
Factors Associated with Platelet Activation-Recent Pharmaceutical Approaches

 , , ,
, , ,
Abstract
:1. Introduction
2. Overview of Platelet Biology and Main Functions
2.1. Platelet Structure and Formation
2.2. Platelet Functions
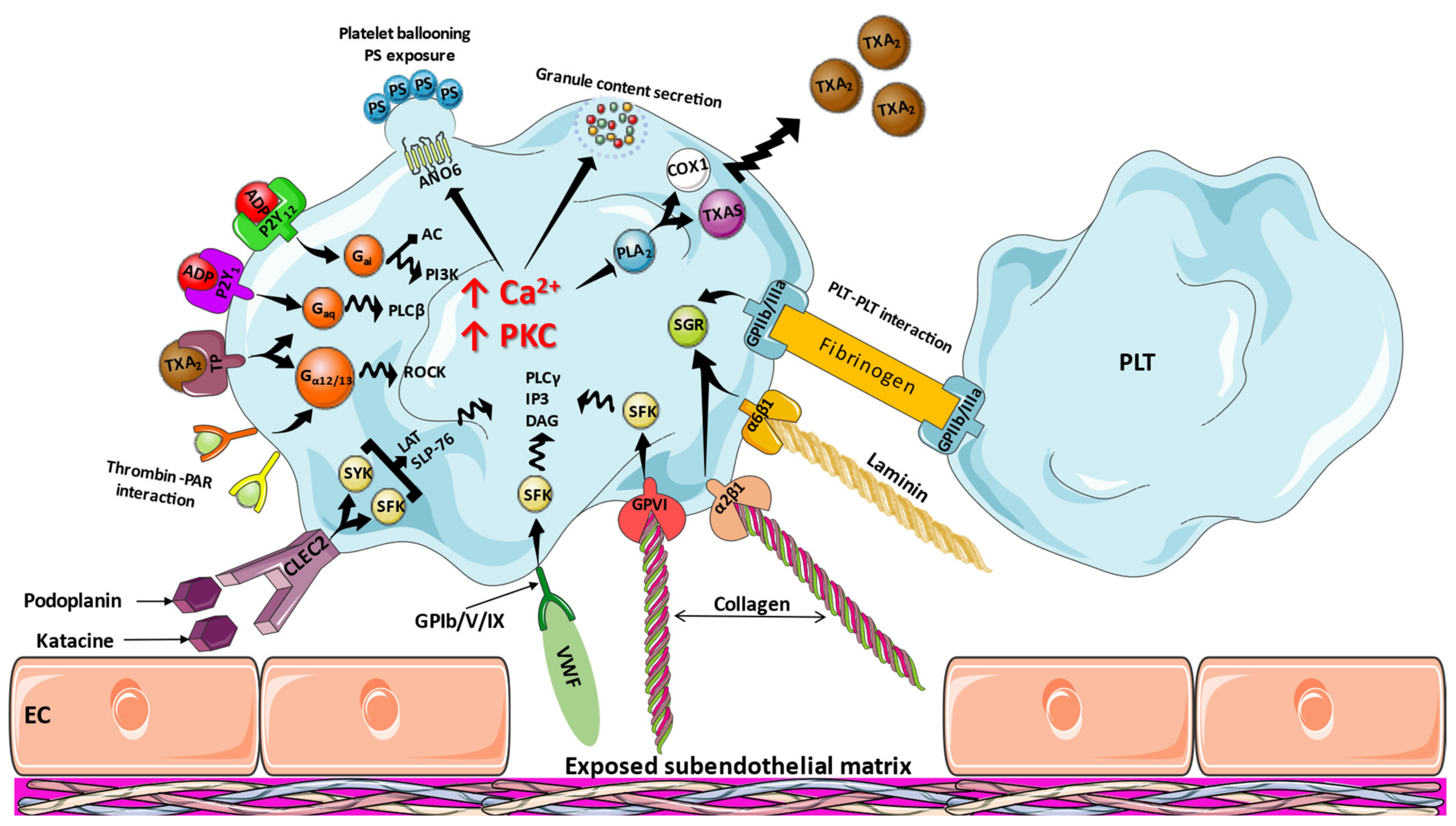
3. Molecular Mechanisms of Platelet Activation
3.1. Inflammation
3.2. Endothelial Dysfunction
3.3. MiRs
4. Therapeutic Approaches
4.1. Antiplatelet Drugs
4.1.1. Aspirin
4.1.2. P2Y12 Inhibitors
4.1.3. Glycoprotein IIb/IIIa Inhibitors
4.1.4. PAR-1 Antagonism
4.1.5. Caplacizumab
4.1.6. Direct Oral Anticoagulants
4.2. Off-Target Antiplatelet Drugs
4.3. Drugs under Investigation
5. Conclusions and Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kuter, D.J. The biology of thrombopoietin and thrombopoietin receptor agonists. Int. J. Hematol. 2013, 98, 10–23. [Google Scholar] [CrossRef] [PubMed]
- Behrens, K.; Alexander, W.S. Cytokine control of megakaryopoiesis. Growth Factors 2018, 36, 89–103. [Google Scholar] [CrossRef] [PubMed]
- Hitchcock, I.S.; Kaushansky, K. Thrombopoietin from beginning to end. Br. J. Haematol. 2014, 165, 259–268. [Google Scholar] [CrossRef] [PubMed]
- Kaser, A.; Brandacher, G.; Steurer, W.; Kaser, S.; Offner, F.A.; Zoller, H.; Theurl, I.; Widder, W.; Molnar, C.; Ludwiczek, O.; et al. Interleukin-6 stimulates thrombopoiesis through thrombopoietin: Role in inflammatory thrombocytosis. Blood 2001, 98, 2720–2725. [Google Scholar] [CrossRef] [Green Version]
- Kanaji, T.; Vo, M.N.; Kanaji, S.; Zarpellon, A.; Shapiro, R.; Morodomi, Y.; Yuzuriha, A.; Eto, K.; Belani, R.; Do, M.H.; et al. Tyrosyl-tRNA synthetase stimulates thrombopoietin-independent hematopoiesis accelerating recovery from thrombocytopenia. Proc. Natl. Acad. Sci. USA 2018, 115, E8228–E8235. [Google Scholar] [CrossRef] [Green Version]
- Noetzli, L.J.; French, S.L.; Machlus, K.R. New Insights Into the Differentiation of Megakaryocytes From Hematopoietic Progenitors. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 1288–1300. [Google Scholar] [CrossRef]
- Boscher, J.; Guinard, I.; Eckly, A.; Lanza, F.; Leon, C. Blood platelet formation at a glance. J. Cell Sci. 2020, 133, jcs244731. [Google Scholar] [CrossRef]
- Schachtner, H.; Calaminus, S.D.; Sinclair, A.; Monypenny, J.; Blundell, M.P.; Leon, C.; Holyoake, T.L.; Thrasher, A.J.; Michie, A.M.; Vukovic, M.; et al. Megakaryocytes assemble podosomes that degrade matrix and protrude through basement membrane. Blood 2013, 121, 2542–2552. [Google Scholar] [CrossRef] [Green Version]
- Eckly, A.; Scandola, C.; Oprescu, A.; Michel, D.; Rinckel, J.Y.; Proamer, F.; Hoffmann, D.; Receveur, N.; Leon, C.; Bear, J.E.; et al. Megakaryocytes use in vivo podosome-like structures working collectively to penetrate the endothelial barrier of bone marrow sinusoids. J. Thromb. Haemost. 2020, 18, 2987–3001. [Google Scholar] [CrossRef]
- Brown, E.; Carlin, L.M.; Nerlov, C.; Lo Celso, C.; Poole, A.W. Multiple membrane extrusion sites drive megakaryocyte migration into bone marrow blood vessels. Life. Sci. Alliance 2018, 1, e201800061. [Google Scholar] [CrossRef]
- Lefrancais, E.; Looney, M.R. Platelet Biogenesis in the Lung Circulation. Physiology 2019, 34, 392–401. [Google Scholar] [CrossRef] [PubMed]
- Ouzegdouh, Y.; Capron, C.; Bauer, T.; Puymirat, E.; Diehl, J.L.; Martin, J.F.; Cramer-Borde, E. The physical and cellular conditions of the human pulmonary circulation enable thrombopoiesis. Exp. Hematol. 2018, 63, 22–27.e3. [Google Scholar] [CrossRef] [PubMed]
- Holinstat, M. Normal platelet function. Cancer Metastasis Rev. 2017, 36, 195–198. [Google Scholar] [CrossRef] [PubMed]
- Tomaiuolo, M.; Brass, L.F.; Stalker, T.J. Regulation of Platelet Activation and Coagulation and Its Role in Vascular Injury and Arterial Thrombosis. Interv. Cardiol. Clin. 2017, 6, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, J.; Li, X.; Shi, X.; Zhu, M.; Wang, J.; Huang, S.; Huang, X.; Wang, H.; Li, L.; Deng, H.; et al. Platelet integrin alphaIIbbeta3: Signal transduction, regulation, and its therapeutic targeting. J. Hematol. Oncol. 2019, 12, 26. [Google Scholar] [CrossRef] [Green Version]
- Hottz, E.D.; Bozza, F.A.; Bozza, P.T. Platelets in Immune Response to Virus and Immunopathology of Viral Infections. Front. Med. 2018, 5, 121. [Google Scholar] [CrossRef]
- McDonald, B.; Dunbar, M. Platelets and Intravascular Immunity: Guardians of the Vascular Space During Bloodstream Infections and Sepsis. Front. Immunol. 2019, 10, 2400. [Google Scholar] [CrossRef]
- Claushuis, T.A.M.; Van Der Veen, A.I.P.; Horn, J.; Schultz, M.J.; Houtkooper, R.H.; Van’t Veer, C.; Van Der Poll, T. Platelet Toll-like receptor expression and activation induced by lipopolysaccharide and sepsis. Platelets 2019, 30, 296–304. [Google Scholar] [CrossRef] [Green Version]
- Badimon, L.; Suades, R.; Fuentes, E.; Palomo, I.; Padro, T. Role of Platelet-Derived Microvesicles As Crosstalk Mediators in Atherothrombosis and Future Pharmacology Targets: A Link between Inflammation, Atherosclerosis, and Thrombosis. Front. Pharm. 2016, 7, 293. [Google Scholar] [CrossRef] [Green Version]
- Louwette, S.; Van Geet, C.; Freson, K. Regulators of G protein signaling: Role in hematopoiesis, megakaryopoiesis and platelet function. J. Thromb. Haemost. 2012, 10, 2215–2222. [Google Scholar] [CrossRef]
- Senis, Y.A.; Mazharian, A.; Mori, J. Src family kinases: At the forefront of platelet activation. Blood 2014, 124, 2013–2024. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Makhoul, S.; Kumm, E.; Zhang, P.; Walter, U.; Jurk, K. The Serine/Threonine Protein Phosphatase 2A (PP2A) Regulates Syk Activity in Human Platelets. Int. J. Mol. Sci. 2020, 21, 8939. [Google Scholar] [CrossRef] [PubMed]
- Moran, L.A.; Di, Y.; Sowa, M.A.; Hermida-Nogueira, L.; Barrachina, M.N.; Martin, E.; Mize, T.H.; Clark, J.C.; Eble, J.A.; Moreira, D.; et al. Katacine is a new ligand of CLEC-2 that acts as a platelet agonist. Thromb. Haemost. 2022. [Google Scholar] [CrossRef] [PubMed]
- Badolia, R.; Inamdar, V.; Manne, B.K.; Dangelmaier, C.; Eble, J.A.; Kunapuli, S.P. Gq pathway regulates proximal C-type lectin-like receptor-2 (CLEC-2) signaling in platelets. J. Biol. Chem. 2017, 292, 14516–14531. [Google Scholar] [CrossRef] [Green Version]
- Navarro-Nunez, L.; Langan, S.A.; Nash, G.B.; Watson, S.P. The physiological and pathophysiological roles of platelet CLEC-2. Thromb. Haemost. 2013, 109, 991–998. [Google Scholar] [CrossRef] [Green Version]
- Koupenova, M.; Ravid, K. Biology of Platelet Purinergic Receptors and Implications for Platelet Heterogeneity. Front. Pharm. 2018, 9, 37. [Google Scholar] [CrossRef] [Green Version]
- Van Kolen, K.; Slegers, H. Integration of P2Y receptor-activated signal transduction pathways in G protein-dependent signalling networks. Purinergic Signal. 2006, 2, 451–469. [Google Scholar] [CrossRef] [Green Version]
- Jin, J.; Mao, Y.; Thomas, D.; Kim, S.; Daniel, J.L.; Kunapuli, S.P. RhoA downstream of G(q) and G(12/13) pathways regulates protease-activated receptor-mediated dense granule release in platelets. Biochem. Pharm. 2009, 77, 835–844. [Google Scholar] [CrossRef] [Green Version]
- Kahn, M.L.; Nakanishi-Matsui, M.; Shapiro, M.J.; Ishihara, H.; Coughlin, S.R. Protease-activated receptors 1 and 4 mediate activation of human platelets by thrombin. J. Clin. Investig. 1999, 103, 879–887. [Google Scholar] [CrossRef] [Green Version]
- Flaumenhaft, R. Molecular basis of platelet granule secretion. Arter. Thromb. Vasc. Biol. 2003, 23, 1152–1160. [Google Scholar] [CrossRef] [Green Version]
- De Jong, J.S.; Dekker, L.R. Platelets and cardiac arrhythmia. Front. Physiol. 2010, 1, 166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernandez, D.I.; Kuijpers, M.J.E.; Heemskerk, J.W.M. Platelet calcium signaling by G-protein coupled and ITAM-linked receptors regulating anoctamin-6 and procoagulant activity. Platelets 2021, 32, 863–871. [Google Scholar] [CrossRef] [PubMed]
- Sagris, M.; Theofilis, P.; Antonopoulos, A.S.; Oikonomou, E.; Paschaliori, C.; Galiatsatos, N.; Tsioufis, K.; Tousoulis, D. Inflammation in Coronary Microvascular Dysfunction. Int. J. Mol. Sci. 2021, 22, 13471. [Google Scholar] [CrossRef] [PubMed]
- Sagris, M.; Theofilis, P.; Antonopoulos, A.S.; Tsioufis, C.; Oikonomou, E.; Antoniades, C.; Crea, F.; Kaski, J.C.; Tousoulis, D. Inflammatory Mechanisms in COVID-19 and Atherosclerosis: Current Pharmaceutical Perspectives. Int. J. Mol. Sci. 2021, 22, 6607. [Google Scholar] [CrossRef]
- Theofilis, P.; Sagris, M.; Oikonomou, E.; Antonopoulos, A.S.; Siasos, G.; Tsioufis, C.; Tousoulis, D. Inflammatory Mechanisms Contributing to Endothelial Dysfunction. Biomedicines 2021, 9, 781. [Google Scholar] [CrossRef]
- Theofilis, P.; Sagris, M.; Antonopoulos, A.S.; Oikonomou, E.; Tsioufis, C.; Tousoulis, D. Inflammatory Mediators of Platelet Activation: Focus on Atherosclerosis and COVID-19. Int. J. Mol. Sci. 2021, 22, 11170. [Google Scholar] [CrossRef]
- Oikonomou, E.; Leopoulou, M.; Theofilis, P.; Antonopoulos, A.S.; Siasos, G.; Latsios, G.; Mystakidi, V.C.; Antoniades, C.; Tousoulis, D. A link between inflammation and thrombosis in atherosclerotic cardiovascular diseases: Clinical and therapeutic implications. Atherosclerosis 2020, 309, 16–26. [Google Scholar] [CrossRef]
- Beaulieu, L.M.; Lin, E.; Mick, E.; Koupenova, M.; Weinberg, E.O.; Kramer, C.D.; Genco, C.A.; Tanriverdi, K.; Larson, M.G.; Benjamin, E.J.; et al. Interleukin 1 receptor 1 and interleukin 1beta regulate megakaryocyte maturation, platelet activation, and transcript profile during inflammation in mice and humans. Arter. Thromb. Vasc. Biol. 2014, 34, 552–564. [Google Scholar] [CrossRef] [Green Version]
- Marta, R.F.; Goette, N.P.; Lev, P.R.; Chazarreta, C.D.; Pirola, C.J.; Molinas, F.C. Normal platelets possess the soluble form of IL-6 receptor. Cytokine 2005, 29, 13–17. [Google Scholar] [CrossRef]
- Regnault, V.; De Maistre, E.; Carteaux, J.P.; Gruel, Y.; Nguyen, P.; Tardy, B.; Lecompte, T. Platelet activation induced by human antibodies to interleukin-8. Blood 2003, 101, 1419–1421. [Google Scholar] [CrossRef] [Green Version]
- Page, M.J.; Bester, J.; Pretorius, E. Interleukin-12 and its procoagulant effect on erythrocytes, platelets and fibrin(ogen): The lesser known side of inflammation. Br. J. Haematol. 2018, 180, 110–117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bester, J.; Pretorius, E. Effects of IL-1beta, IL-6 and IL-8 on erythrocytes, platelets and clot viscoelasticity. Sci. Rep. 2016, 6, 32188. [Google Scholar] [CrossRef] [PubMed]
- Davizon-Castillo, P.; McMahon, B.; Aguila, S.; Bark, D.; Ashworth, K.; Allawzi, A.; Campbell, R.A.; Montenont, E.; Nemkov, T.; D’Alessandro, A.; et al. TNF-alpha-driven inflammation and mitochondrial dysfunction define the platelet hyperreactivity of aging. Blood 2019, 134, 727–740. [Google Scholar] [CrossRef] [PubMed]
- Hottz, E.D.; Monteiro, A.P.; Bozza, F.A.; Bozza, P.T. Inflammasome in platelets: Allying coagulation and inflammation in infectious and sterile diseases? Mediat. Inflamm. 2015, 2015, 435783. [Google Scholar] [CrossRef] [PubMed]
- Brown, G.T.; Narayanan, P.; Li, W.; Silverstein, R.L.; McIntyre, T.M. Lipopolysaccharide stimulates platelets through an IL-1beta autocrine loop. J. Immunol. 2013, 191, 5196–5203. [Google Scholar] [CrossRef] [Green Version]
- Vogel, S.; Kamimura, S.; Arora, T.; Smith, M.L.; Almeida, L.E.F.; Combs, C.A.; Thein, S.L.; Quezado, Z.M.N. NLRP3 inflammasome and bruton tyrosine kinase inhibition interferes with upregulated platelet aggregation and in vitro thrombus formation in sickle cell mice. Biochem. Biophys. Res. Commun. 2021, 555, 196–201. [Google Scholar] [CrossRef]
- Busygina, K.; Jamasbi, J.; Seiler, T.; Deckmyn, H.; Weber, C.; Brandl, R.; Lorenz, R.; Siess, W. Oral Bruton tyrosine kinase inhibitors selectively block atherosclerotic plaque-triggered thrombus formation in humans. Blood 2018, 131, 2605–2616. [Google Scholar] [CrossRef] [Green Version]
- Zucoloto, A.Z.; Jenne, C.N. Platelet-Neutrophil Interplay: Insights Into Neutrophil Extracellular Trap (NET)-Driven Coagulation in Infection. Front. Cardiovasc. Med. 2019, 6, 85. [Google Scholar] [CrossRef] [Green Version]
- Rigg, R.A.; Healy, L.D.; Chu, T.T.; Ngo, A.T.P.; Mitrugno, A.; Zilberman-Rudenko, J.; Aslan, J.E.; Hinds, M.T.; Vecchiarelli, L.D.; Morgan, T.K.; et al. Protease-activated receptor 4 activity promotes platelet granule release and platelet-leukocyte interactions. Platelets 2019, 30, 126–135. [Google Scholar] [CrossRef]
- Seif, K.; Alidzanovic, L.; Tischler, B.; Ibrahim, N.; Zagrapan, B.; Rauscher, S.; Salzmann, M.; Hell, L.; Mauracher, L.M.; Budde, U.; et al. Neutrophil-Mediated Proteolysis of Thrombospondin-1 Promotes Platelet Adhesion and String Formation. Thromb. Haemost. 2018, 118, 2074–2085. [Google Scholar] [CrossRef] [Green Version]
- Quinn, K.L.; Henriques, M.; Tabuchi, A.; Han, B.; Yang, H.; Cheng, W.E.; Tole, S.; Yu, H.; Luo, A.; Charbonney, E.; et al. Human neutrophil peptides mediate endothelial-monocyte interaction, foam cell formation, and platelet activation. Arter. Thromb. Vasc. Biol. 2011, 31, 2070–2079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horn, M.; Bertling, A.; Brodde, M.F.; Muller, A.; Roth, J.; Van Aken, H.; Jurk, K.; Heilmann, C.; Peters, G.; Kehrel, B.E. Human neutrophil alpha-defensins induce formation of fibrinogen and thrombospondin-1 amyloid-like structures and activate platelets via glycoprotein IIb/IIIa. J. Thromb. Haemost. 2012, 10, 647–661. [Google Scholar] [CrossRef] [PubMed]
- Kaiser, P.; Harenberg, J.; Walenga, J.M.; Huhle, G.; Giese, C.; Prechel, M.; Hoppensteadt, D.; Fareed, J. Effects of a heparin-binding protein on blood coagulation and platelet function. Semin. Thromb. Hemost. 2001, 27, 495–502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santilli, F.; Paloscia, L.; Liani, R.; Di Nicola, M.; Di Marco, M.; Lattanzio, S.; La Barba, S.; Pascale, S.; Mascellanti, M.; Davi, G. Circulating myeloid-related protein-8/14 is related to thromboxane-dependent platelet activation in patients with acute coronary syndrome, with and without ongoing low-dose aspirin treatment. J. Am. Heart Assoc. 2014, 3, e000903. [Google Scholar] [CrossRef] [Green Version]
- Liang, X.; Xiu, C.; Liu, M.; Lin, C.; Chen, H.; Bao, R.; Yang, S.; Yu, J. Platelet-neutrophil interaction aggravates vascular in fl ammation and promotes the progression of atherosclerosis by activating the TLR4/NF-kappaB pathway. J. Cell Biochem. 2019, 120, 5612–5619. [Google Scholar] [CrossRef]
- Pircher, J.; Czermak, T.; Ehrlich, A.; Eberle, C.; Gaitzsch, E.; Margraf, A.; Grommes, J.; Saha, P.; Titova, A.; Ishikawa-Ankerhold, H.; et al. Cathelicidins prime platelets to mediate arterial thrombosis and tissue inflammation. Nat. Commun. 2018, 9, 1523. [Google Scholar] [CrossRef] [Green Version]
- Faraday, N.; Schunke, K.; Saleem, S.; Fu, J.; Wang, B.; Zhang, J.; Morrell, C.; Dore, S. Cathepsin G-dependent modulation of platelet thrombus formation in vivo by blood neutrophils. PLoS ONE 2013, 8, e71447. [Google Scholar] [CrossRef] [Green Version]
- Rossaint, J.; Kuhne, K.; Skupski, J.; Van Aken, H.; Looney, M.R.; Hidalgo, A.; Zarbock, A. Directed transport of neutrophil-derived extracellular vesicles enables platelet-mediated innate immune response. Nat. Commun. 2016, 7, 13464. [Google Scholar] [CrossRef]
- Thakur, M.; Evans, B.; Schindewolf, M.; Baumgartner, I.; Doring, Y. Neutrophil Extracellular Traps Affecting Cardiovascular Health in Infectious and Inflammatory Diseases. Cells 2021, 10, 1689. [Google Scholar] [CrossRef]
- Noubouossie, D.F.; Whelihan, M.F.; Yu, Y.B.; Sparkenbaugh, E.; Pawlinski, R.; Monroe, D.M.; Key, N.S. In vitro activation of coagulation by human neutrophil DNA and histone proteins but not neutrophil extracellular traps. Blood 2017, 129, 1021–1029. [Google Scholar] [CrossRef]
- Semeraro, F.; Ammollo, C.T.; Morrissey, J.H.; Dale, G.L.; Friese, P.; Esmon, N.L.; Esmon, C.T. Extracellular histones promote thrombin generation through platelet-dependent mechanisms: Involvement of platelet TLR2 and TLR4. Blood 2011, 118, 1952–1961. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Boer, O.J.; Li, X.; Teeling, P.; Mackaay, C.; Ploegmakers, H.J.; van der Loos, C.M.; Daemen, M.J.; de Winter, R.J.; van der Wal, A.C. Neutrophils, neutrophil extracellular traps and interleukin-17 associate with the organisation of thrombi in acute myocardial infarction. Thromb. Haemost. 2013, 109, 290–297. [Google Scholar] [CrossRef] [PubMed]
- Laridan, E.; Denorme, F.; Desender, L.; Francois, O.; Andersson, T.; Deckmyn, H.; Vanhoorelbeke, K.; De Meyer, S.F. Neutrophil extracellular traps in ischemic stroke thrombi. Ann. Neurol. 2017, 82, 223–232. [Google Scholar] [CrossRef] [PubMed]
- Rivadeneyra, L.; Carestia, A.; Etulain, J.; Pozner, R.G.; Fondevila, C.; Negrotto, S.; Schattner, M. Regulation of platelet responses triggered by Toll-like receptor 2 and 4 ligands is another non-genomic role of nuclear factor-kappaB. Thromb. Res. 2014, 133, 235–243. [Google Scholar] [CrossRef]
- Kalvegren, H.; Skoglund, C.; Helldahl, C.; Lerm, M.; Grenegard, M.; Bengtsson, T. Toll-like receptor 2 stimulation of platelets is mediated by purinergic P2X1-dependent Ca2+ mobilisation, cyclooxygenase and purinergic P2Y1 and P2Y12 receptor activation. Thromb. Haemost. 2010, 103, 398–407. [Google Scholar] [CrossRef]
- Klarstrom Engstrom, K.; Brommesson, C.; Kalvegren, H.; Bengtsson, T. Toll like receptor 2/1 mediated platelet adhesion and activation on bacterial mimetic surfaces is dependent on src/Syk-signaling and purinergic receptor P2X1 and P2Y12 activation. Biointerphases 2014, 9, 041003. [Google Scholar] [CrossRef]
- Lopes Pires, M.E.; Clarke, S.R.; Marcondes, S.; Gibbins, J.M. Lipopolysaccharide potentiates platelet responses via toll-like receptor 4-stimulated Akt-Erk-PLA2 signalling. PLoS ONE 2017, 12, e0186981. [Google Scholar] [CrossRef]
- Stahl, A.L.; Svensson, M.; Morgelin, M.; Svanborg, C.; Tarr, P.I.; Mooney, J.C.; Watkins, S.L.; Johnson, R.; Karpman, D. Lipopolysaccharide from enterohemorrhagic Escherichia coli binds to platelets through TLR4 and CD62 and is detected on circulating platelets in patients with hemolytic uremic syndrome. Blood 2006, 108, 167–176. [Google Scholar] [CrossRef] [Green Version]
- Hally, K.E.; La Flamme, A.C.; Larsen, P.D.; Harding, S.A. Platelet Toll-like receptor (TLR) expression and TLR-mediated platelet activation in acute myocardial infarction. Thromb. Res. 2017, 158, 8–15. [Google Scholar] [CrossRef]
- De Stoppelaar, S.F.; Claushuis, T.A.; Schaap, M.C.; Hou, B.; van der Poll, T.; Nieuwland, R.; van’t Veer, C. Toll-Like Receptor Signalling Is Not Involved in Platelet Response to Streptococcus pneumoniae In Vitro or In Vivo. PLoS ONE 2016, 11, e0156977. [Google Scholar] [CrossRef]
- D’Atri, L.P.; Etulain, J.; Rivadeneyra, L.; Lapponi, M.J.; Centurion, M.; Cheng, K.; Yin, H.; Schattner, M. Expression and functionality of Toll-like receptor 3 in the megakaryocytic lineage. J. Thromb. Haemost. 2015, 13, 839–850. [Google Scholar] [CrossRef] [PubMed]
- Rex, S.; Beaulieu, L.M.; Perlman, D.H.; Vitseva, O.; Blair, P.S.; McComb, M.E.; Costello, C.E.; Freedman, J.E. Immune versus thrombotic stimulation of platelets differentially regulates signalling pathways, intracellular protein-protein interactions, and alpha-granule release. Thromb. Haemost. 2009, 102, 97–110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smolenski, A. Novel roles of cAMP/cGMP-dependent signaling in platelets. J. Thromb. Haemost. 2012, 10, 167–176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szabo, C.; Ischiropoulos, H.; Radi, R. Peroxynitrite: Biochemistry, pathophysiology and development of therapeutics. Nat. Rev. Drug Discov. 2007, 6, 662–680. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, U.R.; Walter, U.; Eigenthaler, M. Taming platelets with cyclic nucleotides. Biochem. Pharm. 2001, 62, 1153–1161. [Google Scholar] [CrossRef]
- Cheng, Y.; Austin, S.C.; Rocca, B.; Koller, B.H.; Coffman, T.M.; Grosser, T.; Lawson, J.A.; FitzGerald, G.A. Role of prostacyclin in the cardiovascular response to thromboxane A2. Science 2002, 296, 539–541. [Google Scholar] [CrossRef]
- Koupenova, M.; Kehrel, B.E.; Corkrey, H.A.; Freedman, J.E. Thrombosis and platelets: An update. Eur. Heart J. 2017, 38, 785–791. [Google Scholar] [CrossRef]
- Watanabe-Kusunoki, K.; Nakazawa, D.; Ishizu, A.; Atsumi, T. Thrombomodulin as a Physiological Modulator of Intravascular Injury. Front. Immunol. 2020, 11, 575890. [Google Scholar] [CrossRef]
- Nightingale, T.; Cutler, D. The secretion of von Willebrand factor from endothelial cells; an increasingly complicated story. J. Thromb. Haemost. 2013, 11 (Suppl. 1), 192–201. [Google Scholar] [CrossRef]
- Blair, P.; Flaumenhaft, R. Platelet alpha-granules: Basic biology and clinical correlates. Blood Rev. 2009, 23, 177–189. [Google Scholar] [CrossRef] [Green Version]
- Wu, M.D.; Atkinson, T.M.; Lindner, J.R. Platelets and von Willebrand factor in atherogenesis. Blood 2017, 129, 1415–1419. [Google Scholar] [CrossRef] [PubMed]
- Dong, J.F.; Moake, J.L.; Nolasco, L.; Bernardo, A.; Arceneaux, W.; Shrimpton, C.N.; Schade, A.J.; McIntire, L.V.; Fujikawa, K.; Lopez, J.A. ADAMTS-13 rapidly cleaves newly secreted ultralarge von Willebrand factor multimers on the endothelial surface under flowing conditions. Blood 2002, 100, 4033–4039. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alphonsus, C.S.; Rodseth, R.N. The endothelial glycocalyx: A review of the vascular barrier. Anaesthesia 2014, 69, 777–784. [Google Scholar] [CrossRef] [PubMed]
- Safiah Mokhtar, S.; Vanhoutte, P.M.; Leung, S.W.S.; Imran Yusof, M.; Wan Sulaiman, W.A.; Zaharil Mat Saad, A.; Suppian, R.; Ghulam Rasool, A.H. Reduced expression of prostacyclin synthase and nitric oxide synthase in subcutaneous arteries of type 2 diabetic patients. Tohoku J. Exp. Med. 2013, 231, 217–222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roy, C.; Tabiasco, J.; Caillon, A.; Delneste, Y.; Merot, J.; Favre, J.; Guihot, A.L.; Martin, L.; Nascimento, D.C.; Ryffel, B.; et al. Loss of vascular expression of nucleoside triphosphate diphosphohydrolase-1/CD39 in hypertension. Purinergic Signal. 2018, 14, 73–82. [Google Scholar] [CrossRef] [PubMed]
- Takahashi-Sato, K.; Murakawa, M.; Kimura, J.; Ito, M.A.; Matsuoka, I. Loss of ectonucleotidases from the coronary vascular bed after ischemia-reperfusion in isolated rat heart. BMC Cardiovasc. Disord. 2013, 13, 53. [Google Scholar] [CrossRef] [Green Version]
- Uchimido, R.; Schmidt, E.P.; Shapiro, N.I. The glycocalyx: A novel diagnostic and therapeutic target in sepsis. Crit. Care 2019, 23, 16. [Google Scholar] [CrossRef] [Green Version]
- Becker, B.F.; Jacob, M.; Leipert, S.; Salmon, A.H.; Chappell, D. Degradation of the endothelial glycocalyx in clinical settings: Searching for the sheddases. Br. J. Clin. Pharm. 2015, 80, 389–402. [Google Scholar] [CrossRef]
- Lukasz, A.; Hillgruber, C.; Oberleithner, H.; Kusche-Vihrog, K.; Pavenstadt, H.; Rovas, A.; Hesse, B.; Goerge, T.; Kumpers, P. Endothelial glycocalyx breakdown is mediated by angiopoietin-2. Cardiovasc. Res. 2017, 113, 671–680. [Google Scholar] [CrossRef] [Green Version]
- Constantinescu, A.A.; Vink, H.; Spaan, J.A. Endothelial cell glycocalyx modulates immobilization of leukocytes at the endothelial surface. Arter. Thromb. Vasc. Biol. 2003, 23, 1541–1547. [Google Scholar] [CrossRef] [Green Version]
- Becker, B.F.; Chappell, D.; Bruegger, D.; Annecke, T.; Jacob, M. Therapeutic strategies targeting the endothelial glycocalyx: Acute deficits, but great potential. Cardiovasc. Res. 2010, 87, 300–310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reitsma, S.; Oude Egbrink, M.G.; Heijnen, V.V.; Megens, R.T.; Engels, W.; Vink, H.; Slaaf, D.W.; Van Zandvoort, M.A. Endothelial glycocalyx thickness and platelet-vessel wall interactions during atherogenesis. Thromb. Haemost. 2011, 106, 939–946. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sempere, L.F.; Azmi, A.S.; Moore, A. microRNA-based diagnostic and therapeutic applications in cancer medicine. Wiley Interdiscip. Rev. RNA 2021, 12, e1662. [Google Scholar] [CrossRef]
- Theofilis, P.; Oikonomou, E.; Vogiatzi, G.; Antonopoulos, A.S.; Siasos, G.; Iliopoulos, D.C.; Perrea, D.; Tsioufis, C.; Tousoulis, D. The impact of proangiogenic microRNA modulation on blood flow recovery following hind limb ischemia. A systematic review and meta-analysis of animal studies. Vasc. Pharm. 2021, 141, 106906. [Google Scholar] [CrossRef] [PubMed]
- Theofilis, P.; Vogiatzi, G.; Oikonomou, E.; Gazouli, M.; Siasos, G.; Katifelis, H.; Perrea, D.; Vavuranakis, M.; Iliopoulos, D.C.; Tsioufis, C.; et al. The Effect of MicroRNA-126 Mimic Administration on Vascular Perfusion Recovery in an Animal Model of Hind Limb Ischemia. Front. Mol. Biosci. 2021, 8, 724465. [Google Scholar] [CrossRef]
- Zhou, S.S.; Jin, J.P.; Wang, J.Q.; Zhang, Z.G.; Freedman, J.H.; Zheng, Y.; Cai, L. miRNAS in cardiovascular diseases: Potential biomarkers, therapeutic targets and challenges. Acta Pharm. Sin. 2018, 39, 1073–1084. [Google Scholar] [CrossRef] [Green Version]
- Choi, J.L.; Li, S.; Han, J.Y. Platelet function tests: A review of progresses in clinical application. Biomed. Res. Int. 2014, 2014, 456569. [Google Scholar] [CrossRef]
- Garcia, A.; Dunoyer-Geindre, S.; Zapilko, V.; Nolli, S.; Reny, J.L.; Fontana, P. Functional Validation of microRNA-126-3p as a Platelet Reactivity Regulator Using Human Haematopoietic Stem Cells. Thromb. Haemost. 2019, 119, 254–263. [Google Scholar] [CrossRef] [Green Version]
- Pordzik, J.; Pisarz, K.; De Rosa, S.; Jones, A.D.; Eyileten, C.; Indolfi, C.; Malek, L.; Postula, M. The Potential Role of Platelet-Related microRNAs in the Development of Cardiovascular Events in High-Risk Populations, Including Diabetic Patients: A Review. Front. Endocrinol. 2018, 9, 74. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Guo, L.Z.; Kim, M.H.; Han, J.Y.; Serebruany, V. Platelet microRNA for predicting acute myocardial infarction. J. Thromb. Thrombolysis lm. 2017, 44, 556–564. [Google Scholar] [CrossRef]
- Landry, P.; Plante, I.; Ouellet, D.L.; Perron, M.P.; Rousseau, G.; Provost, P. Existence of a microRNA pathway in anucleate platelets. Nat. Struct. Mol. Biol. 2009, 16, 961–966. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delgado Lagos, F.; Elgheznawy, A.; Kyselova, A.; Meyer Zu Heringdorf, D.; Ratiu, C.; Randriamboavonjy, V.; Mann, A.W.; Fisslthaler, B.; Siragusa, M.; Fleming, I. Secreted modular calcium-binding protein 1 binds and activates thrombin to account for platelet hyperreactivity in diabetes. Blood 2021, 137, 1641–1651. [Google Scholar] [CrossRef] [PubMed]
- Leierseder, S.; Petzold, T.; Zhang, L.; Loyer, X.; Massberg, S.; Engelhardt, S. MiR-223 is dispensable for platelet production and function in mice. Thromb. Haemost. 2013, 110, 1207–1214. [Google Scholar] [CrossRef]
- Kaudewitz, D.; Skroblin, P.; Bender, L.H.; Barwari, T.; Willeit, P.; Pechlaner, R.; Sunderland, N.P.; Willeit, K.; Morton, A.C.; Armstrong, P.C.; et al. Association of MicroRNAs and YRNAs With Platelet Function. Circ. Res. 2016, 118, 420–432. [Google Scholar] [CrossRef] [PubMed]
- Dahiya, N.; Atreya, C.D. RAP1 Downregulation by miR-320c Reduces Platelet Activation in Ex-vivo Storage. Microrna 2019, 8, 36–42. [Google Scholar] [CrossRef]
- Dahiya, N.; Atreya, C.D. MiR-181a Reduces Platelet Activation via the Inhibition of Endogenous RAP1B. Microrna 2020, 9, 240–246. [Google Scholar] [CrossRef] [PubMed]
- Szilagyi, B.; Fejes, Z.; Poliska, S.; Pocsi, M.; Czimmerer, Z.; Patsalos, A.; Fenyvesi, F.; Rusznyak, A.; Nagy, G.; Kerekes, G.; et al. Reduced miR-26b Expression in Megakaryocytes and Platelets Contributes to Elevated Level of Platelet Activation Status in Sepsis. Int. J. Mol. Sci. 2020, 21, 866. [Google Scholar] [CrossRef] [Green Version]
- Becker, K.C.; Kwee, L.C.; Neely, M.L.; Grass, E.; Jakubowski, J.A.; Fox, K.A.A.; White, H.D.; Gregory, S.G.; Gurbel, P.A.; Carvalho, L.P.; et al. Circulating MicroRNA Profiling in Non-ST Elevated Coronary Artery Syndrome Highlights Genomic Associations with Serial Platelet Reactivity Measurements. Sci. Rep. 2020, 10, 6169. [Google Scholar] [CrossRef]
- Pedersen, O.B.; Hvas, A.M.; Grove, E.L.; Larsen, S.B.; Pasalic, L.; Kristensen, S.D.; Nissen, P.H. Association of whole blood microRNA expression with platelet function and turnover in patients with coronary artery disease. Thromb. Res. 2022, 211, 98–105. [Google Scholar] [CrossRef]
- Garcia, A.; Dunoyer-Geindre, S.; Nolli, S.; Reny, J.L.; Fontana, P. An Ex Vivo and In Silico Study Providing Insights into the Interplay of Circulating miRNAs Level, Platelet Reactivity and Thrombin Generation: Looking beyond Traditional Pharmacogenetics. J. Pers. Med. 2021, 11, 323. [Google Scholar] [CrossRef]
- Tran, J.Q.D.; Pedersen, O.H.; Larsen, M.L.; Grove, E.L.; Kristensen, S.D.; Hvas, A.M.; Nissen, P.H. Platelet microRNA expression and association with platelet maturity and function in patients with essential thrombocythemia. Platelets 2020, 31, 365–372. [Google Scholar] [CrossRef] [PubMed]
- Krammer, T.L.; Mayr, M.; Hackl, M. microRNAs as promising biomarkers of platelet activity in antiplatelet therapy monitoring. Int. J. Mol. Sci. 2020, 21, 3477. [Google Scholar] [CrossRef] [PubMed]
- Willeit, P.; Zampetaki, A.; Dudek, K.; Kaudewitz, D.; King, A.; Kirkby, N.S.; Crosby-Nwaobi, R.; Prokopi, M.; Drozdov, I.; Langley, S.R.; et al. Circulating microRNAs as novel biomarkers for platelet activation. Circ. Res. 2013, 112, 595–600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Braza-Boils, A.; Barwari, T.; Gutmann, C.; Thomas, M.R.; Judge, H.M.; Joshi, A.; Pechlaner, R.; Shankar-Hari, M.; Ajjan, R.A.; Sabroe, I.; et al. Circulating MicroRNA Levels Indicate Platelet and Leukocyte Activation in Endotoxemia Despite Platelet P2Y12 Inhibition. Int. J. Mol. Sci. 2020, 21, 2897. [Google Scholar] [CrossRef] [Green Version]
- Carino, A.; De Rosa, S.; Sorrentino, S.; Polimeni, A.; Sabatino, J.; Caiazzo, G.; Torella, D.; Spaccarotella, C.; Mongiardo, A.; Strangio, A.; et al. Modulation of Circulating MicroRNAs Levels during the Switch from Clopidogrel to Ticagrelor. Biomed. Res. Int. 2016, 2016, 3968206. [Google Scholar] [CrossRef] [Green Version]
- Chyrchel, B.; Toton-Zuranska, J.; Kruszelnicka, O.; Chyrchel, M.; Mielecki, W.; Kolton-Wroz, M.; Wolkow, P.; Surdacki, A. Association of plasma miR-223 and platelet reactivity in patients with coronary artery disease on dual antiplatelet therapy: A preliminary report. Platelets 2015, 26, 593–597. [Google Scholar] [CrossRef]
- Shi, R.; Ge, L.; Zhou, X.; Ji, W.J.; Lu, R.Y.; Zhang, Y.Y.; Zeng, S.; Liu, X.; Zhao, J.H.; Zhang, W.C.; et al. Decreased platelet miR-223 expression is associated with high on-clopidogrel platelet reactivity. Thromb. Res. 2013, 131, 508–513. [Google Scholar] [CrossRef]
- Zhang, Y.Y.; Zhou, X.; Ji, W.J.; Shi, R.; Lu, R.Y.; Li, J.L.; Yang, G.H.; Luo, T.; Zhang, J.Q.; Zhao, J.H.; et al. Decreased circulating microRNA-223 level predicts high on-treatment platelet reactivity in patients with troponin-negative non-ST elevation acute coronary syndrome. J. Thromb. Thrombolysis 2014, 38, 65–72. [Google Scholar] [CrossRef]
- Peng, L.; Liu, J.; Qin, L.; Liu, J.; Xi, S.; Lu, C.; Yin, T. Interaction between platelet-derived microRNAs and CYP2C19*2 genotype on clopidogrel antiplatelet responsiveness in patients with ACS. Thromb. Res. 2017, 157, 97–102. [Google Scholar] [CrossRef]
- Liu, J.; Qin, L.; Wang, Z.; Peng, L.; Liu, J.; Wang, X.; Du, R.; Zou, Y.; Wu, Y.; Yin, T. Platelet-derived miRNAs as determinants of the antiplatelet response in clopidogrel-treated patients with ACS. Thromb. Res. 2020, 186, 71–74. [Google Scholar] [CrossRef]
- Pedersen, O.B.; Grove, E.L.; Kristensen, S.D.; Nissen, P.H.; Hvas, A.M. MicroRNA as Biomarkers for Platelet Function and Maturity in Patients with Cardiovascular Disease. Thromb. Haemost. 2022, 122, 181–195. [Google Scholar] [CrossRef] [PubMed]
- Zheng, S.L.; Roddick, A.J. Association of Aspirin Use for Primary Prevention With Cardiovascular Events and Bleeding Events: A Systematic Review and Meta-analysis. JAMA 2019, 321, 277–287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, H.; Gu, Q.; Niu, H.; Li, X.; Wang, R. Benefits and Risks Associated With Aspirin Use in Patients With Diabetes for the Primary Prevention of Cardiovascular Events and Mortality: A Meta-Analysis. Front. Endocrinol. 2021, 12, 741374. [Google Scholar] [CrossRef] [PubMed]
- Masson, W.; Barbagelata, L.; Lavalle-Cobo, A.; Lobo, M.; Masson, G.; Nogueira, J.P.; Verges, B. Low-doses aspirin in the primary prevention of cardiovascular disease in patients with diabetes: Meta-analysis stratified by baseline cardiovascular risk. Diabetes Metab. Syndr. 2022, 16, 102391. [Google Scholar] [CrossRef]
- Bates, E.R.; Lau, W.C.; Angiolillo, D.J. Clopidogrel-drug interactions. J. Am. Coll. Cardiol. 2011, 57, 1251–1263. [Google Scholar] [CrossRef] [Green Version]
- Condello, F.; Liccardo, G.; Ferrante, G. Clinical Effects of Dual Antiplatelet Therapy or Aspirin Monotherapy after Acute Minor Ischemic Stroke or Transient Ischemic Attack, a Meta-Analysis. Curr. Pharm. Des. 2021, 27, 4140–4146. [Google Scholar] [CrossRef]
- Squizzato, A.; Bellesini, M.; Takeda, A.; Middeldorp, S.; Donadini, M.P. Clopidogrel plus aspirin versus aspirin alone for preventing cardiovascular events. Cochrane Database Syst. Rev. 2017, 12, CD005158. [Google Scholar] [CrossRef]
- Liang, L.R.; Ma, Q.; Feng, L.; Qiu, Q.; Zheng, W.; Xie, W.X. Long-term effect of clopidogrel in patients with and without diabetes: A systematic review and meta-analysis of randomized controlled trials. World J. Diabetes 2020, 11, 137–149. [Google Scholar] [CrossRef]
- Biswas, M.; Kali, S.K. Association of CYP2C19 Loss-of-Function Alleles with Major Adverse Cardiovascular Events of Clopidogrel in Stable Coronary Artery Disease Patients Undergoing Percutaneous Coronary Intervention: Meta-analysis. Cardiovasc. Drugs Ther. 2021, 35, 1147–1159. [Google Scholar] [CrossRef]
- Jafrin, S.; Naznin, N.E.; Reza, M.S.; Aziz, M.A.; Islam, M.S. Risk of stroke in CYP2C19 LoF polymorphism carrier coronary artery disease patients undergoing clopidogrel therapy: An ethnicity-based updated meta-analysis. Eur. J. Intern. Med. 2021, 90, 49–65. [Google Scholar] [CrossRef]
- Biswas, M.; Sukasem, C.; Khatun Kali, M.S.; Ibrahim, B. Effects of the CYP2C19 LoF allele on major adverse cardiovascular events associated with clopidogrel in acute coronary syndrome patients undergoing percutaneous coronary intervention: A meta-analysis. Pharmacogenomics 2022, 23, 207–220. [Google Scholar] [CrossRef] [PubMed]
- Li, J.L.; Fu, Y.; Qin, S.B.; Liang, G.K.; Liu, J.; Nie, X.Y.; Chen, J.; Shi, L.W.; Shao, H.; Lu, Y. Association between P2RY12 gene polymorphisms and adverse clinical events in coronary artery disease patients treated with clopidogrel: A systematic review and meta-analysis. Gene 2018, 657, 69–80. [Google Scholar] [CrossRef] [PubMed]
- Demcsak, A.; Lantos, T.; Balint, E.R.; Hartmann, P.; Vincze, A.; Bajor, J.; Czopf, L.; Alizadeh, H.; Gyongyi, Z.; Marta, K.; et al. PPIs Are Not Responsible for Elevating Cardiovascular Risk in Patients on Clopidogrel-A Systematic Review and Meta-Analysis. Front. Physiol. 2018, 9, 1550. [Google Scholar] [CrossRef] [PubMed]
- Gupta, R.; Malik, A.H.; Briasoulis, A.; Joshi, A.M.; Guthier, D.G.; Popli, T.; Aronow, W.S.; Vyas, A.V.; Patel, N.C.; Ahmad, H.; et al. Comparative Safety and Effectiveness of Loading Doses of P2Y12 Inhibitors in Patients Undergoing Elective PCI: A Network Meta-analysis. Cardiovasc. Drugs Ther. 2021. [Google Scholar] [CrossRef]
- Navarese, E.P.; Khan, S.U.; Kolodziejczak, M.; Kubica, J.; Buccheri, S.; Cannon, C.P.; Gurbel, P.A.; De Servi, S.; Budaj, A.; Bartorelli, A.; et al. Comparative Efficacy and Safety of Oral P2Y12 Inhibitors in Acute Coronary Syndrome: Network Meta-Analysis of 52 816 Patients From 12 Randomized Trials. Circulation 2020, 142, 150–160. [Google Scholar] [CrossRef] [PubMed]
- Farmakis, I.T.; Doundoulakis, I.; Zafeiropoulos, S.; Pagiantza, A.; Apostolidou-Kiouti, F.; Kourti, O.; Kassimis, G.; Haidich, A.B.; Karvounis, H.; Giannakoulas, G. Comparative efficacy and safety of oral P2Y12 inhibitors for patients with chronic kidney disease and acute coronary syndrome: A network meta-analysis. Hell. J. Cardiol. 2022, 63, 40–65. [Google Scholar] [CrossRef]
- Abusnina, W.; Al-Abdouh, A.; Bizanti, A.; Gill, G.; Houssien, A.; Alshebani, Y.; Kanmanthareddy, A.; Dahal, K. Ischemic and bleeding outcomes of potent P2Y12 inhibitor antiplatelet agents versus clopidogrel in elderly patients with acute coronary syndrome: A meta-analysis of randomized trials. Cardiovasc. Revasc. Med. 2021. [Google Scholar] [CrossRef]
- Pereira, N.L.; Rihal, C.; Lennon, R.; Marcus, G.; Shrivastava, S.; Bell, M.R.; So, D.; Geller, N.; Goodman, S.G.; Hasan, A.; et al. Effect of CYP2C19 Genotype on Ischemic Outcomes During Oral P2Y12 Inhibitor Therapy: A Meta-Analysis. JACC Cardiovasc. Interv. 2021, 14, 739–750. [Google Scholar] [CrossRef]
- Galli, M.; Benenati, S.; Franchi, F.; Rollini, F.; Capodanno, D.; Biondi-Zoccai, G.; Vescovo, G.M.; Cavallari, L.H.; Bikdeli, B.; Ten Berg, J.; et al. Comparative effects of guided vs. potent P2Y12 inhibitor therapy in acute coronary syndrome: A network meta-analysis of 61 898 patients from 15 randomized trials. Eur. Heart J. 2021, 43, 959–967. [Google Scholar] [CrossRef]
- Shoji, S.; Kuno, T.; Fujisaki, T.; Takagi, H.; Briasoulis, A.; Deharo, P.; Cuisset, T.; Latib, A.; Kohsaka, S. De-Escalation of Dual Antiplatelet Therapy in Patients With Acute Coronary Syndromes. J. Am. Coll. Cardiol. 2021, 78, 763–777. [Google Scholar] [CrossRef]
- Rey, M.; Kramberg, M.; Hess, P.; Morrison, K.; Ernst, R.; Haag, F.; Weber, E.; Clozel, M.; Baumann, M.; Caroff, E.; et al. The reversible P2Y12 antagonist ACT-246475 causes significantly less blood loss than ticagrelor at equivalent antithrombotic efficacy in rat. Pharm. Res. Perspect. 2017, 5, e00338. [Google Scholar] [CrossRef] [Green Version]
- Crescence, L.; Darbousset, R.; Caroff, E.; Hubler, F.; Riederer, M.A.; Panicot-Dubois, L.; Dubois, C. Selatogrel, a reversible P2Y12 receptor antagonist, has reduced off-target interference with haemostatic factors in a mouse thrombosis model. Thromb. Res. 2021, 200, 133–140. [Google Scholar] [CrossRef] [PubMed]
- Storey, R.F.; Gurbel, P.A.; Ten Berg, J.; Bernaud, C.; Dangas, G.D.; Frenoux, J.M.; Gorog, D.A.; Hmissi, A.; Kunadian, V.; James, S.K.; et al. Pharmacodynamics, pharmacokinetics, and safety of single-dose subcutaneous administration of selatogrel, a novel P2Y12 receptor antagonist, in patients with chronic coronary syndromes. Eur. Heart J. 2020, 41, 3132–3140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sinnaeve, P.; Fahrni, G.; Schelfaut, D.; Spirito, A.; Mueller, C.; Frenoux, J.M.; Hmissi, A.; Bernaud, C.; Ufer, M.; Moccetti, T.; et al. Subcutaneous Selatogrel Inhibits Platelet Aggregation in Patients With Acute Myocardial Infarction. J. Am. Coll. Cardiol. 2020, 75, 2588–2597. [Google Scholar] [CrossRef] [PubMed]
- Schilling, U.; Dingemanse, J.; Dobrow, M.; Baumann, M.; Riederer, M.A.; Juif, P.E.; Ufer, M. Insights from In Vitro and Clinical Data to Guide Transition from the Novel P2Y12 Antagonist Selatogrel to Clopidogrel, Prasugrel, and Ticagrelor. Thromb. Haemost. 2021, 121, 755–766. [Google Scholar] [CrossRef] [PubMed]
- Crescence, L.; Kramberg, M.; Baumann, M.; Rey, M.; Roux, S.; Panicot-Dubois, L.; Dubois, C.; Riederer, M.A. The P2Y12 Receptor Antagonist Selatogrel Dissolves Preformed Platelet Thrombi In Vivo. J. Clin. Med. 2021, 10, 5349. [Google Scholar] [CrossRef]
- Boersma, E.; Harrington, R.A.; Moliterno, D.J.; White, H.; Theroux, P.; Van de Werf, F.; de Torbal, A.; Armstrong, P.W.; Wallentin, L.C.; Wilcox, R.G.; et al. Platelet glycoprotein IIb/IIIa inhibitors in acute coronary syndromes: A meta-analysis of all major randomised clinical trials. Lancet 2002, 359, 189–198. [Google Scholar] [CrossRef]
- Safley, D.M.; Venkitachalam, L.; Kennedy, K.F.; Cohen, D.J. Impact of Glycoprotein IIb/IIIa Inhibition in Contemporary Percutaneous Coronary Intervention for Acute Coronary Syndromes: Insights From the National Cardiovascular Data Registry. JACC Cardiovasc. Interv. 2015, 8, 1574–1582. [Google Scholar] [CrossRef]
- Saleiro, C.; Teixeira, R.; De Campos, D.; Lopes, J.; Oliveiros, B.; Costa, M.; Gonçalves, L. Glycoprotein IIb/IIIa inhibitors for cardiogenic shock complicating acute myocardial infarction: A systematic review, meta-analysis, and meta-regression. J. Intensive Care 2020, 8, 85. [Google Scholar] [CrossRef]
- Ciccone, A.; Motto, C.; Abraha, I.; Cozzolino, F.; Santilli, I. Glycoprotein IIb-IIIa inhibitors for acute ischaemic stroke. Cochrane Database Syst. Rev. 2014, CD005208. [Google Scholar] [CrossRef] [Green Version]
- Zhu, X.; Cao, G. Safety of Glycoprotein IIb-IIIa Inhibitors Used in Stroke-Related Treatment: A Systematic Review and Meta-Analysis. Clin. Appl. Thromb. Hemost. 2020, 26, 1076029620942594. [Google Scholar] [CrossRef] [PubMed]
- Gurbel, P.A.; Jeong, Y.H.; Tantry, U.S. Vorapaxar: A novel protease-activated receptor-1 inhibitor. Expert Opin. Investig. Drugs 2011, 20, 1445–1453. [Google Scholar] [CrossRef] [PubMed]
- Morrow, D.A.; Braunwald, E.; Bonaca, M.P.; Ameriso, S.F.; Dalby, A.J.; Fish, M.P.; Fox, K.A.; Lipka, L.J.; Liu, X.; Nicolau, J.C.; et al. Vorapaxar in the secondary prevention of atherothrombotic events. N. Engl. J. Med. 2012, 366, 1404–1413. [Google Scholar] [CrossRef] [PubMed]
- Tricoci, P.; Huang, Z.; Held, C.; Moliterno, D.J.; Armstrong, P.W.; Van de Werf, F.; White, H.D.; Aylward, P.E.; Wallentin, L.; Chen, E.; et al. Thrombin-receptor antagonist vorapaxar in acute coronary syndromes. N. Engl. J. Med. 2012, 366, 20–33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cavender, M.A.; Scirica, B.M.; Bonaca, M.P.; Angiolillo, D.J.; Dalby, A.J.; Dellborg, M.; Morais, J.; Murphy, S.A.; Ophuis, T.O.; Tendera, M.; et al. Vorapaxar in patients with diabetes mellitus and previous myocardial infarction: Findings from the thrombin receptor antagonist in secondary prevention of atherothrombotic ischemic events-TIMI 50 trial. Circulation 2015, 131, 1047–1053. [Google Scholar] [CrossRef] [Green Version]
- Correa, S.; Bonaca, M.P.; Scirica, B.M.; Murphy, S.A.; Goodrich, E.L.; Morrow, D.A.; O’Donoghue, M.L. Efficacy and safety of more potent antiplatelet therapy with vorapaxar in patients with impaired renal function. J. Thromb. Thrombolysis 2019, 47, 353–360. [Google Scholar] [CrossRef]
- Bonaca, M.P.; Scirica, B.M.; Braunwald, E.; Wiviott, S.D.; O’Donoghue, M.L.; Murphy, S.A.; Morrow, D.A. Coronary stent thrombosis with vorapaxar versus placebo: Results from the TRA 2 degrees P-TIMI 50 trial. J. Am. Coll. Cardiol. 2014, 64, 2309–2317. [Google Scholar] [CrossRef] [Green Version]
- Ulrichts, H.; Silence, K.; Schoolmeester, A.; de Jaegere, P.; Rossenu, S.; Roodt, J.; Priem, S.; Lauwereys, M.; Casteels, P.; Van Bockstaele, F.; et al. Antithrombotic drug candidate ALX-0081 shows superior preclinical efficacy and safety compared with currently marketed antiplatelet drugs. Blood 2011, 118, 757–765. [Google Scholar] [CrossRef] [Green Version]
- Peyvandi, F.; Scully, M.; Kremer Hovinga, J.A.; Cataland, S.; Knobl, P.; Wu, H.; Artoni, A.; Westwood, J.P.; Mansouri Taleghani, M.; Jilma, B.; et al. Caplacizumab for Acquired Thrombotic Thrombocytopenic Purpura. N. Engl. J. Med. 2016, 374, 511–522. [Google Scholar] [CrossRef]
- Scully, M.; Cataland, S.R.; Peyvandi, F.; Coppo, P.; Knobl, P.; Kremer Hovinga, J.A.; Metjian, A.; de la Rubia, J.; Pavenski, K.; Callewaert, F.; et al. Caplacizumab Treatment for Acquired Thrombotic Thrombocytopenic Purpura. N. Engl. J. Med. 2019, 380, 335–346. [Google Scholar] [CrossRef]
- Peyvandi, F.; Scully, M.; Kremer Hovinga, J.A.; Knobl, P.; Cataland, S.; De Beuf, K.; Callewaert, F.; De Winter, H.; Zeldin, R.K. Caplacizumab reduces the frequency of major thromboembolic events, exacerbations and death in patients with acquired thrombotic thrombocytopenic purpura. J. Thromb. Haemost. 2017, 15, 1448–1452. [Google Scholar] [CrossRef] [PubMed]
- Van Loon, J.E.; De Jaegere, P.P.; Ulrichts, H.; Van Vliet, H.H.; De Maat, M.P.; De Groot, P.G.; Simoons, M.L.; Leebeek, F.W. The in vitro effect of the new antithrombotic drug candidate ALX-0081 on blood samples of patients undergoing percutaneous coronary intervention. Thromb. Haemost. 2011, 106, 165–171. [Google Scholar] [CrossRef] [PubMed]
- Momi, S.; Tantucci, M.; Van Roy, M.; Ulrichts, H.; Ricci, G.; Gresele, P. Reperfusion of cerebral artery thrombosis by the GPIb-VWF blockade with the Nanobody ALX-0081 reduces brain infarct size in guinea pigs. Blood 2013, 121, 5088–5097. [Google Scholar] [CrossRef]
- Chen, C.; Kan, Y.; Shi, Z.; Guo, D.; Fu, W.; Li, Y.; Lv, Q.; Li, X.; Si, Y. Low Dose Rivaroxaban for Atherosclerotic Cardiovascular Diseases: A Systematic Review and Meta-analysis. Front. Pharm. 2020, 11, 608247. [Google Scholar] [CrossRef] [PubMed]
- Oi, K.; Shimizu, M.; Natori, T.; Tsuda, K.; Yoshida, M.; Kamada, A.; Ishigaku, Y.; Narumi, S.; Oura, K.; Maeda, T.; et al. Influence of PAR-1 in patients with non-valvular atrial fibrillation: The antiplatelet effect of dabigatran. Thromb. Res. 2021, 201, 123–130. [Google Scholar] [CrossRef] [PubMed]
- Vinholt, P.J.; Nielsen, C.; Soderstrom, A.C.; Brandes, A.; Nybo, M. Dabigatran reduces thrombin-induced platelet aggregation and activation in a dose-dependent manner. J. Thromb. Thrombolysis 2017, 44, 216–222. [Google Scholar] [CrossRef]
- Kim, J.; Jang, H.J.; Schellingerhout, D.; Lee, S.K.; Kim, H.; Kim, Y.D.; Lee, K.Y.; Choi, H.Y.; Cho, H.J.; Jang, S.S.; et al. Short-Term Cessation of Dabigatran Causes a Paradoxical Prothrombotic State. Ann. Neurol. 2021, 89, 444–458. [Google Scholar] [CrossRef]
- Polzin, A.; Dannenberg, L.; Thienel, M.; Orban, M.; Wolff, G.; Hohlfeld, T.; Zeus, T.; Kelm, M.; Petzold, T. Noncanonical Effects of Oral Thrombin and Factor Xa Inhibitors in Platelet Activation and Arterial Thrombosis. Thromb. Haemost. 2021, 121, 122–130. [Google Scholar] [CrossRef]
- Arantes, F.B.B.; Menezes, F.R.; Franci, A.; Barbosa, C.; Dalcoquio, T.F.; Nakashima, C.A.K.; Baracioli, L.M.; Furtado, R.H.M.; Nomelini, Q.S.S.; Ramires, J.A.F.; et al. Influence of Direct Thrombin Inhibitor and Low Molecular Weight Heparin on Platelet Function in Patients with Coronary Artery Disease: A Prospective Interventional Trial. Adv. Ther. 2020, 37, 420–430. [Google Scholar] [CrossRef] [Green Version]
- Jourdi, G.; Bachelot-Loza, C.; Mazoyer, E.; Poirault-Chassac, S.; Duchemin, J.; Fontenay, M.; Gaussem, P. Effect of rivaroxaban and dabigatran on platelet functions: In vitro study. Thromb. Res. 2019, 183, 159–162. [Google Scholar] [CrossRef]
- Shah, B.; Allen, N.; Harchandani, B.; Pillinger, M.; Katz, S.; Sedlis, S.P.; Echagarruga, C.; Samuels, S.K.; Morina, P.; Singh, P.; et al. Effect of Colchicine on Platelet-Platelet and Platelet-Leukocyte Interactions: A Pilot Study in Healthy Subjects. Inflammation 2016, 39, 182–189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cirillo, P.; Taglialatela, V.; Pellegrino, G.; Morello, A.; Conte, S.; Di Serafino, L.; Cimmino, G. Effects of colchicine on platelet aggregation in patients on dual antiplatelet therapy with aspirin and clopidogrel. J. Thromb. Thrombolysis 2020, 50, 468–472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cimmino, G.; Tarallo, R.; Conte, S.; Morello, A.; Pellegrino, G.; Loffredo, F.S.; Cali, G.; De Luca, N.; Golino, P.; Trimarco, B.; et al. Colchicine reduces platelet aggregation by modulating cytoskeleton rearrangement via inhibition of cofilin and LIM domain kinase 1. Vasc. Pharm. 2018, 111, 62–70. [Google Scholar] [CrossRef]
- Pennings, G.J.; Reddel, C.J.; Traini, M.; Campbell, H.; Chen, V.; Kritharides, L. Colchicine inhibits ROS generation in response to glycoprotein VI stimulation. Sci. Rep. 2021, 11, 11965. [Google Scholar] [CrossRef] [PubMed]
- Fiolet, A.T.L.; Opstal, T.S.J.; Mosterd, A.; Eikelboom, J.W.; Jolly, S.S.; Keech, A.C.; Kelly, P.; Tong, D.C.; Layland, J.; Nidorf, S.M.; et al. Efficacy and safety of low-dose colchicine in patients with coronary disease: A systematic review and meta-analysis of randomized trials. Eur. Heart J. 2021, 42, 2765–2775. [Google Scholar] [CrossRef] [PubMed]
- Manfredi, A.A.; Baldini, M.; Camera, M.; Baldissera, E.; Brambilla, M.; Peretti, G.; Maseri, A.; Rovere-Querini, P.; Tremoli, E.; Sabbadini, M.G.; et al. Anti-TNFalpha agents curb platelet activation in patients with rheumatoid arthritis. Ann. Rheum. Dis. 2016, 75, 1511–1520. [Google Scholar] [CrossRef]
- Padfield, G.J.; Din, J.N.; Koushiappi, E.; Mills, N.L.; Robinson, S.D.; Cruden Nle, M.; Lucking, A.J.; Chia, S.; Harding, S.A.; Newby, D.E. Cardiovascular effects of tumour necrosis factor alpha antagonism in patients with acute myocardial infarction: A first in human study. Heart 2013, 99, 1330–1335. [Google Scholar] [CrossRef]
- Nielsen, C.B.; Nielsen, C.; Nybo, M.; Just, S.A.; Vinholt, P.J. The in vitro effect of antirheumatic drugs on platelet function. Platelets 2020, 31, 248–257. [Google Scholar] [CrossRef]
- Canzano, P.; Brambilla, M.; Porro, B.; Cosentino, N.; Tortorici, E.; Vicini, S.; Poggio, P.; Cascella, A.; Pengo, M.F.; Veglia, F.; et al. Platelet and Endothelial Activation as Potential Mechanisms Behind the Thrombotic Complications of COVID-19 Patients. JACC Basic Transl. Sci. 2021, 6, 202–218. [Google Scholar] [CrossRef]
- Ridker, P.M.; Everett, B.M.; Thuren, T.; MacFadyen, J.G.; Chang, W.H.; Ballantyne, C.; Fonseca, F.; Nicolau, J.; Koenig, W.; Anker, S.D.; et al. Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease. N. Engl. J. Med. 2017, 377, 1119–1131. [Google Scholar] [CrossRef]
- DeSena, A.D.; Do, T.; Schulert, G.S. Systemic autoinflammation with intractable epilepsy managed with interleukin-1 blockade. J. Neuroinflamm. 2018, 15, 38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dobie, G.; Kuriri, F.A.; Omar, M.M.A.; Alanazi, F.; Gazwani, A.M.; Tang, C.P.S.; Sze, D.M.; Handunnetti, S.M.; Tam, C.; Jackson, D.E. Ibrutinib, but not zanubrutinib, induces platelet receptor shedding of GPIb-IX-V complex and integrin alphaIIbbeta3 in mice and humans. Blood Adv. 2019, 3, 4298–4311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ninomoto, J.; Mokatrin, A.; Kinoshita, T.; Marimpietri, C.; Barrett, T.D.; Chang, B.Y.; Sukbuntherng, J.; James, D.F.; Crowther, M. Effects of ibrutinib on in vitro platelet aggregation in blood samples from healthy donors and donors with platelet dysfunction. Hematology 2020, 25, 112–117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nicolson, P.L.R.; Nock, S.H.; Hinds, J.; Garcia-Quintanilla, L.; Smith, C.W.; Campos, J.; Brill, A.; Pike, J.A.; Khan, A.O.; Poulter, N.S.; et al. Low-dose Btk inhibitors selectively block platelet activation by CLEC-2. Haematologica 2021, 106, 208–219. [Google Scholar] [CrossRef] [Green Version]
- Diener, J.L.; Daniel Lagasse, H.A.; Duerschmied, D.; Merhi, Y.; Tanguay, J.F.; Hutabarat, R.; Gilbert, J.; Wagner, D.D.; Schaub, R. Inhibition of von Willebrand factor-mediated platelet activation and thrombosis by the anti-von Willebrand factor A1-domain aptamer ARC1779. J. Thromb. Haemost. 2009, 7, 1155–1162. [Google Scholar] [CrossRef]
- Spiel, A.O.; Mayr, F.B.; Ladani, N.; Wagner, P.G.; Schaub, R.G.; Gilbert, J.C.; Jilma, B. The aptamer ARC1779 is a potent and specific inhibitor of von Willebrand Factor mediated ex vivo platelet function in acute myocardial infarction. Platelets 2009, 20, 334–340. [Google Scholar] [CrossRef]
- Gilbert, J.C.; DeFeo-Fraulini, T.; Hutabarat, R.M.; Horvath, C.J.; Merlino, P.G.; Marsh, H.N.; Healy, J.M.; Boufakhreddine, S.; Holohan, T.V.; Schaub, R.G. First-in-human evaluation of anti von Willebrand factor therapeutic aptamer ARC1779 in healthy volunteers. Circulation 2007, 116, 2678–2686. [Google Scholar] [CrossRef]
- Markus, H.S.; McCollum, C.; Imray, C.; Goulder, M.A.; Gilbert, J.; King, A. The von Willebrand inhibitor ARC1779 reduces cerebral embolization after carotid endarterectomy: A randomized trial. Stroke 2011, 42, 2149–2153. [Google Scholar] [CrossRef] [Green Version]
- Sakai, K.; Someya, T.; Harada, K.; Yagi, H.; Matsui, T.; Matsumoto, M. Novel aptamer to von Willebrand factor A1 domain (TAGX-0004) shows total inhibition of thrombus formation superior to ARC1779 and comparable to caplacizumab. Haematologica 2020, 105, 2631–2638. [Google Scholar] [CrossRef]
- Kovacevic, K.D.; Buchtele, N.; Schoergenhofer, C.; Derhaschnig, U.; Gelbenegger, G.; Brostjan, C.; Zhu, S.; Gilbert, J.C.; Jilma, B. The aptamer BT200 effectively inhibits von Willebrand factor (VWF) dependent platelet function after stimulated VWF release by desmopressin or endotoxin. Sci. Rep. 2020, 10, 11180. [Google Scholar] [CrossRef]
- Kovacevic, K.D.; Jilma, B.; Zhu, S.; Gilbert, J.C.; Winter, M.P.; Toma, A.; Hengstenberg, C.; Lang, I.; Kubica, J.; Siller-Matula, J.M. von Willebrand Factor Predicts Mortality in ACS Patients Treated with Potent P2Y12 Antagonists and is Inhibited by Aptamer BT200 Ex Vivo. Thromb. Haemost. 2020, 120, 1282–1290. [Google Scholar] [CrossRef] [PubMed]
- Kovacevic, K.D.; Greisenegger, S.; Langer, A.; Gelbenegger, G.; Buchtele, N.; Pabinger, I.; Petroczi, K.; Zhu, S.; Gilbert, J.C.; Jilma, B. The aptamer BT200 blocks von Willebrand factor and platelet function in blood of stroke patients. Sci. Rep. 2021, 11, 3092. [Google Scholar] [CrossRef] [PubMed]
- Kovacevic, K.D.; Grafeneder, J.; Schorgenhofer, C.; Gelbenegger, G.; Gager, G.; Firbas, C.; Quehenberger, P.; Jilma-Stohlawetz, P.; Bileck, A.; Zhu, S.; et al. The von Willebrand Factor A-1 domain binding aptamer BT200 elevates plasma levels of VWF and Factor VIII: A first-in-human trial. Haematologica 2021. [Google Scholar] [CrossRef] [PubMed]
- Nimjee, S.M.; Dornbos, D., 3rd; Pitoc, G.A.; Wheeler, D.G.; Layzer, J.M.; Venetos, N.; Huttinger, A.; Talentino, S.E.; Musgrave, N.J.; Moody, H.; et al. Preclinical Development of a vWF Aptamer to Limit Thrombosis and Engender Arterial Recanalization of Occluded Vessels. Mol. Ther. 2019, 27, 1228–1241. [Google Scholar] [CrossRef] [PubMed]
- Lei, X.; Reheman, A.; Hou, Y.; Zhou, H.; Wang, Y.; Marshall, A.H.; Liang, C.; Dai, X.; Li, B.X.; Vanhoorelbeke, K.; et al. Anfibatide, a novel GPIb complex antagonist, inhibits platelet adhesion and thrombus formation in vitro and in vivo in murine models of thrombosis. Thromb. Haemost. 2014, 111, 279–289. [Google Scholar] [CrossRef]
- Li, T.T.; Fan, M.L.; Hou, S.X.; Li, X.Y.; Barry, D.M.; Jin, H.; Luo, S.Y.; Kong, F.; Lau, L.F.; Dai, X.R.; et al. A novel snake venom-derived GPIb antagonist, anfibatide, protects mice from acute experimental ischaemic stroke and reperfusion injury. Br. J. Pharm. 2015, 172, 3904–3916. [Google Scholar] [CrossRef]
- Chu, W.; Sun, X.; Zhu, X.; Zhao, Y.C.; Zhang, J.; Kong, Q.; Zhou, L. Blockade of platelet glycoprotein receptor Ib ameliorates blood-brain barrier disruption following ischemic s.stroke via Epac pathway. Biomed. Pharm. 2021, 140, 111698. [Google Scholar] [CrossRef]
- Li, B.X.; Dai, X.; Xu, X.R.; Adili, R.; Neves, M.A.D.; Lei, X.; Shen, C.; Zhu, G.; Wang, Y.; Zhou, H.; et al. In vitro assessment and phase I randomized clinical trial of anfibatide a snake venom derived anti-thrombotic agent targeting human platelet GPIbalpha. Sci. Rep. 2021, 11, 11663. [Google Scholar] [CrossRef]
- Zheng, B.; Li, J.; Jiang, J.; Xiang, D.; Chen, Y.; Yu, Z.; Zeng, H.; Ge, J.; Dai, X.; Liu, J.; et al. Safety and efficacy of a platelet glycoprotein Ib inhibitor for patients with non-ST segment elevation myocardial infarction: A phase Ib/IIa study. Pharmacotherapy 2021, 41, 828–836. [Google Scholar] [CrossRef]
- Fontayne, A.; Meiring, M.; Lamprecht, S.; Roodt, J.; Demarsin, E.; Barbeaux, P.; Deckmyn, H. The humanized anti-glycoprotein Ib monoclonal antibody h6B4-Fab is a potent and safe antithrombotic in a high shear arterial thrombosis model in baboons. Thromb. Haemost. 2008, 100, 670–677. [Google Scholar] [CrossRef]
- Schulz, C.; Penz, S.; Hoffmann, C.; Langer, H.; Gillitzer, A.; Schneider, S.; Brandl, R.; Seidl, S.; Massberg, S.; Pichler, B.; et al. Platelet GPVI binds to collagenous structures in the core region of human atheromatous plaque and is critical for atheroprogression in vivo. Basic Res. Cardiol. 2008, 103, 356–367. [Google Scholar] [CrossRef] [PubMed]
- Massberg, S.; Konrad, I.; Bultmann, A.; Schulz, C.; Munch, G.; Peluso, M.; Lorenz, M.; Schneider, S.; Besta, F.; Muller, I.; et al. Soluble glycoprotein VI dimer inhibits platelet adhesion and aggregation to the injured vessel wall in vivo. FASEB J. 2004, 18, 397–399. [Google Scholar] [CrossRef] [PubMed]
- Goebel, S.; Li, Z.; Vogelmann, J.; Holthoff, H.P.; Degen, H.; Hermann, D.M.; Gawaz, M.; Ungerer, M.; Munch, G. The GPVI-Fc fusion protein Revacept improves cerebral infarct volume and functional outcome in stroke. PLoS ONE 2013, 8, e66960. [Google Scholar] [CrossRef] [PubMed]
- Mojica Munoz, A.K.; Jamasbi, J.; Uhland, K.; Degen, H.; Munch, G.; Ungerer, M.; Brandl, R.; Megens, R.; Weber, C.; Lorenz, R.; et al. Recombinant GPVI-Fc added to single or dual antiplatelet therapy in vitro prevents plaque-induced platelet thrombus formation. Thromb. Haemost. 2017, 117, 1651–1659. [Google Scholar] [CrossRef]
- Degen, H.; Borst, O.; Ziegler, M.; Mojica Munoz, A.K.; Jamasbi, J.; Walker, B.; Gobel, S.; Fassbender, J.; Adler, K.; Brandl, R.; et al. ADPase CD39 Fused to Glycoprotein VI-Fc Boosts Local Antithrombotic Effects at Vascular Lesions. J. Am. Heart Assoc. 2017, 6, e005991. [Google Scholar] [CrossRef]
- Mayer, K.; Hein-Rothweiler, R.; Schupke, S.; Janisch, M.; Bernlochner, I.; Ndrepepa, G.; Sibbing, D.; Gori, T.; Borst, O.; Holdenrieder, S.; et al. Efficacy and Safety of Revacept, a Novel Lesion-Directed Competitive Antagonist to Platelet Glycoprotein VI, in Patients Undergoing Elective Percutaneous Coronary Intervention for Stable Ischemic Heart Disease: The Randomized, Double-blind, Placebo-Controlled ISAR-PLASTER Phase 2 Trial. JAMA Cardiol. 2021, 6, 753–761. [Google Scholar] [CrossRef]
- Groschel, K.; Uphaus, T.; Loftus, I.; Poppert, H.; Diener, H.C.; Zobel, J.; Munch, G. Revacept, an Inhibitor of Platelet Adhesion in Symptomatic Carotid Artery Stenosis: Design and Rationale of a Randomized Phase II Clinical Trial. TH Open 2020, 4, e393–e399. [Google Scholar] [CrossRef]
- Lebozec, K.; Jandrot-Perrus, M.; Avenard, G.; Favre-Bulle, O.; Billiald, P. Design, development and characterization of ACT017, a humanized Fab that blocks platelet’s glycoprotein VI function without causing bleeding risks. MAbs 2017, 9, 945–958. [Google Scholar] [CrossRef] [Green Version]
- Renaud, L.; Lebozec, K.; Voors-Pette, C.; Dogterom, P.; Billiald, P.; Jandrot Perrus, M.; Pletan, Y.; Machacek, M. Population Pharmacokinetic/Pharmacodynamic Modeling of Glenzocimab (ACT017) a Glycoprotein VI Inhibitor of Collagen-Induced Platelet Aggregation. J. Clin. Pharm. 2020, 60, 1198–1208. [Google Scholar] [CrossRef]
- Ahmed, M.U.; Kaneva, V.; Loyau, S.; Nechipurenko, D.; Receveur, N.; Le Bris, M.; Janus-Bell, E.; Didelot, M.; Rauch, A.; Susen, S.; et al. Pharmacological Blockade of Glycoprotein VI Promotes Thrombus Disaggregation in the Absence of Thrombin. Arter. Thromb. Vasc. Biol. 2020, 40, 2127–2142. [Google Scholar] [CrossRef]
- Tsukiji, N.; Osada, M.; Sasaki, T.; Shirai, T.; Satoh, K.; Inoue, O.; Umetani, N.; Mochizuki, C.; Saito, T.; Kojima, S.; et al. Cobalt hematoporphyrin inhibits CLEC-2-podoplanin interaction, tumor metastasis, and arterial/venous thrombosis in mice. Blood Adv. 2018, 2, 2214–2225. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.W.; Hsieh, P.W.; Chang, Y.T.; Lu, M.H.; Huang, T.F.; Chong, K.Y.; Liao, H.R.; Cheng, J.C.; Tseng, C.P. Identification of a novel platelet antagonist that binds to CLEC-2 and suppresses podoplanin-induced platelet aggregation and cancer metastasis. Oncotarget 2015, 6, 42733–42748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carminita, E.; Crescence, L.; Brouilly, N.; Altie, A.; Panicot-Dubois, L.; Dubois, C. DNAse-dependent, NET-independent pathway of thrombus formation in vivo. Proc. Natl. Acad. Sci. USA 2021, 118, e2100561118. [Google Scholar] [CrossRef]
- Li, T.; Wang, C.; Liu, Y.; Li, B.; Zhang, W.; Wang, L.; Yu, M.; Zhao, X.; Du, J.; Zhang, J.; et al. Neutrophil Extracellular Traps Induce Intestinal Damage and Thrombotic Tendency in Inflammatory Bowel Disease. J. Crohns Colitis 2020, 14, 240–253. [Google Scholar] [CrossRef] [PubMed]
- Novotny, J.; Chandraratne, S.; Weinberger, T.; Philippi, V.; Stark, K.; Ehrlich, A.; Pircher, J.; Konrad, I.; Oberdieck, P.; Titova, A.; et al. Histological comparison of arterial thrombi in mice and men and the influence of Cl-amidine on thrombus formation. PLoS ONE 2018, 13, e0190728. [Google Scholar] [CrossRef] [Green Version]
- Clark, S.R.; Ma, A.C.; Tavener, S.A.; McDonald, B.; Goodarzi, Z.; Kelly, M.M.; Patel, K.D.; Chakrabarti, S.; McAvoy, E.; Sinclair, G.D.; et al. Platelet TLR4 activates neutrophil extracellular traps to ensnare bacteria in septic blood. Nat. Med. 2007, 13, 463–469. [Google Scholar] [CrossRef]
- Sun, J.; Zhang, M.; Chen, K.; Chen, B.; Zhao, Y.; Gong, H.; Zhao, X.; Qi, R. Suppression of TLR4 activation by resveratrol is associated with STAT3 and Akt inhibition in oxidized low-density lipoprotein-activated platelets. Eur. J. Pharm. 2018, 836, 1–10. [Google Scholar] [CrossRef]
- Huang, W.C.; Liu, J.C.; Hsia, C.W.; Fong, T.H.; Hsia, C.H.; Tran, O.T.; Velusamy, M.; Yang, C.H.; Sheu, J.R. Pterostilbene, a Dimethylether Analogue of Resveratrol, Possesses High Potency in the Prevention of Platelet Activation in Humans and the Reduction of Vascular Thrombosis in Mice. J. Agric. Food Chem. 2021, 69, 4697–4707. [Google Scholar] [CrossRef]
- Cornelius, D.C.; Travis, O.K.; Tramel, R.W.; Borges-Rodriguez, M.; Baik, C.H.; Greer, M.; Giachelli, C.A.; Tardo, G.A.; Williams, J.M. NLRP3 inflammasome inhibition attenuates sepsis-induced platelet activation and prevents multi-organ injury in cecal-ligation puncture. PLoS ONE 2020, 15, e0234039. [Google Scholar] [CrossRef]
- Wang, Y.; Jiang, H.; Hu, X.; Fu, W. Bone marrow NLRP3 inflammasome-IL-1beta signal regulates post-myocardial infarction megakaryocyte development and platelet production. Biochem. Biophys. Res. Commun. 2021, 585, 96–102. [Google Scholar] [CrossRef]
- Garcia, A.; Dunoyer-Geindre, S.; Nolli, S.; Strassel, C.; Reny, J.L.; Fontana, P. miR-204-5p and Platelet Function Regulation: Insight into a Mechanism Mediated by CDC42 and GPIIbIIIa. Thromb. Haemost. 2021, 121, 1206–1219. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Inflammation | Endothelial Dysfunction | MiRs |
|---|---|---|
| Interleukins | ↓ NO bioavailability | miR-126-3p |
| TNF-α | Thrombomodulin | miR-223 |
| NLRP3 inflammasome | CD39 | miR-320c |
| BTK | CD73 | miR-181 |
| PLT-NEU interactions | PGI2-TXA2 imbalance | miR-26b |
| NETs | VWF | miR-15b-5p |
| TLRs | Endothelial glycocalyx | miR-93 |
| miR-150-5p | ||
| miR-423-3p | ||
| miR-1180-3p | ||
| miR-204-5p |
| Agent | Target | Preclinical/Clinical Evidence |
|---|---|---|
| ARC1779 | VWF (A1 domain) | ↓ VWF-induced platelet activation (in vitro) No effect on ADP-, collagen-, or AA-induced platelet activation (in vitro) ↓ platelet activation, dose- and concentration dependent (in humans) ↓ embolic signals proportional to VWF inhibition in carotid endarterectomy (in humans) ↑ Perioperative bleeding in carotid endarterectomy (in humans) |
| TAGX-004 | VWF (A1 domain) | ↑ antiplatelet activity compared to ARC1779 (in vitro) |
| BT200 | VWF (A1 domain) | ↓ ristocetin-induced platelet aggregation (ex vivo and in humans) ↓ VWF activity in patients with an ACS or stroke (in humans) |
| DTRI-031 | VWF (A1 domain) | ↓ collagen- and ADP-induced platelet activation (ex vivo) Prevention of carotid artery thrombosis (in vivo) Induction of recanalization in carotid artery occlusion (in vivo) |
| Anfibatide | GP1b | ↓ ristocetin- and VWF-induced platelet adhesion and aggregation (in vitro and in humans) ↓ thrombosis in VWF-deficient mice (in vivo) ↓ activation of platelets from patients with an ACS, without any effect on revascularization parameters or myocardial injury markers (in humans) No increases in bleeding |
| h6B4-Fab | GP1b | ↓ cyclic flow reductions in stenosed femoral artery (in vivo) |
| Revacept | GPVI | ↓ platelet aggregation (in vivo) Comparable functional outcomes, infarct size, edema to thrombolytic therapy in cerebral ischemia (in vivo) ↑ platelet inhibition of other antiplatelets when used in combination (in vitro) No difference in death or high-sensitivity cardiac troponin increase in patients undergoing elective PCI for CAD (in humans) No increases in bleeding (in humans) |
| Glenzocimab | GPVI | ↓ collagen-induced platelet activation (ex vivo) Thrombus resolution, enhanced by increased wall shear rate (in vitro) Complete inhibition of platelet activation was noted after infusion, 60% platelet inhibition maintained 12 h post infusion (in humans) No increases in bleeding (in humans) |
| Cobalt hematoporphyrin | CLEC2 | ↓ CLEC2-induced platelet activation (in vitro) Prolonged time to occlusion of injured femoral artery (in vivo) |
| 2CP | CLEC2 | ↓ CLEC2-induced platelet activation (in vitro) |
| AYP1 | CLEC2 | ↓ CLEC2-induced platelet activation (in vitro) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Theofilis, P.; Sagris, M.; Oikonomou, E.; Antonopoulos, A.S.; Tsioufis, K.; Tousoulis, D. Factors Associated with Platelet Activation-Recent Pharmaceutical Approaches. Int. J. Mol. Sci. 2022, 23, 3301. https://doi.org/10.3390/ijms23063301
Theofilis P, Sagris M, Oikonomou E, Antonopoulos AS, Tsioufis K, Tousoulis D. Factors Associated with Platelet Activation-Recent Pharmaceutical Approaches. International Journal of Molecular Sciences. 2022; 23(6):3301. https://doi.org/10.3390/ijms23063301
Chicago/Turabian StyleTheofilis, Panagiotis, Marios Sagris, Evangelos Oikonomou, Alexios S. Antonopoulos, Konstantinos Tsioufis, and Dimitris Tousoulis. 2022. "Factors Associated with Platelet Activation-Recent Pharmaceutical Approaches" International Journal of Molecular Sciences 23, no. 6: 3301. https://doi.org/10.3390/ijms23063301