
Aconitate Decarboxylase 1 Deficiency Exacerbates Mouse Colitis Induced by Dextran Sodium Sulfate

 ,
,  , and
, and 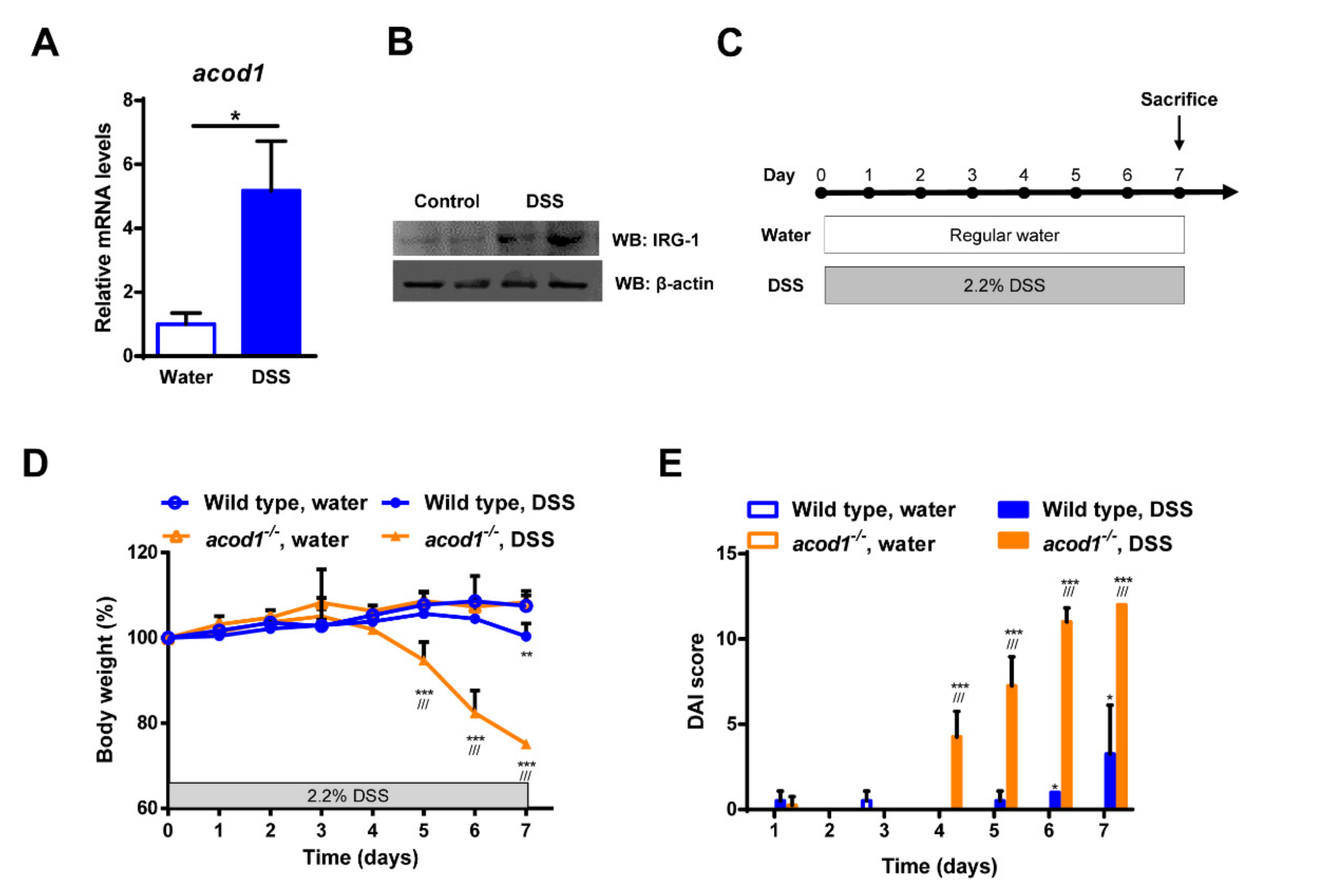{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
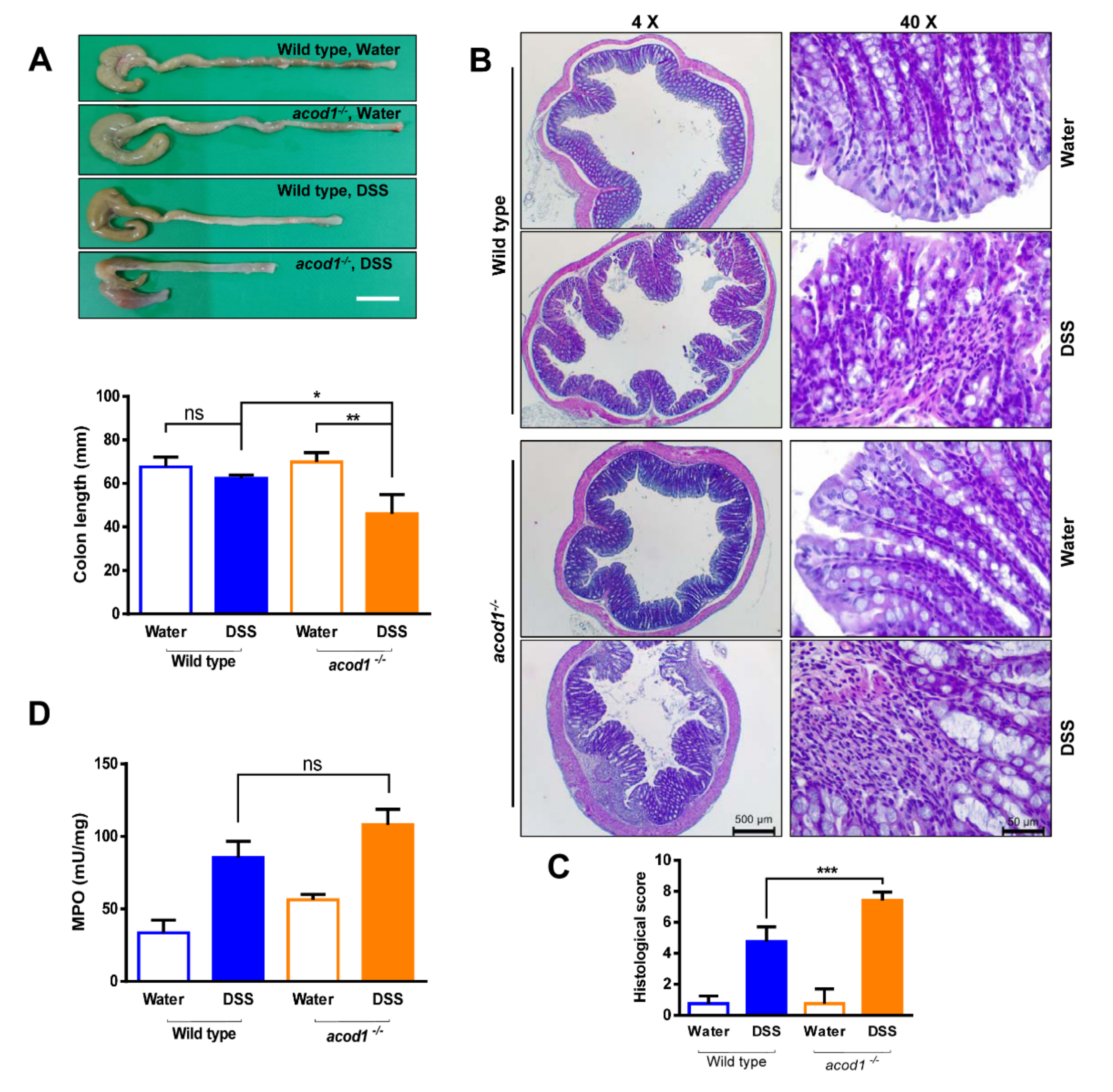
2.1. Acod1 Is Upregulated in DSS-Induced Colitis, and Acod1 Depletion Results in Worsened Symptoms
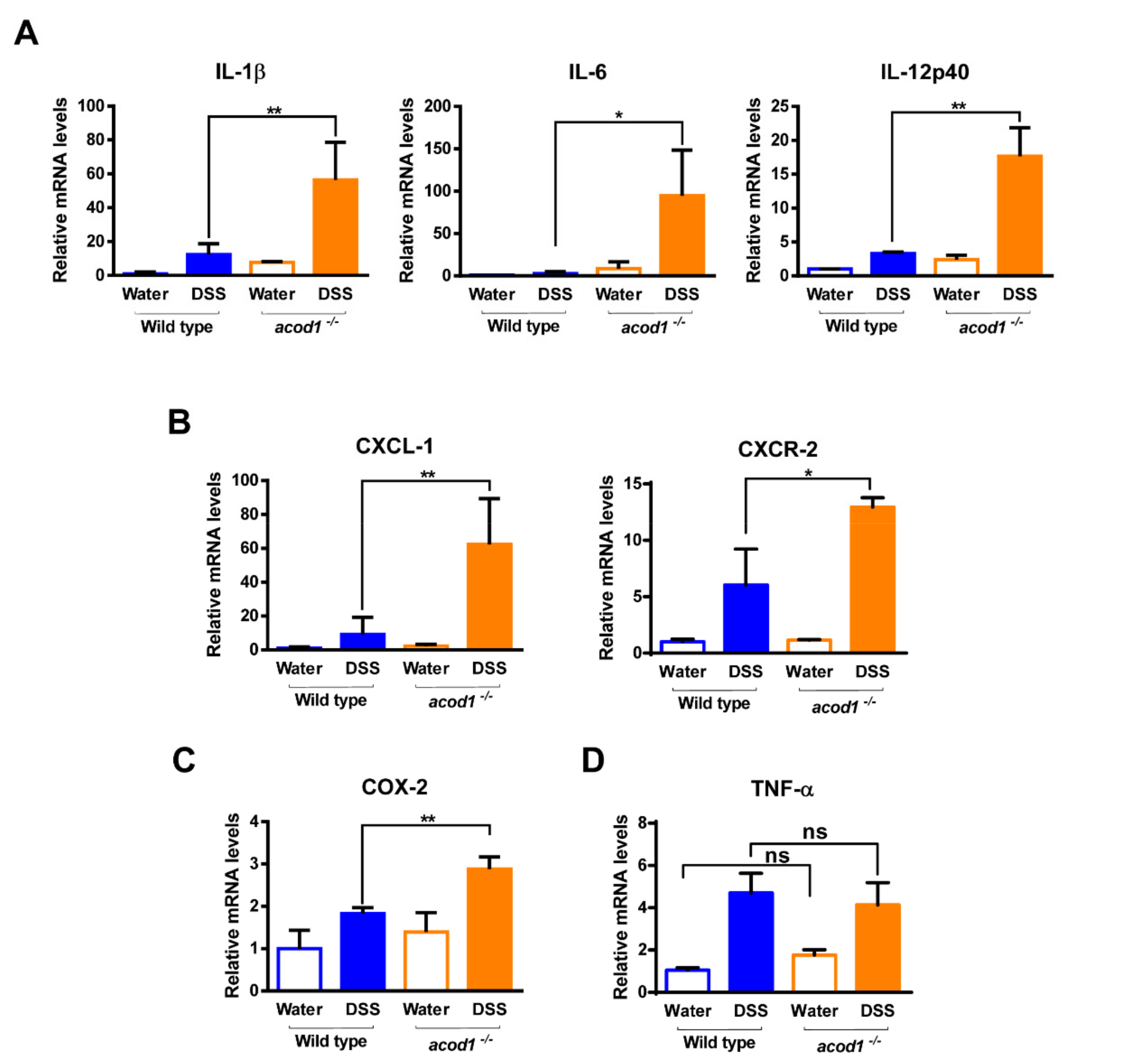
2.2. Acod1 Gene Deletion Increases Inflammatory Cytokine and Chemokine Expression in DSS-Induced Colitis
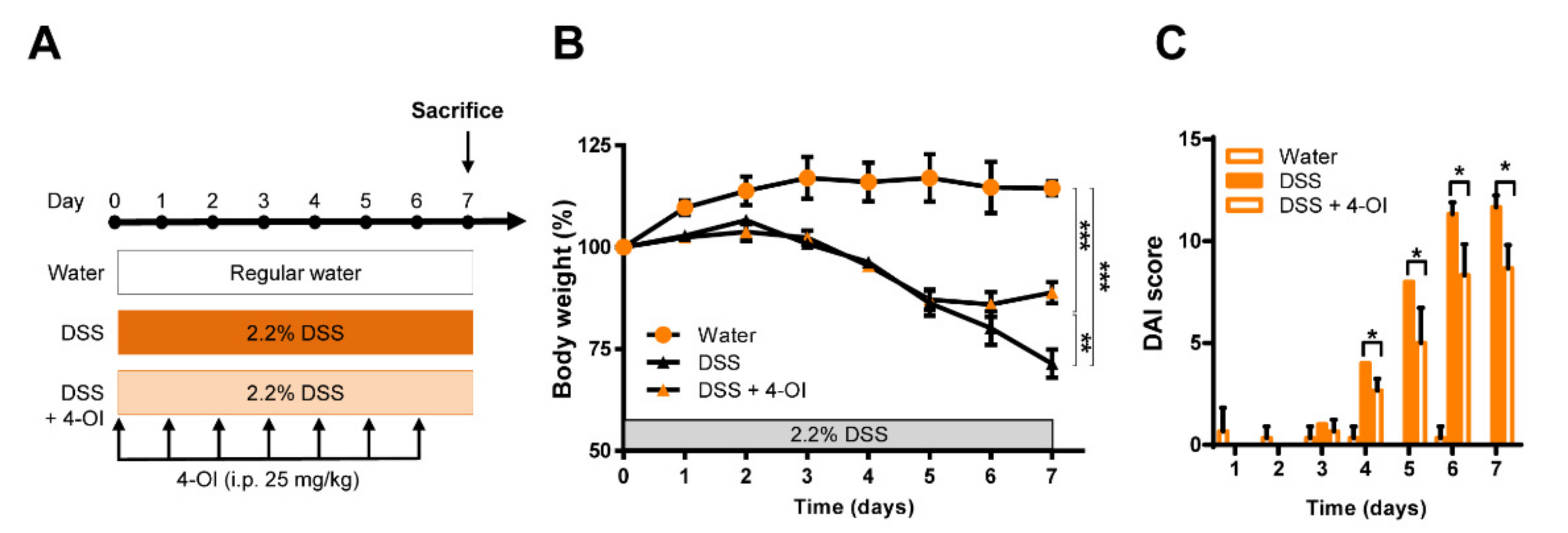
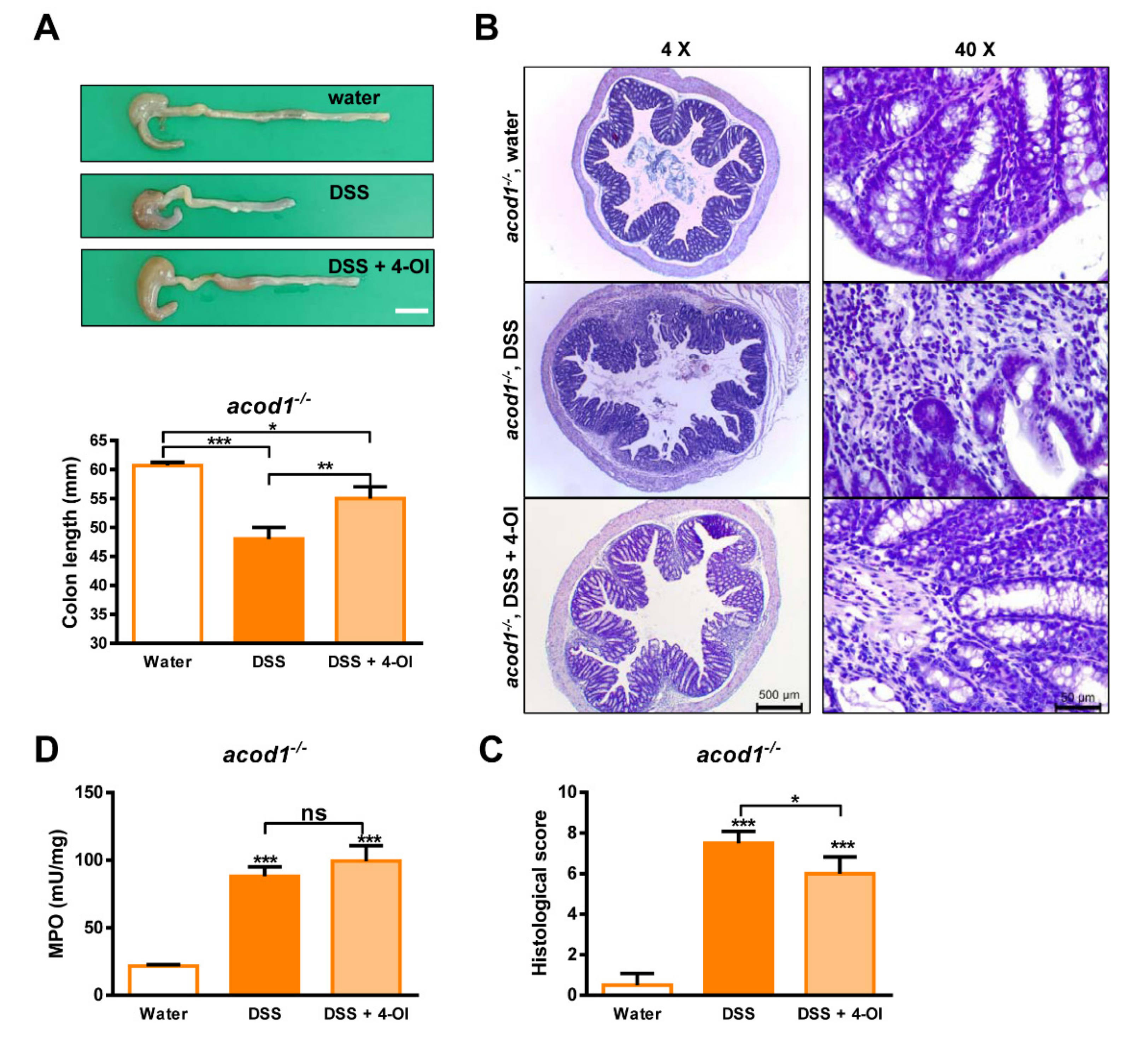
2.3. 4-Octyl Itaconate Alleviates DSS-Induced Colitis in Acod1-Deficient Mice
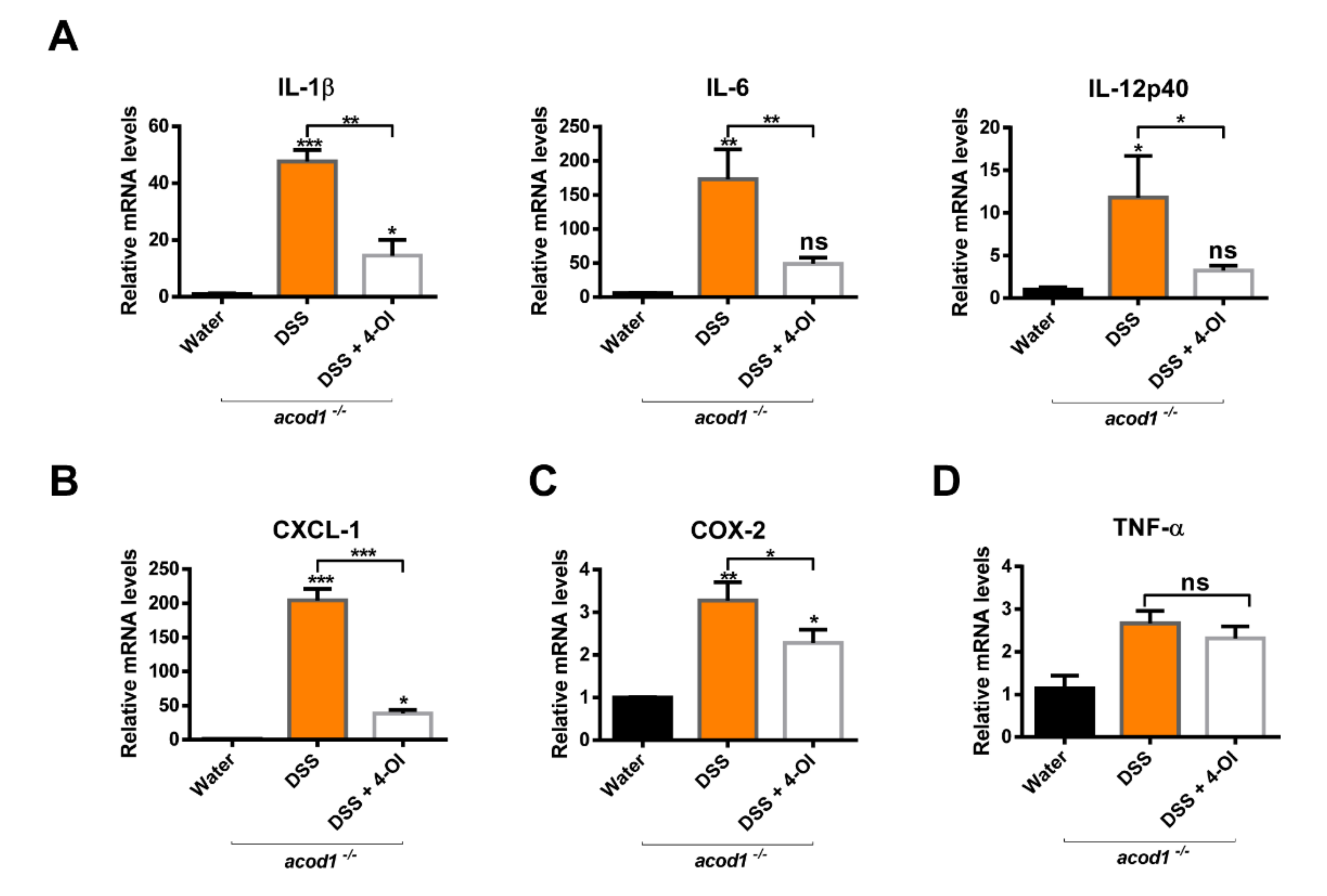
2.4. 4-Octyl Itaconate Diminishes the Expression of Inflammatory Cytokines and Chemokines in an Acod1-Deficient Mouse Model of DSS-Induced Colitis
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. DSS induces Colitis in Mice
4.3. Quantification of mRNA by Real-Time RT–PCR
4.4. Disease Activity Index
4.5. Histological Analysis and Scoring of Colonic Damage
4.6. Determination of Myeloperoxidase (MPO) Activity
4.7. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Calam, C.T.; Oxford, A.E.; Raistrick, H. Studies in the biochemistry of micro-organisms: Itaconic acid, a metabolic product of a strain of Aspergillus terreus Thom. Biochem. J. 1939, 33, 1488–1495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larsen, H.; Eimhjellen, K.E. The mechanism of itaconic acid formation by Aspergillus terreus. 1. The effect of acidity. Biochem. J. 1955, 60, 135–139. [Google Scholar] [CrossRef] [Green Version]
- Strelko, C.L.; Lu, W.; Dufort, F.J.; Seyfried, T.N.; Chiles, T.C.; Rabinowitz, J.D.; Roberts, M.F. Itaconic acid is a mammalian metabolite induced during macrophage activation. J. Am. Chem. Soc. 2011, 133, 16386–16389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michelucci, A.; Cordes, T.; Ghelfi, J.; Pailot, A.; Reiling, N.; Goldmann, O.; Binz, T.; Wegner, A.; Tallam, A.; Rausell, A.; et al. Immune-responsive gene 1 protein links metabolism to immunity by catalyzing itaconic acid production. Proc. Natl. Acad. Sci. USA 2013, 110, 7820–7825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diskin, C.; Palsson-McDermott, E.M. Metabolic Modulation in Macrophage Effector Function. Front. Immunol. 2018, 9, 270. [Google Scholar] [CrossRef] [Green Version]
- Viola, A.; Munari, F.; Sanchez-Rodriguez, R.; Scolaro, T.; Castegna, A. The Metabolic Signature of Macrophage Responses. Front. Immunol. 2019, 10, 1462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cordes, T.; Michelucci, A.; Hiller, K. Itaconic Acid: The Surprising Role of an Industrial Compound as a Mammalian Antimicrobial Metabolite. Annu. Rev. Nutr. 2015, 35, 451–473. [Google Scholar] [CrossRef]
- Lampropoulou, V.; Sergushichev, A.; Bambouskova, M.; Nair, S.; Vincent, E.E.; Loginicheva, E.; Cervantes-Barragan, L.; Ma, X.; Huang, S.C.; Griss, T.; et al. Itaconate Links Inhibition of Succinate Dehydrogenase with Macrophage Metabolic Remodeling and Regulation of Inflammation. Cell Metab. 2016, 24, 158–166. [Google Scholar] [CrossRef] [Green Version]
- Bambouskova, M.; Potuckova, L.; Paulenda, T.; Kerndl, M.; Mogilenko, D.A.; Lizotte, K.; Swain, A.; Hayes, S.; Sheldon, R.D.; Kim, H.; et al. Itaconate confers tolerance to late NLRP3 inflammasome activation. Cell Rep. 2021, 34, 108756. [Google Scholar] [CrossRef]
- Mills, E.L.; Ryan, D.G.; Prag, H.A.; Dikovskaya, D.; Menon, D.; Zaslona, Z.; Jedrychowski, M.P.; Costa, A.S.H.; Higgins, M.; Hams, E.; et al. Itaconate is an anti-inflammatory metabolite that activates Nrf2 via alkylation of KEAP1. Nature 2018, 556, 113–117. [Google Scholar] [CrossRef]
- Baumgart, D.C.; Sandborn, W.J. Inflammatory bowel disease: Clinical aspects and established and evolving therapies. Lancet 2007, 369, 1641–1657. [Google Scholar] [CrossRef]
- Mak, W.Y.; Zhao, M.; Ng, S.C.; Burisch, J. The epidemiology of inflammatory bowel disease: East meets west. J. Gastroenterol. Hepatol. 2020, 35, 380–389. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.H.; Kwon, J.E.; Cho, M.L. Immunological pathogenesis of inflammatory bowel disease. Intest. Res. 2018, 16, 26–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kiesler, P.; Fuss, I.J.; Strober, W. Experimental Models of Inflammatory Bowel Diseases. Cell. Mol. Gastroenterol. Hepatol. 2015, 1, 154–170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sidiq, T.; Yoshihama, S.; Downs, I.; Kobayashi, K.S. Nod2: A Critical Regulator of Ileal Microbiota and Crohn’s Disease. Front. Immunol. 2016, 7, 367. [Google Scholar] [CrossRef]
- Mehto, S.; Jena, K.K.; Nath, P.; Chauhan, S.; Kolapalli, S.P.; Das, S.K.; Sahoo, P.K.; Jain, A.; Taylor, G.A.; Chauhan, S. The Crohn’s Disease Risk Factor IRGM Limits NLRP3 Inflammasome Activation by Impeding Its Assembly and by Mediating Its Selective Autophagy. Mol. Cell 2019, 73, 429–445.e427. [Google Scholar] [CrossRef] [Green Version]
- Pidasheva, S.; Trifari, S.; Phillips, A.; Hackney, J.A.; Ma, Y.; Smith, A.; Sohn, S.J.; Spits, H.; Little, R.D.; Behrens, T.W.; et al. Functional studies on the IBD susceptibility gene IL23R implicate reduced receptor function in the protective genetic variant R381Q. PLoS ONE 2011, 6, e25038. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Li, X.L.; Mei, Y.; Ye, J.C.; Fan, W.; Cheng, G.H.; Zeng, M.S.; Feng, G.K. The anti-inflammatory drug dimethyl itaconate protects against colitis-associated colorectal cancer. J. Mol. Med. 2020, 98, 1457–1466. [Google Scholar] [CrossRef]
- Scheurlen, K.M.; Snook, D.L.; Walter, M.N.; Cook, C.N.; Fiechter, C.R.; Pan, J.; Beal, R.J.; Galandiuk, S. Itaconate and leptin affecting PPARgamma in M2 macrophages: A potential link to early-onset colorectal cancer. Surgery 2021, 171, 650–656. [Google Scholar] [CrossRef]
- Chassaing, B.; Aitken, J.D.; Malleshappa, M.; Vijay-Kumar, M. Dextran sulfate sodium (DSS)-induced colitis in mice. Curr. Protoc. Immunol. 2014, 104, 15.25.1–15.25.14. [Google Scholar] [CrossRef]
- Canton, M.; Sanchez-Rodriguez, R.; Spera, I.; Venegas, F.C.; Favia, M.; Viola, A.; Castegna, A. Reactive Oxygen Species in Macrophages: Sources and Targets. Front. Immunol. 2021, 12, 734229. [Google Scholar] [CrossRef] [PubMed]
- Yi, Z.; Deng, M.; Scott, M.J.; Fu, G.; Loughran, P.A.; Lei, Z.; Li, S.; Sun, P.; Yang, C.; Li, W.; et al. Immune-Responsive Gene 1/Itaconate Activates Nuclear Factor Erythroid 2-Related Factor 2 in Hepatocytes to Protect Against Liver Ischemia-Reperfusion Injury. Hepatology 2020, 72, 1394–1411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sawant, K.V.; Poluri, K.M.; Dutta, A.K.; Sepuru, K.M.; Troshkina, A.; Garofalo, R.P.; Rajarathnam, K. Chemokine CXCL1 mediated neutrophil recruitment: Role of glycosaminoglycan interactions. Sci. Rep. 2016, 6, 33123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, R.; Zhang, P.; Wang, Y.; Tao, K. Itaconate: A Metabolite Regulates Inflammation Response and Oxidative Stress. Oxidative Med. Cell. Longev. 2020, 2020, 5404780. [Google Scholar] [CrossRef] [PubMed]
- Sundaram, K.; Rahman, M.A.; Mitra, S.; Knoell, D.L.; Woodiga, S.A.; King, S.J.; Wewers, M.D. IkappaBzeta Regulates Human Monocyte Pro-Inflammatory Responses Induced by Streptococcus pneumoniae. PLoS ONE 2016, 11, e0161931. [Google Scholar] [CrossRef]
- ElAzzouny, M.; Tom, C.T.; Evans, C.R.; Olson, L.L.; Tanga, M.J.; Gallagher, K.A.; Martin, B.R.; Burant, C.F. Dimethyl Itaconate Is Not Metabolized into Itaconate Intracellularly. J. Biol. Chem. 2017, 292, 4766–4769. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Chen, X.; Zhang, H.; Xiao, J.; Yang, C.; Chen, W.; Wei, Z.; Chen, X.; Liu, J. 4-Octyl Itaconate Alleviates Lipopolysaccharide-Induced Acute Lung Injury in Mice by Inhibiting Oxidative Stress and Inflammation. Drug Des. Dev. Ther. 2020, 14, 5547–5558. [Google Scholar] [CrossRef]
- Xin, Y.; Zou, L.; Lang, S. 4-Octyl itaconate (4-OI) attenuates lipopolysaccharide-induced acute lung injury by suppressing PI3K/Akt/NF-kappaB signaling pathways in mice. Exp. Ther. Med. 2021, 21, 141. [Google Scholar] [CrossRef]
- Shastri, S.; Shinde, T.; Sohal, S.S.; Gueven, N.; Eri, R. Idebenone Protects against Acute Murine Colitis via Antioxidant and Anti-Inflammatory Mechanisms. Int. J. Mol. Sci. 2020, 21, 484. [Google Scholar] [CrossRef] [Green Version]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, H.W.; Yu, A.-R.; Lee, J.W.; Yoon, H.S.; Lee, B.S.; Park, H.-W.; Lee, S.K.; Lee, Y.I.; Whang, J.; Kim, J.-S. Aconitate Decarboxylase 1 Deficiency Exacerbates Mouse Colitis Induced by Dextran Sodium Sulfate. Int. J. Mol. Sci. 2022, 23, 4392. https://doi.org/10.3390/ijms23084392
Kim HW, Yu A-R, Lee JW, Yoon HS, Lee BS, Park H-W, Lee SK, Lee YI, Whang J, Kim J-S. Aconitate Decarboxylase 1 Deficiency Exacerbates Mouse Colitis Induced by Dextran Sodium Sulfate. International Journal of Molecular Sciences. 2022; 23(8):4392. https://doi.org/10.3390/ijms23084392
Chicago/Turabian StyleKim, Ho Won, A-Reum Yu, Ji Won Lee, Hoe Sun Yoon, Byung Soo Lee, Hwan-Woo Park, Sung Ki Lee, Young Ik Lee, Jake Whang, and Jong-Seok Kim. 2022. "Aconitate Decarboxylase 1 Deficiency Exacerbates Mouse Colitis Induced by Dextran Sodium Sulfate" International Journal of Molecular Sciences 23, no. 8: 4392. https://doi.org/10.3390/ijms23084392