
An Overview of the Cardiorenal Protective Mechanisms of SGLT2 Inhibitors

 , , , ,
, , , , 
Abstract
:1. Introduction
2. The Anti-Hyperglycemic Effect of SGLT2-Is
2.1. The Durability of SGLT2-Is
2.2. The Combination Therapy with Other Anti-Hyperglycemic Agents
2.3. The Adverse Effects of SGLT2-I Therapy
3. The Mechanism of Kidney Glucose Reabsorption
3.1. In Healthy Subjects
3.2. In Diabetic Patients
4. Effects of SGLT2-Is on CV and Renal Outcomes: Brief Overview of Results from Main Trials
5. The Renal Effects of SGLT2 Inhibition: Correction of Hyperfiltration, Albuminuria, and Hypoxia
6. Direct Effect of SGLT2-Is on Myocardial Sodium Homeostasis
7. Improvement of Conventional CV and Renal Risk Factors by SGLT2-Is
7.1. Improved Glucose Control
7.2. Loss of Body Weight
7.3. Reduction of Arterial Blood Pressure
7.4. Reduction of Serum Uric Acid
7.5. Effects on Serum Lipids
7.6. SGLT2-Is and Atherosclerosis
8. Metabolic Reprogramming by SGLT2-Is
8.1. Metabolic Effects of SGLT2 Inhibition at Heart Level
8.2. Metabolic Effects of SGLT2 Inhibition on the Kidney
9. Improved Heart Overload by SGLT2-Is: Effect on Diuresis and Vascular Function
9.1. Impact of the Diuretic Effect of SGLT2-Is on Intra- and Extravascular Volumes
9.2. Improvement of Arterial Function and Stiffness by SGLT2-Is
10. Anti-Inflammatory and Anti-Oxidant Effects of SGLT2-Is
10.1. Evidence of Beneficial Anti-Inflammatory, Anti-Oxidant, and Anti-Fibrotic Effects on Heart
10.2. Evidence of Beneficial Anti-Inflammatory, Anti-Oxidant and Anti-Fibrotic Effects on Kidney
11. Modulation of Mitochondrial Function and Autophagy by SGLT2-Is
11.1. Evidence in Heart
11.2. Evidence in Kidney
12. Effects of SGLT2-Is on Erythropoietin and Erythropoiesis
13. Concluding Remarks and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Salvatore, T.; Carbonara, O.; Cozzolino, D.; Torella, R.; Nasti, R.; Lascar, N.; Sasso, F.C. Kidney in diabetes: From organ damage target to therapeutic target. Curr. Drug Metab. 2011, 12, 658–666. [Google Scholar] [CrossRef] [PubMed]
- DeFronzo, R.A.; Hompesch, M.; Kasichayanula, S.; Liu, X.; Hong, Y.; Pfister, M.; Morrow, L.A.; Leslie, B.R.; Boulton, D.W.; Ching, A.; et al. Characterization of renal glucose reabsorption in response to dapagliflozin in healthy subjects and subjects with type 2 diabetes. Diabetes Care 2013, 36, 3169–3176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Merovci, A.; Mari, A.; Solis-Herrera, C.; Xiong, J.; Daniele, G.; Chavez-Velazquez, A.; Tripathy, D.; Urban McCarthy, S.; Abdul-Ghani, M.; DeFronzo, R.A. Dapagliflozin lowers plasma glucose concentration and improves β-cell function. J. Clin. Endocrinol. Metab. 2015, 100, 1927–1932, Erratum in J. Clin. Endocrinol. Metab. 2017, 102, 4662. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Konink, L. Observations sur les proprietes febrifuges de la phloridzine. Bull. Soc. Med. Gand 1836, 1, 75–110. [Google Scholar]
- Chasis, H.; Jolliffe, N.; Smith, H.W. The action of phlorizin on the excretion of glucose, xylose, sucrose, creatinine and urea by man. J. Clin. Investig. 1933, 12, 1083–1090. [Google Scholar] [CrossRef]
- Galiero, R.; Pafundi, P.C.; Nevola, R.; Rinaldi, L.; Acierno, C.; Caturano, A.; Salvatore, T.; Adinolfi, L.E.; Costagliola, C.; Sasso, F.C. The Importance of Telemedicine during COVID-19 Pan-demic: A Focus on Diabetic Retinopathy. J. Diabetes Res. 2020, 2020, 9036847. [Google Scholar] [CrossRef]
- Lombardi, R.; Airaghi, L.; Targher, G.; Serviddio, G.; Maffi, G.; Mantovani, A.; Maffeis, C.; Colecchia, A.; Villani, R.; Rinaldi, L.; et al. Liver fibrosis by FibroScan® independently of established cardiovascular risk parameters associates with macrovascular and microvascular complications in patients with type 2 diabetes. Liver Int. 2020, 40, 347–354. [Google Scholar] [CrossRef]
- Marfella, R.; Sasso, F.C.; Cacciapuoti, F.; Portoghese, M.; Rizzo, M.R.; Siniscalchi, M.; Carbonara, O.; Ferraraccio, F.; Torella, M.; Petrella, A.; et al. Tight glycemic control may increase regenerative potential of myocardium during acute infarction. J. Clin. Endocrinol. Metab. 2012, 97, 933–942. [Google Scholar] [CrossRef] [Green Version]
- Sasso, F.C.; Rinaldi, L.; Lascar, N.; Marrone, A.; Pafundi, P.C.; Adinolfi, L.E.; Marfella, R. Role of Tight Glycemic Control during Acute Coronary Syndrome on CV Outcome in Type 2 Diabetes. J. Diabetes Res. 2018, 2018, 3106056. [Google Scholar] [CrossRef] [Green Version]
- Caturano, A.; Galiero, R.; Pafundi, P.C.; Cesaro, A.; Vetrano, E.; Palmiero, G.; Rinaldi, L.; Salvatore, T.; Marfella, R.; Sardu, C.; et al. Does a strict glycemic control during acute coronary syndrome play a cardioprotective effect? Pathophysiology and clinical evidence. Diabetes Res. Clin. Pract. 2021, 178, 108959. [Google Scholar] [CrossRef]
- Kang, A.; Jardine, M.J. SGLT2 inhibitors may offer benefit beyond diabetes. Nat. Rev. Nephrol. 2021, 17, 83–84. [Google Scholar] [CrossRef] [PubMed]
- Fathi, A.; Vickneson, K.; Singh, J.S. SGLT2-inhibitors; more than just glycosuria and diuresis. Heart Fail Rev. 2021, 26, 623–642. [Google Scholar] [CrossRef] [PubMed]
- Thomas, M.C.; Cherney, D.Z.I. The actions of SGLT2 inhibitors on metabolism, renal function and blood pressure. Diabetologia 2018, 61, 2098–2107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vallon, V.; Thomson, S.C. Targeting renal glucose reabsorption to treat hyperglycaemia: The pleiotropic effects of SGLT2 inhibition. Diabetologia 2017, 60, 215–225. [Google Scholar] [CrossRef]
- Merovci, A.; Solis-Herrera, C.; Daniele, G.; Eldor, R.; Fiorentino, T.V.; Tripathy, D.; Xiong, J.; Perez, Z.; Norton, L.; Abdul-Ghani, M.A.; et al. Dapagliflozin improves muscle insulin sensitivity but enhances endogenous glucose production. J. Clin. Investig. 2014, 124, 509–514, Erratum in J. Clin. Investig. 2014, 124, 2287. [Google Scholar] [CrossRef]
- Hu, S.; Lin, C.; Cai, X.; Zhu, X.; Lv, F.; Nie, L.; Ji, L. The Urinary Glucose Excretion by Sodium-Glucose Cotransporter 2 Inhibitor in Patients With Different Levels of Renal Function: A Systematic Review and Meta-Analysis. Front. Endocrinol. 2022, 12, 814074. [Google Scholar] [CrossRef]
- van Bommel, E.J.; Muskiet, M.H.; Tonneijck, L.; Kramer, M.H.; Nieuwdorp, M.; van Raalte, D.H. SGLT2 Inhibition in the Diabetic Kidney-From Mechanisms to Clinical Outcome. Clin. J. Am. Soc. Nephrol. 2017, 12, 700–710. [Google Scholar] [CrossRef]
- Ferrannini, E.; Muscelli, E.; Frascerra, S.; Baldi, S.; Mari, A.; Heise, T.; Broedl, U.C.; Woerle, H.J. Metabolic response to sodium-glucose cotransporter 2 inhibition in type 2 diabetic patients. J. Clin. Investig. 2014, 124, 499–508, Erratum in J. Clin. Investig. 2014, 124, 1868. [Google Scholar] [CrossRef] [Green Version]
- Foster, D.W. Malonyl-CoA: The regulator of fatty acid synthesis and oxidation. J. Clin. Investig. 2012, 122, 1958–1959. [Google Scholar] [CrossRef] [Green Version]
- Zambrowicz, B.; Freiman, J.; Brown, P.M.; Frazier, K.S.; Turnage, A.; Bronner, J.; Ruff, D.; Shadoan, M.; Banks, P.; Mseeh, F.; et al. LX4211, a dual SGLT1/SGLT2 inhibitor, improved glycemic control in patients with type 2 diabetes in a randomized, placebo-controlled trial. Clin. Pharmacol. Ther. 2012, 92, 158–169. [Google Scholar] [CrossRef]
- Cherukuri, L.; Smith, M.S.; Tayek, J.A. The durability of oral diabetic medications: Time to A1c baseline and a review of common oral medications used by the primary care provider. Endocrinol. Diabetes Metab. J. 2018, 2. Available online: http://researchopenworld.com/wp-content/uploads/2018/07/EDMJ-2018-105-John-A.-Tayek-USA.pdf (accessed on 28 February 2022).
- Vaduganathan, M.; Inzucchi, S.E.; Sattar, N.; Fitchett, D.H.; Ofstad, A.P.; Brueckmann, M.; George, J.T.; Verma, S.; Mattheus, M.; Wanner, C.; et al. Effects of empagliflozin on insulin initiation or intensification in patients with type 2 diabetes and cardiovascular disease: Findings from the EMPA-REG OUTCOME trial. Diabetes Obes. Metab. 2021, 23, 2775–2784. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Chen, S.; Pan, H.; Zou, Y.; Wang, B.; Wang, G.; Zhu, H. Safety and efficiency of SGLT2 inhibitor combining with insulin in subjects with diabetes: Systematic review and meta-analysis of randomized controlled trials. Medicine 2017, 96, e6944, Erratum in Medicine 2017, 96, e7458. [Google Scholar] [CrossRef] [PubMed]
- Mirabelli, M.; Chiefari, E.; Caroleo, P.; Vero, R.; Brunetti, F.S.; Corigliano, D.M.; Arcidiacono, B.; Foti, D.P.; Puccio, L.; Brunetti, A. Long-Term Effectiveness and Safety of SGLT-2 Inhibitors in an Italian Cohort of Patients with Type 2 Diabetes Mellitus. J. Diabetes Res. 2019, 2019, 3971060. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bailey, C.J.; Del Prato, S.; Wei, C.; Reyner, D.; Saraiva, G. Durability of glycaemic control with dapagliflozin, an SGLT2 inhibitor, compared with saxagliptin, a DPP4 inhibitor, in patients with inadequately controlled type 2 diabetes. Diabetes Obes. Metab. 2019, 21, 2564–2569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wysham, C.H.; Lefebvre, P.; Pilon, D.; Lafeuille, M.H.; Emond, B.; Kamstra, R.; Pfeifer, M.; Duh, M.S.; Ingham, M. An investigation into the durability of glycemic control in patients with type II diabetes initiated on canagliflozin or sitagliptin: A real-world analysis of electronic medical records. J. Diabetes Complicat. 2019, 33, 140–147. [Google Scholar] [CrossRef] [PubMed]
- Dimitrakoudis, D.; Vranic, M.; Klip, A. Effects of hyperglycemia on glucose transporters of the muscle: Use of the renal glucose reabsorption inhibitor phlorizin to control glycemia. J. Am. Soc. Nephrol. 1992, 3, 1078–1091. [Google Scholar] [CrossRef]
- Jurczak, M.J.; Lee, H.-Y.; Birkenfeld, A.L.; Jornayvaz, F.R.; Frederick, D.W.; Pongratz, R.L.; Zhao, X.; Moeckel, G.W.; Samuel, V.T.; Whaley, J.; et al. SGLT2 deletion improves glucose homeostasis and preserves pancreatic beta-cell function. Diabetes 2011, 60, 890–898. [Google Scholar] [CrossRef] [Green Version]
- Obata, A.; Kubota, N.; Kubota, T.; Iwamoto, M.; Sato, H.; Sakurai, Y.; Takamoto, I.; Katsuyama, H.; Suzuki, Y.; Fukazawa, M.; et al. Tofogliflozin Improves Insulin Resistance in Skeletal Muscle and Accelerates Lipolysis in Adipose Tissue in Male Mice. Endocrinology 2016, 157, 1029–1042. [Google Scholar] [CrossRef]
- Merovci, A.; Abdul-Ghani, M.; Mari, A.; Solis-Herrera, C.; Xiong, J.; Daniele, G.; Tripathy, D.; DeFronzo, R.A. Effect of Dapagliflozin With and Without Acipimox on Insulin Sensitivity and Insulin Secretion in T2DM Males. J. Clin. Endocrinol. Metab. 2016, 101, 1249–1256. [Google Scholar] [CrossRef] [Green Version]
- Li, D.; Zou, H.; Yin, P.; Li, W.; He, J.; Wang, S.; Huang, L.; Shao, S.; Chen, Y.; Yang, Y.; et al. Durability of glycaemic control in type 2 diabetes: A systematic review and meta-analysis for its association with body weight changes. Diabetes Obes. Metab. 2021, 23, 208–217. [Google Scholar] [CrossRef] [PubMed]
- Meier, J.J.; Nauck, M.A. Glucagon-like peptide 1(GLP-1) in biology and pathology. Diabetes Metab. Res. Rev. 2005, 21, 91–117. [Google Scholar] [CrossRef] [PubMed]
- Frías, J.P.; Guja, C.; Hardy, E.; Ahmed, A.; Dong, F.; Öhman, P.; Jabbour, S.A. Exenatide once weekly plus dapagliflozin once daily versus exenatide or dapagliflozin alone in patients with type 2 diabetes inadequately controlled with metformin monotherapy (DURATION-8): A 28 week, multicentre, double-blind, phase 3, randomised controlled trial. Lancet Diabetes Endocrinol. 2016, 4, 1004–1016, Erratum in Lancet Diabetes Endocrinol. 2017, 5, e8. [Google Scholar] [CrossRef]
- Patoulias, D.; Stavropoulos, K.; Imprialos, K.; Katsimardou, A.; Kalogirou, M.S.; Koutsampasopoulos, K.; Zografou, I.; Papadopoulos, C.; Karagiannis, A.; Doumas, M. Glycemic efficacy and safety of glucagon-like peptide-1 receptor agonist on top of sodium-glucose co-transporter-2 inhibitor treatment compared to sodium-glucose co-transporter-2 inhibitor alone: A systematic review and meta-analysis of randomized controlled trials. Diabetes Res. Clin. Pract. 2019, 158, 107927. [Google Scholar] [CrossRef]
- Li, D.; Shi, W.; Wang, T.; Tang, H. SGLT2 inhibitor plus DPP-4 inhibitor as combination therapy for type 2 diabetes: A systematic review and meta-analysis. Diabetes Obes. Metab. 2018, 20, 1972–1976. [Google Scholar] [CrossRef]
- Min, S.H.; Yoon, J.H.; Hahn, S.; Cho, Y.M. Comparison between SGLT2 inhibitors and DPP4 inhibitors added to insulin therapy in type 2 diabetes: A systematic review with indirect comparison meta-analysis. Diabetes Metab. Res. Rev. 2017, 33, e2818. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Zhao, C.; Ye, Y.; Yu, M.; Qu, X. Prospect of Sodium-Glucose Co-transporter 2 Inhibitors Combined With Insulin for the Treatment of Type 2 Diabetes. Front. Endocrinol. 2020, 11, 190. [Google Scholar] [CrossRef]
- Lu, J.; Tang, L.; Meng, H.; Zhao, J.; Liang, Y. Effects of sodium-glucose cotransporter (SGLT) inhibitors in addition to insulin therapy on glucose control and safety outcomes in adults with type 1 diabetes: A meta-analysis of randomized controlled trials. Diabetes. Metab. Res. Rev. 2019, 35, e3169. [Google Scholar] [CrossRef]
- Forst, T.; Heise, T.; Plum-Morschel, L. Pharmacological Intervention in Type 2 Diabetes Mellitus—A Pathophysiologically Reasoned Approach? Curr. Diabetes Rev. 2016, 12, 429–439. [Google Scholar] [CrossRef]
- Liao, H.W.; Wu, Y.L.; Sue, Y.M.; Lee, M.; Ovbiagele, B. Sodium-glucose cotransporter 2 inhibitor plus pioglitazone vs. pioglitazone alone in patients with diabetes mellitus: A systematic review and meta-analysis of randomized controlled trials. Endocrinol. Diabetes Metab. 2018, 2, e00050. [Google Scholar] [CrossRef] [Green Version]
- Pelletier, R.; Ng, K.; Alkabbani, W.; Labib, Y.; Mourad, N.; Gamble, J.M. Adverse events associated with sodium glucose co-transporter 2 inhibitors: An overview of quantitative systematic reviews. Ther. Adv. Drug Saf. 2021, 12, 2042098621989134. [Google Scholar] [CrossRef] [PubMed]
- Goldenberg, R.M.; Berard, L.D.; Cheng, A.Y.; Gilbert, J.D.; Verma, S.; Woo, V.C.; Yale, J.-F. SGLT2 Inhibitor-associated Diabetic Ketoacidosis: Clinical Review and Recommendations for Prevention and Diagnosis. Clin. Ther. 2016, 38, 2654–2664.e1. [Google Scholar] [CrossRef] [PubMed]
- Bonner, C.; Kerr-Conte, J.J.; Gmyr, V.V.; Queniat, G.G.; Moerman, E.E.; Thévenet, J.J.; Beaucamps, C.C.; Delalleau, N.N.; Popescu, I.I.; Malaisse, W.J.; et al. Inhibition of the glucose transporter SGLT2 with dapagliflozin in pancreatic alpha cells triggers glucagon secretion. Nat. Med. 2015, 21, 512–517. [Google Scholar] [CrossRef] [PubMed]
- Garofalo, C.; Borrelli, S.; Liberti, M.E.; Andreucci, M.; Conte, G.; Minutolo, R. SGLT2 Inhibitors: Nephroprotective Efficacy and Side Effects. Medicina 2019, 55, 268. [Google Scholar] [CrossRef] [Green Version]
- Kamei, J.; Yamamoto, S. Complicated urinary tract infections with diabetes mellitus. J. Infect. Chemother. 2021, 27, 1131–1136. [Google Scholar] [CrossRef]
- Neal, B.; Perkovic, V.; Mahaffey, K.W.; de Zeeuw, D.; Fulcher, G.; Erondu, N.; Shaw, W.; Law, G.; Desai, M.; Matthews, D.R.; et al. Canagliflozin and Cardiovascular and Renal Events in Type 2 Diabetes. N. Engl. J. Med. 2017, 377, 644–657. [Google Scholar] [CrossRef]
- Zinman, B.; Wanner, C.; Lachin, J.M.; Fitchett, D.; Bluhmki, E.; Hantel, S.; Mattheus, M.; Devins, T.; Johansen, O.E.; Woerle, H.J.; et al. Empagliflozin, Cardiovascular Outcomes, and Mortality in Type 2 Diabetes. N. Engl. J. Med. 2015, 373, 2117–2128. [Google Scholar] [CrossRef] [Green Version]
- Wiviott, S.D.; Raz, I.; Bonaca, M.P.; Mosenzon, O.; Kato, E.T.; Cahn, A.; Silverman, M.G.; Zelniker, T.A.; Kuder, J.F.; Murphy, S.A.; et al. Dapagliflozin and Cardiovascular Outcomes in Type 2 Diabetes. N. Engl. J. Med. 2019, 380, 347–357. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.Y.; Singh, S.; Mansour, O.; Baksh, S.; Alexander, G.C. Association Between Sodium-Glucose Cotransporter 2 Inhibitors and Lower Extremity Amputation Among Patients With Type 2 Diabetes. JAMA Intern. Med. 2018, 178, 1190–1198. [Google Scholar] [CrossRef] [Green Version]
- Ueda, P.; Svanström, H.; Melbye, M.; Eliasson, B.; Svensson, A.-M.; Franzén, S.; Gudbjörnsdottir, S.; Hveem, K.; Jonasson, C.; Pasternak, B. Sodium glucose cotransporter 2 inhibitors and risk of serious adverse events: Nationwide register based cohort study. BMJ 2018, 363, k4365. [Google Scholar] [CrossRef] [Green Version]
- Gerich, J.E. Role of the kidney in normal glucose homeostasis and in the hyperglycaemia of diabetes mellitus: Therapeutic implications. Diabet Med. 2010, 27, 136–142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tahrani, A.A.; Barnett, A.H.; Bailey, C.J. SGLT inhibitors in management of diabetes. Lancet Diabetes Endocrinol. 2013, 1, 140–151, Erratum in Lancet Diabetes Endocrinol. 2015, 3, e3. [Google Scholar] [CrossRef]
- DeFronzo, R.A.; Norton, L.; Abdul-Ghani, M. Renal, metabolic and cardiovascular considerations of SGLT2 inhibition. Nat. Rev. Nephrol. 2017, 13, 11–26. [Google Scholar] [CrossRef] [PubMed]
- Scheepers, A.; Joost, H.G.; Schürmann, A. The glucose transporter families SGLT and GLUT: Molecular basis of normal and aberrant function. JPEN J. Parenter Enter. Nutr. 2004, 28, 364–371. [Google Scholar] [CrossRef] [Green Version]
- Poulsen, S.B.; Fenton, R.A.; Rieg, T. Sodium-glucose cotransport. Curr. Opin. Nephrol. Hypertens. 2015, 24, 463–469. [Google Scholar] [CrossRef] [Green Version]
- Hediger, M.A.; Kanai, Y.; You, G.; Nussberger, S. Mammalian ion-coupled solute transporters. J. Physiol. 1995, 482, 7S–17S. [Google Scholar] [CrossRef] [Green Version]
- Wright, E.M.; Loo, D.D.; Hirayama, B.A. Biology of human sodium glucose transporters. Physiol. Rev. 2011, 91, 733–794. [Google Scholar] [CrossRef] [Green Version]
- Coady, M.J.; El Tarazi, A.; Santer, R.; Bissonnette, P.; Sasseville, L.J.; Calado, J.; Lussier, Y.; Dumayne, C.; Bichet, D.G.; Lapointe, J.Y. MAP17 Is a Necessary Activator of Renal Na+/Glucose Cotransporter SGLT2. J. Am. Soc. Nephrol. 2017, 28, 85–93. [Google Scholar] [CrossRef] [Green Version]
- DeFronzo, R.A.; Reeves, W.B.; Awad, A.S. Pathophysiology of diabetic kidney disease: Impact of SGLT2 inhibitors. Nat. Rev. Nephrol. 2021, 17, 319–334. [Google Scholar] [CrossRef] [PubMed]
- Thomson, S.C.; Vallon, V. Renal Effects of Sodium-Glucose Co-Transporter Inhibitors. Am. J. Cardiol. 2019, 124 (Suppl. 1), S28–S35. [Google Scholar] [CrossRef] [Green Version]
- Wilcox, C.S. Antihypertensive and Renal Mechanisms of SGLT2 (Sodium-Glucose Linked Transporter 2) Inhibitors. Hypertension 2020, 75, 894–901. [Google Scholar] [CrossRef] [PubMed]
- Inoue, B.H.; dos Santos, L.; Pessoa, T.D.; Antonio, E.L.; Pacheco, B.P.; Savignano, F.A.; Carraro-Lacroix, L.R.; Tucci, P.J.; Malnic, G.; Girardi, A.C. Increased NHE3 abundance and transport activity in renal proximal tubule of rats with heart failure. Am. J. Physiol. Integr. Comp. Physiol. 2012, 302, R166–R174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Onishi, A.; Fu, Y.; Darshi, M.; Crespo-Masip, M.; Huang, W.; Song, P.; Patel, R.; Kim, Y.C.; Nespoux, J.; Freeman, B.; et al. Effect of renal tubule-specific knockdown of the Na+/H+ exchanger NHE3 in Akita diabetic mice. Am. J. Physiol. Physiol. 2019, 317, F419–F434. [Google Scholar] [CrossRef] [PubMed]
- Pessoa, T.D.; Campos, L.C.; Carraro-Lacroix, L.; Girardi, A.C.; Malnic, G. Functional role of glucose metabolism, osmotic stress, and sodium-glucose cotransporter isoform-mediated transport on Na+/H+ exchanger isoform 3 activity in the renal proximal tubule. J. Am. Soc. Nephrol. 2014, 25, 2028–2039. [Google Scholar] [CrossRef] [Green Version]
- Wright, E.M.; Hirayama, B.A.; Loo, D.F. Active sugar transport in health and disease. J. Intern. Med. 2007, 261, 32–43. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.J.; Lee, Y.J.; Han, H.J. Regulatory mechanisms of Na(+)/glucose cotransporters in renal proximal tubule cells. Kidney Int. 2007, 72, S27–S35. [Google Scholar] [CrossRef] [Green Version]
- Ghezzi, C.; Loo, D.D.F.; Wright, E.M. Physiology of renal glucose handling via SGLT1, SGLT2 and GLUT2. Diabetologia 2018, 61, 2087–2097. [Google Scholar] [CrossRef] [Green Version]
- Jurczak, M.J.; Saini, S.; Ioja, S.; Costa, D.K.; Udeh, N.; Zhao, X.; Whaley, J.M.; Kibbey, R.G. SGLT2 knockout prevents hyperglycemia and is associated with reduced pancreatic β-cell death in genetically obese mice. Islets 2018, 10, 181–189. [Google Scholar] [CrossRef]
- Powell, D.R.; Dacosta, C.M.; Gay, J.; Ding, Z.-M.; Smith, M.; Greer, J.; Doree, D.; Jeter-Jones, S.; Mseeh, F.; Rodriguez, L.A.; et al. Improved glycemic control in mice lacking Sglt1 and Sglt2. Am. J. Physiol. Metab. 2013, 304, E117–E130. [Google Scholar] [CrossRef] [Green Version]
- Song, P.; Huang, W.; Onishi, A.; Patel, R.; Kim, Y.C.; Van Ginkel, C.; Fu, Y.; Freeman, B.; Koepsell, H.; Thomson, S.C.; et al. Knockout of Na+-glucose cotransporter SGLT1 mitigates diabetes-induced upregulation of nitric oxide synthase NOS1 in the macula densa and glomerular hyperfiltration. Am. J. Physiol. Physiol. 2019, 317, F207–F217. [Google Scholar] [CrossRef]
- Vallon, V. The proximal tubule in the pathophysiology of the diabetic kidney. Am. J. Physiol. Integr. Comp. Physiol. 2011, 300, R1009–R1022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Freitas, H.S.; Anhê, G.F.; Melo, K.F.S.; Okamoto, M.M.; Oliveira-Souza, M.; Bordin, S.; Machado, U.F. Na+-Glucose Transporter-2 Messenger Ribonucleic Acid Expression in Kidney of Diabetic Rats Correlates with Glycemic Levels: Involvement of Hepatocyte Nuclear Factor-1α Expression and Activity. Endocrinology 2008, 149, 717–724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vallon, V.; Gerasimova, M.; Rose, M.A.; Masuda, T.; Satriano, J.; Mayoux, E.; Koepsell, H.; Thomson, S.C.; Rieg, T. SGLT2 inhibitor empagliflozin reduces renal growth and albuminuria in proportion to hyperglycemia and prevents glomerular hyperfiltration in diabetic Akita mice. Am. J. Physiol. Physiol. 2014, 306, F194–F204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaur, P.; Behera, B.S.; Singh, S.; Munshi, A. The pharmacological profile of SGLT2 inhibitors: Focus on mechanistic aspects and pharmacogenomics. Eur. J. Pharmacol. 2021, 904, 174169. [Google Scholar] [CrossRef] [PubMed]
- Rabizadeh, S.; Nakhjavani, M.; Esteghamati, A. Cardiovascular and Renal Benefits of SGLT2 Inhibitors: A Narrative Review. Int. J. Endocrinol. Metab. 2019, 17, e84353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vlotides, G.; Mertens, P.R. Sodium-glucose cotransport inhibitors: Mechanisms, metabolic effects and implications for the treatment of diabetic patients with chronic kidney disease: FIGURE 1. Nephrol. Dial. Transplant. 2014, 30, 1272–1276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vallon, V.; Rose, M.; Gerasimova, M.; Satriano, J.; Platt, K.A.; Koepsell, H.; Cunard, R.; Sharma, K.; Thomson, S.C.; Rieg, T. Knockout of Na-glucose transporter SGLT2 attenuates hyperglycemia and glomerular hyperfiltration but not kidney growth or injury in diabetes mellitus. Am. J. Physiol. Physiol. 2013, 304, F156–F167. [Google Scholar] [CrossRef] [Green Version]
- Vallon, V.; Schroth, J.; Satriano, J.; Blantz, R.C.; Thomson, S.C.; Rieg, T. Adenosine A1 Receptors Determine Glomerular Hyperfiltration and the Salt Paradox in Early Streptozotocin Diabetes Mellitus. Nephron Physiol. 2009, 111, p30–p38. [Google Scholar] [CrossRef] [Green Version]
- Vallon, V.; Thomson, S.C. Renal Function in Diabetic Disease Models: The Tubular System in the Pathophysiology of the Diabetic Kidney. Annu. Rev. Physiol. 2012, 74, 351–375. [Google Scholar] [CrossRef] [Green Version]
- Tonneijck, L.; Muskiet, M.; Smits, M.; Van Bommel, E.J.; Heerspink, H.J.L.; Van Raalte, D.H.; Joles, J.A. Glomerular Hyperfiltration in Diabetes: Mechanisms, Clinical Significance, and Treatment. J. Am. Soc. Nephrol. 2017, 28, 1023–1039. [Google Scholar] [CrossRef] [Green Version]
- Evans, R.G.; Harrop, G.K.; Ngo, J.; Ow, P.C.C.; O’Connor, P.M. Basal renal O2 consumption and the efficiency of O2 utilization for Na+ reabsorption. Am. J. Physiol. Physiol. 2014, 306, F551–F560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takiyama, Y.; Haneda, M. Hypoxia in Diabetic Kidneys. BioMed Res. Int. 2014, 2014, 83421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Layton, A.T.; Laghmani, K.; Vallon, V.; Edwards, A. Solute transport and oxygen consumption along the nephrons: Effects of Na+ transport inhibitors. Am. J. Physiol. Physiol. 2016, 311, F1217–F1229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Persson, P.; Palm, F. Hypoxia-inducible factor activation in diabetic kidney disease. Curr. Opin. Nephrol. Hypertens. 2017, 26, 345–350. [Google Scholar] [CrossRef] [PubMed]
- García-Pastor, C.; Benito-Martínez, S.; Moreno-Manzano, V.; Fernández-Martínez, A.B.; De Lucio-Cazaña, F.J. Mechanism and Consequences of The Impaired Hif-1α Response to Hypoxia in Human Proximal Tubular HK-2 Cells Exposed to High Glucose. Sci. Rep. 2019, 9, 15868, Erratum in Sci. Rep. 2020, 10, 8642. [Google Scholar] [CrossRef]
- Basile, D.P.; Donohoe, D.; Roethe, K.; Osborn, J.L. Renal ischemic injury results in permanent damage to peritubular capillaries and influences long-term function. Am. J. Physiol. Physiol. 2001, 281, F887–F899. [Google Scholar] [CrossRef]
- Masarone, M.; Rosato, V.; Aglitti, A.; Bucci, T.; Caruso, R.; Salvatore, T.; Sasso, F.C.; Tripodi, M.F.; Persico, M. Liver biopsy in type 2 diabetes mellitus: Steatohepatitis represents the sole feature of liver damage. PLoS ONE 2017, 12, e0178473. [Google Scholar] [CrossRef]
- Sasso, F.C.; Salvatore, T.; Tranchino, G.; Cozzolino, D.; Caruso, A.A.; Persico, M.; Gentile, S.; Torella, D.; Torella, R. Cochlear dysfunction in type 2 diabetes: A complication independent of neuropathy and acute hyperglycemia. Metabolism 1999, 48, 1346–1350. [Google Scholar] [CrossRef]
- Rizzo, M.R.; Sasso, F.C.; Marfella, R.; Siniscalchi, M.; Paolisso, P.; Carbonara, O.; Capoluongo, M.C.; Lascar, N.; Pace, C.; Sardu, C.; et al. Autonomic dysfunction is associated with brief episodes of atrial fibrillation in type 2 diabetes. J. Diabetes Complicat. 2015, 29, 88–92. [Google Scholar] [CrossRef]
- Galiero, R.; Ricciardi, D.; Pafundi, P.C.; Todisco, V.; Tedeschi, G.; Cirillo, G.; Sasso, F.C. Whole plantar nerve conduction study: A new tool for early diagnosis of peripheral diabetic neuropathy. Diabetes Res. Clin. Pract. 2021, 176, 108856. [Google Scholar] [CrossRef]
- Sasso, F.C.; Pafundi, P.C.; Gelso, A.; Bono, V.; Costagliola, C.; Marfella, R.; Sardu, C.; Rinaldi, L.; Galiero, R.; Acierno, C.; et al. Applicability of telemedicine in the screening of diabetic retinopathy (DR): The first multicentre study in Italy. The No Blind Study. Diabetes/Metab. Res. Rev. 2018, 35, e3113. [Google Scholar] [CrossRef] [PubMed]
- Cannon, C.P.; Pratley, R.; Dagogo-Jack, S.; Mancuso, J.; Huyck, S.; Masiukiewicz, U.; Charbonnel, B.; Frederich, R.; Gallo, S.; Cosentino, F.; et al. Cardiovascular Outcomes with Ertugliflozin in Type 2 Diabetes. N. Engl. J. Med. 2020, 383, 1425–1435. [Google Scholar] [CrossRef] [PubMed]
- Bhatt, D.L.; Szarek, M.; Pitt, B.; Cannon, C.P.; Leiter, L.A.; McGuire, D.K.; Lewis, J.B.; Riddle, M.C.; Inzucchi, S.E.; Kosiborod, M.N.; et al. Sotagliflozin in Patients with Diabetes and Chronic Kidney Disease. N. Engl. J. Med. 2021, 384, 129–139. [Google Scholar] [CrossRef] [PubMed]
- Zelniker, T.A.; Wiviott, S.D.; Raz, I.; Im, K.; Goodrich, E.; Bonaca, M.P.; Mosenzon, O.; Kato, E.; Cahn, A.; Furtado, R.H.M.; et al. SGLT2 inhibitors for primary and secondary prevention of cardiovascular and renal outcomes in type 2 diabetes: A systematic review and meta-analysis of cardiovascular outcome trials. Lancet 2019, 393, 31–39, Erratum in Lancet 2019, 393, 30. [Google Scholar] [CrossRef]
- Cosentino, F.; Grant, P.J.; Aboyans, V.; Bailey, C.J.; Ceriello, A.; Delgado, V.; Federici, M.; Filippatos, G.; Grobbee, D.E.; Hansen, T.B.; et al. 2019 ESC Guidelines on diabetes, pre-diabetes, and cardiovascular diseases developed in collaboration with the EASD. Eur. Heart J. 2020, 41, 255–323, Erratum in Eur. Heart J. 2020, 41, 4317. [Google Scholar] [CrossRef] [Green Version]
- Verma, S.; McMurray, J.J.V.; Cherney, D.Z.I. The Metabolodiuretic Promise of Sodium-Dependent Glucose Cotransporter 2 Inhibition. JAMA Cardiol. 2017, 2, 939–940. [Google Scholar] [CrossRef]
- Sasso, F.C.; Chiodini, P.; Carbonara, O.; De Nicola, L.; Conte, G.; Salvatore, T.; Nasti, R.; Marfella, R.; Gallo, C.; Signoriello, S.; et al. High cardiovascular risk in patients with Type 2 diabetic nephropathy: The predictive role of albuminuria and glomerular filtration rate. The NID-2 Prospective Cohort Study. Nephrol. Dial. Transplant. 2011, 27, 2269–2274. [Google Scholar] [CrossRef] [Green Version]
- Sasso, F.C.; De Nicola, L.; Carbonara, O.; Nasti, R.; Minutolo, R.; Salvatore, T.; Conte, G.; Torella, R. Cardiovascular Risk Factors and Disease Management in Type 2 Diabetic Patients With Diabetic Nephropathy. Diabetes Care 2006, 29, 498–503. [Google Scholar] [CrossRef] [Green Version]
- Minutolo, R.; Sasso, F.C.; Chiodini, P.; Cianciaruso, B.; Carbonara, O.; Zamboli, P.; Tirino, G.; Pota, A.; Torella, R.; Conte, G.; et al. Management of cardiovascular risk factors in advanced type 2 diabetic nephropathy: A comparative analysis in nephrology, diabetology and primary care settings. J. Hypertens. 2006, 24, 1655–1661. [Google Scholar] [CrossRef]
- McMurray, J.J.V.; Solomon, S.D.; Inzucchi, S.E.; Køber, L.; Kosiborod, M.N.; Martinez, F.A.; Ponikowski, P.; Sabatine, M.S.; Anand, I.S.; Bělohlávek, J.; et al. Dapagliflozin in Patients with Heart Failure and Reduced Ejection Fraction. N. Engl. J. Med. 2019, 381, 1995–2008. [Google Scholar] [CrossRef] [Green Version]
- Packer, M.; Anker, S.D.; Butler, J.; Filippatos, G.; Pocock, S.J.; Carson, P.; Januzzi, J.; Verma, S.; Tsutsui, H.; Brueckmann, M.; et al. Cardiovascular and Renal Outcomes with Empagliflozin in Heart Failure. N. Engl. J. Med. 2020, 383, 1413–1424. [Google Scholar] [CrossRef] [PubMed]
- Bhatt, D.L.; Szarek, M.; Steg, P.G.; Cannon, C.P.; Leiter, L.A.; McGuire, D.K.; Lewis, J.B.; Riddle, M.C.; Voors, A.A.; Metra, M.; et al. Sotagliflozin in Patients with Diabetes and Recent Worsening Heart Failure. N. Engl. J. Med. 2021, 384, 117–128. [Google Scholar] [CrossRef] [PubMed]
- Nassif, M.E.; Windsor, S.L.; Tang, F.; Khariton, Y.; Husain, M.; Inzucchi, S.E.; Mc-Guire, D.K.; Pitt, B.; Scirica, B.M.; Austin, B.; et al. Dapagliflozin Effects on Biomarkers, Symptoms, and Functional Status in Patients With Heart Failure With Reduced Ejection Fraction. Circulation 2019, 140, 1463–1476. [Google Scholar] [CrossRef] [PubMed]
- Abraham, W.T.; Lindenfeld, J.; Ponikowski, P.; Agostoni, P.; Butler, J.; Desai, A.S.; Filippatos, G.; Gniot, J.; Fu, M.; Gullestad, L.; et al. Effect of empagliflozin on exercise ability and symptoms in heart failure patients with reduced and preserved ejection fraction, with and without type 2 diabetes. Eur. Heart J. 2020, 42, 700–710. [Google Scholar] [CrossRef] [PubMed]
- Perkovic, V.; Jardine, M.J.; Neal, B.; Bompoint, S.; Heerspink, H.J.L.; Charytan, D.M.; Edwards, R.; Agarwal, R.; Bakris, G.; Bull, S.; et al. Canagliflozin and Renal Outcomes in Type 2 Diabetes and Nephropathy. N. Engl. J. Med. 2019, 380, 2295–2306. [Google Scholar] [CrossRef] [Green Version]
- Heerspink, H.J.L.; Stefánsson, B.V.; Correa-Rotter, R.; Chertow, G.M.; Greene, T.; Hou, F.-F.; Mann, J.F.E.; McMurray, J.J.V.; Lindberg, M.; Rossing, P.; et al. Dapagliflozin in Patients with Chronic Kidney Disease. N. Engl. J. Med. 2020, 383, 1436–1446. [Google Scholar] [CrossRef]
- Pollock, C.; Stefánsson, B.; Reyner, D.; Rossing, P.; Sjöström, C.D.; Wheeler, D.C.; Langkilde, A.M.; Heerspink, H.J.L. Albuminuria-lowering effect of dapagliflozin alone and in combination with saxagliptin and effect of dapagliflozin and saxagliptin on glycaemic control in patients with type 2 diabetes and chronic kidney disease (DELIGHT): A randomised, double-blind, placebo-controlled trial. Lancet Diabetes Endocrinol. 2019, 7, 429–441. [Google Scholar] [CrossRef]
- Fioretto, P.; Del Prato, S.; Buse, J.B.; Goldenberg, R.; Giorgino, F.; Reyner, D.; Langkilde, A.M.; Sjöström, C.D.; Sartipy, P. On Behalf of the DERIVE Study Investigators Efficacy and safety of dapagliflozin in patients with type 2 diabetes and moderate renal impairment (chronic kidney disease stage 3A): The DERIVE Study. Diabetes, Obes. Metab. 2018, 20, 2532–2540, Erratum in Diabetes Obes. Metab. 2019, 21, 203. [Google Scholar] [CrossRef] [Green Version]
- Cherney, D.Z.I.; Dekkers, C.C.J.; Barbour, S.J.; Cattran, D.; Gafor, A.H.A.; Greasley, P.J.; Laverman, G.D.; Lim, S.K.; Di Tanna, G.L.; Reich, H.N.; et al. Effects of the SGLT2 inhibitor dapagliflozin on proteinuria in non-diabetic patients with chronic kidney disease (DIAMOND): A randomised, double-blind, crossover trial. Lancet Diabetes Endocrinol. 2020, 8, 582–593, Erratum in Lancet Diabetes Endocrinol. 2020, 8, 582–593. [Google Scholar] [CrossRef]
- McDonagh, T.A.; Metra, M.; Adamo, M.; Gardner, R.S.; Baumbach, A.; Böhm, M.; Burri, H.; Butler, J.; Čelutkienė, J.; Chioncel, O.; et al. 2021 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure: Developed by the Task Force for the Diagnosis and Treatment of Acute and Chronic Heart Failure of the European Society of Cardiology (ESC) With the Special Contribution of the Heart Failure Association (HFA) of the ESC. Eur. Heart J. 2021, 42, 3599–3726, Erratum in Eur. Heart J. 2021, 5.3, 3620–3623. [Google Scholar] [CrossRef]
- Yancy, C.W.; Januzzi, J.L.; Allen, L.A.; Butler, J.; Davis, L.L.; Fonarow, G.C.; Ibrahim, N.E.; Jessup, M.; Lindenfeld, J.; Maddox, T.M.; et al. 2017 ACC Expert Consensus Decision Pathway for Optimization of Heart Failure Treatment: Answers to 10 Pivotal Issues About Heart Failure With Reduced Ejection Fraction. J. Am. Coll. Cardiol. 2018, 71, 201–230, Erratum in Eur. Heart J. 2018, 72, 2549. [Google Scholar] [CrossRef] [PubMed]
- American Diabetes Association Professional Practice, Committee; American Diabetes Association Professional Practice, Committee; Draznin, B.; Aroda, V.R.; Bakris, G.; Benson, G.; Brown, F.M.; Freeman, R.; Green, J.; Huang, E.; et al. Cardiovascular Disease and Risk Management: Standards of Medical Care in Diabetes-2022. Diabetes Care 2022, 45 (Suppl. 1), S144–S174. [Google Scholar] [CrossRef] [PubMed]
- American Diabetes Association Professional Practice, Committee; American Diabetes Association Professional Practice, Committee; Draznin, B.; Aroda, V.R.; Bakris, G.; Benson, G.; Brown, F.M.; Freeman, R.; Green, J.; Huang, E.; et al. Chronic Kidney Disease and Risk Management: Standards of Medical Care in Diabetes-2022. Diabetes Care 2022, 45 (Suppl. 1), S175–S184. [Google Scholar] [CrossRef]
- Anker, S.D.; Butler, J.; Filippatos, G.; Ferreira, J.P.; Bocchi, E.; Böhm, M.; Brunner–La Rocca, H.-P.; Choi, D.-J.; Chopra, V.; Chuquiure-Valenzuela, E.; et al. Empagliflozin in Heart Failure with a Preserved Ejection Fraction. N. Engl. J. Med. 2021, 385, 1451–1461. [Google Scholar] [CrossRef] [PubMed]
- Packer, M.; Butler, J.; Zannad, F.; Filippatos, G.; Ferreira, J.P.; Pocock, S.J.; Carson, P.; Anand, I.; Doehner, W.; Haass, M.; et al. Effect of Empagliflozin on Worsening Heart Failure Events in Patients With Heart Failure and Preserved Ejection Fraction: EMPEROR-Preserved Trial. Circulation 2021, 144, 1284–1294. [Google Scholar] [CrossRef] [PubMed]
- Anker, S.D.; Butler, J.; Filippatos, G.; Khan, M.S.; Ferreira, J.P.; Bocchi, E.; Böhm, M.; Rocca, H.P.B.; Choi, D.; Chopra, V.; et al. Baseline characteristics of patients with heart failure with preserved ejection fraction in the EMPEROR-Preserved trial. Eur. J. Heart Fail. 2020, 22, 2383–2392. [Google Scholar] [CrossRef] [PubMed]
- Brenner, B.M. Hemodynamically mediated glomerular injury and the progressive nature of kidney disease. Kidney Int. 1983, 23, 647–655. [Google Scholar] [CrossRef] [Green Version]
- Thomson, S.C.; Rieg, T.; Miracle, C.; Mansoury, H.; Whaley, J.; Vallon, V.; Singh, P. Acute and chronic effects of SGLT2 blockade on glomerular and tubular function in the early diabetic rat. Am. J. Physiol. Integr. Comp. Physiol. 2012, 302, R75–R83. [Google Scholar] [CrossRef] [Green Version]
- Terami, N.; Ogawa, D.; Tachibana, H.; Hatanaka, T.; Wada, J.; Nakatsuka, A.; Eguchi, J.; Horiguchi, C.S.; Nishii, N.; Yamada, H.; et al. Long-Term Treatment with the Sodium Glucose Cotransporter 2 Inhibitor, Dapagliflozin, Ameliorates Glucose Homeostasis and Diabetic Nephropathy in db/db Mice. PLoS ONE 2014, 9, e100777. [Google Scholar] [CrossRef]
- Cherney, D.Z.; Perkins, B.A.; Soleymanlou, N.; Maione, M.; Lai, V.; Lee, A.; Fagan, N.M.; Woerle, H.J.; Johansen, O.E.; Broedl, U.C.; et al. Renal Hemodynamic Effect of Sodium-Glucose Cotransporter 2 Inhibition in Patients With Type 1 Diabetes Mellitus. Circulation 2014, 129, 587–597. [Google Scholar] [CrossRef] [Green Version]
- Škrtić, M.; Yang, G.; Perkins, B.A.; Soleymanlou, N.; Lytvyn, Y.; Von Eynatten, M.; Woerle, H.J.; Johansen, O.E.; Broedl, U.C.; Hach, T.; et al. Characterisation of glomerular haemodynamic responses to SGLT2 inhibition in patients with type 1 diabetes and renal hyperfiltration. Diabetologia 2014, 57, 2599–2602, Erratum in Diabetologia 2017, 60, 1159–1160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kidokoro, K.; Cherney, D.Z.I.; Bozovic, A.; Nagasu, H.; Satoh, M.; Kanda, E.; Sasaki, T.; Kashihara, N. Evaluation of Glomerular Hemodynamic Function by Empagliflozin in Diabetic Mice Using In Vivo Imaging. Circulation 2019, 140, 303–315. [Google Scholar] [CrossRef] [PubMed]
- van Bommel, E.J.; Muskiet, M.; van Baar, M.J.; Tonneijck, L.; Smits, M.; Emanuel, A.L.; Bozovic, A.; Danser, A.J.; Geurts, F.; Hoorn, E.J.; et al. The renal hemodynamic effects of the SGLT2 inhibitor dapagliflozin are caused by post-glomerular vasodilatation rather than pre-glomerular vasoconstriction in metformin-treated patients with type 2 diabetes in the randomized, double-blind RED trial. Kidney Int. 2020, 97, 202–212, Erratum in Kidney Int. 2020, 97, 1061. [Google Scholar] [CrossRef] [PubMed]
- Heerspink, H.J.L.; Perkins, B.A.; Fitchett, D.H.; Husain, M.; Cherney, D.Z.I. Sodium Glucose Cotransporter 2 Inhibitors in the Treatment of Diabetes Mellitus. Circulation 2016, 134, 752–772. [Google Scholar] [CrossRef]
- Wanner, C.; Heerspink, H.J.; Zinman, B.; Inzucchi, S.E.; Koitka-Weber, A.; Mattheus, M.; Hantel, S.; Woerle, H.-J.; Broedl, U.C.; von Eynatten, M.; et al. Empagliflozin and Kidney Function Decline in Patients with Type 2 Diabetes: A Slope Analysis from the EMPA-REG OUTCOME Trial. J. Am. Soc. Nephrol. 2018, 29, 2755–2769. [Google Scholar] [CrossRef] [Green Version]
- De Nicola, L.; Gabbai, F.B.; Garofalo, C.; Conte, G.; Minutolo, R. Nephroprotection by SGLT2 Inhibition: Back to the Future? J. Clin. Med. 2020, 9, 2243. [Google Scholar] [CrossRef]
- Heerspink, H.J.; Kosiborod, M.; Inzucchi, S.E.; Cherney, D.Z. Renoprotective effects of sodium-glucose cotransporter-2 inhibitors. Kidney Int. 2018, 94, 26–39. [Google Scholar] [CrossRef]
- Cherney, D.Z.I.; Zinman, B.; Inzucchi, S.E.; Koitka-Weber, A.; Mattheus, M.; von Eynatten, M.; Wanner, C. Effects of empagliflozin on the urinary albumin-to-creatinine ratio in patients with type 2 diabetes and established cardiovascular disease: An exploratory analysis from the EMPA-REG OUTCOME randomised, placebo-controlled trial. Lancet Diabetes Endocrinol. 2017, 5, 610–621. [Google Scholar] [CrossRef]
- Wanner, C.; Inzucchi, S.E.; Lachin, J.M.; Fitchett, D.; Von Eynatten, M.; Mattheus, M.; Johansen, O.E.; Woerle, H.J.; Broedl, U.C.; Zinman, B. Empagliflozin and Progression of Kidney Disease in Type 2 Diabetes. N. Engl. J. Med. 2016, 375, 323–334. [Google Scholar] [CrossRef]
- Giugliano, D.; De Nicola, L.; Maiorino, M.I.; Bellastella, G.; Garofalo, C.; Chiodini, P.; Ceriello, A.; Esposito, K. Preventing major adverse cardiovascular events by SGLT-2 inhibition in patients with type 2 diabetes: The role of kidney. Cardiovasc. Diabetol. 2020, 19, 35. [Google Scholar] [CrossRef] [Green Version]
- Steffes, M.W.; Schmidt, D.; Mccrery, R.; Basgen, J.M.; International Diabetic Nephropathy Study Group. Glomerular cell number in normal subjects and in type 1 diabetic patients. Kidney Int. 2001, 59, 2104–2113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pagtalunan, M.E.; Miller, P.L.; Jumping-Eagle, S.; Nelson, R.G.; Myers, B.D.; Rennke, H.G.; Coplon, N.S.; Sun, L.; Meyer, T.W. Podocyte loss and progressive glomerular injury in type II diabetes. J. Clin. Investig. 1997, 99, 342–348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cassis, P.; Locatelli, M.; Cerullo, D.; Corna, D.; Buelli, S.; Zanchi, C.; Villa, S.; Morigi, M.; Remuzzi, G.; Benigni, A.; et al. SGLT2 inhibitor dapagliflozin limits podocyte damage in proteinuric nondiabetic nephropathy. JCI Insight 2018, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanaka, S.; Tanaka, T.; Nangaku, M. Hypoxia as a key player in the AKI-to-CKD transition. Am. J. Physiol. Physiol. 2014, 307, F1187–F1195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hesp, A.C.; Schaub, J.A.; Prasad, P.V.; Vallon, V.; Laverman, G.D.; Bjornstad, P.; van Raalte, D.H. The role of renal hypoxia in the pathogenesis of diabetic kidney disease: A promising target for newer renoprotective agents including SGLT2 inhibitors? Kidney Int. 2020, 98, 579–589. [Google Scholar] [CrossRef]
- Ganz, M.B.; Hawkins, K.; Reilly, R.F. High glucose induces the activity and expression of Na(+)/H(+) exchange in glomerular mesangial cells. Am. J. Physiol. Physiol. 2000, 278, F91–F96. [Google Scholar] [CrossRef]
- Chen, J.; Williams, S.; Ho, S.; Loraine, H.; Hagan, D.; Whaley, J.M.; Feder, J.N. Quantitative PCR tissue expression profiling of the human SGLT2 gene and related family members. Diabetes Ther. 2010, 1, 57–92. [Google Scholar] [CrossRef] [Green Version]
- Chen-Izu, Y.; Shaw, R.M.; Pitt, G.S.; Yarov-Yarovoy, V.; Sack, J.T.; Abriel, H.; Aldrich, R.W.; Belardinelli, L.; Cannell, M.B.; Catterall, W.A.; et al. Na+ channel function, regulation, structure, trafficking and sequestration. J. Physiol. 2015, 593, 1347–1360. [Google Scholar] [CrossRef] [Green Version]
- Trum, M.; Riechel, J.; Wagner, S. Cardioprotection by SGLT2 Inhibitors—Does It All Come Down to Na+? Int. J. Mol. Sci. 2021, 22, 7976. [Google Scholar] [CrossRef]
- Bertero, E.; Prates Roma, L.; Ameri, P.; Maack, C. Cardiac effects of SGLT2 inhibitors: The sodium hypothesis. Cardiovasc. Res. 2018, 114, 12–18. [Google Scholar] [CrossRef] [Green Version]
- Mustroph, J.; Neef, S.; Maier, L.S. CaMKII as a target for arrhythmia suppression. Pharmacol. Ther. 2017, 176, 22–31. [Google Scholar] [CrossRef] [PubMed]
- Mustroph, J.; Sag, C.M.; Bähr, F.; Schmidtmann, A.-L.; Gupta, S.N.; Dietz, A.; Islam, M.T.; Lücht, C.M.; Beuthner, B.E.; Pabel, S.; et al. Loss of CASK Accelerates Heart Failure Development. Circ. Res. 2021, 128, 1139–1155. [Google Scholar] [CrossRef] [PubMed]
- Pabel, S.; Hamdani, N.; Luedde, M.; Sossalla, S. SGLT2 Inhibitors and Their Mode of Action in Heart Failure—Has the Mystery Been Unravelled? Curr. Heart Fail. Rep. 2021, 18, 315–328. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.; Li, G.; Huang, C.L.-H.; Lei, M.; Wu, L. Late sodium current associated cardiac electrophysiological and mechanical dysfunction. Pflügers Arch.-Eur. J. Physiol. 2017, 470, 461–469. [Google Scholar] [CrossRef] [PubMed]
- Baartscheer, A.; Schumacher, C.A.; Van Borren, M.M.G.J.; Belterman, C.N.W.; Coronel, R.; Fiolet, J.W.T. Increased Na+/H+-exchange activity is the cause of increased [Na+]i and underlies disturbed calcium handling in the rabbit pressure and volume overload heart failure model. Cardiovasc. Res. 2003, 57, 1015–1024. [Google Scholar] [CrossRef] [Green Version]
- Packer, M. Activation and Inhibition of Sodium-Hydrogen Exchanger Is a Mechanism That Links the Pathophysiology and Treatment of Diabetes Mellitus With That of Heart Failure. Circulation 2017, 136, 1548–1559. [Google Scholar] [CrossRef]
- Studer, R.; Reinecke, H.; Bilger, J.; Eschenhagen, T.; Böhm, M.; Hasenfuss, G.; Just, H.; Holtz, J.; Drexler, H. Gene expression of the cardiac Na(+)-Ca2+ exchanger in end-stage human heart failure. Circ. Res. 1994, 75, 443–453. [Google Scholar] [CrossRef] [Green Version]
- Sayour, A.A.; Oláh, A.; Ruppert, M.; Barta, B.A.; Horváth, E.M.; Benke, K.; Pólos, M.; Hartyánszky, I.; Merkely, B.; Radovits, T. Characterization of left ventricular myocardial sodium-glucose cotransporter 1 expression in patients with end-stage heart failure. Cardiovasc. Diabetol. 2020, 19, 159. [Google Scholar] [CrossRef]
- Baartscheer, A.; Schumacher, C.A.; Wust, R.C.; Fiolet, J.W.T.; Stienen, G.; Coronel, R.; Zuurbier, C.J. Empagliflozin decreases myocardial cytoplasmic Na+ through inhibition of the cardiac Na+/H+ exchanger in rats and rabbits. Diabetologia 2016, 60, 568–573. [Google Scholar] [CrossRef] [Green Version]
- Uthman, L.; Baartscheer, A.; Bleijlevens, B.; Schumacher, C.A.; Fiolet, J.W.T.; Koeman, A.; Jancev, M.; Hollmann, M.W.; Weber, N.C.; Coronel, R.; et al. Class effects of SGLT2 inhibitors in mouse cardiomyocytes and hearts: Inhibition of Na+/H+ exchanger, lowering of cytosolic Na+ and vasodilation. Diabetologia 2017, 61, 722–726. [Google Scholar] [CrossRef] [Green Version]
- Trum, M.; Riechel, J.; Lebek, S.; Pabel, S.; Sossalla, S.T.; Hirt, S.; Arzt, M.; Maier, L.S.; Wagner, S. Empagliflozin inhibits Na+ /H+ exchanger activity in human atrial cardiomyocytes. ESC Heart Fail. 2020, 7, 4429–4437. [Google Scholar] [CrossRef] [PubMed]
- Philippaert, K.; Kalyaanamoorthy, S.; Fatehi, M.; Long, W.; Soni, S.; Byrne, N.J.; Barr, A.; Singh, J.; Wong, J.; Palechuk, T.; et al. Cardiac Late Sodium Channel Current Is a Molecular Target for the Sodium/Glucose Cotransporter 2 Inhibitor Empagliflozin. Circulation 2021, 143, 2188–2204. [Google Scholar] [CrossRef] [PubMed]
- Lapuerta, P.; Zambrowicz, B.; Strumph, P.; Sands, A. Development of sotagliflozin, a dual sodium-dependent glucose transporter 1/2 inhibitor. Diabetes Vasc. Dis. Res. 2015, 12, 101–110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mustroph, J.; Wagemann, O.; Lücht, C.M.; Trum, M.; Hammer, K.; Sag, C.M.; Lebek, S.; Tarnowski, D.; Reinders, J.; Perbellini, F.; et al. Empagliflozin reduces Ca/calmodulin-dependent kinase II activity in isolated ventricular cardiomyocytes. ESC Heart Fail. 2018, 5, 642–648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maejima, Y. SGLT2 Inhibitors Play a Salutary Role in Heart Failure via Modulation of the Mitochondrial Function. Front. Cardiovasc. Med. 2020, 6, 186. [Google Scholar] [CrossRef] [PubMed]
- Sasso, F.C.; Pafundi, P.C.; Simeon, V.; De Nicola, L.; Chiodini, P.; Galiero, R.; Rinaldi, L.; Nevola, R.; Salvatore, T.; Sardu, C.; et al. Efficacy and durability of multifactorial intervention on mortality and MACEs: A randomized clinical trial in type-2 diabetic kidney disease. Cardiovasc. Diabetol. 2021, 20, 145. [Google Scholar] [CrossRef]
- Abdul-Ghani, M.; Del Prato, S.; Chilton, R.; DeFronzo, R.A. SGLT2 Inhibitors and Cardiovascular Risk: Lessons Learned From the EMPA-REG OUTCOME Study. Diabetes Care 2016, 39, 717–725. [Google Scholar] [CrossRef] [Green Version]
- Butler, J.; Hamo, C.E.; Filippatos, G.; Pocock, S.J.; Bernstein, R.A.; Brueckmann, M.; Cheung, A.K.; George, J.T.; Green, J.B.; Januzzi, J.L.; et al. The potential role and rationale for treatment of heart failure with sodium-glucose co-transporter 2 inhibitors. Eur. J. Heart Fail. 2017, 19, 1390–1400. [Google Scholar] [CrossRef] [Green Version]
- Inzucchi, S.E.; Zinman, B.; Fitchett, D.; Wanner, C.; Ferrannini, E.; Schumacher, M.; Schmoor, C.; Ohneberg, K.; Johansen, O.E.; George, J.T.; et al. How Does Empagliflozin Reduce Cardiovascular Mortality? Insights From a Mediation Analysis of the EMPA-REG OUTCOME Trial. Diabetes Care 2017, 41, 356–363. [Google Scholar] [CrossRef] [Green Version]
- Koye, D.N.; Magliano, D.; Nelson, R.G.; Pavkov, M.E. The Global Epidemiology of Diabetes and Kidney Disease. Adv. Chronic Kidney Dis. 2018, 25, 121–132. [Google Scholar] [CrossRef]
- Maeda, S.; Matsui, T.; Takeuchi, M.; Yamagishi, S.-I. Sodium-glucose cotransporter 2-mediated oxidative stress augments advanced glycation end products-induced tubular cell apoptosis. Diabetes Metab. Res. Rev. 2013, 29, 406–412. [Google Scholar] [CrossRef] [PubMed]
- Ferrannini, G.; Hach, T.; Crowe, S.; Sanghvi, A.; Hall, K.D.; Ferrannini, E. Energy Balance After Sodium–Glucose Cotransporter 2 Inhibition. Diabetes Care 2015, 38, 1730–1735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sa-Nguanmoo, P.; Tanajak, P.; Kerdphoo, S.; Jaiwongkam, T.; Pratchayasakul, W.; Chattipakorn, N.; Chattipakorn, S.C. SGLT2-inhibitor and DPP-4 inhibitor improve brain function via attenuating mitochondrial dysfunction, insulin resistance, inflammation, and apoptosis in HFD-induced obese rats. Toxicol. Appl. Pharmacol. 2017, 333, 43–50. [Google Scholar] [CrossRef] [PubMed]
- Millar, P.; Pathak, N.; Parthsarathy, V.; Bjourson, A.J.; O’Kane, M.; Pathak, V.; Moffett, R.C.; Flatt, P.R.; Gault, V.A. Metabolic and neuroprotective effects of dapagliflozin and liraglutide in diabetic mice. J. Endocrinol. 2017, 234, 255–267. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Yang, J.; Chen, X.; Qiu, F.; Li, J. Effects of Sodium-glucose Cotransporter 2 Inhibitor Monotherapy on Weight Changes in Patients With Type 2 Diabetes Mellitus: A Bayesian Network Meta-analysis. Clin. Ther. 2019, 41, 322–334.e11. [Google Scholar] [CrossRef] [PubMed]
- Bolinder, J.; Ljunggren, Ö.; Kullberg, J.; Johansson, L.; Wilding, J.; Langkilde, A.M.; Sugg, J.; Parikh, S. Effects of Dapagliflozin on Body Weight, Total Fat Mass, and Regional Adipose Tissue Distribution in Patients with Type 2 Diabetes Mellitus with Inadequate Glycemic Control on Metformin. J. Clin. Endocrinol. Metab. 2012, 97, 1020–1031. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, E.; Wilding, J.P.; Barber, T.M.; Alam, U.; Cuthbertson, D.J. Weight loss variability with SGLT2 inhibitors and GLP-1 receptor agonists in type 2 diabetes mellitus and obesity: Mechanistic possibilities. Obes. Rev. 2019, 20, 816–828. [Google Scholar] [CrossRef] [Green Version]
- Zheng, R.; Zhou, D.; Zhu, Y. The long-term prognosis of cardiovascular disease and all-cause mortality for metabolically healthy obesity: A systematic review and meta-analysis. J. Epidemiol. Community Health 2016, 70, 1024–1031. [Google Scholar] [CrossRef]
- Foster, M.C.; Hwang, S.-J.; Larson, M.; Lichtman, J.H.; Parikh, N.I.; Vasan, R.S.; Levy, D.; Fox, C.S. Overweight, Obesity, and the Development of Stage 3 CKD: The Framingham Heart Study. Am. J. Kidney Dis. 2008, 52, 39–48. [Google Scholar] [CrossRef] [Green Version]
- Hsu, C.-Y.; McCulloch, C.E.; Iribarren, C.; Darbinian, J.; Go, A.S. Body Mass Index and Risk for End-Stage Renal Disease. Ann. Intern. Med. 2006, 144, 21–28. [Google Scholar] [CrossRef]
- Tamura, K.; Wakui, H.; Azushima, K.; Uneda, K.; Umemura, S. Circadian blood pressure rhythm as a possible key target of SGLT2 inhibitors used for the treatment of Type 2 diabetes. Hypertens. Res. 2016, 39, 396–398. [Google Scholar] [CrossRef] [PubMed]
- Rahman, A.; Hitomi, H.; Nishiyama, A. Cardioprotective effects of SGLT2 inhibitors are possibly associated with normalization of the circadian rhythm of blood pressure. Hypertens. Res. 2017, 40, 535–540. [Google Scholar] [CrossRef] [PubMed]
- Georgianos, P.I.; Agarwal, R. Ambulatory Blood Pressure Reduction With SGLT-2 Inhibitors: Dose-Response Meta-analysis and Comparative Evaluation With Low-Dose Hydrochlorothiazide. Diabetes Care 2019, 42, 693–700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Majewski, C.; Bakris, G.L. Blood Pressure Reduction: An Added Benefit of Sodium–Glucose Cotransporter 2 Inhibitors in Patients With Type 2 Diabetes. Diabetes Care 2015, 38, 429–430. [Google Scholar] [CrossRef] [Green Version]
- Puglisi, S.; Rossini, A.; Poli, R.; Dughera, F.; Pia, A.; Terzolo, M.; Reimondo, G. Effects of SGLT2 Inhibitors and GLP-1 Receptor Agonists on Renin-Angiotensin-Aldosterone System. Front. Endocrinol. 2021, 12, 738848. [Google Scholar] [CrossRef]
- Cefalu, W.T.; Stenlöf, K.; Leiter, L.A.; Wilding, J.; Blonde, L.; Polidori, D.; Xie, J.; Sullivan, D.; Usiskin, K.; Canovatchel, W.; et al. Effects of canagliflozin on body weight and relationship to HbA1c and blood pressure changes in patients with type 2 diabetes. Diabetologia 2015, 58, 1183–1187. [Google Scholar] [CrossRef]
- Sjöström, C.D.; Johansson, P.; Ptaszynska, A.; List, J.; Johnsson, E. Dapagliflozin lowers blood pressure in hypertensive and non-hypertensive patients with type 2 diabetes. Diabetes Vasc. Dis. Res. 2015, 12, 352–358. [Google Scholar] [CrossRef] [Green Version]
- Dimova, R.; Tankova, T. Does SGLT2 Inhibition Affect Sympathetic Nerve Activity in Type 2 Diabetes? Horm. Metab. Res. 2020, 53, 75–84. [Google Scholar] [CrossRef]
- Sugiyama, S.; Jinnouchi, H.; Kurinami, N.; Hieshima, K.; Yoshida, A.; Jinnouchi, K.; Nishimura, H.; Suzuki, T.; Miyamoto, F.; Kajiwara, K.; et al. The SGLT2 Inhibitor Dapagliflozin Significantly Improves the Peripheral Microvascular Endothelial Function in Patients with Uncontrolled Type 2 Diabetes Mellitus. Intern. Med. 2018, 57, 2147–2156. [Google Scholar] [CrossRef] [Green Version]
- Chilton, R.; Tikkanen, I.; Cannon, C.P.; Crowe, S.; Woerle, H.J.; Broedl, U.C.; Johansen, O.E. Effects of empagliflozin on blood pressure and markers of arterial stiffness and vascular resistance in patients with type 2 diabetes. Diabetes Obes. Metab. 2015, 17, 1180–1193. [Google Scholar] [CrossRef]
- Benham, J.L.; Booth, J.E.; Sigal, R.J.; Daskalopoulou, S.S.; Leung, A.A.; Rabi, D.M. Systematic review and meta-analysis: SGLT2 inhibitors, blood pressure and cardiovascular outcomes. Int. J. Cardiol. Heart Vasc. 2021, 33, 100725. [Google Scholar] [CrossRef] [PubMed]
- Guo, M.; Ding, J.; Li, J.; Wang, J.; Zhang, T.; Liu, C.; Huang, W.; Long, Y.; Gao, C.; Xu, Y. SGLT2 inhibitors and risk of stroke in patients with type 2 diabetes: A systematic review and meta-analysis. Diabetes Obes. Metab. 2018, 20, 1977–1982. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Jardine, M.J.; Li, Q.; Neuen, B.L.; Cannon, C.P.; de Zeeuw, D.; Edwards, R.; Levin, A.; Mahaffey, K.W.; Perkovic, V.; et al. Effect of SGLT2 Inhibitors on Stroke and Atrial Fibrillation in Diabetic Kidney Disease. Stroke 2021, 52, 1545–1556. [Google Scholar] [CrossRef] [PubMed]
- Scheen, A.; Delanaye, P. Effects of reducing blood pressure on renal outcomes in patients with type 2 diabetes: Focus on SGLT2 inhibitors and EMPA-REG OUTCOME. Diabetes Metab. 2017, 43, 99–109. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Chen, Y.; Xu, W.; Lu, N.; Cao, J.; Yu, S. Effects of intensive blood pressure lowering on mortality and cardiovascular and renal outcomes in type 2 diabetic patients: A meta-analysis. PLoS ONE 2019, 14, e0215362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kimura, Y.; Tsukui, D.; Kono, H. Uric Acid in Inflammation and the Pathogenesis of Atherosclerosis. Int. J. Mol. Sci. 2021, 22, 12394. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, A.; Garcia-Arroyo, F.; Sasai, F.; Rodriguez-Iturbe, B.; Sanchez-Lozada, L.G.; Lanaspa, M.A.; Johnson, R.J. Mini Review: Reappraisal of Uric Acid in Chronic Kidney Disease. Am. J. Nephrol. 2021, 52, 837–844. [Google Scholar] [CrossRef]
- Zhao, Y.; Xu, L.; Tian, D.; Xia, P.; Zheng, H.; Wang, L.; Chen, L. Effects of sodium-glucose co-transporter 2 (SGLT2) inhibitors on serum uric acid level: A meta-analysis of randomized controlled trials. Diabetes Obes. Metab. 2017, 20, 458–462. [Google Scholar] [CrossRef]
- Novikov, A.; Fu, Y.; Huang, W.; Freeman, B.; Patel, R.; Van Ginkel, C.; Koepsell, H.; Busslinger, M.; Onishi, A.; Nespoux, J.; et al. SGLT2 inhibition and renal urate excretion: Role of luminal glucose, GLUT9, and URAT1. Am. J. Physiol. Physiol. 2019, 316, F173–F185. [Google Scholar] [CrossRef]
- Calapkulu, M.; Cander, S.; Gul, O.O.; Ersoy, C. Lipid profile in type 2 diabetic patients with new dapagliflozin treatment; actual clinical experience data of six months retrospective lipid profile from single center. Diabetes Metab. Syndr. Clin. Res. Rev. 2019, 13, 1031–1034. [Google Scholar] [CrossRef]
- Inagaki, N.; Goda, M.; Yokota, S.; Maruyama, N.; Iijima, H. Effects of Baseline Blood Pressure and Low-Density Lipoprotein Cholesterol on Safety and Efficacy of Canagliflozin in Japanese Patients with Type 2 Diabetes Mellitus. Adv. Ther. 2015, 32, 1085–1103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Halimi, S.; Vergès, B. Adverse effects and safety of SGLT-2 inhibitors. Diabetes Metab. 2014, 40 (Suppl. 1), S28–S34. [Google Scholar] [CrossRef]
- Szekeres, Z.; Toth, K.; Szabados, E. The Effects of SGLT2 Inhibitors on Lipid Metabolism. Metabolites 2021, 11, 87. [Google Scholar] [CrossRef]
- Hayashi, T.; Fukui, T.; Nakanishi, N.; Yamamoto, S.; Tomoyasu, M.; Osamura, A.; Ohara, M.; Yamamoto, T.; Ito, Y.; Hirano, T. Dapagliflozin decreases small dense low-density lipoprotein-cholesterol and increases high-density lipoprotein 2-cholesterol in patients with type 2 diabetes: Comparison with sitagliptin. Cardiovasc. Diabetol. 2017, 16, 8, Erratum in Cardiovasc. Diabetol. 2017, 16, 149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamijo, Y.; Ishii, H.; Yamamoto, T.; Kobayashi, K.; Asano, H.; Miake, S.; Kanda, E.; Urata, H.; Yoshida, M. Potential Impact on Lipoprotein Subfractions in Type 2 Diabetes. Clin. Med. Insights: Endocrinol. Diabetes 2019, 12, 1179551419866811. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Xu, J.; Wu, M.; Xu, B.; Kang, L. Empagliflozin protects against atherosclerosis progression by modulating lipid profiles and sympathetic activity. Lipids Health Dis. 2021, 20, 5. [Google Scholar] [CrossRef]
- Chen, M.-B.; Wang, H.; Cui, W.-Y.; Xu, H.-L.; Zheng, Q.-H. Effect of SGLT inhibitors on weight and lipid metabolism at 24 weeks of treatment in patients with diabetes mellitus. Medicine 2021, 100, e24593. [Google Scholar] [CrossRef]
- Filippas-Ntekouan, S.; Tsimihodimos, V.; Filippatos, T.; Dimitriou, T.; Elisaf, M. SGLT-2 inhibitors: Pharmacokinetics characteristics and effects on lipids. Expert Opin. Drug Metab. Toxicol. 2018, 14, 1113–1121. [Google Scholar] [CrossRef]
- Lazarte, J.; Kanagalingam, T.; Hegele, R.A. Lipid effects of sodium-glucose cotransporter 2 inhibitors. Curr. Opin. Lipidol. 2021, 32, 183–190. [Google Scholar] [CrossRef]
- Liu, Z.; Ma, X.; Ilyas, I.; Zheng, X.; Luo, S.; Little, P.J.; Kamato, D.; Sahebkar, A.; Wu, W.; Weng, J.; et al. Impact of sodium glucose cotransporter 2 (SGLT2) inhibitors on atherosclerosis: From pharmacology to pre-clinical and clinical therapeutics. Theranostics 2021, 11, 4502–4515. [Google Scholar] [CrossRef]
- Barraclough, J.Y.; Patel, S.; Yu, J.; Neal, B.; Arnott, C. The Role of Sodium Glucose Cotransporter-2 Inhibitors in Atherosclerotic Cardiovascular Disease: A Narrative Review of Potential Mechanisms. Cells 2021, 10, 2699. [Google Scholar] [CrossRef]
- Daniele, G.; Xiong, J.; Solis-Herrera, C.; Merovci, A.; Eldor, R.; Tripathy, D.; DeFronzo, R.A.; Norton, L.; Abdul-Ghani, M. Dapagliflozin Enhances Fat Oxidation and Ketone Production in Patients With Type 2 Diabetes. Diabetes Care 2016, 39, 2036–2041. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferrannini, E.; Baldi, S.; Frascerra, S.; Astiarraga, B.; Heise, T.; Bizzotto, R.; Mari, A.; Pieber, T.R.; Muscelli, E. Shift to Fatty Substrate Utilization in Response to Sodium–Glucose Cotransporter 2 Inhibition in Subjects Without Diabetes and Patients With Type 2 Diabetes. Diabetes 2016, 65, 1190–1195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferrannini, E.; Baldi, S.; Frascerra, S.; Astiarraga, B.; Barsotti, E.; Clerico, A.; Muscelli, E. Renal Handling of Ketones in Response to Sodium–Glucose Cotransporter 2 Inhibition in Patients With Type 2 Diabetes. Diabetes Care 2017, 40, 771–776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Osataphan, S.; Macchi, C.; Singhal, G.; Chimene-Weiss, J.; Sales, V.; Kozuka, C.; Dreyfuss, J.; Pan, H.; Tangcharoenpaisan, Y.; Morningstar, J.; et al. SGLT2 inhibition reprograms systemic metabolism via FGF21-dependent and -independent mechanisms. JCI Insight 2019, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tezze, C.; Romanello, V.; Sandri, M. FGF21 as Modulator of Metabolism in Health and Disease. Front. Physiol. 2019, 10, 419. [Google Scholar] [CrossRef] [PubMed]
- Avogaro, A.; Fadini, G.P.; Del Prato, S. Reinterpreting Cardiorenal Protection of Renal Sodium–Glucose Cotransporter 2 Inhibitors via Cellular Life History Programming. Diabetes Care 2020, 43, 501–507. [Google Scholar] [CrossRef]
- Aubert, G.; Martin, O.J.; Horton, J.L.; Lai, L.; Vega, R.B.; Leone, T.C.; Koves, T.; Gardell, S.J.; Krüger, M.; Hoppel, C.L.; et al. The Failing Heart Relies on Ketone Bodies as a Fuel. Circulation 2016, 133, 698–705, Erratum in Circulation 2018, 138, e422. [Google Scholar] [CrossRef]
- Dutka, M.; Bobiński, R.; Ulman-Włodarz, I.; Hajduga, M.; Bujok, J.; Pająk, C.; Ćwiertnia, M. Sodium glucose cotransporter 2 inhibitors: Mechanisms of action in heart failure. Heart Fail. Rev. 2020, 26, 603–622. [Google Scholar] [CrossRef]
- Hattori, Y. Beneficial effects on kidney during treatment with sodium-glucose cotransporter 2 inhibitors: Proposed role of ketone utilization. Heart Fail. Rev. 2021, 26, 947–952. [Google Scholar] [CrossRef]
- Bedi, K.C., Jr.; Snyder, N.W.; Brandimarto, J.; Aziz, M.; Mesaros, C.; Worth, A.J.; Wang, L.L.; Javaheri, A.; Blair, I.A.; Margulies, K.B.; et al. Evidence for Intramyocardial Disruption of Lipid Metabolism and Increased Myocardial Ketone Utilization in Advanced Human Heart Failure. Circulation 2016, 133, 706–716. [Google Scholar] [CrossRef] [Green Version]
- Sun, H.; Olson, K.C.; Gao, C.; Prosdocimo, D.A.; Zhou, M.; Wang, Z.; Jeyaraj, D.; Youn, J.-Y.; Ren, S.; Liu, Y.; et al. Catabolic Defect of Branched-Chain Amino Acids Promotes Heart Failure. Circulation 2016, 133, 2038–2049. [Google Scholar] [CrossRef] [PubMed]
- Kappel, B.A.; Lehrke, M.; Schütt, K.; Artati, A.; Adamski, J.; Lebherz, C.; Marx, N. Effect of Empagliflozin on the Metabolic Signature of Patients With Type 2 Diabetes Mellitus and Cardiovascular Disease. Circulation 2017, 136, 969–972. [Google Scholar] [CrossRef] [PubMed]
- Ferrannini, E.; Mark, M.; Mayoux, E. CV Protection in the EMPA-REG OUTCOME Trial: A “Thrifty Substrate” Hypothesis. Diabetes Care 2016, 39, 1108–1114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mudaliar, S.; Alloju, S.; Henry, R.R. Can a Shift in Fuel Energetics Explain the Beneficial Cardiorenal Outcomes in the EMPA-REG OUTCOME Study? A Unifying Hypothesis. Diabetes Care 2016, 39, 1115–1122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verma, S.; Rawat, S.; Ho, K.L.; Wagg, C.S.; Zhang, L.; Teoh, H.; Dyck, J.E.; Uddin, G.M.; Oudit, G.Y.; Mayoux, E.; et al. Empagliflozin Increases Cardiac Energy Production in Diabetes. JACC: Basic Transl. Sci. 2018, 3, 575–587. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, R.; Møller, N.; Gormsen, L.C.; Tolbod, L.P.; Hansson, N.H.; Sorensen, J.; Harms, H.J.; Frøkiær, J.; Eiskjaer, H.; Jespersen, N.R.; et al. Cardiovascular Effects of Treatment With the Ketone Body 3-Hydroxybutyrate in Chronic Heart Failure Patients. Circulation 2019, 139, 2129–2141. [Google Scholar] [CrossRef]
- Adingupu, D.D.; Göpel, S.O.; Grönros, J.; Behrendt, M.; Sotak, M.; Miliotis, T.; Dahlqvist, U.; Gan, L.-M.; Jönsson-Rylander, A.-C. SGLT2 inhibition with empagliflozin improves coronary microvascular function and cardiac contractility in prediabetic ob/ob−/− mice. Cardiovasc. Diabetol. 2019, 18, 16. [Google Scholar] [CrossRef] [Green Version]
- Santos-Gallego, C.G.; Requena-Ibanez, J.A.; Antonio, R.S.; Ishikawa, K.; Watanabe, S.; Picatoste, B.; Flores, E.; Garcia-Ropero, A.; Sanz, J.; Hajjar, R.J.; et al. Empagliflozin Ameliorates Adverse Left Ventricular Remodeling in Nondiabetic Heart Failure by Enhancing Myocardial Energetics. J. Am. Coll. Cardiol. 2019, 73, 1931–1944. [Google Scholar] [CrossRef]
- Oh, C.-M.; Cho, S.; Jang, J.-Y.; Kim, H.; Chun, S.; Choi, M.; Park, S.; Ko, Y.-G. Cardioprotective Potential of an SGLT2 Inhibitor Against Doxorubicin-Induced Heart Failure. Korean Circ. J. 2019, 49, 1183–1195. [Google Scholar] [CrossRef] [Green Version]
- Shimazu, T.; Hirschey, M.D.; Newman, J.; He, W.; Shirakawa, K.; Le Moan, N.; Grueter, C.A.; Lim, H.; Saunders, L.R.; Stevens, R.D.; et al. Suppression of Oxidative Stress by β-Hydroxybutyrate, an Endogenous Histone Deacetylase Inhibitor. Science 2013, 339, 211–214. [Google Scholar] [CrossRef] [Green Version]
- Youm, Y.-H.; Nguyen, K.Y.; Grant, R.W.; Goldberg, E.L.; Bodogai, M.; Kim, D.; D’Agostino, D.; Planavsky, N.; Lupfer, C.; Kanneganti, T.-D.; et al. The ketone metabolite β-hydroxybutyrate blocks NLRP3 inflammasome–mediated inflammatory disease. Nat. Med. 2015, 21, 263–269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.R.; Lee, S.-G.; Kim, S.H.; Kim, J.H.; Choi, E.; Cho, W.; Rim, J.; Hwang, I.; Lee, C.J.; Lee, M.; et al. SGLT2 inhibition modulates NLRP3 inflammasome activity via ketones and insulin in diabetes with cardiovascular disease. Nat. Commun. 2020, 11, 2127. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Chen, X.; Zhang, Y.; George, J.; Cobbs, A.; Wang, G.; Li, L.; Emmett, N. Kidney Injury Molecule-1 Is Upregulated in Renal Lipotoxicity and Mediates Palmitate-Induced Tubular Cell Injury and Inflammatory Response. Int. J. Mol. Sci. 2019, 20, 3406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Little, J.R.; Spitzer, J.J. Uptake of ketone bodies by dog kidney in vivo. Am. J. Physiol. Content 1971, 221, 679–683. [Google Scholar] [CrossRef] [PubMed]
- Fukao, T.; Song, X.-Q.; Mitchell, G.A.; Yamaguchi, S.; Sukegawa, K.; Or, T.; Kondo, N. Enzymes of Ketone Body Utilization in Human Tissues: Protein and Messenger RNA Levels of Succinyl-Coenzyme A (CoA):3-Ketoacid CoA Transferase and Mitochondrial and Cytosolic Acetoacetyl-CoA Thiolases. Pediatric Res. 1997, 42, 498–502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nosadini, R.; Trevisan, R.; Fioretto, P.; Semplicini, A.; Samà, B.; Velussi, M.; Da Campo, G.L.; Avogaro, A.; Vizzaccaro, A.; Donadon, V.; et al. Kidney Hemodynamics After Ketone Body and Amino Acid Infusion in Normal and IDDM Subjects. Diabetes 1989, 38, 75–83. [Google Scholar] [CrossRef]
- Zhang, D.; Yang, H.; Kong, X.; Wang, K.; Mao, X.; Yan, X.; Wang, Y.; Liu, S.; Zhang, X.; Li, J.; et al. Proteomics analysis reveals diabetic kidney as a ketogenic organ in type 2 diabetes. Am. J. Physiol. Metab. 2011, 300, E287–E295. [Google Scholar] [CrossRef]
- Verma, S.; McMurray, J.J.V. SGLT2 inhibitors and mechanisms of cardiovascular benefit: A state-of-the-art review. Diabetologia 2018, 61, 2108–2117. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; LaRocque, L.M.; Efe, O.; Wang, J.; Sands, J.M.; Klein, J.D. Effect of Dapagliflozin Treatment on Fluid and Electrolyte Balance in Diabetic Rats. Am. J. Med. Sci. 2016, 352, 517–523. [Google Scholar] [CrossRef] [Green Version]
- Heerspink, H.J.L.; de Zeeuw, D.; Wie, L.; Leslie, B.; List, J. Dapagliflozin a glucose-regulating drug with diuretic properties in subjects with type 2 diabetes. Diabetes Obes. Metab. 2013, 15, 853–862. [Google Scholar] [CrossRef] [Green Version]
- Sha, S.; Polidori, D.; Heise, T.; Natarajan, J.; Farrell, K.; Wang, S.-S.; Sica, D.; Rothenberg, P.; Plum-Mörschel, L. Effect of the sodium glucose co-transporter 2 inhibitor canagliflozin on plasma volume in patients with type 2 diabetes mellitus. Diabetes Obes. Metab. 2014, 16, 1087–1095. [Google Scholar] [CrossRef] [PubMed]
- Griffin, M.; Rao, V.S.; Ivey-Miranda, J.; Fleming, J.; Mahoney, D.; Maulion, C.; Suda, N.; Siwakoti, K.; Ahmad, T.; Jacoby, D.; et al. Empagliflozin in Heart Failure. Circulation 2020, 142, 1028–1039. [Google Scholar] [CrossRef] [PubMed]
- Karg, M.V.; Bosch, A.; Kannenkeril, D.; Striepe, K.; Ott, C.; Schneider, M.P.; Boemke-Zelch, F.; Linz, P.; Nagel, A.M.; Titze, J.; et al. SGLT-2-inhibition with dapagliflozin reduces tissue sodium content: A randomised controlled trial. Cardiovasc. Diabetol. 2018, 17, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Packer, M.; Anker, S.D.; Butler, J.; Filippatos, G.; Ferreira, J.P.; Pocock, S.J.; Sattar, N.; Brueckmann, M.; Jamal, W.; Cotton, D.; et al. Empagliflozin in Patients With Heart Failure, Reduced Ejection Fraction, and Volume Overload. J. Am. Coll. Cardiol. 2021, 77, 1381–1392. [Google Scholar] [CrossRef] [PubMed]
- Yasui, A.; Lee, G.; Hirase, T.; Kaneko, T.; Kaspers, S.; Von Eynatten, M.; Okamura, T. Empagliflozin Induces Transient Diuresis Without Changing Long-Term Overall Fluid Balance in Japanese Patients With Type 2 Diabetes. Diabetes Ther. 2018, 9, 863–871. [Google Scholar] [CrossRef] [Green Version]
- Heise, T.; Jordan, J.; Wanner, C.; Heer, M.; Macha, S.; Mattheus, M.; Lund, S.S.; Woerle, H.J.; Broedl, U.C. Pharmacodynamic Effects of Single and Multiple Doses of Empagliflozin in Patients With Type 2 Diabetes. Clin. Ther. 2016, 38, 2265–2276. [Google Scholar] [CrossRef] [PubMed]
- Schnermann, J. Juxtaglomerular cell complex in the regulation of renal salt excretion. Am. J. Physiol. Integr. Comp. Physiol. 1998, 274, R263–R279. [Google Scholar] [CrossRef] [PubMed]
- Francis, G.S.; Siegel, R.M.; Goldsmith, S.R.; Olivari, M.T.; Levine, T.B.; Cohn, J.N. Acute Vasoconstrictor Response to Intravenous Furosemide in Patients with Chronic Congestive Heart Failure. Ann. Intern. Med. 1985, 103, 1–6. [Google Scholar] [CrossRef]
- Škrtić, M.; Cherney, D.Z. Sodium–glucose cotransporter-2 inhibition and the potential for renal protection in diabetic nephropathy. Curr. Opin. Nephrol. Hypertens. 2015, 24, 96–103. [Google Scholar] [CrossRef]
- Hallow, K.M.; Helmlinger, G.; Greasley, P.J.; McMurray, J.J.V.; Boulton, D.W. Why do SGLT2 inhibitors reduce heart failure hospitalization? A differential volume regulation hypothesis. Diabetes Obes. Metab. 2017, 20, 479–487. [Google Scholar] [CrossRef] [Green Version]
- Jensen, J.; Omar, M.; Kistorp, C.; Tuxen, C.; Gustafsson, I.; Køber, L.; Gustafsson, F.; Faber, J.; Malik, M.E.; Fosbøl, E.L.; et al. Effects of empagliflozin on estimated extracellular volume, estimated plasma volume, and measured glomerular filtration rate in patients with heart failure (Empire HF Renal): A prespecified substudy of a double-blind, randomised, placebo-controlled trial. Lancet Diabetes Endocrinol. 2020, 9, 106–116. [Google Scholar] [CrossRef]
- Cohen, N.D.; Gutman, S.; Briganti, E.; Taylor, A. Effects of empagliflozin treatment on cardiac function and structure in patients with type 2 diabetes: A cardiac magnetic resonance study. Intern. Med. J. 2019, 49, 1006–1010. [Google Scholar] [CrossRef] [PubMed]
- Bosch, A.; Ott, C.; Jung, S.; Striepe, K.; Karg, M.V.; Kannenkeril, D.; Dienemann, T.; Schmieder, R.E. How does empagliflozin improve arterial stiffness in patients with type 2 diabetes mellitus? Sub analysis of a clinical trial. Cardiovasc. Diabetol. 2019, 18, 44. [Google Scholar] [CrossRef] [PubMed]
- Lunder, M.; Janić, M.; Japelj, M.; Juretič, A.; Janež, A.; Šabovič, M. Empagliflozin on top of metformin treatment improves arterial function in patients with type 1 diabetes mellitus. Cardiovasc. Diabetol. 2018, 17, 153. [Google Scholar] [CrossRef] [PubMed]
- Irace, C.; Cutruzzolà, A.; Parise, M.; Fiorentino, R.; Frazzetto, M.; Gnasso, C.; Casciaro, F.; Gnasso, A. Effect of empagliflozin on brachial artery shear stress and endothelial function in subjects with type 2 diabetes: Results from an exploratory study. Diabetes Vasc. Dis. Res. 2020, 17, 1479164119883540. [Google Scholar] [CrossRef] [Green Version]
- Solini, A.; Giannini, L.; Seghieri, M.; Vitolo, E.; Taddei, S.; Ghiadoni, L.; Bruno, R.M. Dapagliflozin acutely improves endothelial dysfunction, reduces aortic stiffness and renal resistive index in type 2 diabetic patients: A pilot study. Cardiovasc. Diabetol. 2017, 16, 138. [Google Scholar] [CrossRef] [Green Version]
- Solini, A.; Seghieri, M.; Giannini, L.; Biancalana, E.; Parolini, F.; Rossi, C.; Dardano, A.; Taddei, S.; Ghiadoni, L.; Bruno, R.M. The Effects of Dapagliflozin on Systemic and Renal Vascular Function Display an Epigenetic Signature. J. Clin. Endocrinol. Metab. 2019, 104, 4253–4263. [Google Scholar] [CrossRef]
- Pfeifer, M.; Townsend, R.R.; Davies, M.J.; Vijapurkar, U.; Ren, J. Effects of canagliflozin, a sodium glucose co-transporter 2 inhibitor, on blood pressure and markers of arterial stiffness in patients with type 2 diabetes mellitus: A post hoc analysis. Cardiovasc. Diabetol. 2017, 16, 29. [Google Scholar] [CrossRef] [Green Version]
- Katakami, N.; Mita, T.; Yoshii, H.; Shiraiwa, T.; Yasuda, T.; Okada, Y.; Torimoto, K.; Umayahara, Y.; Kaneto, H.; Osonoi, T.; et al. Effect of tofogliflozin on arterial stiffness in patients with type 2 diabetes: Prespecified sub-analysis of the prospective, randomized, open-label, parallel-group comparative UTOPIA trial. Cardiovasc. Diabetol. 2021, 20, 4. [Google Scholar] [CrossRef]
- Aroor, A.R.; Das, N.A.; Carpenter, A.J.; Habibi, J.; Jia, G.; Ramirez-Perez, F.; Martinez-Lemus, L.; Manrique-Acevedo, C.M.; Hayden, M.R.; Duta, C.; et al. Glycemic control by the SGLT2 inhibitor empagliflozin decreases aortic stiffness, renal resistivity index and kidney injury. Cardiovasc. Diabetol. 2018, 17, 108. [Google Scholar] [CrossRef]
- Lee, D.M.; Battson, M.L.; Jarrell, D.K.; Hou, S.; Ecton, K.E.; Weir, T.L.; Gentile, C.L. SGLT2 inhibition via dapagliflozin improves generalized vascular dysfunction and alters the gut microbiota in type 2 diabetic mice. Cardiovasc. Diabetol. 2018, 17, 62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Shin, S.E.; Seo, M.S.; An, J.R.; Choi, I.-W.; Jung, W.-K.; Firth, A.L.; Lee, D.-S.; Yim, M.-J.; Choi, G.; et al. The anti-diabetic drug dapagliflozin induces vasodilation via activation of PKG and Kv channels. Life Sci. 2018, 197, 46–55. [Google Scholar] [CrossRef] [PubMed]
- Striepe, K.; Jumar, A.; Ott, C.; Karg, M.V.; Schneider, M.P.; Kannenkeril, D.; Schmieder, R.E. Effects of the Selective Sodium-Glucose Cotransporter 2 Inhibitor Empagliflozin on Vascular Function and Central Hemodynamics in Patients With Type 2 Diabetes Mellitus. Circulation 2017, 136, 1167–1169. [Google Scholar] [CrossRef] [PubMed]
- Weber, T.; Wassertheurer, S.; Rammer, M.; Haiden, A.; Hametner, B.; Eber, B. Wave Reflections, Assessed With a Novel Method for Pulse Wave Separation, Are Associated With End-Organ Damage and Clinical Outcomes. Hypertension 2012, 60, 534–541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Durante, W.; Behnammanesh, G.; Peyton, K.J. Effects of Sodium-Glucose Co-Transporter 2 Inhibitors on Vascular Cell Function and Arterial Remodeling. Int. J. Mol. Sci. 2021, 22, 8786. [Google Scholar] [CrossRef]
- Bray, J.J.; Foster-Davies, H.; Stephens, J.W. A systematic review examining the effects of sodium-glucose cotransporter-2 inhibitors (SGLT2is) on biomarkers of inflammation and oxidative stress. Diabetes Res. Clin. Pract. 2020, 168. [Google Scholar] [CrossRef]
- Nabrdalik-Leśniak, D.; Nabrdalik, K.; Sedlaczek, K.; Główczyński, P.; Kwiendacz, H.; Sawczyn, T.; Hajzler, W.; Drożdż, K.; Hendel, M.; Irlik, K.; et al. Influence of SGLT2 Inhibitor Treatment on Urine Antioxidant Status in Type 2 Diabetic Patients: A Pilot Study. Oxidative Med. Cell. Longev. 2021, 2021, 5593589. [Google Scholar] [CrossRef]
- Hotamisligil, G.S. Inflammation and metabolic disorders. Nature 2006, 444, 860–867. [Google Scholar] [CrossRef]
- Garvey, W.T.; Van Gaal, L.; Leiter, L.A.; Vijapurkar, U.; List, J.; Cuddihy, R.; Ren, J.; Davies, M.J. Effects of canagliflozin versus glimepiride on adipokines and inflammatory biomarkers in type 2 diabetes. Metabolism 2018, 85, 32–37. [Google Scholar] [CrossRef] [Green Version]
- Benetti, E.; Mastrocola, R.; Vitarelli, G.; Cutrin, J.C.; Nigro, D.; Chiazza, F.; Mayoux, E.; Collino, M.; Fantozzi, R. Empagliflozin Protects against Diet-Induced NLRP-3 Inflammasome Activation and Lipid Accumulation. J. Pharmacol. Exp. Ther. 2016, 359, 45–53. [Google Scholar] [CrossRef]
- Xu, L.; Nagata, N.; Chen, G.; Nagashimada, M.; Zhuge, F.; Ni, Y.; Sakai, Y.; Kaneko, S.; Ota, T. Empagliflozin reverses obesity and insulin resistance through fat browning and alternative macrophage activation in mice fed a high-fat diet. BMJ Open Diabetes Res. Care 2019, 7, e000783. [Google Scholar] [CrossRef] [PubMed]
- Kusaka, H.; Koibuchi, N.; Hasegawa, Y.; Ogawa, H.; Kim-Mitsuyama, S. Empagliflozin lessened cardiac injury and reduced visceral adipocyte hypertrophy in prediabetic rats with metabolic syndrome. Cardiovasc. Diabetol. 2016, 15, 157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patel, V.B.; Shah, S.; Verma, S.; Oudit, G.Y. Epicardial adipose tissue as a metabolic transducer: Role in heart failure and coronary artery disease. Heart Fail Rev. 2017, 22, 889–902. [Google Scholar] [CrossRef] [PubMed]
- Salvatore, T.; Galiero, R.; Caturano, A.; Vetrano, E.; Rinaldi, L.; Coviello, F.; Di Martino, A.; Albanese, G.; Colantuoni, S.; Medicamento, G.; et al. Dysregulated Epicardial Adipose Tissue as a Risk Factor and Potential Therapeutic Target of Heart Failure with Preserved Ejection Fraction in Diabetes. Biomolecules 2022, 12, 176. [Google Scholar] [CrossRef] [PubMed]
- Zibadi, S.; Cordova, F.; Slack, E.H.; Watson, R.R.; Larson, D.F. Leptin’s regulation of obesity-induced cardiac extracellular matrix remodeling. Cardiovasc. Toxicol. 2011, 11, 325–333. [Google Scholar] [CrossRef]
- Packer, M. Do sodium-glucose co-transporter-2 inhibitors prevent heart failure with a preserved ejection fraction by counterbalancing the effects of leptin? A novel hypothesis. Diabetes Obes. Metab. 2018, 20, 1361–1366. [Google Scholar] [CrossRef]
- Hao, Z.; Huang, X.; Shao, H.; Tian, F. Effects of dapagliflozin on serum uric acid levels in hospitalized type 2 diabetic patients with inadequate glycemic control: A randomized controlled trial. Ther. Clin. Risk Manag. 2018, ume14, 2407–2413. [Google Scholar] [CrossRef] [Green Version]
- Braga, T.T.; Forni, M.F.; Correa-Costa, M.; Ramos, R.N.; Barbuto, J.A.M.; Branco, P.; Castoldi, A.; Hiyane, M.I.; Davanso, M.R.; Latz, E.; et al. Soluble Uric Acid Activates the NLRP3 Inflammasome. Sci. Rep. 2017, 7, 39884. [Google Scholar] [CrossRef]
- Prattichizzo, F.; De Nigris, V.; Micheloni, S.; La Sala, L.; Ceriello, A. Increases in circulating levels of ketone bodies and cardiovascular protection with SGLT2 inhibitors: Is low-grade inflammation the neglected component? Diabetes Obes. Metab. 2018, 20, 2515–2522. [Google Scholar] [CrossRef]
- Yaribeygi, H.; Atkin, S.L.; Butler, A.E.; Sahebkar, A. Sodium–glucose cotransporter inhibitors and oxidative stress: An update. J. Cell. Physiol. 2018, 234, 3231–3237. [Google Scholar] [CrossRef]
- Salvatore, T.; Caturano, A.; Galiero, R.; Di Martino, A.; Albanese, G.; Vetrano, E.; Sardu, C.; Marfella, R.; Rinaldi, L.; Sasso, F.C. Cardiovascular Benefits from Gliflozins: Effects on Endothelial Function. Biomedicines 2021, 9, 1356. [Google Scholar] [CrossRef] [PubMed]
- Tahara, A.; Kurosaki, E.; Yokono, M.; Yamajuku, D.; Kihara, R.; Hayashizaki, Y.; Takasu, T.; Imamura, M.; Li, Q.; Tomiyama, H.; et al. Effects of SGLT2 selective inhibitor ipragliflozin on hyperglycemia, hyperlipidemia, hepatic steatosis, oxidative stress, inflammation, and obesity in type 2 diabetic mice. Eur. J. Pharmacol. 2013, 715, 246–255. [Google Scholar] [CrossRef] [PubMed]
- Tahara, A.; Kurosaki, E.; Yokono, M.; Yamajuku, D.; Kihara, R.; Hayashizaki, Y.; Takasu, T.; Imamura, M.; Li, Q.; Tomiyama, H.; et al. Effects of sodium-glucose cotransporter 2 selective inhibitor ipragliflozin on hyperglycaemia, oxidative stress, inflammation and liver injury in streptozotocin-induced type 1 diabetic rats. J. Pharm. Pharmacol. 2014, 66, 975–987. [Google Scholar] [CrossRef] [PubMed]
- Salim, H.M.; Fukuda, D.; Yagi, S.; Soeki, T.; Shimabukuro, M.; Sata, M. Glycemic Control with Ipragliflozin, a Novel Selective SGLT2 Inhibitor, Ameliorated Endothelial Dysfunction in Streptozotocin-Induced Diabetic Mouse. Front. Cardiovasc. Med. 2016, 3, 43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oelze, M.; Kröller-Schön, S.; Welschof, P.; Jansen, T.; Hausding, M.; Mikhed, Y.; Stamm, P.; Mader, M.; Zinßius, E.; Agdauletova, S.; et al. The Sodium-Glucose Co-Transporter 2 Inhibitor Empagliflozin Improves Diabetes-Induced Vascular Dysfunction in the Streptozotocin Diabetes Rat Model by Interfering with Oxidative Stress and Glucotoxicity. PLoS ONE 2014, 9, e112394. [Google Scholar] [CrossRef]
- Steven, S.; Oelze, M.; Hanf, A.; Kröller-Schön, S.; Kashani, F.; Roohani, S.; Welschof, P.; Kopp, M.; Gödtel-Armbrust, U.; Xia, N.; et al. The SGLT2 inhibitor empagliflozin improves the primary diabetic complications in ZDF rats. Redox Biol. 2017, 13, 370–385. [Google Scholar] [CrossRef]
- Shigiyama, F.; Kumashiro, N.; Miyagi, M.; Ikehara, K.; Kanda, E.; Uchino, H.; Hirose, T. Effectiveness of dapagliflozin on vascular endothelial function and glycemic control in patients with early-stage type 2 diabetes mellitus: DEFENCE study. Cardiovasc. Diabetol. 2017, 16, 84. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, A.; Shimabukuro, M.; Machii, N.; Teragawa, H.; Okada, Y.; Shima, K.R.; Takamura, T.; Taguchi, I.; Hisauchi, I.; Toyoda, S.; et al. Effect of Empagliflozin on Endothelial Function in Patients With Type 2 Diabetes and Cardiovascular Disease: Results from the Multicenter, Randomized, Placebo-Controlled, Double-Blind EMBLEM Trial. Diabetes Care 2019, 42, e159–e161. [Google Scholar] [CrossRef] [Green Version]
- Iannantuoni, F.; De Marañon, A.M.; Diaz-Morales, N.; Falcon, R.; Bañuls, C.; Abad-Jimenez, Z.; Victor, V.M.; Hernandez-Mijares, A.; Rovira-Llopis, S. The SGLT2 Inhibitor Empagliflozin Ameliorates the Inflammatory Profile in Type 2 Diabetic Patients and Promotes an Antioxidant Response in Leukocytes. J. Clin. Med. 2019, 8, 1814. [Google Scholar] [CrossRef] [Green Version]
- Lee, N.; Heo, Y.J.; Choi, S.-E.; Jeon, J.Y.; Han, S.J.; Kim, D.J.; Kang, Y.; Lee, K.W.; Kim, H.J. Anti-inflammatory Effects of Empagliflozin and Gemigliptin on LPS-Stimulated Macrophage via the IKK/NF-κB, MKK7/JNK, and JAK2/STAT1 Signalling Pathways. J. Immunol. Res. 2021, 2021, 9944880. [Google Scholar] [CrossRef]
- Heymans, S.; Hirsch, E.; Anker, S.D.; Aukrust, P.; Balligand, J.-L.; Cohen-Tervaert, J.W.; Drexler, H.; Filippatos, G.; Felix, S.B.; Gullestad, L.; et al. Inflammation as a therapeutic target in heart failure? A scientific statement from the Translational Research Committee of the Heart Failure Association of the European Society of Cardiology. Eur. J. Heart Fail. 2009, 11, 119–129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, Y.; Bajaj, M.; Yang, H.-C.; Perez-Polo, J.R.; Birnbaum, Y. SGLT-2 Inhibition with Dapagliflozin Reduces the Activation of the Nlrp3/ASC Inflammasome and Attenuates the Development of Diabetic Cardiomyopathy in Mice with Type 2 Diabetes. Further Augmentation of the Effects with Saxagliptin, a DPP4 Inhibitor. Cardiovasc. Drugs Ther. 2017, 31, 119–132. [Google Scholar] [CrossRef] [PubMed]
- Byrne, N.J.; Matsumura, N.; Maayah, Z.H.; Ferdaoussi, M.; Takahara, S.; Darwesh, A.M.; Levasseur, J.L.; Jahng, J.W.S.; Vos, D.; Parajuli, N.; et al. Empagliflozin Blunts Worsening Cardiac Dysfunction Associated With Reduced NLRP3 (Nucleotide-Binding Domain-Like Receptor Protein 3) Inflammasome Activation in Heart Failure. Circ. Heart Fail. 2020, 13, e006277. [Google Scholar] [CrossRef] [PubMed]
- Koyani, C.N.; Plastira, I.; Sourij, H.; Hallström, S.; Schmidt, A.; Rainer, P.P.; Bugger, H.; Frank, S.; Malle, E.; von Lewinski, D. Empagliflozin protects heart from inflammation and energy depletion via AMPK activation. Pharmacol. Res. 2020, 158, 104870. [Google Scholar] [CrossRef]
- Chen, H.; Tran, D.; Yang, H.-C.; Nylander, S.; Birnbaum, Y.; Ye, Y. Dapagliflozin and Ticagrelor Have Additive Effects on the Attenuation of the Activation of the NLRP3 Inflammasome and the Progression of Diabetic Cardiomyopathy: An AMPK–mTOR Interplay. Cardiovasc. Drugs Ther. 2020, 34, 443–461. [Google Scholar] [CrossRef]
- Kohlhaas, M.; Liu, T.; Knopp, A.; Zeller, T.; Ong, M.F.; Böhm, M.; O’Rourke, B.; Maack, C. Elevated Cytosolic Na+ Increases Mitochondrial Formation of Reactive Oxygen Species in Failing Cardiac Myocytes. Circulation 2010, 121, 1606–1613. [Google Scholar] [CrossRef] [Green Version]
- Li, C.; Zhang, J.; Xue, M.; Li, X.; Han, F.; Liu, X.; Xu, L.; Lu, Y.; Cheng, Y.; Li, T.; et al. SGLT2 inhibition with empagliflozin attenuates myocardial oxidative stress and fibrosis in diabetic mice heart. Cardiovasc. Diabetol. 2019, 18, 15. [Google Scholar] [CrossRef]
- Hasan, R.; Lasker, S.; Hasan, A.; Zerin, F.; Zamila, M.; Chowdhury, F.I.; Nayan, S.I.; Rahman, M.; Khan, F.; Subhan, N.; et al. Canagliflozin attenuates isoprenaline-induced cardiac oxidative stress by stimulating multiple antioxidant and anti-inflammatory signaling pathways. Sci. Rep. 2020, 10, 14459. [Google Scholar] [CrossRef]
- Lee, T.-M.; Chang, N.-C.; Lin, S.-Z. Dapagliflozin, a selective SGLT2 Inhibitor, attenuated cardiac fibrosis by regulating the macrophage polarization via STAT3 signaling in infarcted rat hearts. Free Radic. Biol. Med. 2017, 104, 298–310. [Google Scholar] [CrossRef]
- Lee, H.-C.; Shiou, Y.-L.; Jhuo, S.-J.; Chang, C.-Y.; Liu, P.-L.; Jhuang, W.-J.; Dai, Z.-K.; Chen, W.-Y.; Chen, Y.-F.; Lee, A.-S. The sodium–glucose co-transporter 2 inhibitor empagliflozin attenuates cardiac fibrosis and improves ventricular hemodynamics in hypertensive heart failure rats. Cardiovasc. Diabetol. 2019, 18, 45. [Google Scholar] [CrossRef]
- Kang, S.; Verma, S.; Hassanabad, A.F.; Teng, G.; Belke, D.D.; Dundas, J.A.; Guzzardi, D.G.; Svystonyuk, D.A.; Pattar, S.S.; Park, D.S.; et al. Direct Effects of Empagliflozin on Extracellular Matrix Remodelling in Human Cardiac Myofibroblasts: Novel Translational Clues to Explain EMPA-REG OUTCOME Results. Can. J. Cardiol. 2019, 36, 543–553. [Google Scholar] [CrossRef] [Green Version]
- Sun, X.; Han, F.; Lu, Q.; Li, X.; Ren, D.; Zhang, J.; Han, Y.; Xiang, Y.K.; Li, J. Empagliflozin Ameliorates Obesity-Related Cardiac Dysfunction by Regulating Sestrin2-Mediated AMPK-mTOR Signaling and Redox Homeostasis in High-Fat Diet–Induced Obese Mice. Diabetes 2020, 69, 1292–1305. [Google Scholar] [CrossRef] [PubMed]
- Franssen, C.; Chen, S.; Unger, A.; Korkmaz, H.I.; De Keulenaer, G.W.; Tschöpe, C.; Leite-Moreira, A.F.; Musters, R.; Niessen, H.W.; Linke, W.A.; et al. Myocardial Microvascular Inflammatory Endothelial Activation in Heart Failure With Preserved Ejection Fraction. JACC Heart Fail. 2016, 4, 312–324. [Google Scholar] [CrossRef] [PubMed]
- Juni, R.P.; Kuster, D.W.; Goebel, M.; Helmes, M.; Musters, R.J.; van der Velden, J.; Koolwijk, P.; Paulus, W.J.; van Hinsbergh, V.W. Cardiac Microvascular Endothelial Enhancement of Cardiomyocyte Function Is Impaired by Inflammation and Restored by Empagliflozin. JACC Basic Transl. Sci. 2019, 4, 575–591. [Google Scholar] [CrossRef] [PubMed]
- Hamdani, N.; Bishu, K.G.; Von Frieling-Salewsky, M.; Redfield, M.M.; Linke, W.A. Deranged myofilament phosphorylation and function in experimental heart failure with preserved ejection fraction. Cardiovasc. Res. 2012, 97, 464–471. [Google Scholar] [CrossRef] [Green Version]
- Pabel, S.; Wagner, S.; Bollenberg, H.; Bengel, P.; Kovács, A.; Schach, C.; Tirilomis, P.; Mustroph, J.; Renner, A.; Gummert, J.; et al. Empagliflozin directly improves diastolic function in human heart failure. Eur. J. Heart Fail. 2018, 20, 1690–1700. [Google Scholar] [CrossRef]
- Xue, M.; Li, T.; Wang, Y.; Chang, Y.; Cheng, Y.; Lu, Y.; Liu, X.; Xu, L.; Li, X.; Yu, X.; et al. Empagliflozin prevents cardiomyopathy via sGC-cGMP-PKG pathway in type 2 diabetes mice. Clin. Sci. 2019, 133, 1705–1720. [Google Scholar] [CrossRef] [Green Version]
- Kolijn, D.; Pabel, S.; Tian, Y.; Lódi, M.; Herwig, M.; Carrizzo, A.; Zhazykbayeva, S.; Kovács, Á.; Fülöp, G..; Falcão-Pires, I.; et al. Empagliflozin improves endothelial and cardiomyocyte function in human heart failure with preserved ejection fraction via reduced pro-inflammatory-oxidative pathways and protein kinase Gα oxidation. Cardiovasc. Res. 2020, 117, 495–507. [Google Scholar] [CrossRef]
- Yaribeygi, H.; Butler, A.E.; Atkin, S.L.; Katsiki, N.; Sahebkar, A. Sodium–glucose cotransporter 2 inhibitors and inflammation in chronic kidney disease: Possible molecular pathways. J. Cell. Physiol. 2018, 234, 223–230. [Google Scholar] [CrossRef] [Green Version]
- Ishibashi, Y.; Matsui, T.; Yamagishi, S. Tofogliflozin, A Highly Selective Inhibitor of SGLT2 Blocks Proinflammatory and Proapoptotic Effects of Glucose Overload on Proximal Tubular Cells Partly by Suppressing Oxidative Stress Generation. Horm. Metab. Res. 2015, 48, 191–195. [Google Scholar] [CrossRef]
- Xu, J.; Kitada, M.; Ogura, Y.; Liu, H.; Koya, D. Dapagliflozin Restores Impaired Autophagy and Suppresses Inflammation in High Glucose-Treated HK-2 Cells. Cells 2021, 10, 1457. [Google Scholar] [CrossRef] [PubMed]
- Das, N.A.; Carpenter, A.J.; Belenchia, A.; Aroor, A.R.; Noda, M.; Siebenlist, U.; Chandrasekar, B.; DeMarco, V.G. Empagliflozin reduces high glucose-induced oxidative stress and miR-21-dependent TRAF3IP2 induction and RECK suppression, and inhibits human renal proximal tubular epithelial cell migration and epithelial-to-mesenchymal transition. Cell Signal. 2019, 68, 109506. [Google Scholar] [CrossRef] [PubMed]
- Satou, R.; Cypress, M.W.; Woods, T.C.; Katsurada, A.; Dugas, C.M.; Fonseca, V.A.; Navar, L.G. Blockade of sodium-glucose cotransporter 2 suppresses high glucose-induced angiotensinogen augmentation in renal proximal tubular cells. Am. J. Physiol. Physiol. 2020, 318, F67–F75. [Google Scholar] [CrossRef] [PubMed]
- Birnbaum, Y.; Bajaj, M.; Yang, H.-C.; Ye, Y. Combined SGLT2 and DPP4 Inhibition Reduces the Activation of the Nlrp3/ASC Inflammasome and Attenuates the Development of Diabetic Nephropathy in Mice with Type 2 Diabetes. Cardiovasc. Drugs Ther. 2018, 32, 135–145. [Google Scholar] [CrossRef]
- Wu, R.; Liu, X.; Yin, J.; Wu, H.; Cai, X.; Wang, N.; Qian, Y.; Wang, F. IL-6 receptor blockade ameliorates diabetic nephropathy via inhibiting inflammasome in mice. Metabolism 2018, 83, 18–24. [Google Scholar] [CrossRef]
- Hasan, R.; Lasker, S.; Hasan, A.; Zerin, F.; Zamila, M.; Parvez, F.; Rahman, M.; Khan, F.; Subhan, N.; Alam, A. Canagliflozin ameliorates renal oxidative stress and inflammation by stimulating AMPK–Akt–eNOS pathway in the isoprenaline-induced oxidative stress model. Sci. Rep. 2020, 10, 14659. [Google Scholar] [CrossRef]
- Jaikumkao, K.; Pongchaidecha, A.; Chueakula, N.; Thongnak, L.; Wanchai, K.; Chatsudthipong, V.; Chattipakorn, N.; Lungkaphin, A. Dapagliflozin, a sodium-glucose co-transporter-2 inhibitor, slows the progression of renal complications through the suppression of renal inflammation, endoplasmic reticulum stress and apoptosis in prediabetic rats. Diabetes Obes. Metab. 2018, 20, 2617–2626. [Google Scholar] [CrossRef]
- Elkazzaz, S.K.; Khodeer, D.M.; El Fayoumi, H.M.; Moustafa, Y.M. Role of sodium glucose cotransporter type 2 inhibitors dapagliflozin on diabetic nephropathy in rats; Inflammation, angiogenesis and apoptosis. Life Sci. 2021, 280, 119018. [Google Scholar] [CrossRef]
- Shiraki, A.; Kotooka, N.; Komoda, H.; Hirase, T.; Oyama, J.-I.; Node, K. Pentraxin-3 regulates the inflammatory activity of macrophages. Biochem. Biophys. Rep. 2016, 5, 290–295. [Google Scholar] [CrossRef] [Green Version]
- Huang, F.; Zhao, Y.; Wang, Q.; Hillebrands, J.-L.; Born, J.V.D.; Ji, L.; An, T.; Qin, G. Dapagliflozin Attenuates Renal Tubulointerstitial Fibrosis Associated With Type 1 Diabetes by Regulating STAT1/TGFβ1 Signaling. Front. Endocrinol. 2019, 10, 441. [Google Scholar] [CrossRef] [Green Version]
- Heerspink, H.J.L.; Perco, P.; Mulder, S.; Leierer, J.; Hansen, M.K.; Heinzel, A.; Mayer, G. Canagliflozin reduces inflammation and fibrosis biomarkers: A potential mechanism of action for beneficial effects of SGLT2 inhibitors in diabetic kidney disease. Diabetologia 2019, 62, 1154–1166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mulder, S.; Hammarstedt, A.; Nagaraj, S.B.; Nair, V.; Ju, W.; Hedberg, J.; Greasley, P.J.; Eriksson, J.W.; Oscarsson, J.; Heerspink, H.J.L. A metabolomics-based molecular pathway analysis of how the sodium-glucose co-transporter-2 inhibitor dapagliflozin may slow kidney function decline in patients with diabetes. Diabetes Obes. Metab. 2020, 22, 1157–1166. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Liu, Y.; Dorn, G.W. Mitochondrial Fusion is Essential for Organelle Function and Cardiac Homeostasis. Circ. Res. 2011, 109, 1327–1331. [Google Scholar] [CrossRef] [PubMed]
- Ong, S.-B.; Subrayan, S.; Lim, S.Y.; Yellon, D.M.; Davidson, S.M.; Hausenloy, D.J. Inhibiting Mitochondrial Fission Protects the Heart Against Ischemia/Reperfusion Injury. Circulation 2010, 121, 2012–2022. [Google Scholar] [CrossRef] [Green Version]
- Onishi, M.; Yamano, K.; Sato, M.; Matsuda, N.; Okamoto, K. Molecular mechanisms and physiological functions of mitophagy. EMBO J. 2021, 40, e104705. [Google Scholar] [CrossRef]
- Durak, A.; Olgar, Y.; Degirmenci, S.; Akkus, E.; Tuncay, E.; Turan, B. A SGLT2 inhibitor dapagliflozin suppresses prolonged ventricular-repolarization through augmentation of mitochondrial function in insulin-resistant metabolic syndrome rats. Cardiovasc. Diabetol. 2018, 17, 144. [Google Scholar] [CrossRef]
- Zhou, H.; Wang, S.; Zhu, P.; Hu, S.; Chen, Y.; Ren, J. Empagliflozin rescues diabetic myocardial microvascular injury via AMPK-mediated inhibition of mitochondrial fission. Redox Biol. 2017, 15, 335–346. [Google Scholar] [CrossRef]
- Mizuno, M.; Kuno, A.; Yano, T.; Miki, T.; Oshima, H.; Sato, T.; Nakata, K.; Kimura, Y.; Tanno, M.; Miura, T. Empagliflozin normalizes the size and number of mitochondria and prevents reduction in mitochondrial size after myocardial infarction in diabetic hearts. Physiol. Rep. 2018, 6, e13741. [Google Scholar] [CrossRef]
- Sciarretta, S.; Maejima, Y.; Zablocki, D.; Sadoshima, J. The Role of Autophagy in the Heart. Annu. Rev. Physiol. 2018, 80, 1–26. [Google Scholar] [CrossRef]
- Packer, M. Autophagy stimulation and intracellular sodium reduction as mediators of the cardioprotective effect of sodium–glucose cotransporter 2 inhibitors. Eur. J. Heart Fail. 2020, 22, 618–628. [Google Scholar] [CrossRef] [Green Version]
- Xu, C.; Wang, W.; Zhong, J.; Lei, F.; Xu, N.; Zhang, Y.; Xie, W. Canagliflozin exerts anti-inflammatory effects by inhibiting intracellular glucose metabolism and promoting autophagy in immune cells. Biochem. Pharmacol. 2018, 152, 45–59. [Google Scholar] [CrossRef] [PubMed]
- Aragón-Herrera, A.; Feijóo-Bandín, S.; Santiago, M.O.; Barral, L.; Campos-Toimil, M.; Gil-Longo, J.; Pereira, T.M.C.; García-Caballero, T.; Rodríguez-Segade, S.; Rodríguez, J.; et al. Empagliflozin reduces the levels of CD36 and cardiotoxic lipids while improving autophagy in the hearts of Zucker diabetic fatty rats. Biochem. Pharmacol. 2019, 170, 113677. [Google Scholar] [CrossRef] [PubMed]
- Jiang, K.; Xu, Y.; Wang, D.; Chen, F.; Tu, Z.; Qian, J.; Xu, S.; Xu, Y.; Hwa, J.; Li, J.; et al. Cardioprotective mechanism of SGLT2 inhibitor against myocardial infarction is through reduction of autosis. Protein Cell 2021. online ahead of print. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.-Y.; Chen, C.-C.; Lin, M.-H.; Su, H.-T.; Ho, M.-Y.; Yeh, J.-K.; Tsai, M.-L.; Hsieh, I.-C.; Wen, M.-S. TLR9 Binding to Beclin 1 and Mitochondrial SIRT3 by a Sodium-Glucose Co-Transporter 2 Inhibitor Protects the Heart from Doxorubicin Toxicity. Biology 2020, 9, 369. [Google Scholar] [CrossRef]
- Zhan, M.; Brooks, C.; Liu, F.; Sun, L.; Dong, Z. Mitochondrial dynamics: Regulatory mechanisms and emerging role in renal pathophysiology. Kidney Int. 2013, 83, 568–581. [Google Scholar] [CrossRef] [Green Version]
- Takagi, S.; Li, J.; Takagaki, Y.; Kitada, M.; Nitta, K.; Takasu, T.; Kanasaki, K.; Koya, D. Ipragliflozin improves mitochondrial abnormalities in renal tubules induced by a high-fat diet. J. Diabetes Investig. 2018, 9, 1025–1032. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.H.; Kim, S.H.; Kang, J.M.; Heo, J.H.; Kim, D.-J.; Park, S.H.; Sung, M.; Kim, J.; Oh, J.; Yang, D.H.; et al. Empagliflozin attenuates diabetic tubulopathy by improving mitochondrial fragmentation and autophagy. Am. J. Physiol. Physiol. 2019, 317, F767–F780. [Google Scholar] [CrossRef]
- Tang, C.; Livingston, M.J.; Liu, Z.; Dong, Z. Autophagy in kidney homeostasis and disease. Nat. Rev. Nephrol. 2020, 16, 489–508. [Google Scholar] [CrossRef]
- Umino, H.; Hasegawa, K.; Minakuchi, H.; Muraoka, H.; Kawaguchi, T.; Kanda, T.; Tokuyama, H.; Wakino, S.; Itoh, H. High Basolateral Glucose Increases Sodium-Glucose Cotransporter 2 and Reduces Sirtuin-1 in Renal Tubules through Glucose Transporter-2 Detection. Sci. Rep. 2018, 8, 6791. [Google Scholar] [CrossRef] [Green Version]
- Packer, M. Role of Impaired Nutrient and Oxygen Deprivation Signaling and Deficient Autophagic Flux in Diabetic CKD Development: Implications for Understanding the Effects of Sodium-Glucose Cotransporter 2-Inhibitors. J. Am. Soc. Nephrol. 2020, 31, 907–919. [Google Scholar] [CrossRef]
- Korbut, A.I.; Taskaeva, I.S.; Bgatova, N.P.; Muraleva, N.A.; Orlov, N.B.; Dashkin, M.V.; Khotskina, A.; Zavyalov, E.L.; Konenkov, V.I.; Klein, T.; et al. SGLT2 Inhibitor Empagliflozin and DPP4 Inhibitor Linagliptin Reactivate Glomerular Autophagy in db/db Mice, a Model of Type 2 Diabetes. Int. J. Mol. Sci. 2020, 21, 2987. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sano, M.; Goto, S. Possible Mechanism of Hematocrit Elevation by Sodium Glucose Cotransporter 2 Inhibitors and Associated Beneficial Renal and Cardiovascular Effects. Circulation 2019, 139, 1985–1987. [Google Scholar] [CrossRef] [PubMed]
- Sano, M.; Takei, M.; Shiraishi, Y.; Suzuki, Y. Increased Hematocrit During Sodium-Glucose Cotransporter 2 Inhibitor Therapy Indicates Recovery of Tubulointerstitial Function in Diabetic Kidneys. J. Clin. Med. Res. 2016, 8, 844–847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thiele, K.; Rau, M.; Hartmann, N.K.; Möllmann, J.; Jankowski, J.; Böhm, M.; Keszei, A.P.; Marx, N.; Lehrke, M. Effects of empagliflozin on erythropoiesis in patients with type 2 diabetes: Data from a randomized, placebo-controlled study. Diabetes Obes. Metab. 2021, 23, 2814–2818. [Google Scholar] [CrossRef]
- Haase, V.H. Hypoxia-inducible factors in the kidney. Am. J. Physiol. Physiol. 2006, 291, F271–F281. [Google Scholar] [CrossRef]
- O’Neill, J.; Fasching, A.; Pihl, L.; Patinha, D.; Franzén, S.; Palm, F. Acute SGLT inhibition normalizes O2 tension in the renal cortex but causes hypoxia in the renal medulla in anaesthetized control and diabetic rats. Am. J. Physiol. Physiol. 2015, 309, F227–F234. [Google Scholar] [CrossRef] [Green Version]
- Takaori, K.; Nakamura, J.; Yamamoto, S.; Nakata, H.; Sato, Y.; Takase, M.; Nameta, M.; Yamamoto, T.; Economides, A.; Kohno, K.; et al. Severity and Frequency of Proximal Tubule Injury Determines Renal Prognosis. J. Am. Soc. Nephrol. 2015, 27, 2393–2406. [Google Scholar] [CrossRef] [Green Version]
- Symeonidis, A.; Kouraklis-Symeonidis, A.; Psiroyiannis, A.; Leotsinidis, M.; Kyriazopoulou, V.; Vassilakos, P.; Vagenakis, A.; Zoumbos, N. Inappropriately low erythropoietin response for the degree of anemia in patients with noninsulin-dependent diabetes mellitus. Ann. Hematol. 2005, 85, 79–85. [Google Scholar] [CrossRef]
- Ghanim, H.; Abuaysheh, S.; Hejna, J.; Green, K.; Batra, M.; Makdissi, A.; Chaudhuri, A.; Dandona, P. Dapagliflozin Suppresses Hepcidin And Increases Erythropoiesis. J. Clin. Endocrinol. Metab. 2020, 105, e1056–e1063. [Google Scholar] [CrossRef]
- Mazer, C.D.; Hare, G.M.; Connelly, P.W.; Gilbert, R.E.; Shehata, N.; Quan, A.; Teoh, H.; Leiter, L.A.; Zinman, B.; Jüni, P.; et al. Effect of Empagliflozin on Erythropoietin Levels, Iron Stores, and Red Blood Cell Morphology in Patients with Type 2 Diabetes Mellitus and Coronary Artery Disease. Circulation 2020, 141, 704–707. [Google Scholar] [CrossRef]
- Hare, G.M.T.; Zhang, Y.; Chin, K.; Thai, K.; Jacobs, E.; Cazorla-Bak, M.P.; Nghiem, L.; Wilson, D.F.; Vinogradov, S.A.; Connelly, K.A.; et al. Impact of sodium glucose linked cotransporter-2 inhibition on renal microvascular oxygen tension in a rodent model of diabetes mellitus. Physiol. Rep. 2021, 9, e14890. [Google Scholar] [CrossRef] [PubMed]
- Maruyama, T.; Takashima, H.; Oguma, H.; Nakamura, Y.; Ohno, M.; Utsunomiya, K.; Furukawa, T.; Tei, R.; Abe, M. Canagliflozin Improves Erythropoiesis in Diabetes Patients with Anemia of Chronic Kidney Disease. Diabetes Technol. Ther. 2019, 21, 713–720. [Google Scholar] [CrossRef] [PubMed]
- Oshima, M.; Neuen, B.L.; Jardine, M.J.; Bakris, G.; Edwards, R.; Levin, A.; Mahaffey, K.W.; Neal, B.; Pollock, C.; Rosenthal, N.; et al. Effects of canagliflozin on anaemia in patients with type 2 diabetes and chronic kidney disease: A post-hoc analysis from the CREDENCE trial. Lancet Diabetes Endocrinol. 2020, 8, 903–914. [Google Scholar] [CrossRef]
- Yanai, H.; Katsuyayama, H. A Possible Mechanism for Renoprotective Effect of Sodium-Glucose Cotransporter 2 Inhibitor: Elevation of Erythropoietin Production. J. Clin. Med. Res. 2017, 9, 178–179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verma, S.; Mazer, C.D.; Yan, A.T.; Mason, T.; Garg, V.; Teoh, H.; Zuo, F.; Quan, A.; Farkouh, M.E.; Fitchett, D.H.; et al. Effect of Empagliflozin on Left Ventricular Mass in Patients With Type 2 Diabetes Mellitus and Coronary Artery Disease. Circulation 2019, 140, 1693–1702. [Google Scholar] [CrossRef] [PubMed]
- Fitchett, D.; Inzucchi, S.E.; Zinman, B.; Wanner, C.; Schumacher, M.; Schmoor, C.; Ohneberg, K.; Ofstad, A.P.; Salsali, A.; George, J.T.; et al. Mediators of the improvement in heart failure outcomes with empagliflozin in the EMPA-REG OUTCOME trial. ESC Heart Fail. 2021, 8, 4517–4527. [Google Scholar] [CrossRef] [PubMed]




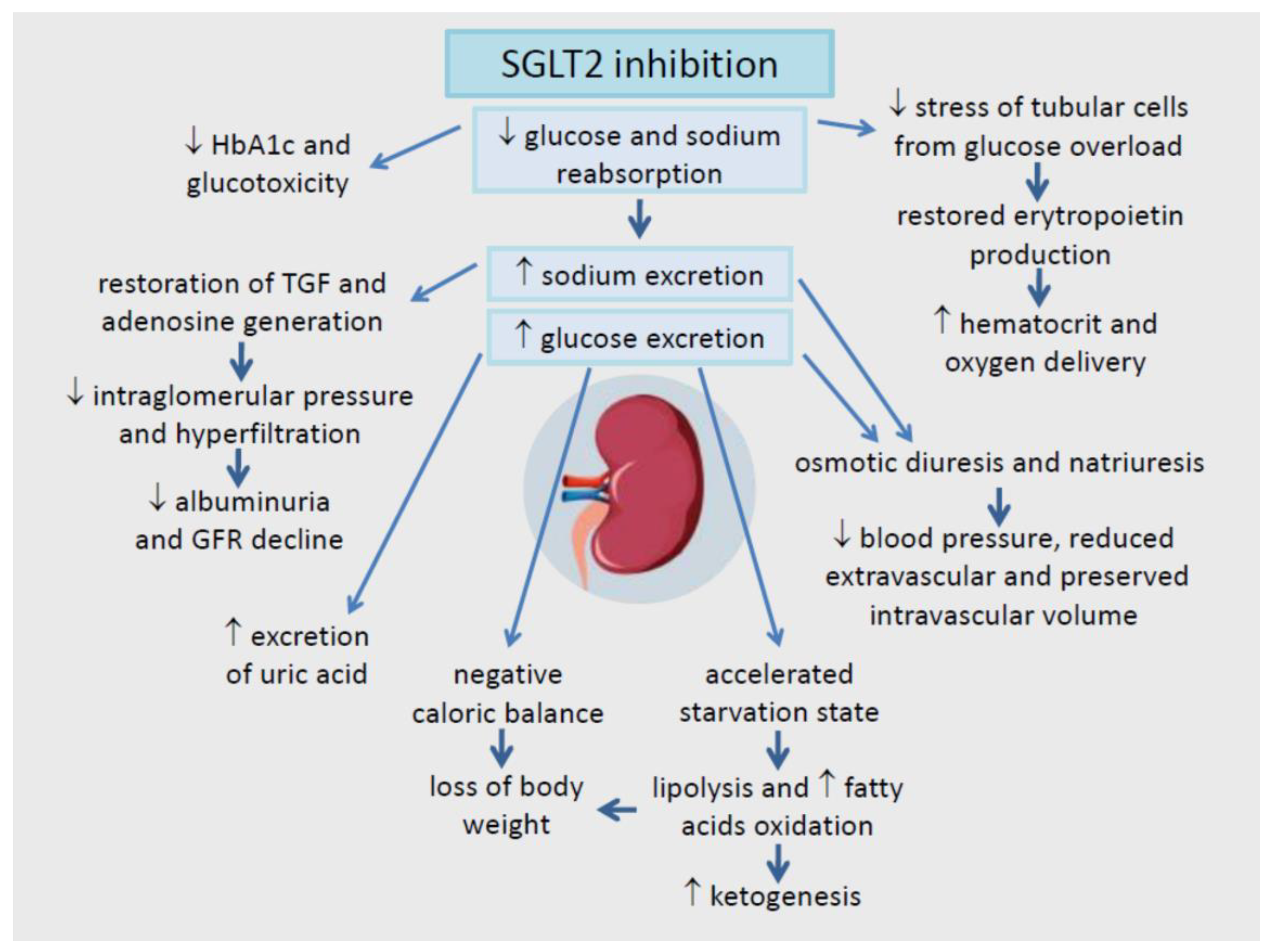{kind=link}
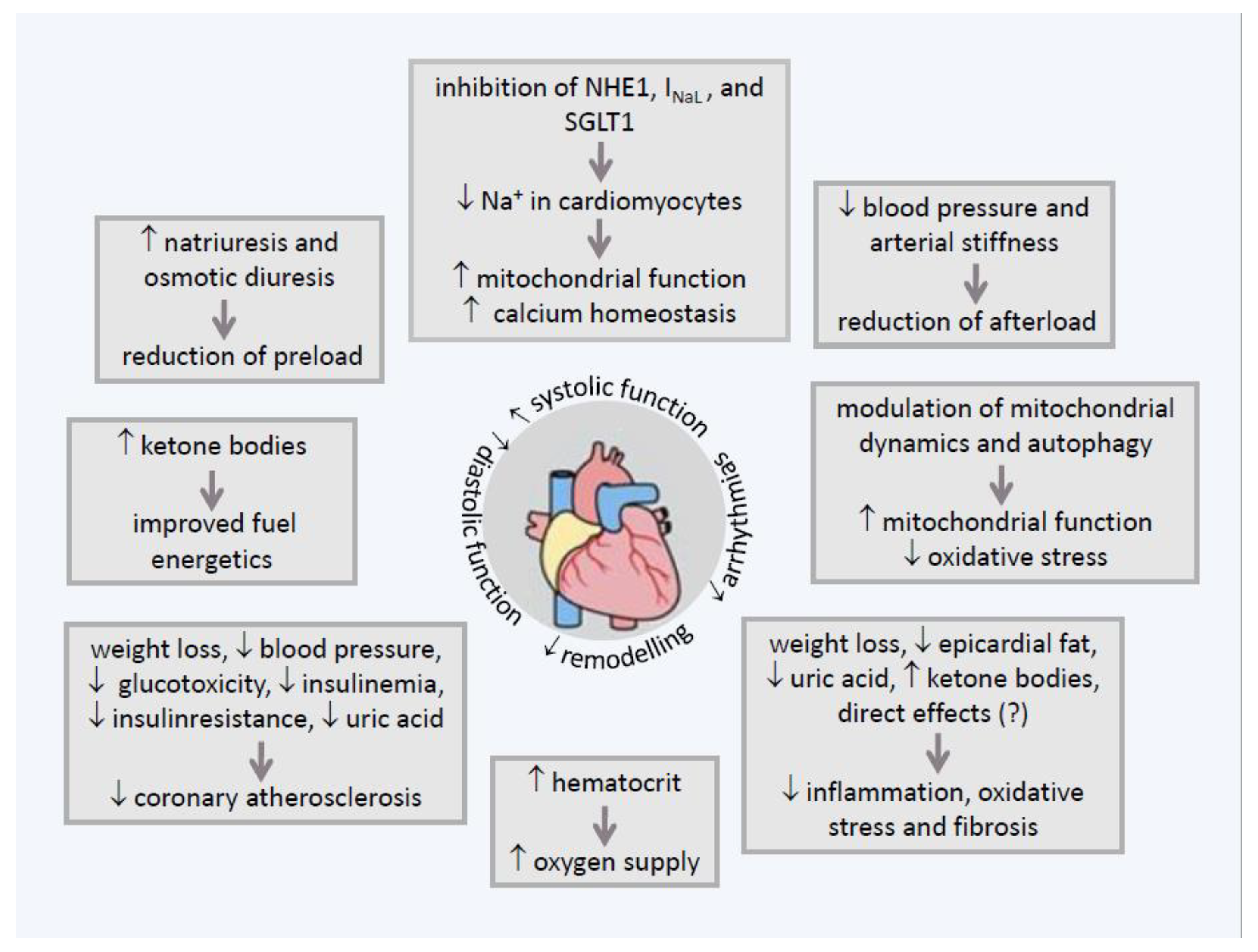{kind=link}
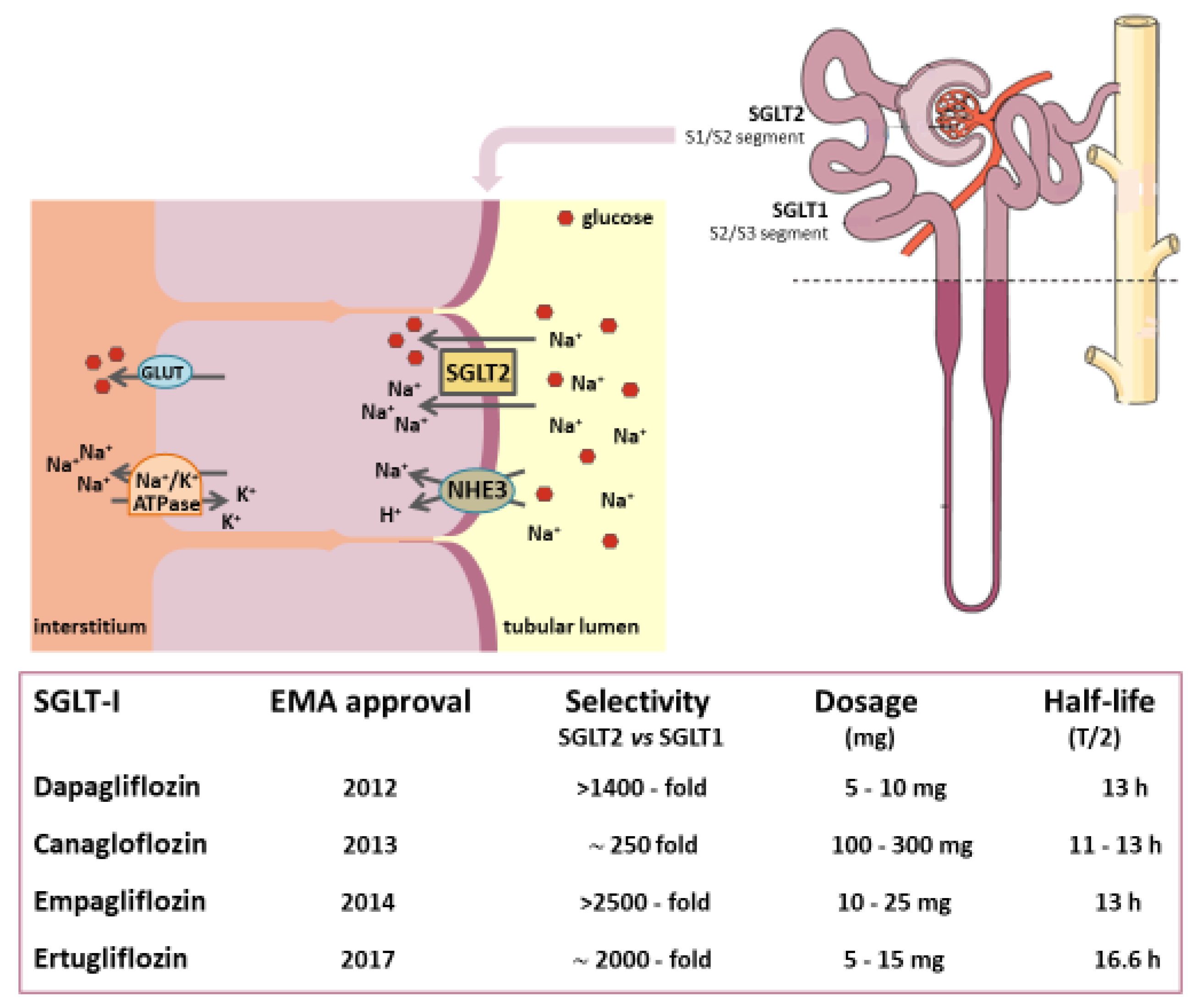{kind=link}
| Drug | Trial (Ref.) | Patients | Follow-Up (Median) | Outcomes | Hazard Ratio (95% CI) |
|---|---|---|---|---|---|
| Empagliflozin | EMPAREG OUTCOME study [47] | 7020 T2DM patients at high risk for CV events and an eGFR ≥ 30 mL/min/1.73 m2 | 3.1 years (2.6 years of treatment) | composite of death from CV causes, nonfatal myocardial infarction, or nonfatal stroke | 0.86 (0.74–0.99) |
| empagliflozin 10 mg vs. empagliflozin 25 mg vs. matching placebo | death from cardiovascular causes nonfatal myocardial infarction nonfatal stroke | 0.62 (0.49–0.77) 0.87 (0.70–1.09) 1.24 (0.92–1.56) | |||
| hospitalization for heart failure | 0.65 (0.50–0.85) | ||||
| death from any cause | 0.68 (0.57–0.82) | ||||
| EMPEROR-Reduced [101] | 3730 diabetic or not diabetic patients with class II, III, or IV HF and EF ≤ 40% | 16 months | cardiovascular death or hospitalization for worsening heart failure | 0.75 (0.65–0.86) | |
| empagliflozin 10 mg vs. placebo (in addition to recommended therapy) | hospitalization for heart failure | 0.70 (0.58–0.85) | |||
| EMPEROR-Preserved [114] | 26.2 months | cardiovascular death or hospitalization for worsening heart failure | 0.79 (0.69–0.90) | ||
| 5988 diabetic or not diabetic patients with class II-IV HF and EF > 40% | hospitalization for heart failure | 0.73 (0.61–0.88) | |||
| empagliflozin 10 mg vs. placebo (in addition to usual therapy) | |||||
| EMPERIAL [104] | 12 weeks | change in 6-minute walk test distance | ns | ||
| patients with HFrE (EF ≤ 40%, n = 312) or with HFpEF (EF > 40%, n = 315) | KCCQ-TSS (Kansas City Cardiomyopathy Questionnaire Total Symptom Score) | ns | |||
| empagliflozin 10 mg vs. placebo | CHQ-SAS (Chronic Heart Failure Questionnaire Self-Administered Standardized format) dyspnoea score | ns | |||
| Canagliflozin | CANVAS study [46] | 10,142 participants with T2DM and high CV risk 100 mg (with an optional increase to 300 mg) vs. placebo | 188.2 weeks | composite of death from CV causes, nonfatal myocardial infarction, or nonfatal stroke | 0.86 (0.75–0.97) |
| Dapagliflozin | DECLARE-TIMI 58 [48] | 17,160 T2DM patients at high risk for CV events (only 7% of patients had an eGFR < 60 mL/min/1.73 m2) | 4.2 years | composite of CV death, myocardial infarction, or ischemic stroke | 0.93 (0.84–1.03) |
| dapagliflozin 10 mg vs. placebo | CV death or hospitalization for HF | 0.83 (0.73–0.95) | |||
| hospitalization for HF | 0.73 (0.61–0.88) | ||||
| DAPA-HF Trial [100] | 4304 diabetic (68%) or not diabetic patients with class II-IV HF | 18.2 months | worsening HF (hospitalization or urgent visit resulting in IV therapy for HF) or CV death | 0.74 (0.65–0.85) | |
| dapagliflozin 10 mg vs. placebo | first worsening HF event | 0.70 (0.59–0.83) | |||
| (in addition to recommended therapy) | CV death | 0.83 (0.71–0.97) | |||
| DEFINE HF [103] | 263 diabetic or not diabetic patient with class II-III HF | 12 weeks | mean NT-proBNP | ns | |
| dapagliflozin 10 mg vs. placebo | % of patients with ameliorated functional status | 1.8 (1.03–3.06) | |||
| Sotagliflozin | SCORED [93] | 10,584 T2DM patients with CKD and CV risk sotagliflozin 200–400 mg vs. placebo | 16 months | composite of CV death, hospitalization fo HF, and urgent visit for HF | 0.74 (0.63–0.88) |
| SOLOIST-WHF [102] | 1222 T2DM patients recently hospitalized for worsening HF sotagliflozin 200–400 mg vs. placebo | 9 months | CV deaths and hospitalization or urgent visits for HF | 0.67 (0.52–0.85) | |
| CV death | 0.84 (0.58–1.22) | ||||
| death from any cause | 0.82 (0.59–1.14) | ||||
| Ertugliflozin | VERTIS CV study [92] | 8246 T2DM patients with established CV disease and an eGFR ≥ 30 mL/min/ 1.73 m2 | 3 years | composite of CV death, myocardial infarction, or ischemic stroke | 0.97 (0.85–1.11) |
| ertugliflozin 5 or 15 mg vs. placebo | death from CV causes or hospitalization for HF | 0.88 (0.75–1.03) | |||
| death from CV causes | 0.92 (0.77–1.11) |
| Drug | Trial (Ref.) | Patients | Follow-Up (Median) | Outcomes | Hazard Ratio (95% CI) |
|---|---|---|---|---|---|
| Empagliflozin | EMPAREG OUTCOME study [47] | 7020 T2DM patients with high risk for CV events and eGFR ≥ 30 mL/min/1.73 m2 | 3.1 years (2.6 years of treatment) | incident or worsening nephropathy | 0.61 (0.53–0.70) |
| progression to macroalbuminuria | 0.65 (0.54–0.72) | ||||
| empagliflozin 10 mg vs. | doubling of the serum creatinine level | 0.56 (0.39–0.79) | |||
| empagliflozin 25 mg vs. placebo | initiation of renal-replacement therapy | 0.45 (0.21–0.97) | |||
| post hoc composite of doubling of serum creatinine, renal replacement therapy, or death for renal causes | 0.54 (0.40–0.75) | ||||
| incident albuminuria | 0.95 (0.87–1.04) | ||||
| Canagliflozin | CANVAS–R study [46] | 5812 T2DM patients with high risk for CV events and eGFR > 30 mL/min/1.73 m2 | 126.1 weeks | lower progression of albuminuria | 0.73 (0.67–0.79) |
| canagliflozin 100 or 300 mg vs. placebo | composite of 40% reduction in eGFR, renal replacement therapy, or death from renal causes | 0.60 (0.47–0.77) | |||
| CREDENCE study [105] | 4401 T2DM patients with albuminuric CKD (eGFR of 30 to < 90 mL/min/1.73 m2) | 2.62 years | composite of ESRD (dialysis, transplantation, or sustained eGFR < 15 mL/min/1.73 m2), doubling of serum creatinine, or death from renal or CV causes | 0.70 (0.59–0.82) | |
| canagliflozin 100 mg vs. placebo | composite of ESRD, a doubling of the creatinine level, or death from renal causes | 0.66 (0.53–0.81) | |||
| composite of cardiovascular death, myocardial infarction, or stroke | 0.80 (0.67–0.95) | ||||
| hospitalization for heart failure | 0.61 (0.47–0.80) | ||||
| Dapagliflozin | DECLARE-TIMI 58 [48] | 7160 T2DM patients at high risk for CV events (only 7% with eGFR < 60 mL/min/1.73 m2) dapagliflozin 10 mg vs. placebo | 4.2 years | composite of ≥40% reduction in eGFR, new ESRD, or death from renal or CV causes | 0.76 (0.67–0.87) |
| DAPA-CKD study [106] | 4304 diabetic (68%) or not diabetic patients suffering from CKD (UACR 200–5000 mg/g and eGFR 25–75 mL/min/1.73 m2) | 2.4 years | composite of ≥50% sustained decline in eGFR or ESRD or CV or renal death | 0.56 (0.45–0.69) | |
| dapagliflozin 10 mg vs. placebo | composite of CV death and hospitalization for heart failure | 0.71 (0.55–0.92) | |||
| DELIGHT study [107] | 461 T2DM patients with albuminuria (UACR 30–3500 mg/g) and eGFR of 25–75 mL/min/1.73 m2, treated with ACE-Is or ARBs | 24 weeks | variation of albumin-to-creatinine ratio | −21.0% for dapagliflozin (p = 0.011) | |
| dapagliflozin 10 mg vs. dapagliflozin 10 mg–saxagliptin 2.5 mg vs. placebo | −38.0% for dapagliflozin + saxagliptin (<0.0001) | ||||
| DERIVE study [108] | 321 T2DM patients with CKD in stage 3A (eGFR of 45–59 mL/min/1.73 m2) dapagliflozin 10 mg vs. placebo | 24 weeks | change from baseline in urine eGFR | decrease at week 4 with a trend to recovery at weeks 12 and 24 eGFR similar to placebo after a 3 week period without treatment | |
| DIAMOND study [109] | 53 non-diabetic patients with CKD (24-h urinary protein excretion > 500 mg and ≤3500 mg, eGFR ≥ 25 mL/min/1.73 m2) on stable RAS blockade 27 received dapagliflozin 10 mg then placebo 26 received placebo then dapagliflozin 10 mg | cross-over trial (6 weeks for each treatment and washout period) | mean proteinuria measured GFR | no significant change from baseline change with dapagliflozin at week 6 by −6.6 mL/min/1.73 m2 (−9.0 to −4.2; p < 0.0001) (fully reversible within 6 weeks after dapagliflozin discontinuation) | |
| Ertugliflozin | VERTIS CV study [92] | 8246 T2DM patients with established CV disease and eGFR ≥ 30 mL/min/1.73 m2 ertugliflozin 5 or 15 mg vs. placebo | 3 years | composite of death from renal causes, renal replacement therapy, or doubling of serum creatinine | 0.81 (0.63–1.04) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Salvatore, T.; Galiero, R.; Caturano, A.; Rinaldi, L.; Di Martino, A.; Albanese, G.; Di Salvo, J.; Epifani, R.; Marfella, R.; Docimo, G.; et al. An Overview of the Cardiorenal Protective Mechanisms of SGLT2 Inhibitors. Int. J. Mol. Sci. 2022, 23, 3651. https://doi.org/10.3390/ijms23073651
Salvatore T, Galiero R, Caturano A, Rinaldi L, Di Martino A, Albanese G, Di Salvo J, Epifani R, Marfella R, Docimo G, et al. An Overview of the Cardiorenal Protective Mechanisms of SGLT2 Inhibitors. International Journal of Molecular Sciences. 2022; 23(7):3651. https://doi.org/10.3390/ijms23073651
Chicago/Turabian StyleSalvatore, Teresa, Raffaele Galiero, Alfredo Caturano, Luca Rinaldi, Anna Di Martino, Gaetana Albanese, Jessica Di Salvo, Raffaella Epifani, Raffaele Marfella, Giovanni Docimo, and et al. 2022. "An Overview of the Cardiorenal Protective Mechanisms of SGLT2 Inhibitors" International Journal of Molecular Sciences 23, no. 7: 3651. https://doi.org/10.3390/ijms23073651