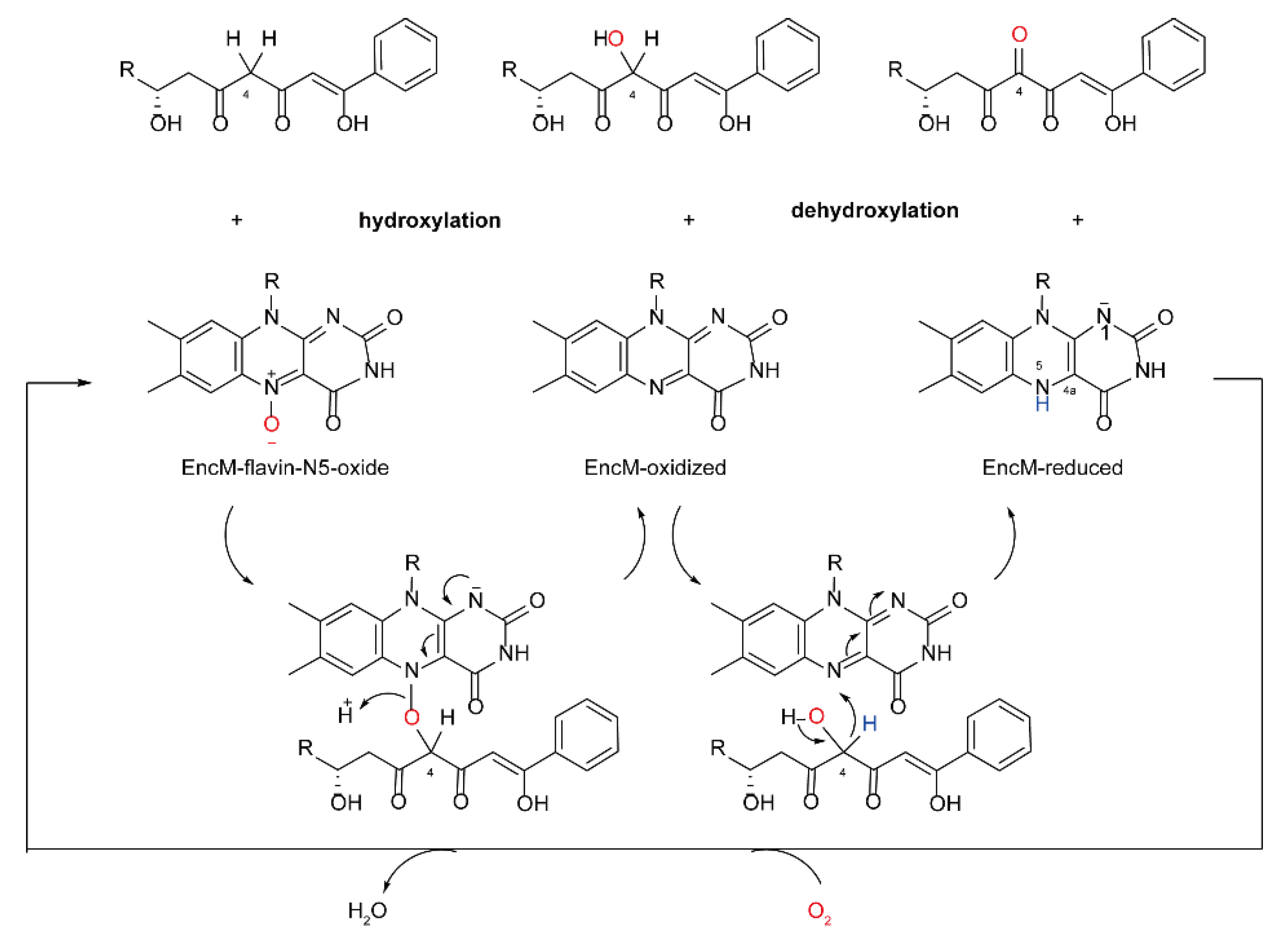
2.3. The Catalytic Mechanism for Internal FMOs
3. The Similarities and Differences between FMOs and CYP450
4. The Substrate Specificity of FMOs
5. Reactions Catalyzed by FMOs in Natural Product Biosynthesis
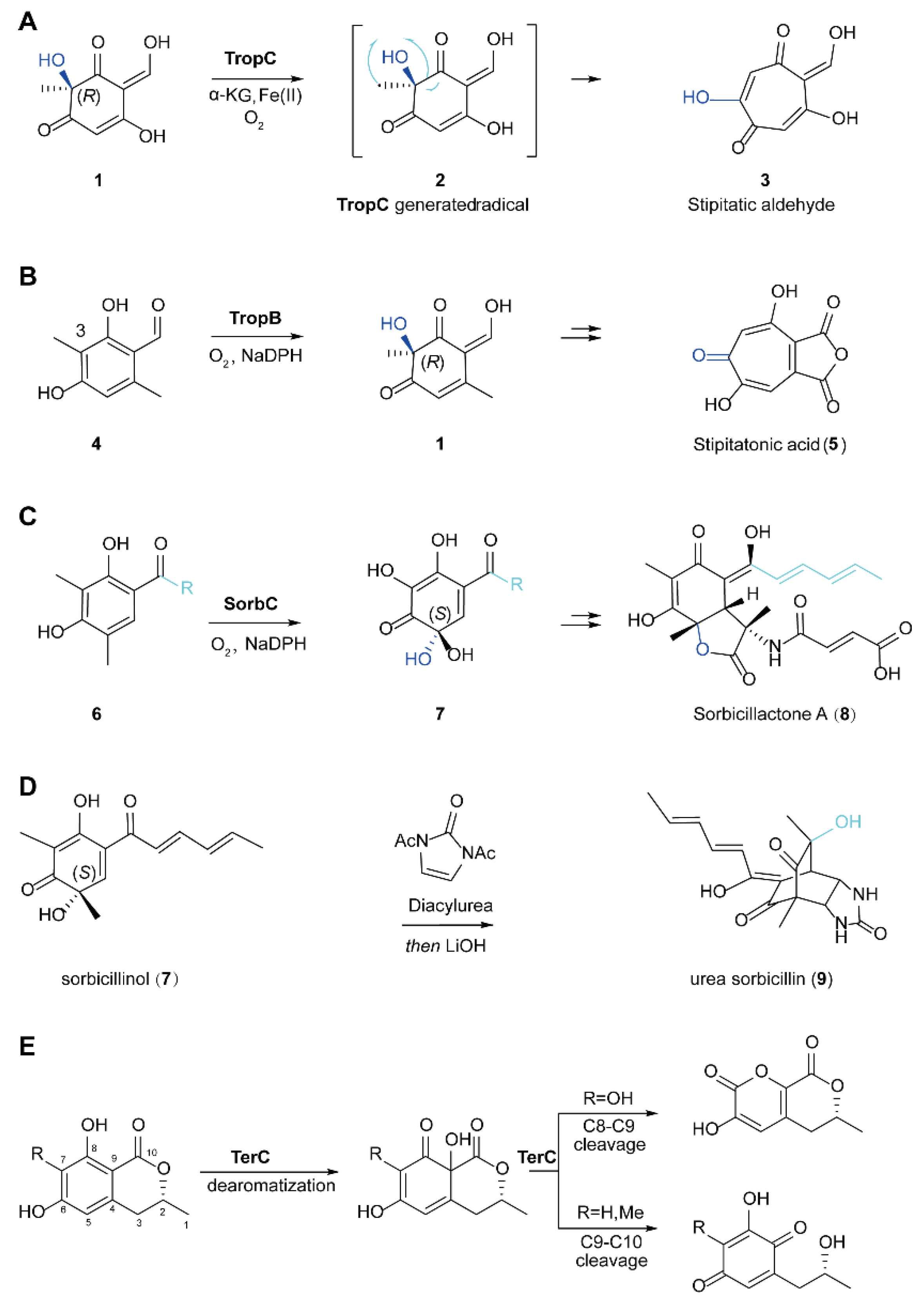
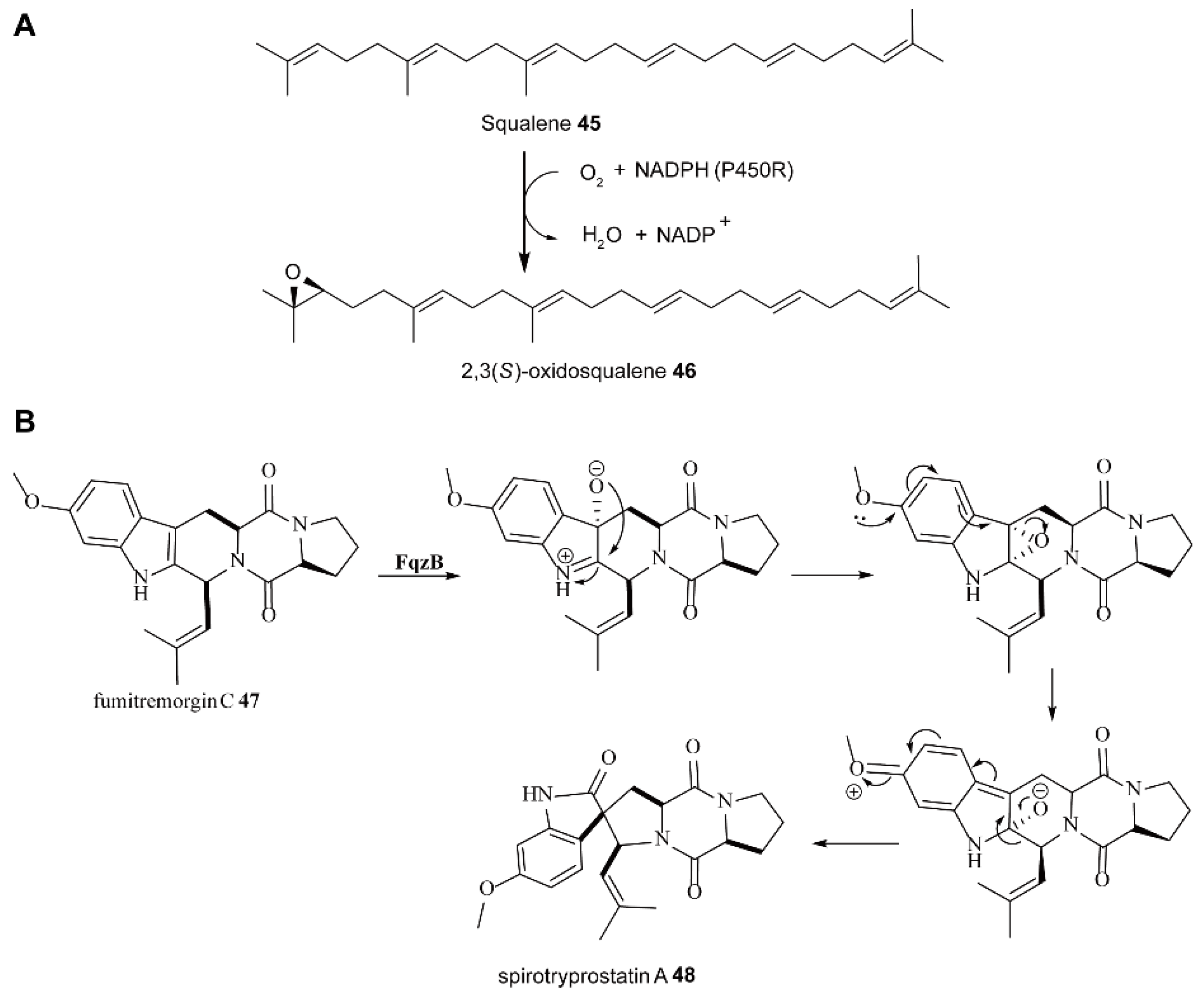
5.1. The Biosynthesis of Natural Products through Dearomatization Catalyzed by FMOs
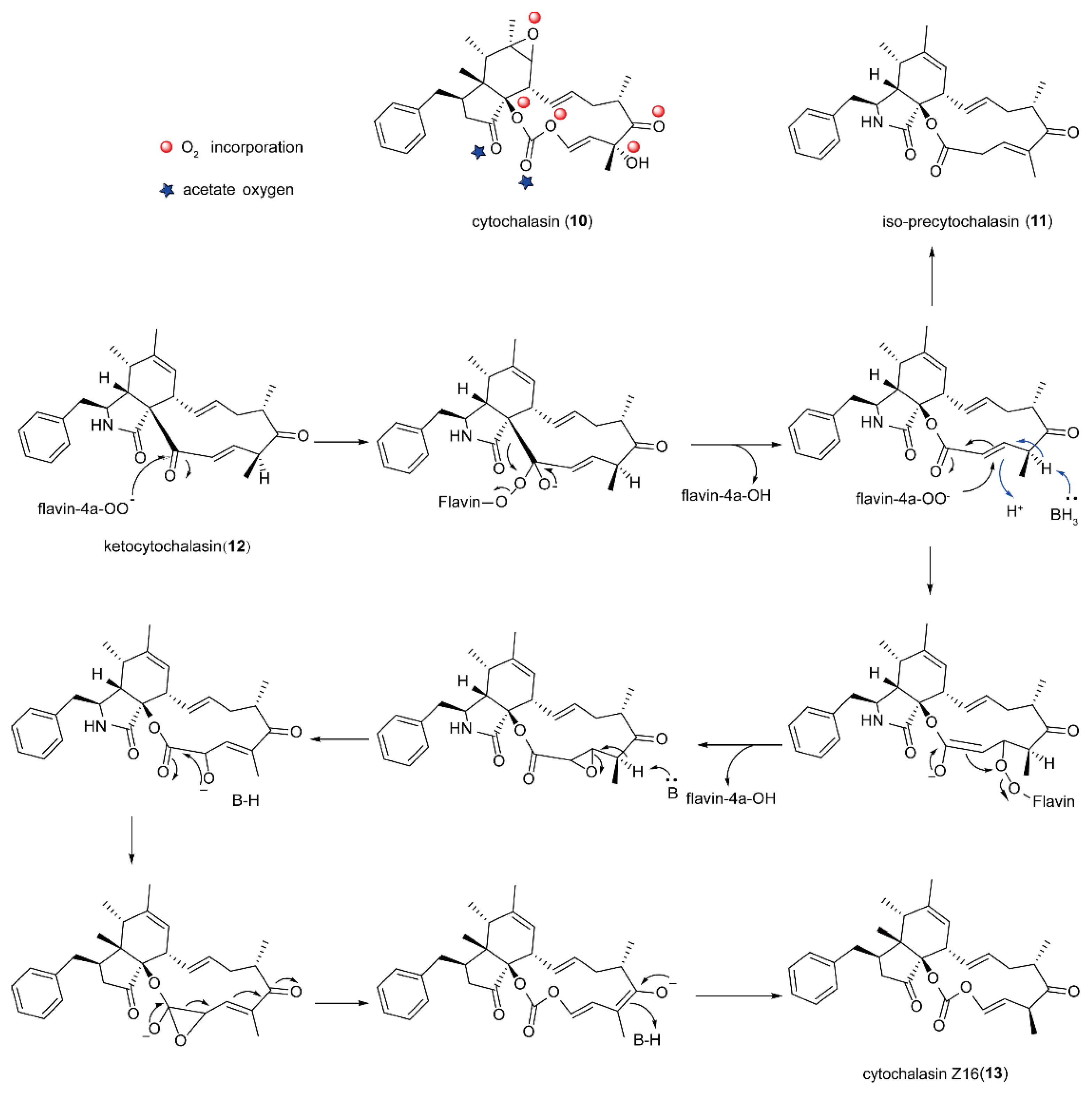
5.2. The Biosynthesis of Cytochalasin Natural Products Catalyzed by FMOs
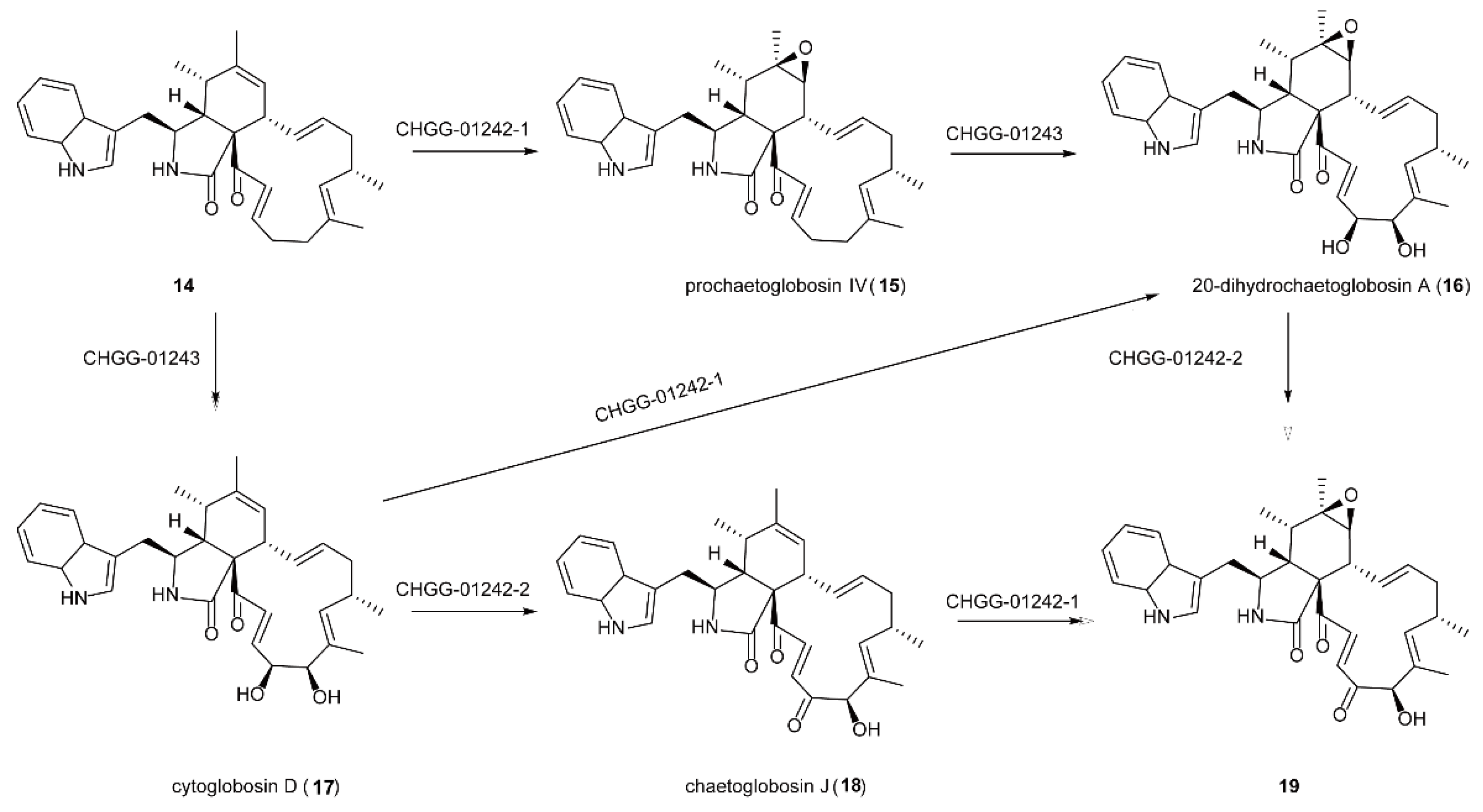
5.3. The Synthesis of Natural Products of Chaetoglobosin A Catalyzed by FMOs
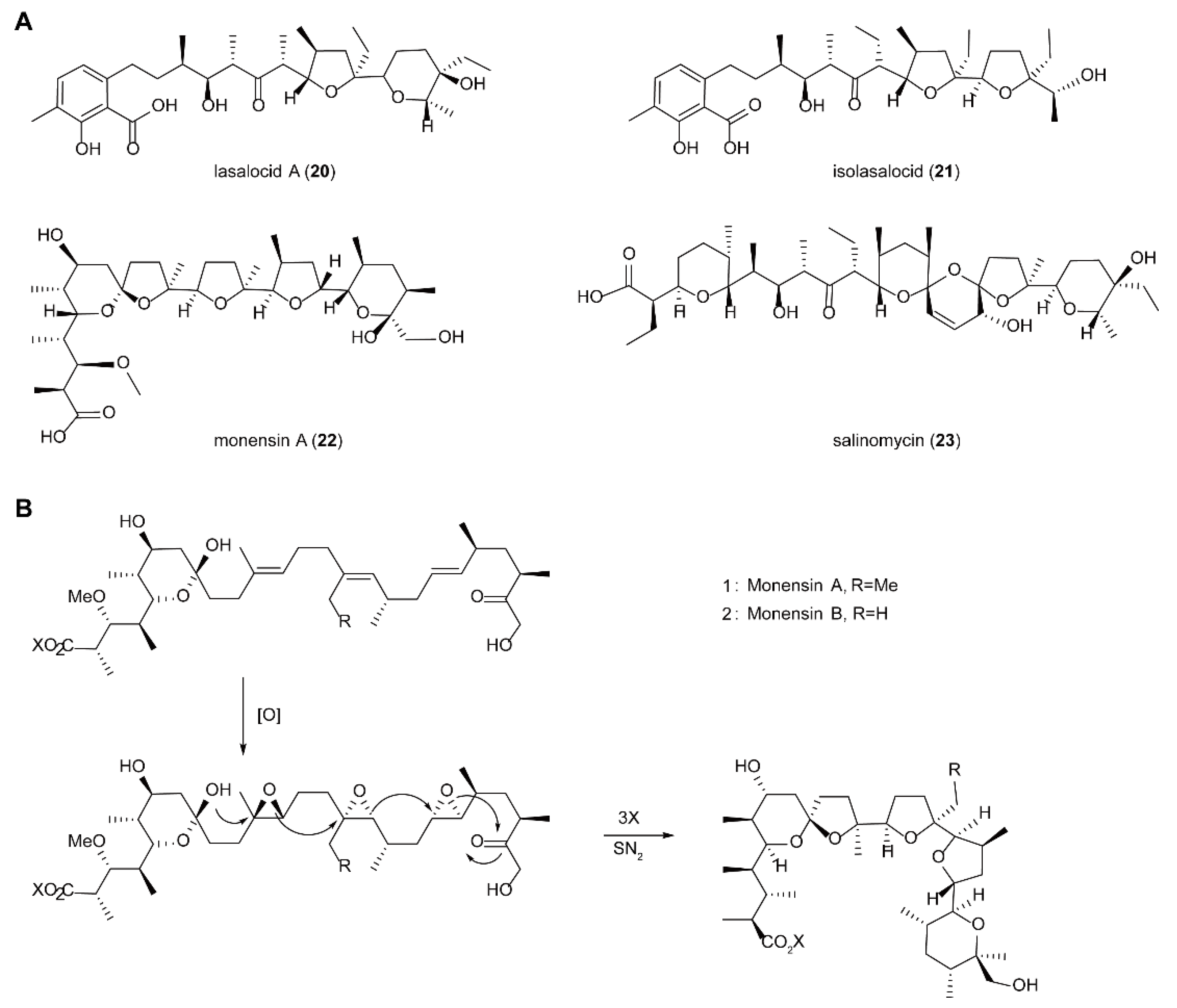
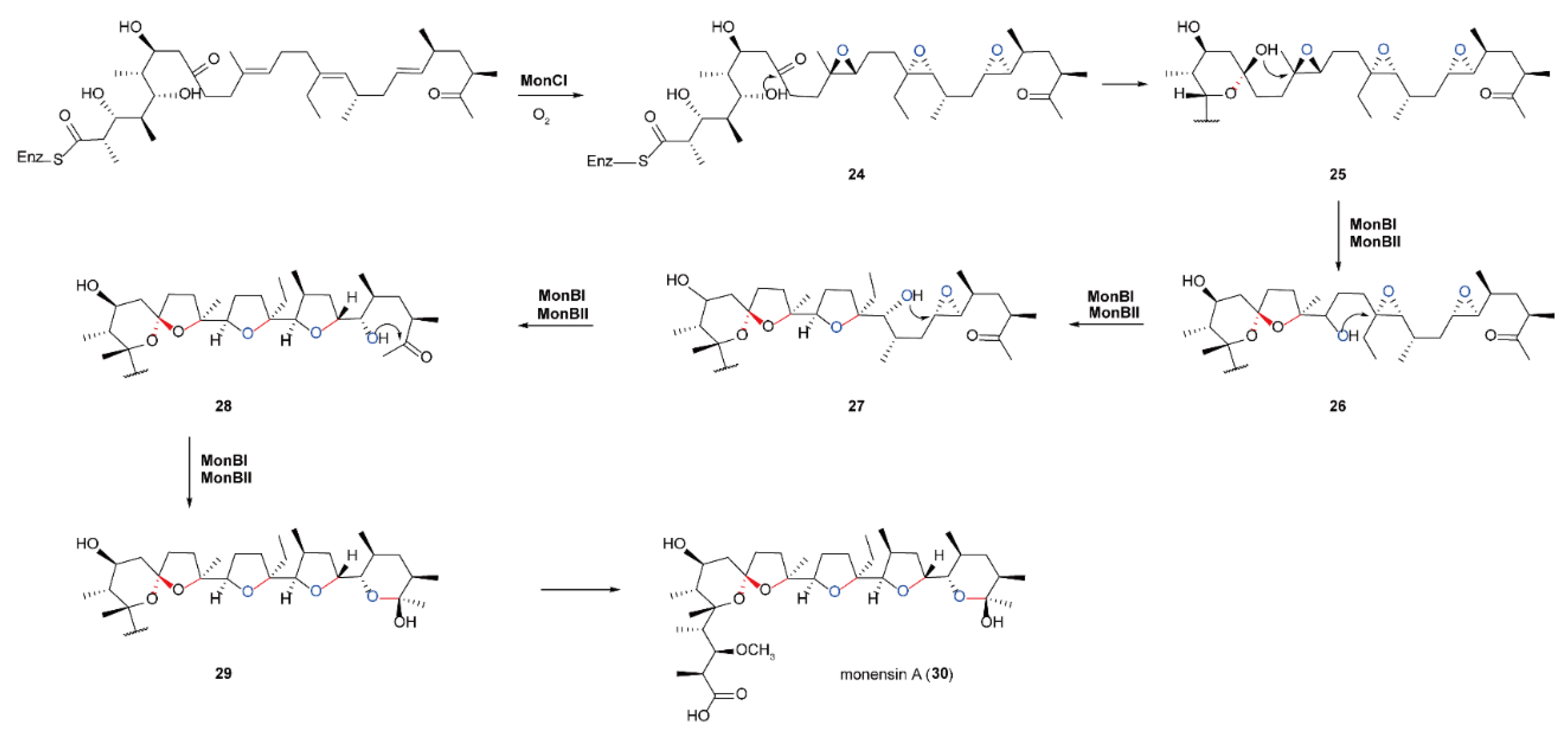
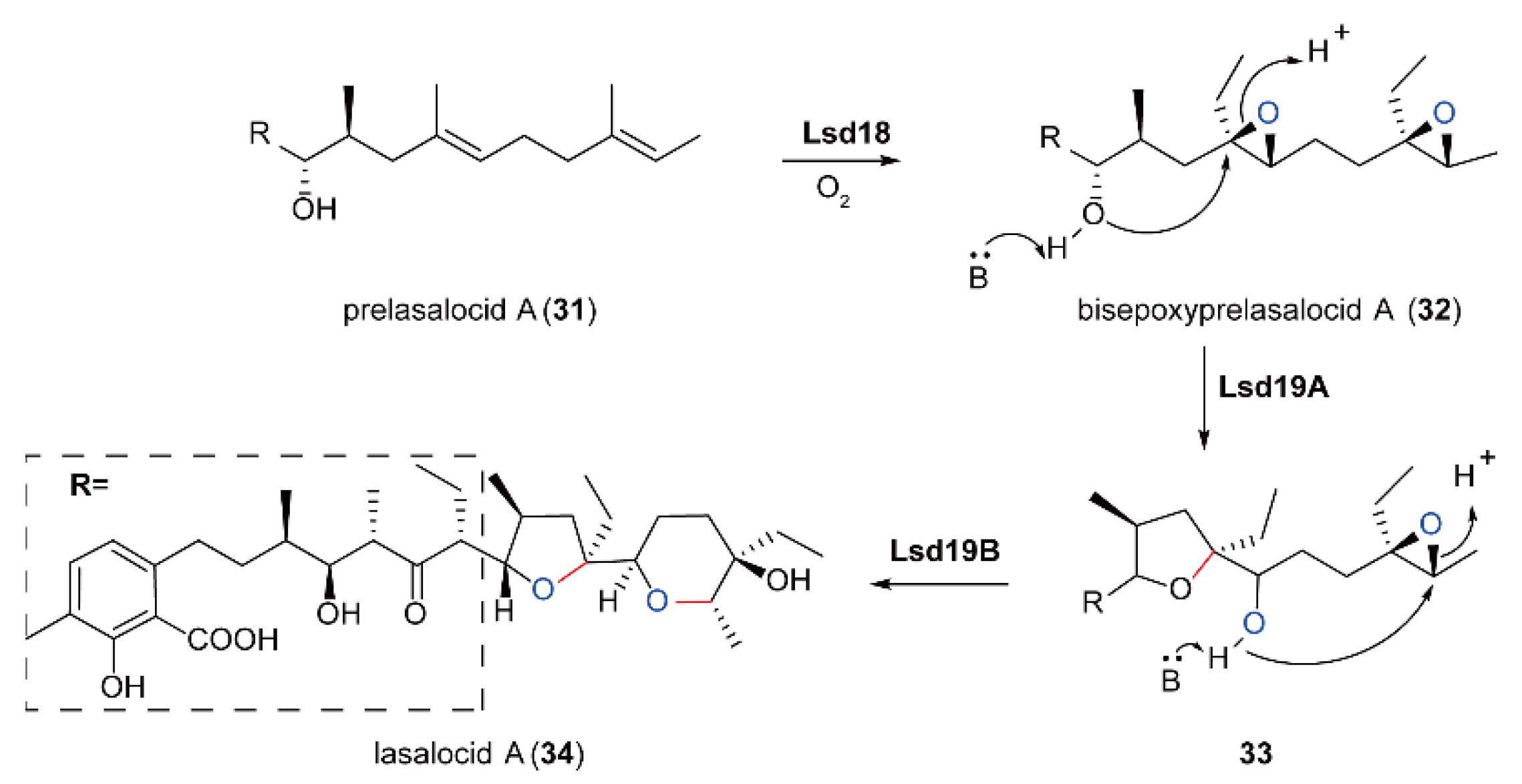
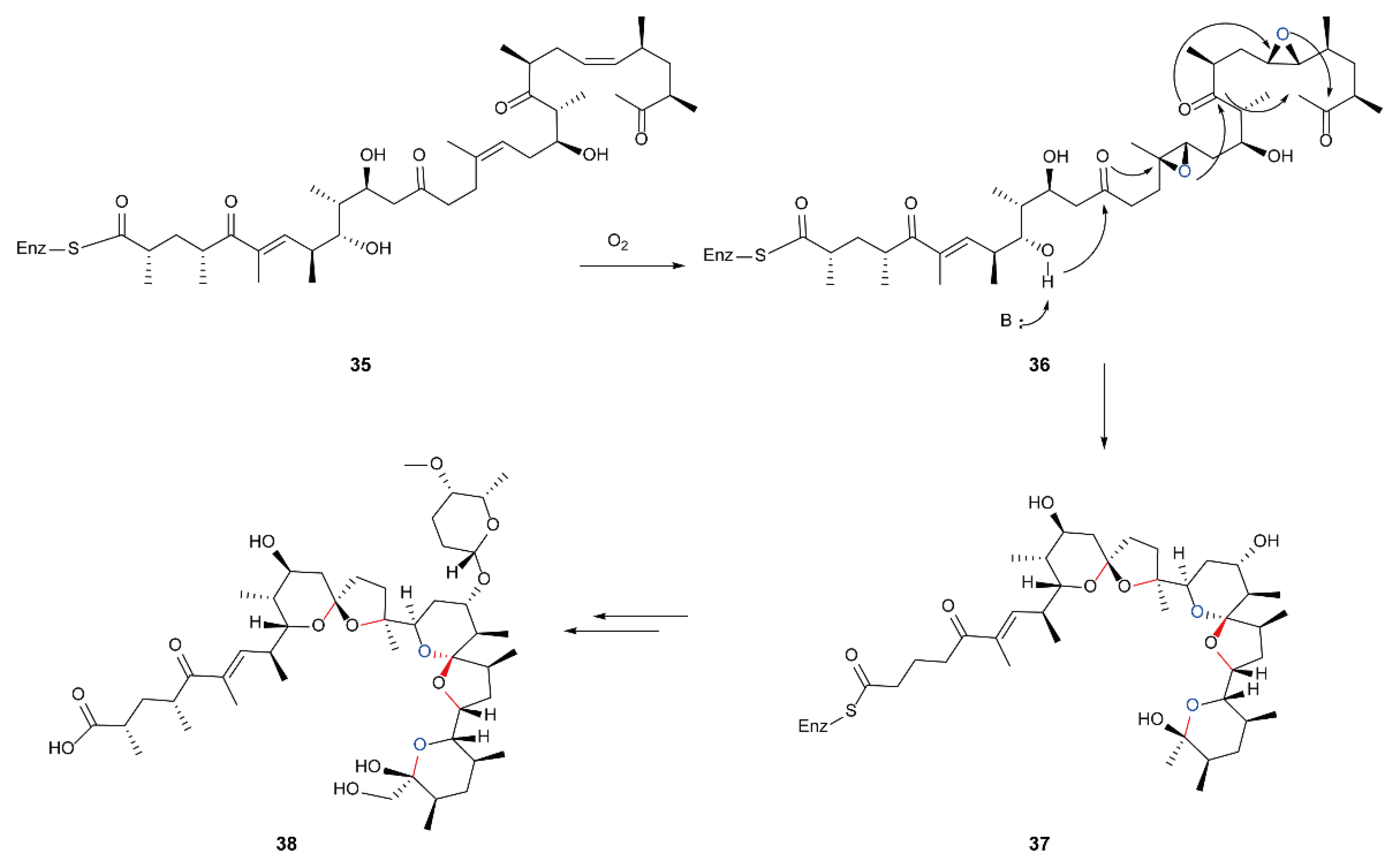
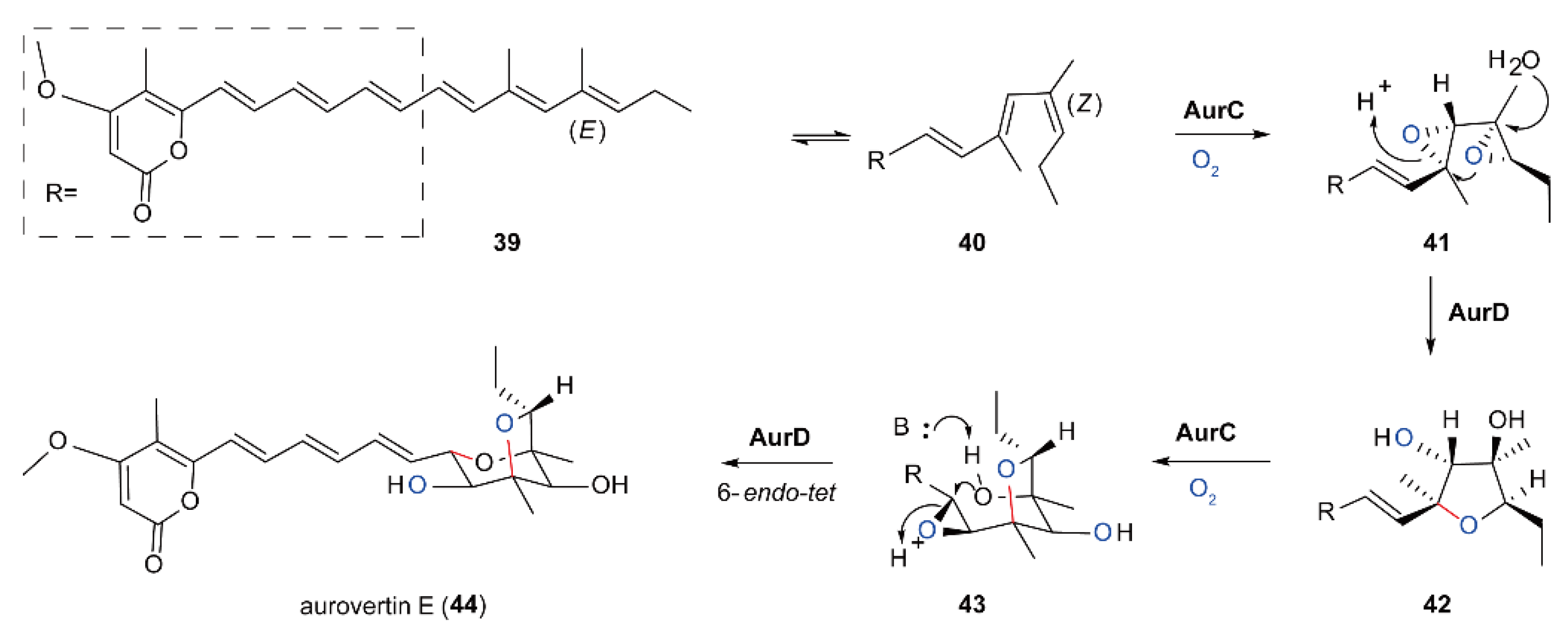
5.4. Biosynthesis of Polyether Natural Products Catalyzed by FMOs
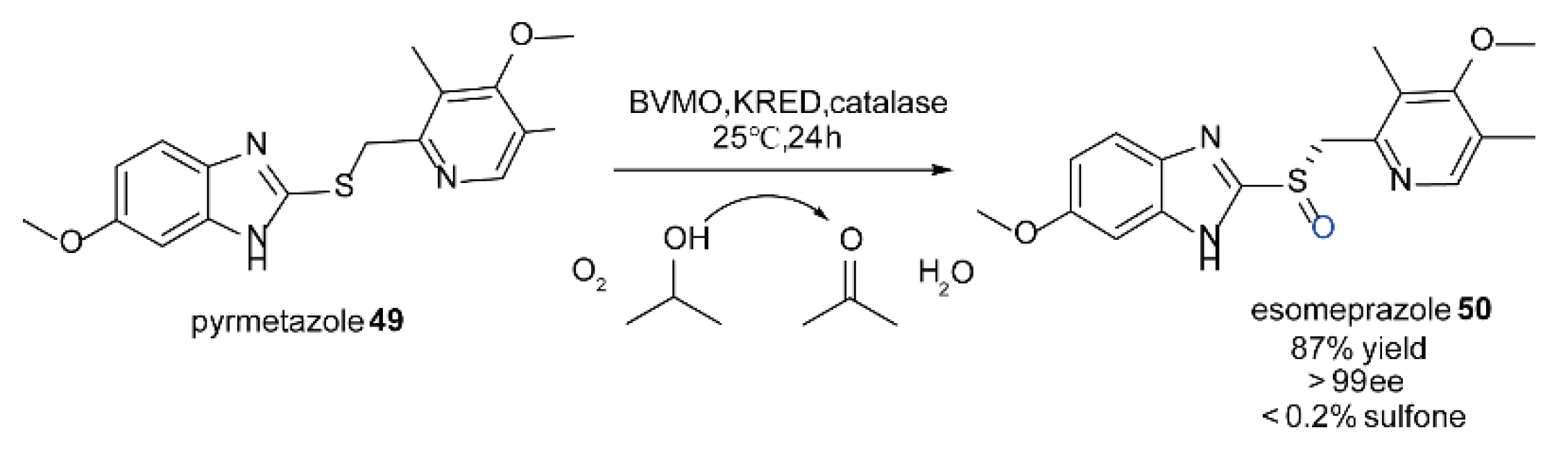
5.5. The Synthesis of Chiral Sulfoxide Compounds Catalyzed by FMOs
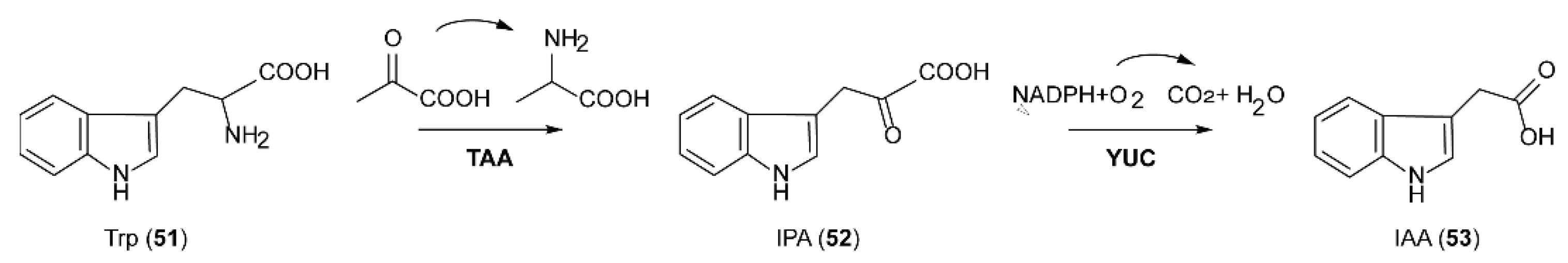
5.6. The Biosynthesis of Auxin Natural Products Catalyzed by FMOs
6. Outlook
Author Contributions
Funding
Conflicts of Interest
References
- De Gonzalo, G.; Mihovilovic, M.D.; Fraaije, M.W. Recent developments in the application of Baeyer-Villiger monooxygenases as biocatalysts. Chembiochem 2010, 11, 2208–2231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pickstone, J.M. Weatherall. In Search of a Cure. A history of pharmaceutical discovery. Br. J. Hist. Sci 1992, 24, 493–504. [Google Scholar] [CrossRef]
- Di Nardo, G.; Gilardi, G. Natural Compounds as Pharmaceuticals: The Key Role of Cytochromes P450 Reactivity. Trends Biochem. Sci. 2020, 45, 511–525. [Google Scholar] [CrossRef] [PubMed]
- Teufel, R. Flavin-catalyzed redox tailoring reactions in natural product biosynthesis. Arch. Biochem. Biophys. 2017, 632, 20–27. [Google Scholar] [CrossRef] [PubMed]
- Walsh, C.T. The chemical versatility of natural-product assembly lines. Acc. Chem. Res. 2008, 41, 4–10. [Google Scholar] [CrossRef] [PubMed]
- Sundaram, S.; Hertweck, C. On-line enzymatic tailoring of polyketides and peptides in thiotemplate systems. Curr. Opin. Chem. Biol. 2016, 31, 82–94. [Google Scholar] [CrossRef]
- Que, L., Jr.; Tolman, W.B. Biologically inspired oxidation catalysis. Nature 2008, 455, 333–340. [Google Scholar] [CrossRef]
- Walsh, C.T. A chemocentric view of the natural product inventory. Nat. Chem. Biol. 2015, 11, 620–624. [Google Scholar] [CrossRef]
- Pickens, L.B.; Tang, Y.; Chooi, Y.H. Metabolic engineering for the production of natural products. Annu. Rev. Chem. Biomol. Eng. 2011, 2, 211–236. [Google Scholar] [CrossRef] [Green Version]
- Wang, P.; Gao, X.; Tang, Y. Complexity generation during natural product biosynthesis using redox enzymes. Curr. Opin. Chem. Biol. 2012, 16, 362–369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thodberg, S.; Neilson, E.J.H. The “Green” FMOs: Diversity, Functionality and Application of Plant Flavoproteins. Catalysts 2020, 10, 329. [Google Scholar] [CrossRef] [Green Version]
- Walsh, C.T.; Tang, Y. Natural Product Biosynthesis; Royal Society of Chemistry: London, UK, 2017. [Google Scholar]
- Chaiyen, P.; Fraaije, M.W.; Mattevi, A. The enigmatic reaction of flavins with oxygen. Trends Biochem. Sci. 2012, 37, 373–380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romero, E.; Gomez Castellanos, J.R.; Gadda, G.; Fraaije, M.W.; Mattevi, A. Same Substrate, Many Reactions: Oxygen Activation in Flavoenzymes. Chem. Rev. 2018, 118, 1742–1769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paul, C.E.; Eggerichs, D.; Westphal, A.H.; Tischler, D.; van Berkel, W.J.H. Flavoprotein monooxygenases: Versatile biocatalysts. Biotechnol. Adv. 2021, 51, 107712. [Google Scholar] [CrossRef]
- Basaran, R.; Can Eke, B. Flavin Containing Monooxygenases and Metabolism of Xenobiotics. Turk. J. Pharm. Sci. 2017, 14, 90–94. [Google Scholar] [CrossRef] [PubMed]
- Van Berkel, W.; Kamerbeek, N.; Fraaije, M. Flavoprotein monooxygenases, a diverse class of oxidative biocatalysts. J. Biotechnol. 2006, 124, 670–689. [Google Scholar] [CrossRef] [Green Version]
- Sucharitakul, J.; Tinikul, R.; Chaiyen, P. Mechanisms of reduced flavin transfer in the two-component flavin-dependent monooxygenases. Arch. Biochem. Biophys. 2014, 555–556, 33–46. [Google Scholar] [CrossRef]
- Huijbers, M.M.; Montersino, S.; Westphal, A.H.; Tischler, D.; van Berkel, W.J. Flavin dependent monooxygenases. Arch. Biochem. Biophys. 2014, 544, 2–17. [Google Scholar] [CrossRef]
- Sucharitakul, J.; Phongsak, T.; Entsch, B.; Svasti, J.; Chaiyen, P.; Ballou, D.P. Kinetics of a two-component p-hydroxyphenylacetate hydroxylase explain how reduced flavin is transferred from the reductase to the oxygenase. Biochemistry 2007, 46, 8611–8623. [Google Scholar] [CrossRef]
- Tinikul, R.; Pitsawong, W.; Sucharitakul, J.; Nijvipakul, S.; Ballou, D.P.; Chaiyen, P. The transfer of reduced flavin mononucleotide from LuxG oxidoreductase to luciferase occurs via free diffusion. Biochemistry 2013, 52, 6834–6843. [Google Scholar] [CrossRef]
- Montersino, S.; Tischler, D.; Gassner, G.T.; van Berkel, W.J.H. Catalytic and Structural Features of Flavoprotein Hydroxylases and Epoxidases. Adv. Synth. Catal. 2011, 353, 2301–2319. [Google Scholar] [CrossRef]
- Olucha, J.; Meneely, K.M.; Chilton, A.S.; Lamb, A.L. Two structures of an N-hydroxylating flavoprotein monooxygenase: Ornithine hydroxylase from Pseudomonas aeruginosa. J. Biol. Chem. 2011, 286, 31789–31798. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pazmiño, D.E.T.; Dudek, H.M.; Fraaije, M.W. Baeyer–Villiger monooxygenases: Recent advances and future challenges. Curr. Opin. Chem. Biol. 2010, 14, 138–144. [Google Scholar] [CrossRef]
- Teufel, R.; Miyanaga, A.; Michaudel, Q.; Stull, F.; Louie, G.; Noel, J.P.; Baran, P.S.; Palfey, B.; Moore, B.S. Flavin-mediated dual oxidation controls an enzymatic Favorskii-type rearrangement. Nature 2013, 503, 552–556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teufel, R.; Stull, F.; Meehan, M.J.; Michaudel, Q.; Dorrestein, P.C.; Palfey, B.; Moore, B.S. Biochemical establishment and characterization of EncM’s flavin-N5-oxide cofactor. J. Am. Chem. Soc. 2015, 137, 8078–8085. [Google Scholar] [CrossRef] [Green Version]
- Klingenberg, M. Pigments of rat liver microsomes. Arch. Biochem. Biophys. 1958, 75, 376–386. [Google Scholar] [CrossRef]
- Zerbe, K.; Woithe, K.; Li, D.B.; Vitali, F.; Bigler, L.; Robinson, J.A. An oxidative phenol coupling reaction catalyzed by OxyB, a cytochrome P450 from the vancomycin-producing microorganism. Angew. Chem. Int. Ed. 2004, 43, 6709–6713. [Google Scholar] [CrossRef]
- Isin, E.M.; Guengerich, F.P. Complex reactions catalyzed by cytochrome P450 enzymes. Biochim. Biophys. Acta Gen. Subj. 2007, 1770, 314–329. [Google Scholar] [CrossRef]
- Urlacher, V.B.; Girhard, M. Cytochrome P450 monooxygenases: An update on perspectives for synthetic application. Trends Biotechnol. 2012, 30, 26–36. [Google Scholar] [CrossRef]
- Poulsen, L.L.; Ziegler, D.M. Multisubstrate flavin-containing monooxygenases: Applications of mechanism to specificity. Chem. Biol. Interact. 1995, 96, 57–73. [Google Scholar] [CrossRef]
- Guo, W.-X.A.; Poulsen, L.L.; Ziegler, D.M. Use of thiocarbamides as selective substrate probes for isoforms of flavin-containing monooxygenases. Biochem. Pharmacol. 1992, 44, 2029–2037. [Google Scholar] [CrossRef]
- Motika, M.S.; Zhang, J.; Cashman, J.R. Flavin-containing monooxygenase 3 and human disease. Expert Opin. Drug Metab. Toxicol. 2007, 3, 831–845. [Google Scholar] [CrossRef] [PubMed]
- Padyana, A.K.; Gross, S.; Jin, L.; Cianchetta, G.; Narayanaswamy, R.; Wang, F.; Wang, R.; Fang, C.; Lv, X.; Biller, S.A. Structure and inhibition mechanism of the catalytic domain of human squalene epoxidase. Nat. Commun. 2019, 10, 97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montemiglio, L.C.; Parisi, G.; Scaglione, A.; Sciara, G.; Savino, C.; Vallone, B. Functional analysis and crystallographic structure of clotrimazole bound OleP, a cytochrome P450 epoxidase from Streptomyces antibioticus involved in oleandomycin biosynthesis. Biochim. Biophys. Acta Gen. Subj. 2016, 1860, 465–475. [Google Scholar] [CrossRef]
- Taylor, K.L.; Ziegler, D.M. Studies on substrate specificity of the hog liver flavin-containing monooxygenase. Anionic organic sulfur compounds. Biochem. Pharm. 1987, 36, 141–146. [Google Scholar] [CrossRef]
- Hvattum, E.; Bergseth, S.; Pedersen, C.N.; Bremer, J.; Aarsland, A.; Berge, R.K. Microsomal oxidation of dodecylthioacetic acid (a 3-thia fatty acid) in rat liver. Biochem. Pharm. 1991, 41, 945–953. [Google Scholar] [CrossRef]
- Attar, M.; Dong, D.; Ling, K.H.; Tang-Liu, D.D. Cytochrome P450 2C8 and flavin-containing monooxygenases are involved in the metabolism of tazarotenic acid in humans. Drug Metab. Dispos. 2003, 31, 476–481. [Google Scholar] [CrossRef] [Green Version]
- Cashman, J.R.; Park, S.B.; Yang, Z.C.; Wrighton, S.A.; Jacob, P., 3rd; Benowitz, N.L. Metabolism of nicotine by human liver microsomes: Stereoselective formation of trans-nicotine N′-oxide. Chem. Res. Toxicol. 1992, 5, 639–946. [Google Scholar] [CrossRef]
- Duescher, R.J.; Lawton, M.P.; Philpot, R.M.; Elfarra, A.A. Flavin-containing monooxygenase (FMO)-dependent metabolism of methionine and evidence for FMO3 being the major FMO involved in methionine sulfoxidation in rabbit liver and kidney microsomes. J. Biol. Chem. 1994, 269, 17525–17530. [Google Scholar] [CrossRef]
- Elfarra, A.A. Potential role of the flavin-containing monooxygenases in the metabolism of endogenous compounds. Chem. Biol. Interact. 1995, 96, 47–55. [Google Scholar] [CrossRef]
- Ohmiya, Y.; Mehendale, H.M. Species differences in pulmonary N-oxidation of chlorpromazine and imipramine. Pharmacology 1984, 28, 289–295. [Google Scholar] [CrossRef]
- Nagata, T.; Williams, D.E.; Ziegler, D. Substrate specificities of rabbit lung and porcine liver flavin-containing monooxygenases: Differences due to substrate size. Chem. Res. Toxicol. 1990, 3, 372–376. [Google Scholar] [CrossRef] [PubMed]
- Shehin-Johnson, S.E.; Williams, D.E.; Larsen-Su, S.; Stresser, D.M.; Hines, R.N. Tissue-specific expression of flavin-containing monooxygenase (FMO) forms 1 and 2 in the rabbit. J. Pharmacol. Exp. Ther. 1995, 272, 1293–1299. [Google Scholar] [PubMed]
- Kim, Y.M.; Ziegler, D.M. Size limits of thiocarbamides accepted as substrates by human flavin-containing monooxygenase 1. Drug Metab. Dispos. 2000, 28, 1003–1006. [Google Scholar] [PubMed]
- Damani, L.A.; Pool, W.F.; Crooks, P.A.; Kaderlik, R.K.; Ziegler, D.M. Stereoselectivity in the N’-oxidation of nicotine isomers by flavin-containing monooxygenase. Mol. Pharmacol. 1988, 33, 702–705. [Google Scholar]
- Devereux, T.R.; Fouts, J.R. N-oxidation and demethylation of N,N-dimethylaniline by rabbit liver and lung microsomes. Effects of age and metals. Chem. Biol. Interact. 1974, 8, 91–105. [Google Scholar] [CrossRef]
- Devereux, T.R.; Philpot, R.M.; Fouts, J.R. The effect of Hg2+ on rabbit hepatic and pulmonary solubilized, partially purified N, N-dimethylaniline N-oxidases. Chem.-Biol. Interact. 1977, 18, 277–287. [Google Scholar] [CrossRef]
- Sabourin, P.J.; Hodgson, E. Characterization of the purified microsomal FAD-containing monooxygenase from mouse and pig liver. Chem Biol Interact 1984, 51, 125–139. [Google Scholar] [CrossRef]
- Lawton, M.P.; Kronbach, T.; Johnson, E.F.; Philpot, R.M. Properties of expressed and native flavin-containing monooxygenases: Evidence of multiple forms in rabbit liver and lung. Mol. Pharmacol. 1991, 40, 692. [Google Scholar]
- Krueger, S.K.; Siddens, L.K.; Henderson, M.C.; Andreasen, E.A.; Tanguay, R.L.; Pereira, C.B.; Cabacungan, E.T.; Hines, R.N.; Ardlie, K.G.; Williams, D.E. Haplotype and functional analysis of four flavin-containing monooxygenase isoform 2 (FMO2) polymorphisms in Hispanics. Pharm. Genom. 2005, 15, 245–256. [Google Scholar] [CrossRef] [Green Version]
- Newman, D.J.; Cragg, G.M. Natural products as sources of new drugs over the 30 years from 1981 to 2010. J. Nat. Prod. 2012, 75, 311–335. [Google Scholar] [CrossRef] [Green Version]
- Waltenberger, B.; Mocan, A.; Šmejkal, K.; Heiss, E.H.; Atanasov, A.G. Natural products to counteract the epidemic of cardiovascular and metabolic disorders. Molecules 2016, 21, 807. [Google Scholar] [CrossRef] [PubMed]
- Tintore, M.; Vidal-Jordana, A.; Sastre-Garriga, J. Treatment of multiple sclerosis—success from bench to bedside. Nat. Rev. Neurology 2019, 15, 53–58. [Google Scholar] [CrossRef]
- Lachance, H.; Wetzel, S.; Kumar, K.; Waldmann, H. Charting, navigating, and populating natural product chemical space for drug discovery. J. Med. Chem. 2012, 55, 5989–6001. [Google Scholar] [CrossRef] [PubMed]
- Gertz, M.; Renz, M.; Zhou, X.; Hoel, E.; Ku, W.-S.; Voisard, A.; Zhang, C.; Chen, H.; Tang, L.; Huang, Y. Advances in Spatial and Temporal Databases: 15th International Symposium SSTD 2017, Arlington, VA, USA, 21–23 August 2017; Springer: Berlin/Heidelberg, Germany, 2017; Volume 10411. [Google Scholar]
- Mascotti, M.L.; Juri Ayub, M.; Furnham, N.; Thornton, J.M.; Laskowski, R.A. Chopping and Changing: The Evolution of the Flavin-dependent Monooxygenases. J. Mol. Biol. 2016, 428, 3131–3146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reis, R.A.; Li, H.; Johnson, M.; Sobrado, P. New frontiers in flavin-dependent monooxygenases. Arch. Biochem. Biophys. 2021, 699, 108765. [Google Scholar] [CrossRef]
- Chen, K.; Arnold, F.H. Engineering new catalytic activities in enzymes. Nat. Catal. 2020, 3, 203–213. [Google Scholar] [CrossRef]
- Zhang, X.; King-Smith, E.; Dong, L.-B.; Yang, L.-C.; Rudolf, J.D.; Shen, B.; Renata, H. Divergent synthesis of complex diterpenes through a hybrid oxidative approach. Science 2020, 369, 799–806. [Google Scholar] [CrossRef]
- McGrath, N.A.; Bartlett, E.S.; Sittihan, S.; Njardarson, J.T. A Concise Ring-Expansion Route to the Compact Core of Platensimycin. Angew. Chem. Int. Ed. 2009, 48, 8543–8546. [Google Scholar] [CrossRef]
- Kung, J.W.; Baumann, S.; von Bergen, M.; Müller, M.; Hagedoorn, P.-L.; Hagen, W.R.; Boll, M. Reversible biological Birch reduction at an extremely low redox potential. J. Am. Chem. Soc. 2010, 132, 9850–9856. [Google Scholar] [CrossRef]
- Pinkerton, D.M.; Banwell, M.G.; Willis, A.C. Chemoenzymatic access to versatile epoxyquinol synthons. Org. Lett. 2009, 11, 4290–4293. [Google Scholar] [CrossRef]
- Levin, S.; Nani, R.R.; Reisman, S.E. Rapid Assembly of the Salvileucalin B Norcaradiene Core. Org. Lett. 2010, 12, 780–783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davison, J.; Al Fahad, A.; Cai, M.; Song, Z.; Yehia, S.Y.; Lazarus, C.M.; Bailey, A.M.; Simpson, T.J.; Cox, R.J. Genetic, molecular, and biochemical basis of fungal tropolone biosynthesis. Proc. Natl. Acad. Sci. USA 2012, 109, 7642–7647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, N.; Song, W.; Schienebeck, C.M.; Zhang, M.; Tang, W. Synthesis of Naturally Occurring Tropones and Tropolones. Tetrahedron 2014, 70, 9281–9305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barbier, M.; Barton, D.H.; Devys, M.; Topgi, R.S. A simple synthesis of the tropone nucleus. J. Chem. Soc. Chem. Commun. 1984, 12, 743–744. [Google Scholar] [CrossRef]
- Doyon, T.J.; Skinner, K.; Yang, D.; Mallik, L.; Wymore, T.; Koutmos, M.; Zimmerman, P.M.; Narayan, A. Radical Tropolone Biosynthesis. Organ. Chem. 2020. preprint. [Google Scholar] [CrossRef]
- Baker Dockrey, S.A.; Lukowski, A.L.; Becker, M.R.; Narayan, A.R.H. Biocatalytic site- and enantioselective oxidative dearomatization of phenols. Nat. Chem. 2018, 10, 119–125. [Google Scholar] [CrossRef] [PubMed]
- Cox, R. Oxidative rearrangements during fungal biosynthesis. Nat. Prod. Rep. 2014, 31, 1405–1424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chakrabarty, S.; Romero, E.O.; Pyser, J.B.; Yazarians, J.A.; Narayan, A.R.H. Chemoenzymatic Total Synthesis of Natural Products. Acc. Chem. Res. 2021, 54, 1374–1384. [Google Scholar] [CrossRef] [PubMed]
- Shu, X.; Wei, G.; Qiao, Y.; Zhang, K.; Zhang, J.; Ai, G.; Tang, M.C.; Zhang, Y.; Gao, S.S. TerC Is a Multifunctional and Promiscuous Flavoprotein Monooxygenase That Catalyzes Bimodal Oxidative Transformations. Org. Lett. 2021, 23, 8947–8951. [Google Scholar] [CrossRef]
- Cabrera, G.M.; Butler, M.; Rodriguez, M.A.; Godeas, A.; Haddad, R.; Eberlin, M.N. A sorbicillinoid urea from an intertidal Paecilomyces marquandii. J. Nat. Prod. 2006, 69, 1806–1808. [Google Scholar] [CrossRef] [PubMed]
- Scherlach, K.; Boettger, D.; Remme, N.; Hertweck, C. The chemistry and biology of cytochalasans. Nat. Prod. Rep. 2010, 27, 869–886. [Google Scholar] [CrossRef]
- Smith, E.L.; Austen, B.M.; Blumenthal, K.M.; Nyc, J.F. 5 Glutamate dehydrogenases. In The Enzymes; Elsevier: Amsterdam, The Netherlands, 1975; Volume 11, pp. 293–367. [Google Scholar]
- Binder, M.; Tamm, C. The cytochalasans: A new class of biologically active microbial metabolites. Angew. Chem. Int. Ed. Engl. 1973, 12, 370–380. [Google Scholar] [CrossRef] [PubMed]
- Flanagan, M.D.; Lin, S. Cytochalasins block actin filament elongation by binding to high affinity sites associated with F-actin. J. Biol. Chem. 1980, 255, 835–838. [Google Scholar] [CrossRef]
- Robert, J.L.; Tamm, C. Biosynthesis of cytochalasans. Part 5. The incorporation of deoxaphomin into cytochalasin B (phomin). Helv. Chim. Acta 1975, 58, 2501–2504. [Google Scholar] [CrossRef] [PubMed]
- Walsh, C.T.; Wencewicz, T.A. Flavoenzymes: Versatile catalysts in biosynthetic pathways. Nat. Prod. Rep. 2013, 30, 175–200. [Google Scholar] [CrossRef] [Green Version]
- Rebehmed, J.; Alphand, V.; De Berardinis, V.; de Brevern, A.G. Evolution study of the Baeyer–Villiger monooxygenases enzyme family: Functional importance of the highly conserved residues. Biochimie 2013, 95, 1394–1402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baily, M.N.; Taylor, J.B.; Hoover Institution on War Revolution and Peace; Brookings Institution. Across the Great Divide: New Perspectives on the Financial Crisis; Hoover Institution Press, Stanford University: Stanford, CA, USA, 2014; p. vi, 409p. [Google Scholar]
- Ebrahimi, K.S.; Ansari, M.; Hosseyni Moghaddam, M.S.; Ebrahimi, Z.; Salehi, Z.; Shahlaei, M.; Moradi, S. In silico investigation on the inhibitory effect of fungal secondary metabolites on RNA dependent RNA polymerase of SARS-CoV-II: A docking and molecular dynamic simulation study. Comput. Biol. Med. 2021, 135, 104613. [Google Scholar] [CrossRef]
- Kuhn, D.; Kholiq, M.A.; Heinzle, E.; Bühler, B.; Schmid, A. Intensification and economic and ecological assessment of a biocatalytic oxyfunctionalization process. Green Chem. 2010, 12, 815–827. [Google Scholar] [CrossRef]
- Kuhn, D.; Bühler, B.; Schmid, A. Production host selection for asymmetric styrene epoxidation: Escherichia coli vs. solvent-tolerant Pseudomonas. J. Ind. Microbiol. Biotechnol. 2012, 39, 1125–1133. [Google Scholar] [CrossRef]
- Silverton, J.; Akiyama, T.; Kabuto, C.; Sekita, S.; Yoshihira, K.; Natori, S. X-ray analysis of chaetoglobosin A, an indol-3-yl-cytochalasan from Chaetomium globosum. Tetrahedron Lett. 1976, 17, 1349–1350. [Google Scholar] [CrossRef]
- Borges, W.S.; Mancilla, G.; Guimaraes, D.O.; Durán-Patrón, R.; Collado, I.G.; Pupo, M.T. Azaphilones from the endophyte Chaetomium globosum. J. Nat. Prod. 2011, 74, 1182–1187. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, M.; Koyama, K.; Natori, S. Four new azaphilones from Chaetomium globosum var. flavo-viridae. Chem. Pharm. Bull. 1990, 38, 625–628. [Google Scholar] [CrossRef] [Green Version]
- Jiao, W.; Feng, Y.; Blunt, J.W.; Cole, A.L.; Munro, M.H. Chaetoglobosins Q, R, and T, Three Further New Metabolites from Chaetomium g lobosum. J. Nat. Prod. 2004, 67, 1722–1725. [Google Scholar] [CrossRef] [PubMed]
- Tang, M.C.; Zou, Y.; Watanabe, K.; Walsh, C.T.; Tang, Y. Oxidative Cyclization in Natural Product Biosynthesis. Chem. Rev. 2017, 117, 5226–5333. [Google Scholar] [CrossRef] [PubMed]
- Tena, S.M.; Nanda, J.; Díaz-Oltra, S.; Chotera, A.; Ashkenasy, G.; Escuder, B. Emergent catalytic behavior of self-assembled low molecular weight peptide-based aggregates and hydrogels. Chem. A Eur. J. 2016, 22, 6687–6694. [Google Scholar] [CrossRef]
- Bloch, P.; Tamm, C. Isolation and structure of pseurotin A, a microbial metabolite of Pseudeurotium ovalis Stolk with an unusual heterospirocyclic system. Helv. Chim. Acta 1981, 64, 304–315. [Google Scholar] [CrossRef]
- Breitenstein, W.; Chexal, K.K.; Mohr, P.; Tamm, C. Pseurotin B, C, D, and E. Further new metabolites of Pseudeurotium ovalis Stolk. Helv. Chim. Acta 1981, 64, 379–388. [Google Scholar] [CrossRef]
- Bloch, P.; Tamm, C.; Bollinger, P.; Petcher, T.J.; Weber, H.P. Pseurotin, a New Metabolite of Pseudeurotium ovalis STOLK Having an Unusual Hetero-Spirocyclic System.(Preliminary Communication). Helv. Chim. Acta 1976, 59, 133–137. [Google Scholar] [CrossRef]
- Yamada, T.; Imai, E.; Nakatuji, K.; Numata, A.; Tanaka, R. Cephalimysin A, a potent cytotoxic metabolite from an Aspergillus species separated from a marine fish. Tetrahedron Lett. 2007, 48, 6294–6296. [Google Scholar] [CrossRef]
- Aparicio, J.F.; Caffrey, P.; Marsden, A.; Staunton, J.; Leadlay, P.F. Limited proteolysis and active-site studies of the first multienzyme component of the erythromycin-producing polyketide synthase. J. Biol. Chem. 1994, 269, 8524–8528. [Google Scholar] [CrossRef]
- Caffrey, P.; Bevitt, D.J.; Staunton, J.; Leadlay, P.F. Identification of DEBS 1, DEBS 2 and DEBS 3, the multienzyme polypeptides of the erythromycin-producing polyketide synthase from Saccharopolyspora erythraea. FEBS Lett. 1992, 304, 225–228. [Google Scholar] [CrossRef] [Green Version]
- Cane, D.E.; Celmer, W.D.; Westley, J.W. Unified stereochemical model of polyether antibiotic structure and biogenesis. J. Am. Chem. Soc. 1983, 105, 3594–3600. [Google Scholar] [CrossRef]
- Leadlay, P.; Staunton, J.; Oliynyk, M.; Bisang, C.; Cortes, J.; Frost, E.; Hughes-Thomas, Z.; Jones, M.; Kendrew, S.; Lester, J. Engineering of complex polyketide biosynthesis—insights from sequencing of the monensin biosynthetic gene cluster. J. Ind. Microbiol. Biotechnol. 2001, 27, 360–367. [Google Scholar] [CrossRef] [PubMed]
- Callaway, T.; Edrington, T.; Rychlik, J.; Genovese, K.; Poole, T.; Jung, Y.S.; Bischoff, K.; Anderson, R.; Nisbet, D.J. Ionophores: Their use as ruminant growth promotants and impact on food safety. Curr. Issues Intest. Microbiol. 2003, 4, 43–51. [Google Scholar] [PubMed]
- Gumila, C.; Ancelin, M.-L.; Delort, A.-M.; Jeminet, G.; Vial, H.J. Characterization of the potent in vitro and in vivo antimalarial activities of ionophore compounds. Antimicrob. Agents Chemother. 1997, 41, 523–529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakamura, M.; Kunimoto, S.; Takahashi, Y.; Naganawa, H.; Sakaue, M.; Inoue, S.; Ohno, T.; Takeuchi, T. Inhibitory effects of polyethers on human immunodeficiency virus replication. Antimicrob. Agents Chemother. 1992, 36, 492–494. [Google Scholar] [CrossRef] [Green Version]
- Fuchs, D.; Heinold, A.; Opelz, G.; Daniel, V.; Naujokat, C. Salinomycin induces apoptosis and overcomes apoptosis resistance in human cancer cells. Biochem. Bioph. Res. Co. 2009, 390, 743–749. [Google Scholar] [CrossRef] [PubMed]
- Minami, A.; Oguri, H.; Watanabe, K.; Oikawa, H. Biosynthetic machinery of ionophore polyether lasalocid: Enzymatic construction of polyether skeleton. Curr. Opin. Chem. Biol. 2013, 17, 555–561. [Google Scholar] [CrossRef] [PubMed]
- Bhatt, A.; Stark, C.B.; Harvey, B.M.; Gallimore, A.R.; Demydchuk, Y.A.; Spencer, J.B.; Staunton, J.; Leadlay, P.F. Accumulation of an E, E, E-triene by the monensin-producing polyketide synthase when oxidative cyclization is blocked. Angew. Chem. 2005, 117, 7237–7240. [Google Scholar] [CrossRef]
- Gallimore, A.R.; Stark, C.B.; Bhatt, A.; Harvey, B.M.; Demydchuk, Y.; Bolanos-Garcia, V.; Fowler, D.J.; Staunton, J.; Leadlay, P.F.; Spencer, J.B. Evidence for the role of the monB genes in polyether ring formation during monensin biosynthesis. Chem. Biol. 2006, 13, 453–460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Westley, J.; Evans, R.; Williams, T.; Stempel, A. Structure of antibiotic X-537A. J. Chem. Soc. D Chem. Commun. 1970, 2, 71–72. [Google Scholar] [CrossRef]
- Chen, H.; Gertz, M.; Hoel, E.; Huang, Y.; Ku, W.-S.; Lu, C.-T.; Ravada, S.; Renz, M.; Tang, L.; Voisard, A.; et al. Advances in Spatial and Temporal Databases: 15th International Symposium, SSTD 2017, Arlington, VA, USA, August 21–23, 2017, Proceedings. In Information Systems and Applications, Incl Internet/Web, and HCI 10411, 1st ed.; Springer International Publishing: Springer: Cham, Switzerland, 2017; p. 1, online resource (XIV, 454 pages 206 illustrations). [Google Scholar]
- Johnson, K.M.; Swenson, L.; Opipari, A.W., Jr.; Reuter, R.; Zarrabi, N.; Fierke, C.A.; Börsch, M.; Glick, G.D. Mechanistic basis for differential inhibition of the F1Fo-ATPase by aurovertin. Biopolym. Orig. Res. Biomol. 2009, 91, 830–840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, T.-C.; Chang, H.-Y.; Hsu, C.-H.; Kuo, W.-H.; Chang, K.-J.; Juan, H.-F. Targeting therapy for breast carcinoma by ATP synthase inhibitor aurovertin B. J. Proteome Res. 2008, 7, 1433–1444. [Google Scholar] [CrossRef] [PubMed]
- Mao, X.-M.; Zhan, Z.-J.; Grayson, M.N.; Tang, M.-C.; Xu, W.; Li, Y.-Q.; Yin, W.-B.; Lin, H.-C.; Chooi, Y.-H.; Houk, K. Efficient biosynthesis of fungal polyketides containing the dioxabicyclo-octane ring system. J. Am. Chem. Soc. 2015, 137, 11904–11907. [Google Scholar] [CrossRef] [Green Version]
- Matsushita, T.; Kishimoto, S.; Hara, K.; Hashimoto, H.; Watanabe, K. Structural and Functional Analyses of a Spiro-Carbon-Forming, Highly Promiscuous Epoxidase from Fungal Natural Product Biosynthesis. Biochemistry 2020, 59, 4787–4792. [Google Scholar] [CrossRef]
- Wohlgemuth, R. Tools for selective enzyme reaction steps in the synthesis of laboratory chemicals. Eng. Life Sci. 2006, 6, 577–583. [Google Scholar] [CrossRef]
- Van Beilen, J.B.; Duetz, W.A.; Schmid, A.; Witholt, B. Practical issues in the application of oxygenases. Trends Biotechnol. 2003, 21, 170–177. [Google Scholar] [CrossRef]
- Stewart, J.D. Cyclohexanone monooxygenase: A useful asymmetric Baeyer-Villiger reactions. Curr. Org. Chem. 1998, 2, 195–216. [Google Scholar] [CrossRef]
- Roberts, S.M.; Wan, P.W. enzyme-catalysed Baeyer–Villiger oxidations. J. Mol. Catal. B Enzym. 1998, 4, 111–136. [Google Scholar] [CrossRef]
- Walsh, C.T.; Chen, Y.C.J. Enzymic Baeyer–Villiger oxidations by flavin-dependent monooxygenases. Angew. Chem. Int. Ed. Engl. 1988, 27, 333–343. [Google Scholar] [CrossRef]
- Mihovilovic, M.D.; Kapitán, P. Regiodivergent Baeyer–Villiger oxidation of fused ketone substrates by recombinant whole-cells expressing two monooxygenases from Brevibacterium. Tetrahedron Lett. 2004, 45, 2751–2754. [Google Scholar] [CrossRef]
- Kamerbeek, N.M.; Janssen, D.B.; van Berkel, W.J.; Fraaije, M.W. Baeyer–Villiger monooxygenases, an emerging family of flavin-dependent biocatalysts. Adv. Synth. Catal. 2003, 345, 667–678. [Google Scholar] [CrossRef] [Green Version]
- Wichmann, R.; Vasic-Racki, D. Cofactor regeneration at the lab scale. Technol. Transf. Biotechnol. 2005, 92, 225–260. [Google Scholar]
- Hollmann, F.; Taglieber, A.; Schulz, F.; Reetz, M.T. A light-driven stereoselective biocatalytic oxidation. Angew. Chem. 2007, 119, 2961–2964. [Google Scholar] [CrossRef]
- Hollmann, F.; Hofstetter, K.; Schmid, A. Non-enzymatic regeneration of nicotinamide and flavin cofactors for monooxygenase catalysis. TRENDS Biotechnol. 2006, 24, 163–171. [Google Scholar] [CrossRef] [PubMed]
- Van Der Donk, W.A.; Zhao, H. Recent developments in pyridine nucleotide regeneration. Curr. Opin. Biotechnol. 2003, 14, 421–426. [Google Scholar] [CrossRef]
- Walton, A.Z.; Stewart, J.D. An Efficient Enzymatic Baeyer–Villiger Oxidation by Engineered Escherichiacoli Cells under Non-Growing Conditions. Biotechnol. Prog. 2002, 18, 262–268. [Google Scholar] [CrossRef] [PubMed]
- Hilker, I.; Alphand, V.; Wohlgemuth, R.; Furstoss, R. Microbial transformations, 56. Preparative scale asymmetric Baeyer–Villiger oxidation using a highly productive “Two-in-One” resin-based in situ SFPR concept. Adv. Synth. Catal. 2004, 346, 203–214. [Google Scholar] [CrossRef]
- Gutiérrez, M.C.; Furstoss, R.; Alphand, V. Microbiological Transformations 60. Enantioconvergent Baeyer–Villiger Oxidation via a Combined Whole Cells and Ionic Exchange Resin-Catalysed Dynamic Kinetic Resolution Process. Adv. Synth. Catal. 2005, 347, 1051–1059. [Google Scholar] [CrossRef]
- Walton, A.Z.; Stewart, J.D. Understanding and improving NADPH-dependent reactions by nongrowing Escherichia coli cells. Biotechnol. Prog. 2004, 20, 403–411. [Google Scholar] [CrossRef] [PubMed]
- Rudroff, F.; Alphand, V.; Furstoss, R.; Mihovilovic, M.D. Optimizing fermentation conditions of recombinant Escherichia coli expressing cyclopentanone monooxygenase. Org. Process Res. Dev. 2006, 10, 599–604. [Google Scholar] [CrossRef]
- Torres Pazmiño, D.E.; Snajdrova, R.; Baas, B.J.; Ghobrial, M.; Mihovilovic, M.D.; Fraaije, M.W. Self-sufficient Baeyer–Villiger monooxygenases: Effective coenzyme regeneration for biooxygenation by fusion engineering. Angew. Chem. 2008, 120, 2307–2310. [Google Scholar] [CrossRef] [Green Version]
- Pazmiño, D.T.; Riebel, A.; de Lange, J.; Rudroff, F.; Mihovilovic, M.D.; Fraaije, M.W. Efficient biooxidations catalyzed by a new generation of self-sufficient Baeyer-Villiger monooxygenases. ChemBioChem 2009, 10, 2595–2598. [Google Scholar] [CrossRef] [Green Version]
- Donoghue, N.A.; Norris, D.B.; Trudgill, P.W. The purification and properties of cyclohexanone oxygenase from Nocardia globerula CL1 and Acinetobacter NCIB 9871. Eur. J. Biochem. 1976, 63, 175–192. [Google Scholar] [CrossRef]
- Branchaud, B.P.; Walsh, C.T. Functional group diversity in enzymic oxygenation reactions catalyzed by bacterial flavin-containing cyclohexanone oxygenase. J. Am. Chem. Soc. 1985, 107, 2153–2161. [Google Scholar] [CrossRef]
- Bong, Y.K.; Song, S.; Nazor, J.; Vogel, M.; Widegren, M.; Smith, D.; Collier, S.J.; Wilson, R.; Palanivel, S.M.; Narayanaswamy, K.; et al. Baeyer-Villiger Monooxygenase-Mediated Synthesis of Esomeprazole As an Alternative for Kagan Sulfoxidation. J. Org. Chem. 2018, 83, 7453–7458. [Google Scholar] [CrossRef]
- Zhang, Y.; Wu, Y.-Q.; Xu, N.; Zhao, Q.; Yu, H.-L.; Xu, J.-H. Engineering of Cyclohexanone Monooxygenase for the Enantioselective Synthesis of (S)-Omeprazole. ACS Sustain. Chem. Eng. 2019, 7, 7218–7226. [Google Scholar] [CrossRef]
- Xu, N.; Zhu, J.; Wu, Y.-Q.; Zhang, Y.; Xia, J.-Y.; Zhao, Q.; Lin, G.-Q.; Yu, H.-L.; Xu, J.-H. Enzymatic Preparation of the Chiral (S)-Sulfoxide Drug Esomeprazole at Pilot-Scale Levels. Org. Process Res. Dev. 2020, 24, 1124–1130. [Google Scholar] [CrossRef]
- Cheng, Y.; Dai, X.; Zhao, Y. Auxin biosynthesis by the YUCCA flavin monooxygenases controls the formation of floral organs and vascular tissues in Arabidopsis. Gene Dev. 2006, 20, 1790–1799. [Google Scholar] [CrossRef] [Green Version]
- Cheng, Y.; Dai, X.; Zhao, Y. Auxin synthesized by the YUCCA flavin monooxygenases is essential for embryogenesis and leaf formation in Arabidopsis. Plant Cell 2007, 19, 2430–2439. [Google Scholar] [CrossRef] [Green Version]
- Tao, Y.; Ferrer, J.-L.; Ljung, K.; Pojer, F.; Hong, F.; Long, J.A.; Li, L.; Moreno, J.E.; Bowman, M.E.; Ivans, L.J. Rapid synthesis of auxin via a new tryptophan-dependent pathway is required for shade avoidance in plants. Cell 2008, 133, 164–176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y. Auxin biosynthesis: A simple two-step pathway converts tryptophan to indole-3-acetic acid in plants. Mol. Plant 2012, 5, 334–338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Won, C.; Shen, X.; Mashiguchi, K.; Zheng, Z.; Dai, X.; Cheng, Y.; Kasahara, H.; Kamiya, Y.; Chory, J.; Zhao, Y. Conversion of tryptophan to indole-3-acetic acid by TRYPTOPHAN AMINOTRANSFERASES OF ARABIDOPSIS and YUCCAs in Arabidopsis. Proc. Natl. Acad. Sci. USA 2011, 108, 185–203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uc-Chuc, M.A.; Perez-Hernandez, C.; Galaz-Avalos, R.M.; Brito-Argaez, L.; Aguilar-Hernandez, V.; Loyola-Vargas, V.M. YUCCA-Mediated Biosynthesis of the Auxin IAA Is Required during the Somatic Embryogenic Induction Process in Coffea canephora. Int. J. Mol. Sci. 2020, 21, 4751. [Google Scholar] [CrossRef]
- Schmidt, B.M.; Ribnicky, D.M.; Lipsky, P.E.; Raskin, I. Revisiting the ancient concept of botanical therapeutics. Nat. Chem. Biol. 2007, 3, 360–366. [Google Scholar] [CrossRef]
- Schmidt, B.; Ribnicky, D.M.; Poulev, A.; Logendra, S.; Cefalu, W.T.; Raskin, I. A natural history of botanical therapeutics. Metabolism 2008, 57, S3–S9. [Google Scholar] [CrossRef] [Green Version]
- Hansen, C.C.; Sørensen, M.; Veiga, T.A.; Zibrandtsen, J.F.; Heskes, A.M.; Olsen, C.E.; Boughton, B.A.; Møller, B.L.; Neilson, E.H. Reconfigured cyanogenic glucoside biosynthesis in Eucalyptus cladocalyx involves a cytochrome P450 CYP706C55. Plant Physiol. 2018, 178, 1081–1095. [Google Scholar] [CrossRef] [Green Version]
- Yamaguchi, T.; Yamamoto, K.; Asano, Y. Identification and characterization of CYP79D16 and CYP71AN24 catalyzing the first and second steps in L-phenylalanine-derived cyanogenic glycoside biosynthesis in the Japanese apricot, Prunus mume Sieb. et Zucc. Plant Mol. Biol. 2014, 86, 215–223. [Google Scholar] [CrossRef]
- Toplak, M.; Matthews, A.; Teufel, R. The devil is in the details: The chemical basis and mechanistic versatility of flavoprotein monooxygenases. Arch. Biochem. Biophys. 2021, 698, 108732. [Google Scholar] [CrossRef]
- Smanski, M.J.; Zhou, H.; Claesen, J.; Shen, B.; Fischbach, M.A.; Voigt, C.A. Synthetic biology to access and expand nature’s chemical diversity. Nat. Rev. Microbiol. 2016, 14, 135–149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
















Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Deng, Y.; Zhou, Q.; Wu, Y.; Chen, X.; Zhong, F. Properties and Mechanisms of Flavin-Dependent Monooxygenases and Their Applications in Natural Product Synthesis. Int. J. Mol. Sci. 2022, 23, 2622. https://doi.org/10.3390/ijms23052622
Deng Y, Zhou Q, Wu Y, Chen X, Zhong F. Properties and Mechanisms of Flavin-Dependent Monooxygenases and Their Applications in Natural Product Synthesis. International Journal of Molecular Sciences. 2022; 23(5):2622. https://doi.org/10.3390/ijms23052622
Chicago/Turabian StyleDeng, Yaming, Quan Zhou, Yuzhou Wu, Xi Chen, and Fangrui Zhong. 2022. "Properties and Mechanisms of Flavin-Dependent Monooxygenases and Their Applications in Natural Product Synthesis" International Journal of Molecular Sciences 23, no. 5: 2622. https://doi.org/10.3390/ijms23052622