
Pyruvate Dehydrogenase Kinase Inhibition by Dichloroacetate in Melanoma Cells Unveils Metabolic Vulnerabilities

 and
and
Abstract
:1. Introduction
2. Results
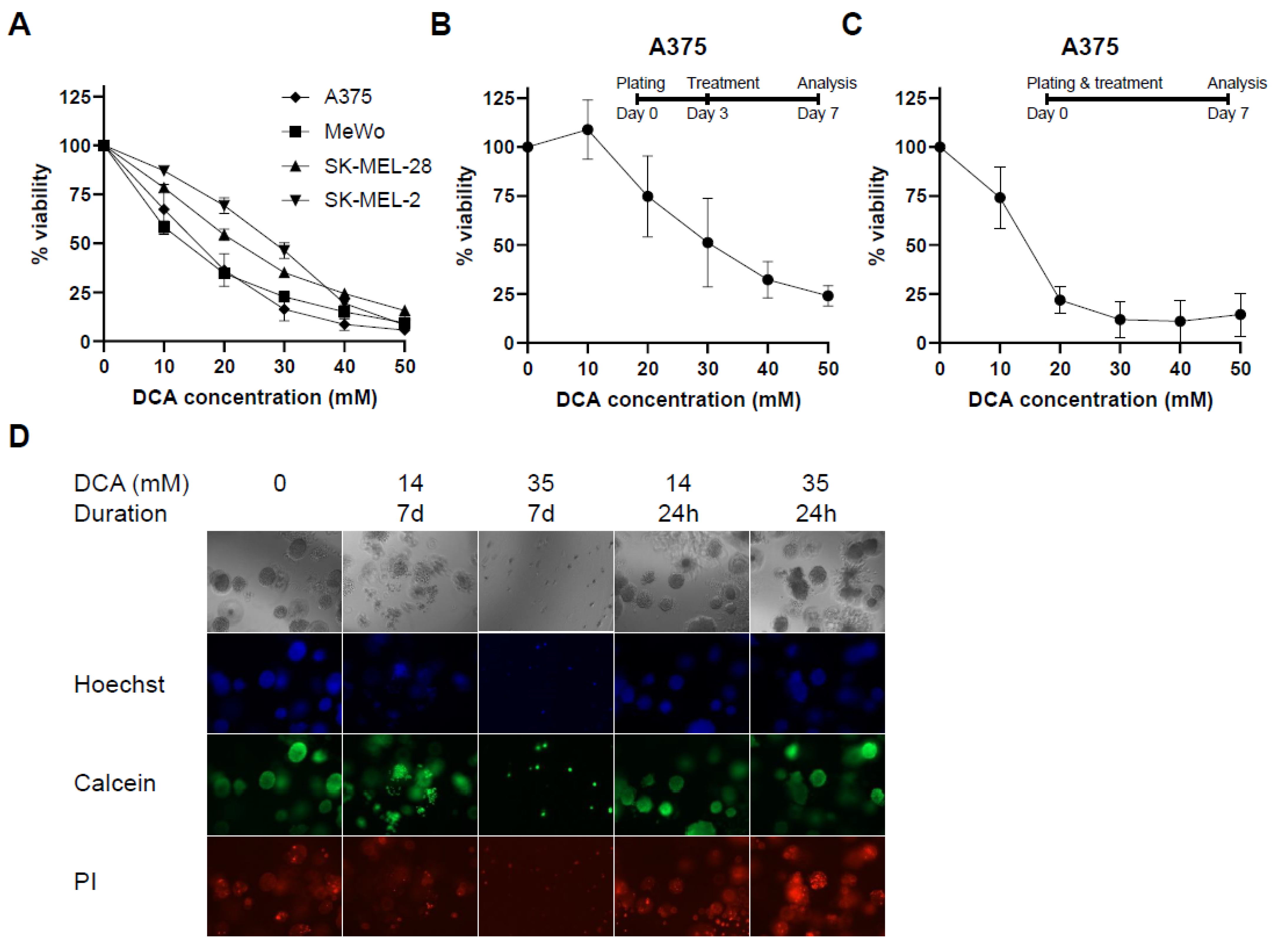
2.1. PDK Inhibition Inhibits Proliferation in 2D and 3D Culture
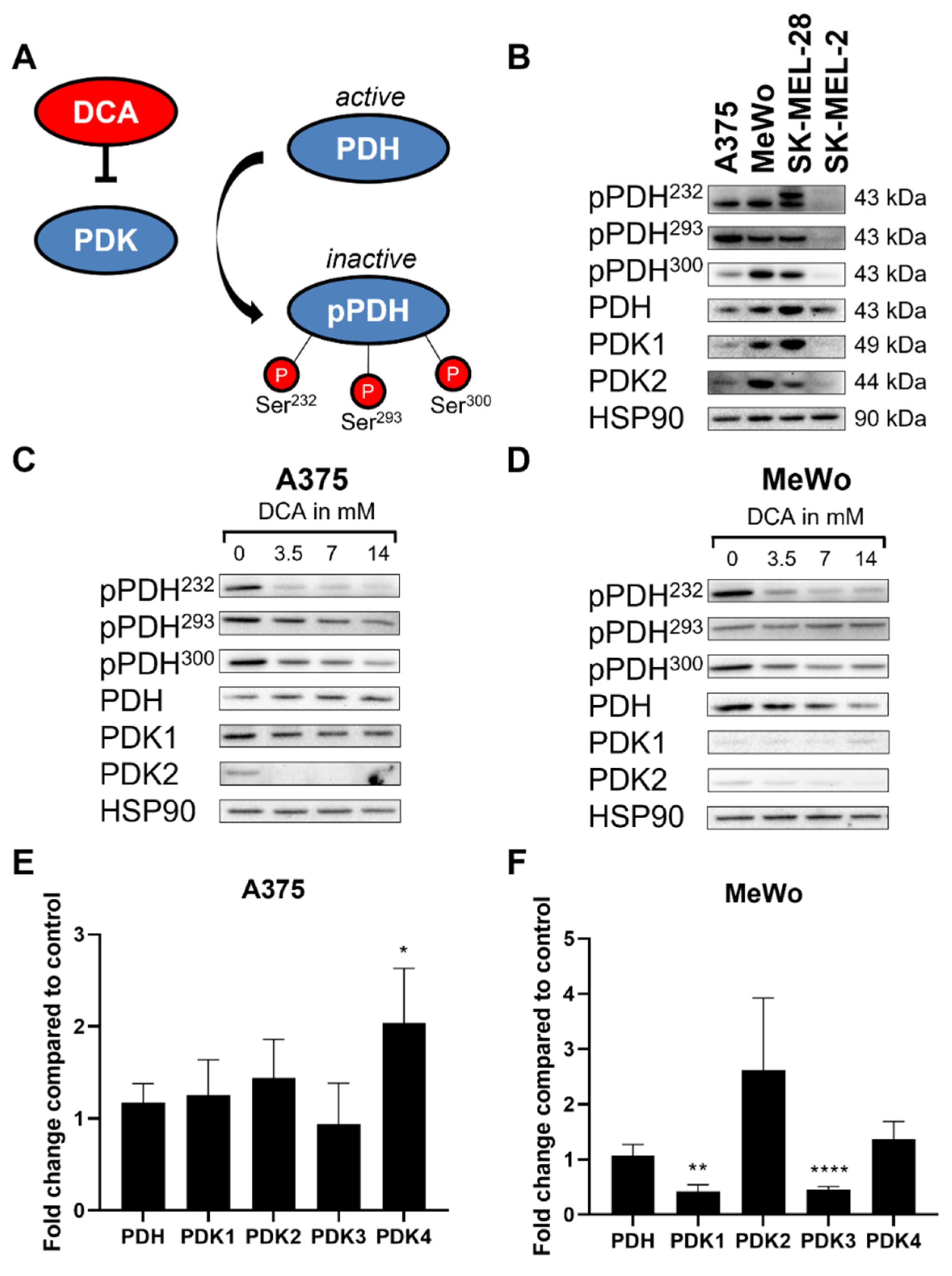
2.2. DCA Decreases PDH Phosphorylation in Melanoma Cell Lines
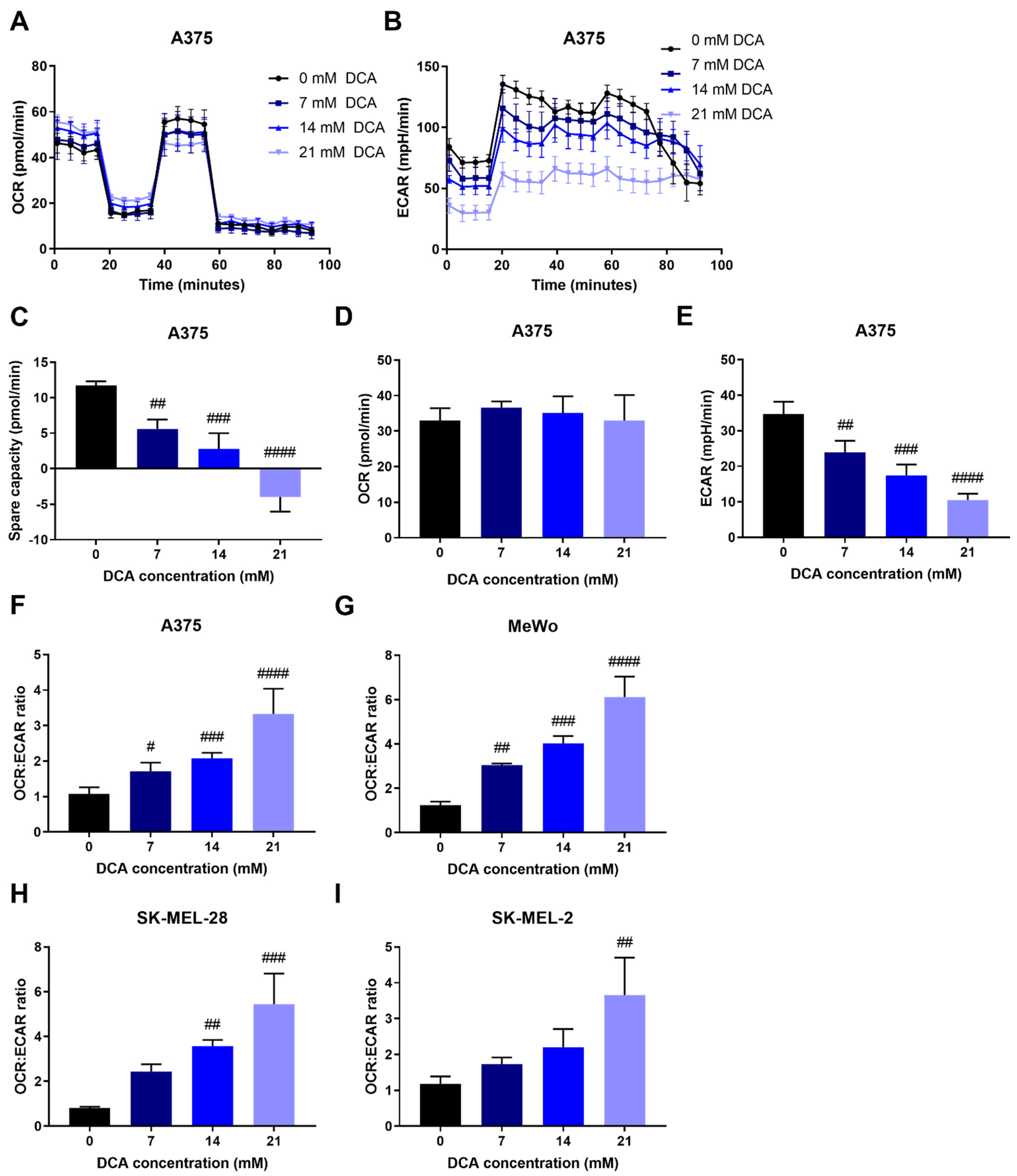
2.3. DCA Decreases Glycolysis in Melanoma Cells
2.4. Effect of AZD7545 Treatment on Viability, Protein Levels, RNA Levels and OCR:ECAR Ratio
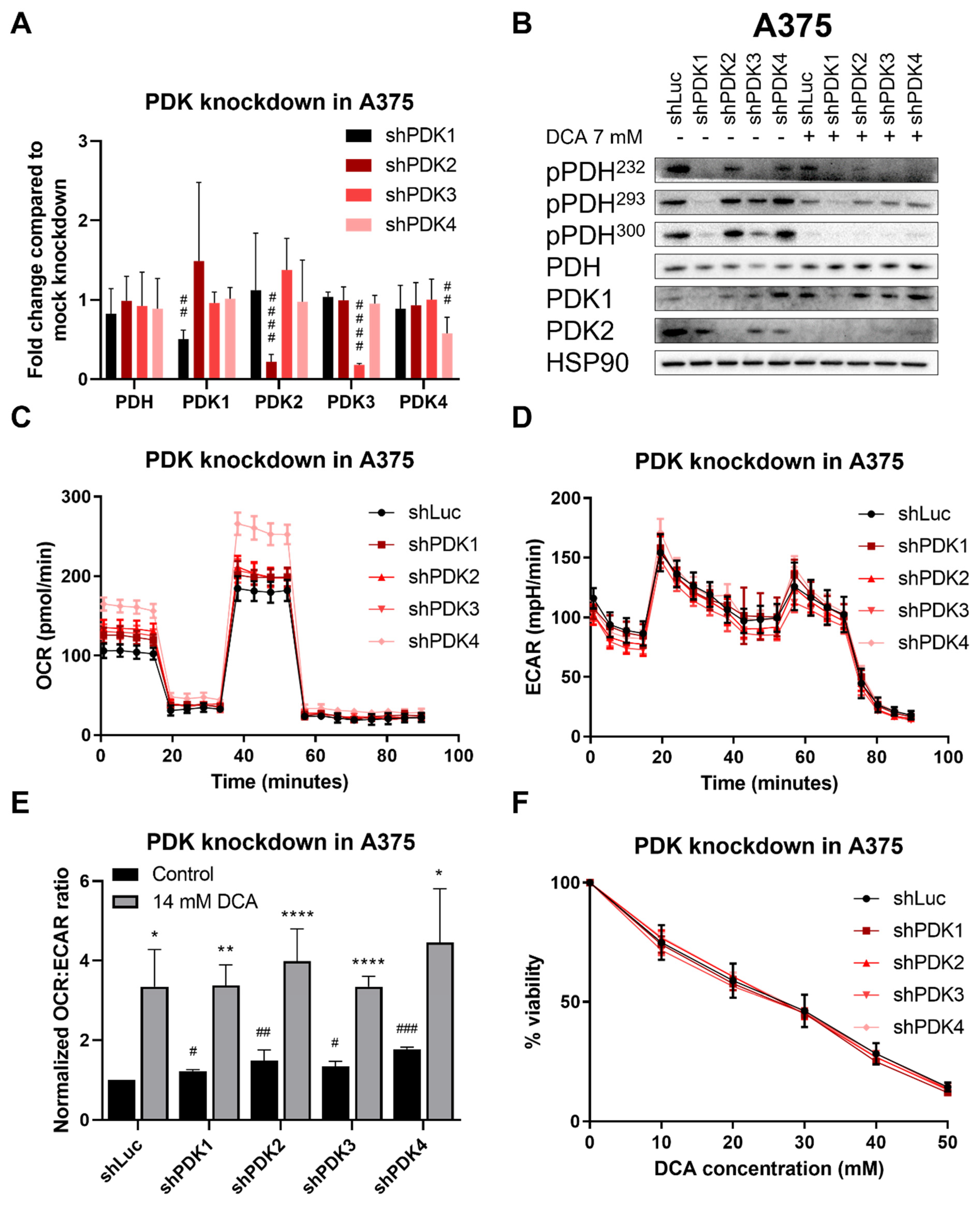
2.5. Effect of PDK Knockdown Is Similar to That of DCA on PDH Phosphorylation and OCR:ECAR Ratio
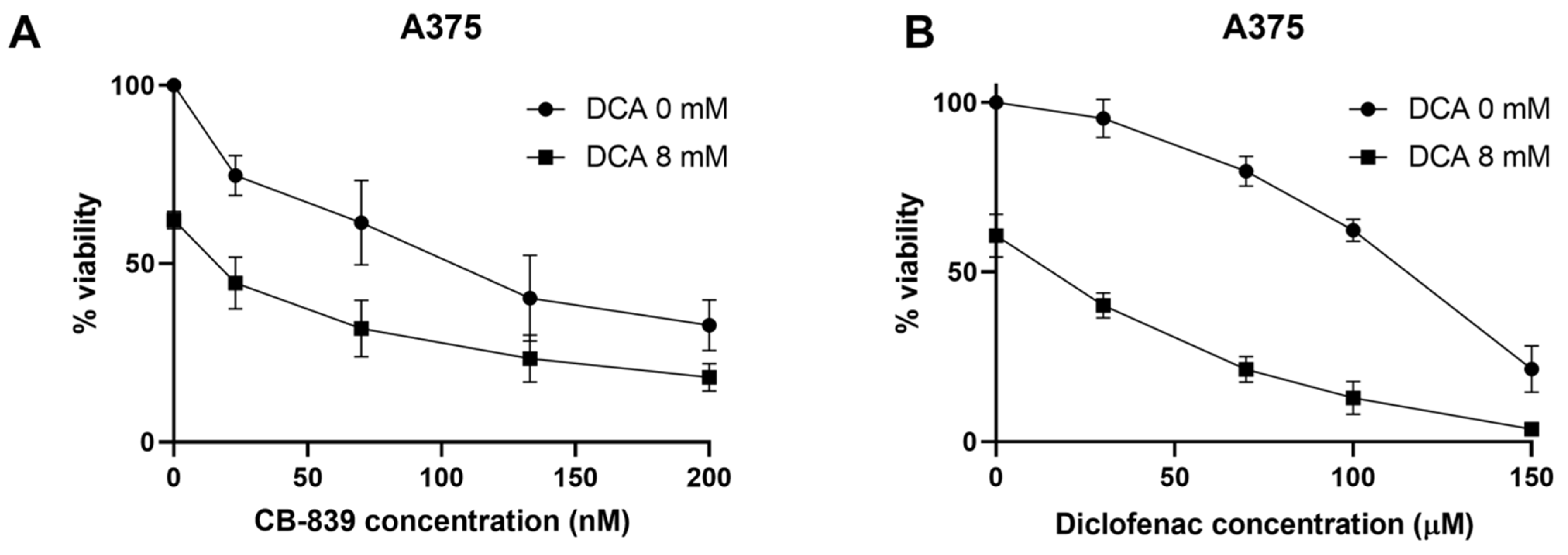
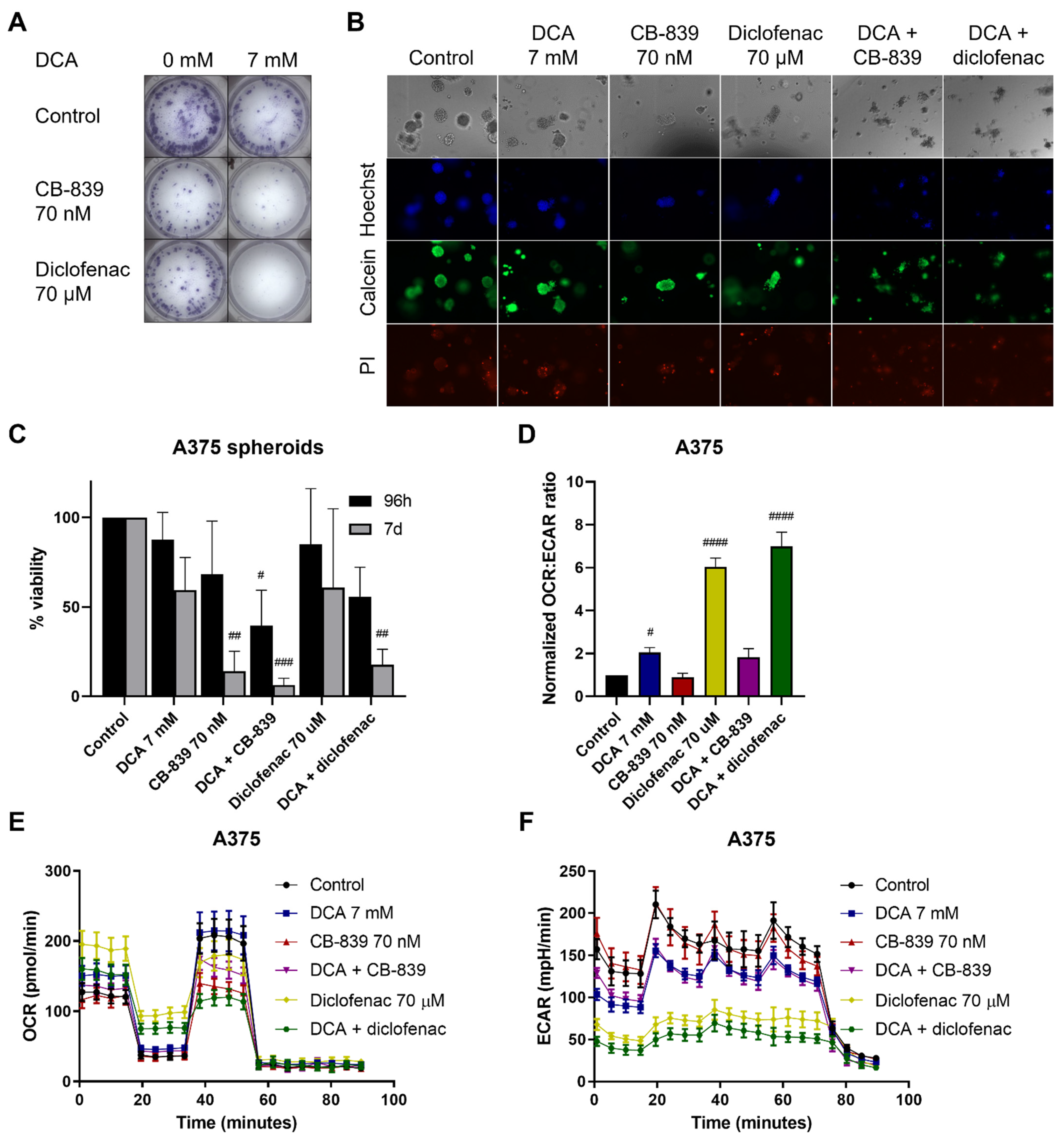
2.6. DCA Synergizes with Other Metabolic Inhibitors
3. Discussion
4. Materials and Methods
4.1. Cell Lines and Materials
4.2. Spheroid Culture
4.3. Cell Viability
4.4. Immunoblotting
4.5. PCR
4.6. Seahorse Analysis
4.7. Generation of PDK Knockdown Cell Lines
4.8. Combination Treatments
4.9. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Luebker, S.A.; Koepsell, S.A. Diverse Mechanisms of BRAF Inhibitor Resistance in Melanoma Identified in Clinical and Preclinical Studies. Front. Oncol. 2019, 9, 268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robert, C.; Grob, J.J.; Stroyakovskiy, D.; Karaszewska, B.; Hauschild, A.; Levchenko, E.; Chiarion Sileni, V.; Schachter, J.; Garbe, C.; Bondarenko, I.; et al. Five-Year Outcomes with Dabrafenib plus Trametinib in Metastatic Melanoma. N. Engl. J. Med. 2019, 381, 626–636. [Google Scholar] [CrossRef]
- Abildgaard, C.; Guldberg, P. Molecular Drivers of Cellular Metabolic Reprogramming in Melanoma. Trends Mol. Med. 2015, 21, 164–171. [Google Scholar] [CrossRef]
- Ratnikov, B.I.; Scott, D.A.; Osterman, A.L.; Smith, J.W.; Ronai, Z.A. Metabolic Rewiring in Melanoma. Oncogene 2017, 36, 147–157. [Google Scholar] [CrossRef] [Green Version]
- Shi, H.; Hugo, W.; Kong, X.; Hong, A.; Koya, R.C.; Moriceau, G.; Chodon, T.; Guo, R.; Johnson, D.B.; Dahlman, K.B.; et al. Acquired Resistance and Clonal Evolution in Melanoma during BRAF Inhibitor Therapy. Cancer Discov. 2014, 4, 80–93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; de Oliveira, R.L.; Huijberts, S.; Bosdriesz, E.; Pencheva, N.; Brunen, D.; Bosma, A.; Song, J.Y.; Zevenhoven, J.; Los-de Vries, G.T.; et al. An Acquired Vulnerability of Drug-Resistant Melanoma with Therapeutic Potential. Cell 2018, 173, 1413–1425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baenke, F.; Chaneton, B.; Smith, M.; van den Broek, N.; Hogan, K.; Tang, H.; Viros, A.; Martin, M.; Galbraith, L.; Girotti, M.R.; et al. Resistance to BRAF Inhibitors Induces Glutamine Dependency in Melanoma Cells. Mol. Oncol. 2016, 10, 73–84. [Google Scholar] [CrossRef]
- Sheta, E.A.; Trout, H.; Gildea, J.J.; Harding, M.A.; Theodorescu, D. Cell Density Mediated Pericellular Hypoxia Leads to Induction of HIF-1a via Nitric Oxide and Ras/MAP Kinase Mediated Signaling Pathways. Oncogene 2001, 20, 7624–7634. [Google Scholar] [CrossRef] [Green Version]
- Pavlova, N.N.; Thompson, C.B. The Emerging Hallmarks of Cancer Metabolism. Cell Metab. 2016, 23, 27–47. [Google Scholar] [CrossRef] [Green Version]
- Scott, D.A.; Richardson, A.D.; Filipp, F.V.; Knutzen, C.A.; Chiang, G.G.; Ronai, Z.A.; Osterman, A.L.; Smith, J.W. Comparative Metabolic Flux Profiling of Melanoma Cell Lines: Beyond the Warburg Effect. J. Biol. Chem. 2011, 286, 42626–42634. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.W.; Tchernyshyov, I.; Semenza, G.L.; Dang, C.V. HIF-1-Mediated Expression of Pyruvate Dehydrogenase Kinase: A Metabolic Switch Required for Cellular Adaptation to Hypoxia. Cell Metab. 2006, 3, 177–185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papandreou, I.; Cairns, R.A.; Fontana, L.; Lim, A.L.; Denko, N.C. HIF-1 Mediates Adaptation to Hypoxia by Actively Downregulating Mitochondrial Oxygen Consumption. Cell Metab. 2006, 3, 187–197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saunier, E.; Benelli, C.; Bortoli, S. The Pyruvate Dehydrogenase Complex in Cancer: An Old Metabolic Gatekeeper Regulated by New Pathways and Pharmacological Agents. Int. J. Cancer 2016, 138, 809–817. [Google Scholar] [CrossRef] [PubMed]
- Pópulo, H.; Caldas, R.; Lopes, J.M.; Pardal, J.; Máximo, V.; Soares, P. Overexpression of Pyruvate Dehydrogenase Kinase Supports Dichloroacetate as a Candidate for Cutaneous Melanoma Therapy. Expert Opin. Ther. Targets 2015, 19, 733–745. [Google Scholar] [CrossRef] [PubMed]
- Stacpoole, P.W.; Gilbert, L.R.; Neiberger, R.E.; Carney, P.R.; Valenstein, E.; Theriaque, D.W.; Shuster, J.J. Evaluation of Long-Term Treatment of Children with Congenital Lactic Acidosis with Dichloroacetate. Pediatrics 2008, 121, e1223–e1228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morten, K.J.; Beattie, P.; Brown, G.K.; Matthews, P.M. Dichloroacetate Stabilizes the Mutant E1alpha Subunit in Pyruvate Dehydrogenase Deficiency. Neurology 1999, 53, 612–616. [Google Scholar] [CrossRef]
- Chu, Q.S.-C.; Sangha, R.; Spratlin, J.; Vos, L.J.; Mackey, J.R.; McEwan, A.J.B.; Venner, P.; Michelakis, E.D. A Phase I Open-Labeled, Single-Arm, Dose-Escalation, Study of Dichloroacetate (DCA) in Patients with Advanced Solid Tumors. Investig. New Drugs 2015, 33, 603–610. [Google Scholar] [CrossRef]
- Dunbar, E.M.; Coats, B.S.; Shroads, A.L.; Langaee, T.; Lew, A.; Forder, J.R.; Shuster, J.J.; Wagner, D.A.; Stacpoole, P.W. Phase 1 Trial of Dichloroacetate (DCA) in Adults with Recurrent Malignant Brain Tumors. Investig. New Drugs 2014, 32, 452–464. [Google Scholar] [CrossRef] [Green Version]
- Michelakis, E.D.; Sutendra, G.; Dromparis, P.; Webster, L.; Haromy, A.; Niven, E.; Maguire, C.; Gammer, T.L.; Mackey, J.R.; Fulton, D.; et al. Metabolic Modulation of Glioblastoma with Dichloroacetate. Sci. Transl. Med. 2010, 2, 31ra34. [Google Scholar] [CrossRef] [Green Version]
- Garon, E.B.; Christofk, H.R.; Hosmer, W.; Britten, C.D.; Bahng, A.; Crabtree, M.J.; Hong, C.S.; Kamranpour, N.; Pitts, S.; Kabbinavar, F.; et al. Dichloroacetate Should Be Considered with Platinum-Based Chemotherapy in Hypoxic Tumors Rather than as a Single Agent in Advanced Non-Small Cell Lung Cancer. J. Cancer Res. Clin. Oncol. 2014, 140, 443–452. [Google Scholar] [CrossRef] [Green Version]
- Kato, M.; Li, J.; Chuang, J.L.; Chuang, D.T. Distinct Structural Mechanisms for Inhibition of Pyruvate Dehydrogenase Kinase Isoforms by AZD7545, Dichloroacetate, and Radicicol. Structure 2007, 15, 992–1004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Eu, J.Q.; Kong, L.R.; Wang, L.; Lim, Y.C.; Goh, B.C.; Wong, A.L.A. Targeting Metabolism in Cancer Cells and the Tumour Microenvironment for Cancer Therapy. Molecules 2020, 25, 4831. [Google Scholar] [CrossRef] [PubMed]
- Kouidhi, S.; ben Ayed, F.; Elgaaied, A.B. Targeting Tumor Metabolism: A New Challenge to Improve Immunotherapy. Front. Immunol. 2018, 9, 353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stacpoole, P.W. Therapeutic Targeting of the Pyruvate Dehydrogenase Complex/Pyruvate Dehydrogenase Kinase (PDC/PDK) Axis in Cancer. J. Natl. Cancer Inst. 2017, 109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murray, K.; Dickson, A.J. Dichloroacetate Inhibits Glutamine Oxidation by Decreasing Pyruvate Availability for Transamination. Metab. Clin. Exp. 1997, 46, 268–272. [Google Scholar] [CrossRef]
- Renner, K.; Bruss, C.; Schnell, A.; Koehl, G.; Becker, H.M.; Fante, M.; Menevse, A.-N.; Kauer, N.; Blazquez, R.; Hacker, L.; et al. Restricting Glycolysis Preserves T Cell Effector Functions and Augments Checkpoint Therapy. Cell Rep. 2019, 29, 135–150.e9. [Google Scholar] [CrossRef] [Green Version]
- Abildgaard, C.; Dahl, C.; Basse, A.L.; Ma, T.; Guldberg, P. Bioenergetic Modulation with Dichloroacetate Reduces the Growth of Melanoma Cells and Potentiates Their Response to BRAFV600E Inhibition. J. Transl. Med. 2014, 12, 247. [Google Scholar] [CrossRef] [Green Version]
- Gottfried, E.; Lang, S.A.; Renner, K.; Bosserhoff, A.; Gronwald, W.; Rehli, M.; Einhell, S.; Gedig, I.; Singer, K.; Seilbeck, A.; et al. New Aspects of an Old Drug—Diclofenac Targets MYC and Glucose Metabolism in Tumor Cells. PLoS ONE 2013, 8, e66987. [Google Scholar] [CrossRef] [Green Version]
- Stockwin, L.H.; Yu, S.X.; Borgel, S.; Hancock, C.; Wolfe, T.L.; Phillips, L.R.; Hollingshead, M.G.; Newton, D.L. Sodium Dichloroacetate Selectively Targets Cells with Defects in the Mitochondrial ETC. Int. J. Cancer 2010, 127, 2510–2519. [Google Scholar] [CrossRef]
- Škorja Milić, N.; Dolinar, K.; Miš, K.; Matkovič, U.; Bizjak, M.; Pavlin, M.; Podbregar, M.; Pirkmajer, S. Suppression of Pyruvate Dehydrogenase Kinase by Dichloroacetate in Cancer and Skeletal Muscle Cells Is Isoform Specific and Partially Independent of HIF-1α. Int. J. Mol. Sci. 2021, 22, 8610. [Google Scholar] [CrossRef]
- Tso, S.C.; Qi, X.; Gui, W.J.; Wu, C.Y.; Chuang, J.L.; Wernstedt-Asterholm, I.; Morlock, L.K.; Owens, K.R.; Scherer, P.E.; Williams, N.S.; et al. Structure-Guided Development of Specific Pyruvate Dehydrogenase Kinase Inhibitors Targeting the ATP-Binding Pocket. J. Biol. Chem. 2014, 289, 4432–4443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, A.U.H.; Allende-Vega, N.; Gitenay, D.; Gerbal-Chaloin, S.; Gondeau, C.; Vo, D.N.; Belkahla, S.; Orecchioni, S.; Talarico, G.; Bertolini, F.; et al. The PDK1 Inhibitor Dichloroacetate Controls Cholesterol Homeostasis Through the ERK5/MEF2 Pathway. Sci. Rep. 2017, 7, 10654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, W.; Hu, X.; Zhou, W.; Tam, K.Y. Liquid Chromatography-Tandem Mass Spectrometry Method Revealed That Lung Cancer Cells Exhibited Distinct Metabolite Profiles upon the Treatment with Different Pyruvate Dehydrogenase Kinase Inhibitors. J. Proteome Res. 2018, 17, 3012–3021. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Li, X.; Ni, Z.; Zhang, Y.; Zeng, Y.; Yan, X.; Huang, Y.; He, J.; Lyu, X.; Wu, Y.; et al. Dichloroacetate and Metformin Synergistically Suppress the Growth of Ovarian Cancer Cells. Oncotarget 2016, 7, 59458–59470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vogl, D.T.; Younes, A.; Stewart, K.; Orford, K.W.; Bennett, M.; Siegel, D.; Berdeja, J.G. Phase 1 Study of CB-839, a First-in-Class, Glutaminase Inhibitor in Patients with Multiple Myeloma and Lymphoma. Blood 2015, 126, 3059. [Google Scholar] [CrossRef]
- Schoonjans, C.A.; Joudiou, N.; Brusa, D.; Corbet, C.; Feron, O.; Gallez, B. Acidosis-Induced Metabolic Reprogramming in Tumor Cells Enhances the Anti-Proliferative Activity of the PDK Inhibitor Dichloroacetate. Cancer Lett. 2020, 470, 18–28. [Google Scholar] [CrossRef] [PubMed]
- Fowler, P.D.; Shadforth, M.F.; Crook, P.R.; John, V.A. Plasma and Synovial Fluid Concentrations of Diclofenac Sodium and Its Major Hydroxylated Metabolites during Long-Term Treatment of Rheumatoid Arthritis. Eur. J. Clin. Pharmacol. 1983, 25, 389–394. [Google Scholar] [CrossRef]
- Ayyanathan, K.; Kesaraju, S.; Dawson-Scully, K.; Weissbach, H. Combination of Sulindac and Dichloroacetate Kills Cancer Cells via Oxidative Damage. PLoS ONE 2012, 7, e39949. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ullah, M.S.; Davies, A.J.; Halestrap, A.P. The Plasma Membrane Lactate Transporter MCT4, but Not MCT1, Is Up-Regulated by Hypoxia through a HIF-1α-Dependent Mechanism. J. Biol. Chem. 2006, 281, 9030–9037. [Google Scholar] [CrossRef] [Green Version]
- Curtis, N.J.; Mooney, L.; Hopcroft, L.; Michopoulos, F.; Whalley, N.; Zhong, H.; Murray, C.; Logie, A.; Revill, M.; Byth, K.F.; et al. Pre-Clinical Pharmacology of AZD3965, a Selective Inhibitor of MCT1: DLBCL, NHL and Burkitt’s Lymphoma Anti-Tumor Activity. Oncotarget 2017, 8, 69219–69236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larkin, J.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.-J.; Rutkowski, P.; Lao, C.D.; Cowey, C.L.; Schadendorf, D.; Wagstaff, J.; Dummer, R.; et al. Five-Year Survival with Combined Nivolumab and Ipilimumab in Advanced Melanoma. N. Engl. J. Med. 2019, 381, 1535–1546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amoedo, N.D.; Obre, E.; Rossignol, R. Drug Discovery Strategies in the Field of Tumor Energy Metabolism: Limitations by Metabolic Flexibility and Metabolic Resistance to Chemotherapy. Biochim. Biophys. Acta (BBA)-Bioenerg. 2017, 1858, 674–685. [Google Scholar] [CrossRef] [PubMed]
- Quinn, W.J.; Jiao, J.; TeSlaa, T.; Stadanlick, J.; Wang, Z.; Wang, L.; Akimova, T.; Angelin, A.; Schäfer, P.M.; Cully, M.D.; et al. Lactate Limits T Cell Proliferation via the NAD(H) Redox State. Cell Rep. 2020, 33, 108500. [Google Scholar] [CrossRef] [PubMed]
- Varghese, S.; Pramanik, S.; Williams, L.J.; Hodges, H.R.; Hudgens, C.W.; Fischer, G.M.; Luo, C.K.; Knighton, B.; Tan, L.; Lorenzi, P.L.; et al. The Glutaminase Inhibitor CB-839 (Telaglenastat) Enhances the Antimelanoma Activity of T-Cell-Mediated Immunotherapies. Mol. Cancer Ther. 2020, 20, 500–511. [Google Scholar] [CrossRef] [PubMed]
- Lyssiotis, C.A.; Kimmelman, A.C. Metabolic Interactions in the Tumor Microenvironment. Trends Cell Biol. 2017, 27, 863–875. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ianevski, A.; Giri, A.K.; Aittokallio, T. SynergyFinder 2.0: Visual Analytics of Multi-Drug Combination Synergies. Nucleic Acids Res. 2020, 48, W488–W493. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Line | DCA IC50 (mM) | AZD7545 IC50 (μM) |
|---|---|---|
| A375 | 14.9 ± 1.0 | 35.0 ± 4.1 |
| MeWo | 13.3 ± 0.6 | 89.3 ± 5.0 |
| SK-MEL-28 | 20.0 ± 1.4 | >100 |
| SK-MEL-2 | 27.3 ± 1.7 | >100 |
| Cell Line | CB-839 IC50 (nM) | Diclofenac IC50 (μM) | Metformin IC50 (mM) | Vemurafenib IC50 (nM) |
|---|---|---|---|---|
| A375 | 99.4 ± 37.7 | 112.7 ± 4.1 | 3.0 ± 0.8 | 70.7 ± 22.7 |
| MeWo | 7.9 ± 0.9 | 146.0 ± 27.0 | 7.5 ± 2.6 | N/A |
| SK-MEL-28 | N/A | 134.0 ± 4.6 | N/A | 82.0 ± 25.4 |
| SK-MEL-2 | 139.2 ± 34.8 | 133.8 ± 13.6 | N/A | N/A |
| Drug | A375 | MeWo | SK-MEL-28 | SK-MEL-2 |
|---|---|---|---|---|
| CB-839 | 20.3 | 17.7 | N/A | 31.7 |
| Diclofenac | 27.4 | 21.5 | 25.1 | 27.0 |
| Metformin | 12.4 | 23.1 | N/A | N/A |
| Vemurafenib | 26.0 | N/A | 23.8 | N/A |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tiersma, J.F.; Evers, B.; Bakker, B.M.; Jalving, M.; de Jong, S. Pyruvate Dehydrogenase Kinase Inhibition by Dichloroacetate in Melanoma Cells Unveils Metabolic Vulnerabilities. Int. J. Mol. Sci. 2022, 23, 3745. https://doi.org/10.3390/ijms23073745
Tiersma JF, Evers B, Bakker BM, Jalving M, de Jong S. Pyruvate Dehydrogenase Kinase Inhibition by Dichloroacetate in Melanoma Cells Unveils Metabolic Vulnerabilities. International Journal of Molecular Sciences. 2022; 23(7):3745. https://doi.org/10.3390/ijms23073745
Chicago/Turabian StyleTiersma, Jiske F., Bernard Evers, Barbara M. Bakker, Mathilde Jalving, and Steven de Jong. 2022. "Pyruvate Dehydrogenase Kinase Inhibition by Dichloroacetate in Melanoma Cells Unveils Metabolic Vulnerabilities" International Journal of Molecular Sciences 23, no. 7: 3745. https://doi.org/10.3390/ijms23073745