
Yeast as a Model to Find New Drugs and Drug Targets for VPS13-Dependent Neurodegenerative Diseases


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
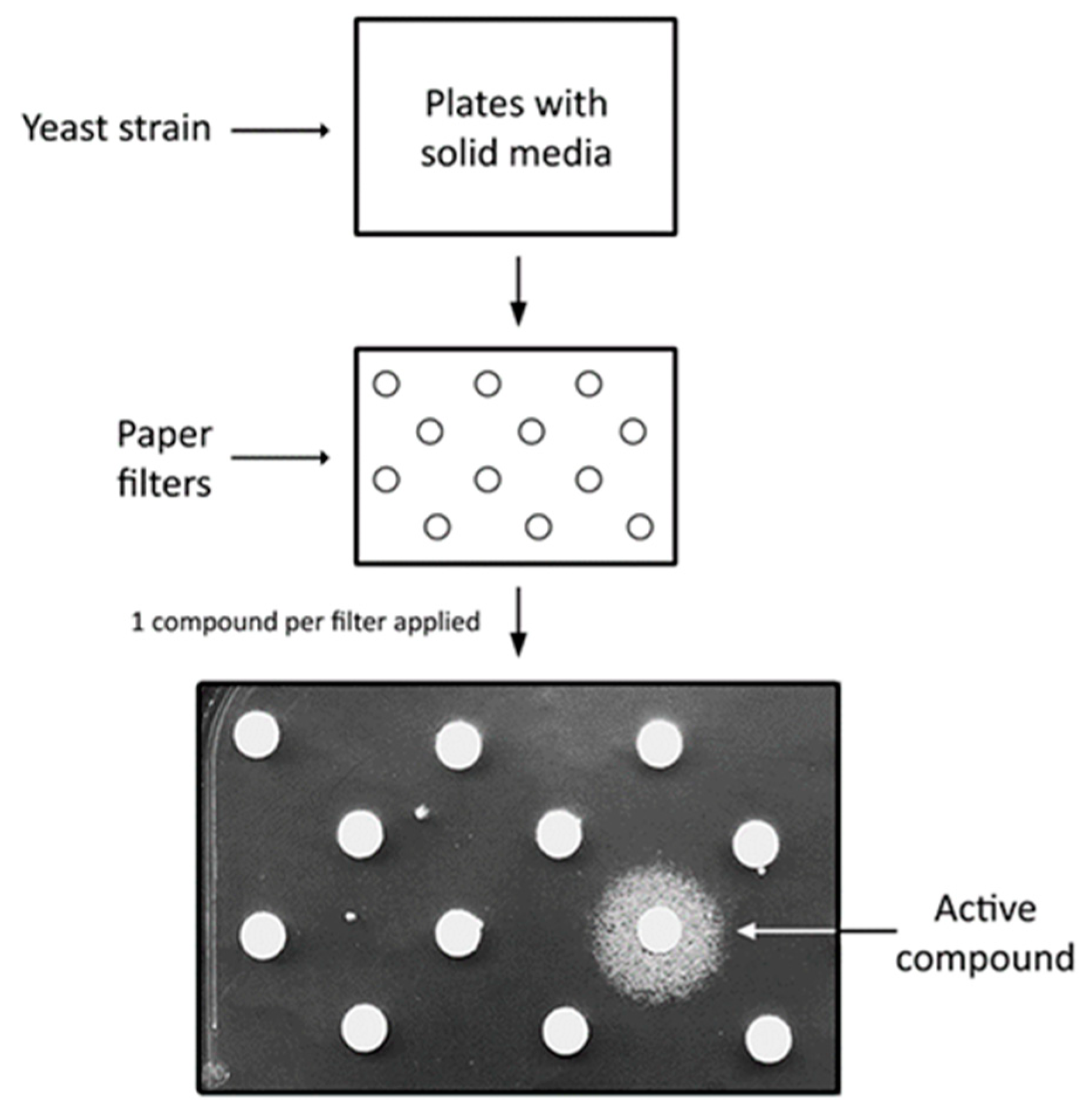
2. Saccharomyces cerevisiae as a Disease Model and Simple Platform for High Throughput Screens

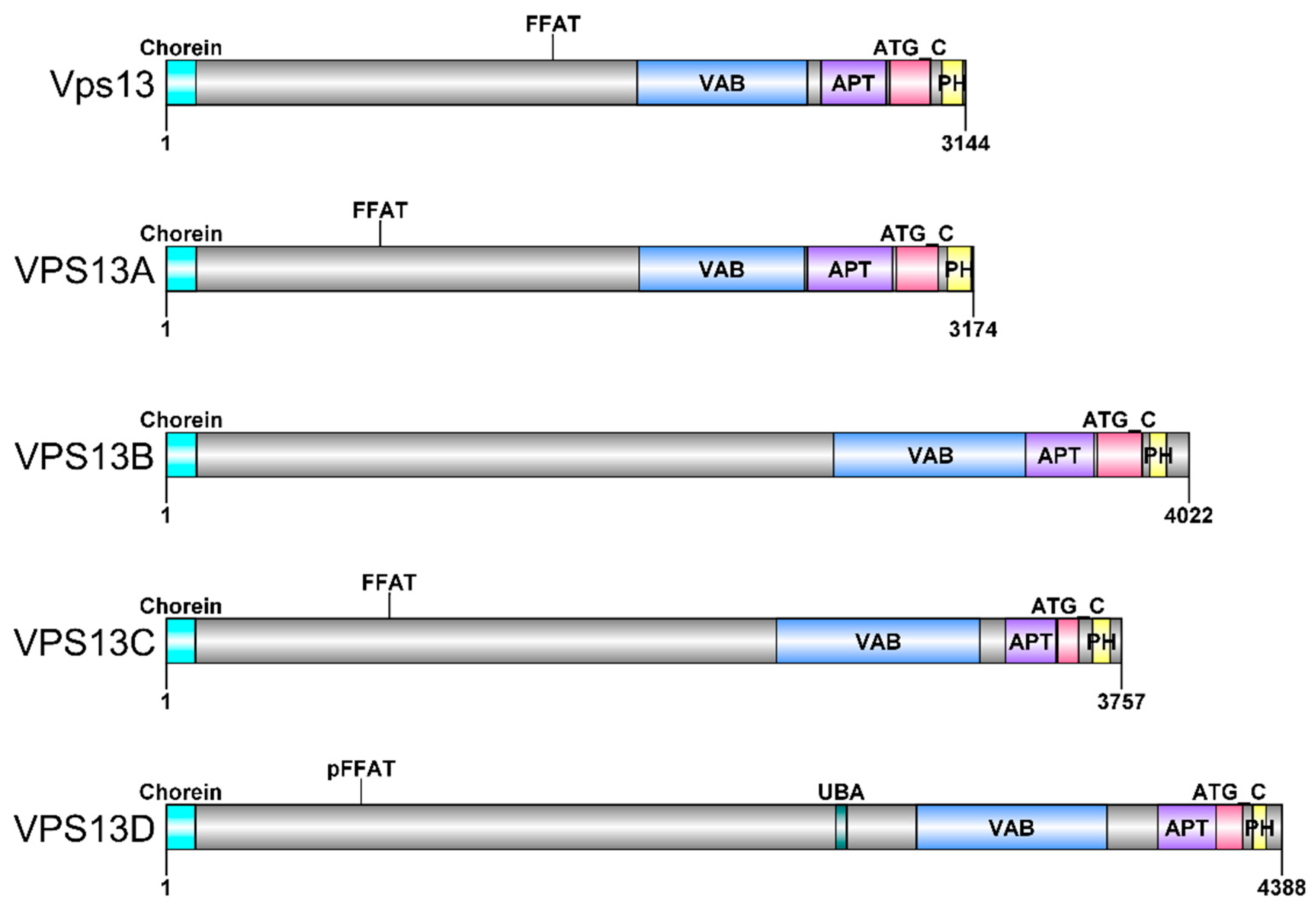
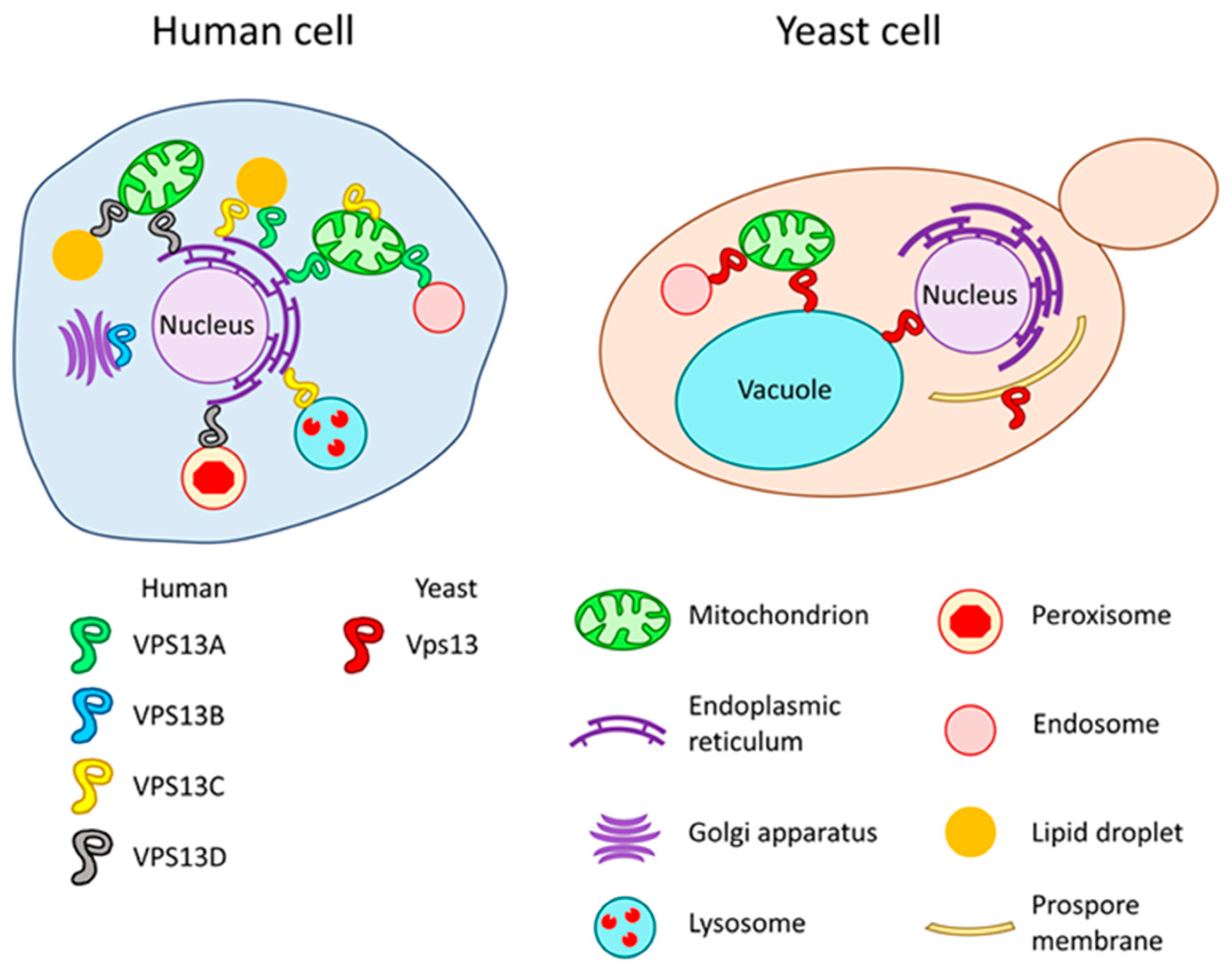
3. VPS13 Gene and Vps13 Protein in Saccharomyces cerevisiae Yeast
4. VPS13 Genes and VPS13 Proteins in Human and Related Diseases
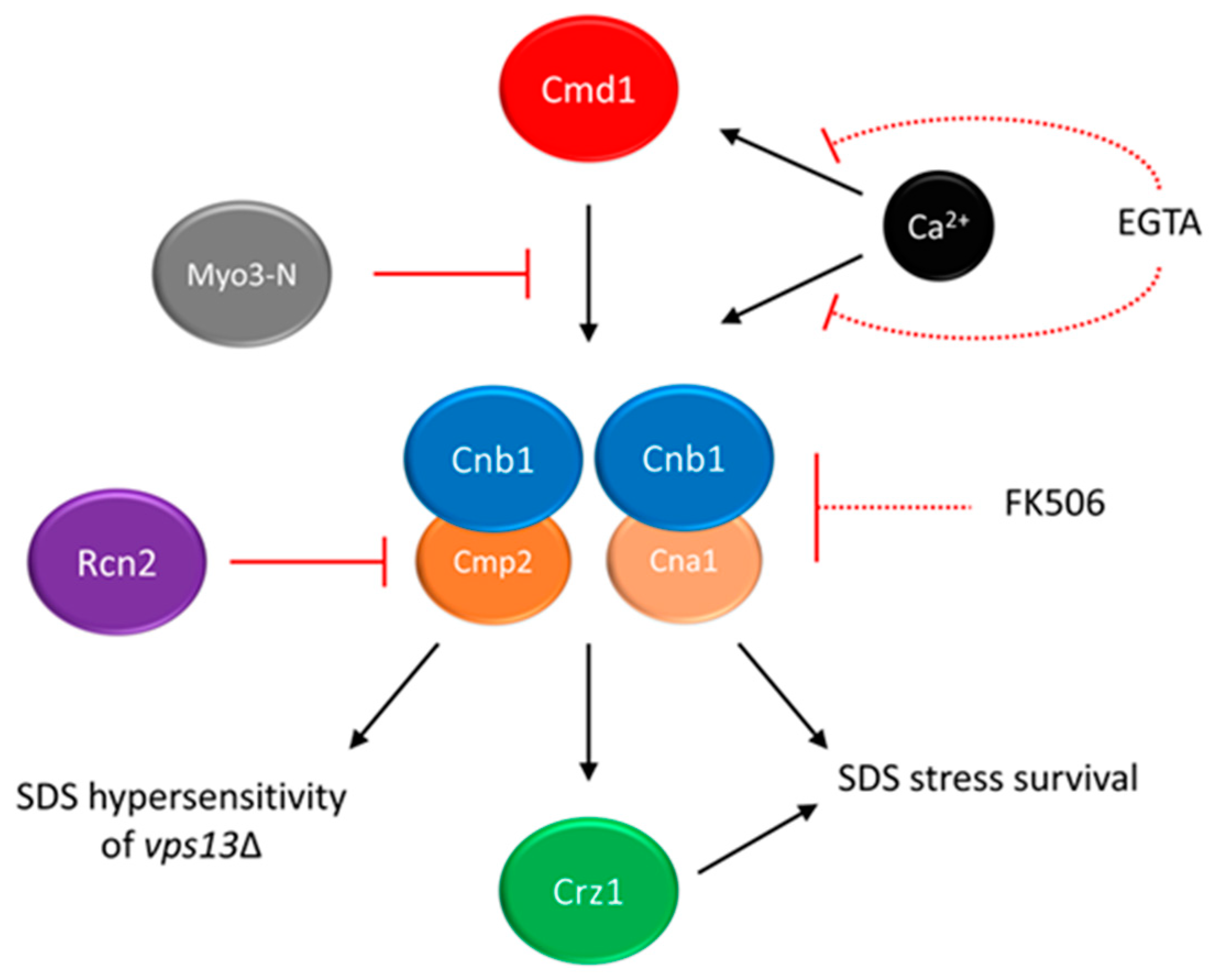
5. Calcium Signalling as a Potential Target for Drug Intervention in VPS13-Dependent Neurodegenerative Diseases
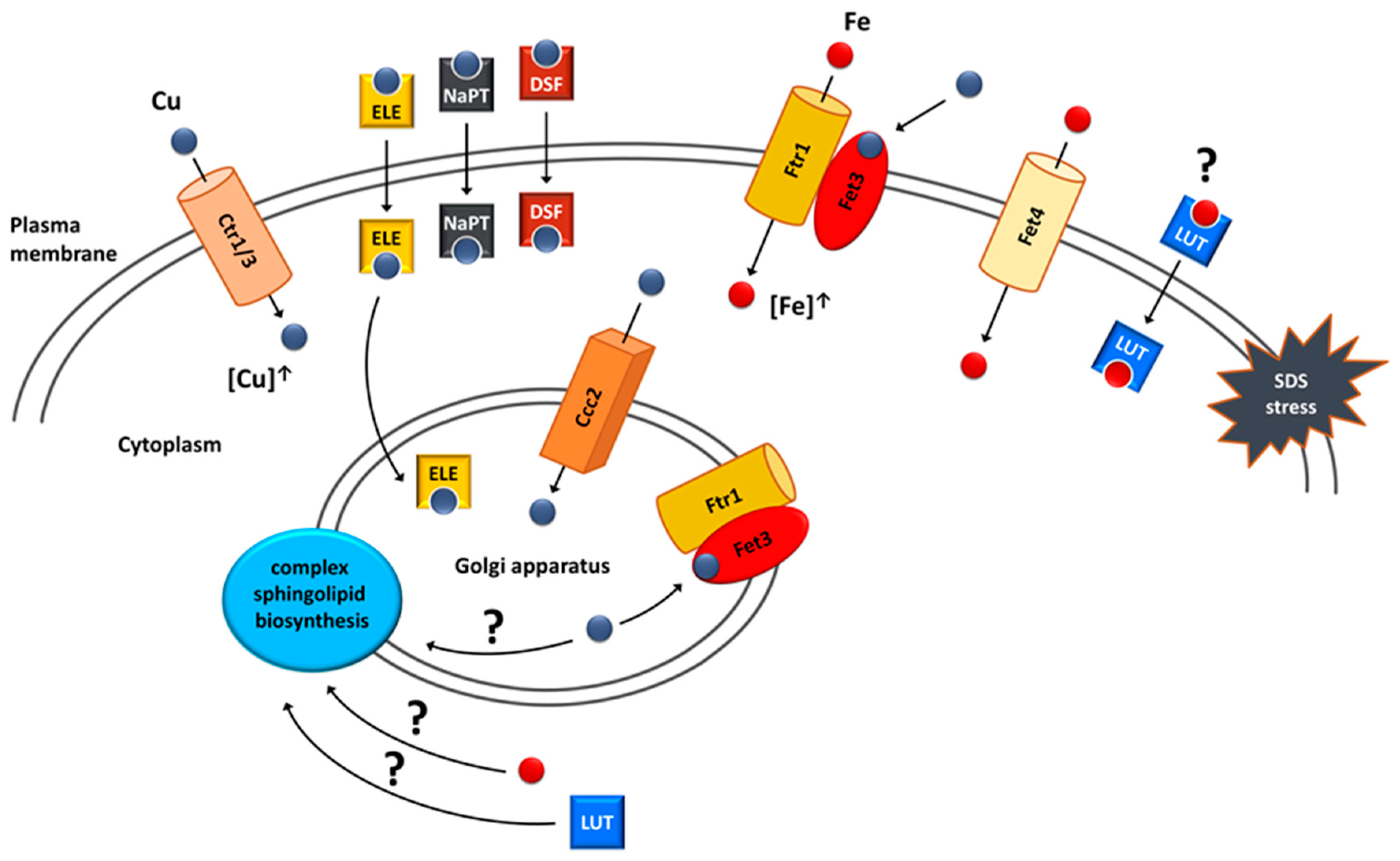
6. Copper and Iron Homeostasis as a Potential Target for Treatment of VPS13-Dependent Neurodegenerative Diseases
7. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Roessler, H.I.; Knoers, N.V.A.M.; van Haelst, M.M.; van Haaften, G. Drug Repurposing for Rare Diseases. Trends Pharmacol. Sci. 2021, 42, 255–267. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, K.; Balasubramaniam, S.; Choy, Y.S.; Fietz, M.; Fu, A.; Jin, D.K.; Kim, O.H.; Kosuga, M.; Kwun, Y.H.; Inwood, A.; et al. Overcoming the barriers to diagnosis of Morquio A syndrome. Orphanet J. Rare Dis. 2014, 9, 192. [Google Scholar] [CrossRef] [Green Version]
- Blöß, S.; Klemann, C.; Rother, A.K.; Mehmecke, S.; Schumacher, U.; Mücke, U.; Mücke, M.; Stieber, C.; Klawonn, F.; Kortum, X.; et al. Diagnostic needs for rare diseases and shared prediagnostic phenomena: Results of a German-wide expert Delphi survey. PLoS ONE 2017, 12, e0172532. [Google Scholar] [CrossRef] [Green Version]
- Bouwman, M.G.; Teunissen, Q.G.; Wijburg, F.A.; Linthorst, G.E. ‘Doctor Google’ ending the diagnostic odyssey in lysosomal storage disorders: Parents using internet search engines as an efficient diagnostic strategy in rare diseases. Arch. Dis. Child. 2010, 95, 642–644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hallal, C.; Kieling, C.O.; Nunes, D.L.; Ferreira, C.T.; Peterson, G.; Barros, S.G.; Arruda, C.A.; Fraga, J.C.; Goldani, H.A. Diagnosis, misdiagnosis, and associated diseases of achalasia in children and adolescents: A twelve-year single center experience. Pediatr. Surg. Int. 2012, 28, 1211–1217. [Google Scholar] [CrossRef] [PubMed]
- Kishnani, P.S.; Amartino, H.M.; Lindberg, C.; Miller, T.M.; Wilson, A.; Keutzer, J.; Advisors, P.R.B.o. Timing of diagnosis of patients with Pompe disease: Data from the Pompe registry. Am. J. Med. Genet. A 2013, 161A, 2431–2443. [Google Scholar] [CrossRef] [PubMed]
- Prencipe, N.; Floriani, I.; Guaraldi, F.; Di Giacomo, S.V.; Cannavo, S.; Arnaldi, G.; Berton, A.; Torri, V.; Spinello, M.; Arvat, E.; et al. ACROSCORE: A new and simple tool for the diagnosis of acromegaly, a rare and underdiagnosed disease. Clin. Endocrinol. 2016, 84, 380–385. [Google Scholar] [CrossRef]
- Guffon, N.; Heron, B.; Chabrol, B.; Feillet, F.; Montauban, V.; Valayannopoulos, V. Diagnosis, quality of life, and treatment of patients with Hunter syndrome in the French healthcare system: A retrospective observational study. Orphanet J. Rare Dis. 2015, 10, 43. [Google Scholar] [CrossRef]
- Pierucci, P.; Lenato, G.M.; Suppressa, P.; Lastella, P.; Triggiani, V.; Valerio, R.; Comelli, M.; Salvante, D.; Stella, A.; Resta, N.; et al. A long diagnostic delay in patients with Hereditary Haemorrhagic Telangiectasia: A questionnaire-based retrospective study. Orphanet J. Rare Dis. 2012, 7, 33. [Google Scholar] [CrossRef] [Green Version]
- Dziurdzik, S.K.; Bean, B.D.M.; Davey, M.; Conibear, E. A VPS13D spastic ataxia mutation disrupts the conserved adaptor-binding site in yeast Vps13. Hum. Mol. Genet. 2020, 29, 635–648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, J.S.; Hollingsworth, N.M.; Neiman, A.M. Genetic Dissection of Vps13 Regulation in Yeast Using Disease Mutations from Human Orthologs. Int. J. Mol. Sci. 2021, 22, 6200. [Google Scholar] [CrossRef] [PubMed]
- Rzepnikowska, W.; Kaminska, J.; Kabzińska, D.; Kochański, A. Pathogenic Effect of GDAP1 Gene Mutations in a Yeast Model. Genes 2020, 11, 310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rzepnikowska, W.; Flis, K.; Muñoz-Braceras, S.; Menezes, R.; Escalante, R.; Zoladek, T. Yeast and other lower eukaryotic organisms for studies of Vps13 proteins in health and disease. Traffic 2017, 18, 711–719. [Google Scholar] [CrossRef] [Green Version]
- Dautant, A.; Meier, T.; Hahn, A.; Tribouillard-Tanvier, D.; di Rago, J.P.; Kucharczyk, R. ATP Synthase Diseases of Mitochondrial Genetic Origin. Front. Physiol. 2018, 9, 329. [Google Scholar] [CrossRef]
- Strynatka, K.A.; Gurrola-Gal, M.C.; Berman, J.N.; McMaster, C.R. How Surrogate and Chemical Genetics in Model Organisms Can Suggest Therapies for Human Genetic Diseases. Genetics 2018, 208, 833–851. [Google Scholar] [CrossRef] [Green Version]
- Oliveira, A.V.; Vilaça, R.; Santos, C.N.; Costa, V.; Menezes, R. Exploring the power of yeast to model aging and age-related neurodegenerative disorders. Biogerontology 2017, 18, 3–34. [Google Scholar] [CrossRef]
- Sampaio-Marques, B.; Guedes, A.; Vasilevskiy, I.; Gonçalves, S.; Outeiro, T.F.; Winderickx, J.; Burhans, W.C.; Ludovico, P. α-Synuclein toxicity in yeast and human cells is caused by cell cycle re-entry and autophagy degradation of ribonucleotide reductase 1. Aging Cell 2019, 18, e12922. [Google Scholar] [CrossRef] [Green Version]
- Ceccatelli Berti, C.; di Punzio, G.; Dallabona, C.; Baruffini, E.; Goffrini, P.; Lodi, T.; Donnini, C. The Power of Yeast in Modelling Human Nuclear Mutations Associated with Mitochondrial Diseases. Genes 2021, 12, 300. [Google Scholar] [CrossRef]
- Lasserre, J.P.; Dautant, A.; Aiyar, R.S.; Kucharczyk, R.; Glatigny, A.; Tribouillard-Tanvier, D.; Rytka, J.; Blondel, M.; Skoczen, N.; Reynier, P.; et al. Yeast as a system for modeling mitochondrial disease mechanisms and discovering therapies. Dis. Models Mech. 2015, 8, 509–526. [Google Scholar] [CrossRef] [Green Version]
- Su, X.; Dautant, A.; Godard, F.; Bouhier, M.; Zoladek, T.; Kucharczyk, R.; di Rago, J.P.; Tribouillard-Tanvier, D. Molecular Basis of the Pathogenic Mechanism Induced by the m.9191T>C Mutation in Mitochondrial ATP6 Gene. Int. J. Mol. Sci. 2020, 21, 5083. [Google Scholar] [CrossRef] [PubMed]
- Yu, R.; Nielsen, J. Yeast systems biology in understanding principles of physiology underlying complex human diseases. Curr. Opin. Biotechnol. 2020, 63, 63–69. [Google Scholar] [CrossRef] [PubMed]
- Parkhitko, A.A.; Filine, E.; Mohr, S.E.; Moskalev, A.; Perrimon, N. Targeting metabolic pathways for extension of lifespan and healthspan across multiple species. Ageing Res. Rev. 2020, 64, 101188. [Google Scholar] [CrossRef] [PubMed]
- Sampaio-Marques, B.; Burhans, W.C.; Ludovico, P. Yeast at the Forefront of Research on Ageing and Age-Related Diseases. Prog. Mol. Subcell. Biol. 2019, 58, 217–242. [Google Scholar] [CrossRef]
- Ishikawa, T. Saccharomyces cerevisiae in neuroscience: How unicellular organism helps to better understand prion protein? Neural Regen. Res. 2021, 16, 489–495. [Google Scholar] [CrossRef] [PubMed]
- Chernova, T.A.; Chernoff, Y.O.; Wilkinson, K.D. Yeast Models for Amyloids and Prions: Environmental Modulation and Drug Discovery. Molecules 2019, 24, 3388. [Google Scholar] [CrossRef] [Green Version]
- Cervelli, T.; Lodovichi, S.; Bellè, F.; Galli, A. Yeast-based assays for the functional characterization of cancer-associated variants of human DNA repair genes. Microb. Cell 2020, 7, 162–174. [Google Scholar] [CrossRef] [PubMed]
- Duronio, R.J.; O’Farrell, P.H.; Sluder, G.; Su, T.T. Sophisticated lessons from simple organisms: Appreciating the value of curiosity-driven research. Dis. Models Mech. 2017, 10, 1381–1389. [Google Scholar] [CrossRef] [Green Version]
- Kachroo, A.H.; Laurent, J.M.; Yellman, C.M.; Meyer, A.G.; Wilke, C.O.; Marcotte, E.M. Evolution. Systematic humanization of yeast genes reveals conserved functions and genetic modularity. Science 2015, 348, 921–925. [Google Scholar] [CrossRef] [Green Version]
- Skjørringe, T.; Amstrup Pedersen, P.; Salling Thorborg, S.; Nissen, P.; Gourdon, P.; Birk Møller, L. Characterization of ATP7A missense mutants suggests a correlation between intracellular trafficking and severity of Menkes disease. Sci. Rep. 2017, 7, 757. [Google Scholar] [CrossRef]
- Ponnandai Shanmugavel, K.; Petranovic, D.; Wittung-Stafshede, P. Probing functional roles of Wilson disease protein (ATP7B) copper-binding domains in yeast. Metallomics 2017, 9, 981–988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shanmugavel, K.P.; Kumar, R.; Li, Y.; Wittung-Stafshede, P. Wilson disease missense mutations in ATP7B affect metal-binding domain structural dynamics. Biometals 2019, 32, 875–885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cervelli, T.; Galli, A. Yeast as a Tool to Understand the Significance of Human Disease-Associated Gene Variants. Genes 2021, 12, 1303. [Google Scholar] [CrossRef] [PubMed]
- Tardiff, D.F.; Jui, N.T.; Khurana, V.; Tambe, M.A.; Thompson, M.L.; Chung, C.Y.; Kamadurai, H.B.; Kim, H.T.; Lancaster, A.K.; Caldwell, K.A.; et al. Yeast reveal a “druggable” Rsp5/Nedd4 network that ameliorates α-synuclein toxicity in neurons. Science 2013, 342, 979–983. [Google Scholar] [CrossRef] [Green Version]
- van Leeuwen, J.; Pons, C.; Boone, C.; Andrews, B.J. Mechanisms of suppression: The wiring of genetic resilience. Bioessays 2017, 39, 42. [Google Scholar] [CrossRef]
- Chung, C.Y.; Khurana, V.; Auluck, P.K.; Tardiff, D.F.; Mazzulli, J.R.; Soldner, F.; Baru, V.; Lou, Y.; Freyzon, Y.; Cho, S.; et al. Identification and rescue of α-synuclein toxicity in Parkinson patient-derived neurons. Science 2013, 342, 983–987. [Google Scholar] [CrossRef] [Green Version]
- Aiyar, R.S.; Bohnert, M.; Duvezin-Caubet, S.; Voisset, C.; Gagneur, J.; Fritsch, E.S.; Couplan, E.; von der Malsburg, K.; Funaya, C.; Soubigou, F.; et al. Mitochondrial protein sorting as a therapeutic target for ATP synthase disorders. Nat. Commun. 2014, 5, 5585. [Google Scholar] [CrossRef] [Green Version]
- Duffy, S.; Fam, H.K.; Wang, Y.K.; Styles, E.B.; Kim, J.H.; Ang, J.S.; Singh, T.; Larionov, V.; Shah, S.P.; Andrews, B.; et al. Overexpression screens identify conserved dosage chromosome instability genes in yeast and human cancer. Proc. Natl. Acad. Sci. USA 2016, 113, 9967–9976. [Google Scholar] [CrossRef] [Green Version]
- Wong, L.H.; Unciti-Broceta, A.; Spitzer, M.; White, R.; Tyers, M.; Harrington, L. A yeast chemical genetic screen identifies inhibitors of human telomerase. Chem. Biol. 2013, 20, 333–340. [Google Scholar] [CrossRef] [Green Version]
- Stafa, K.; Trancikova, A.; Webber, P.J.; Glauser, L.; West, A.B.; Moore, D.J. GTPase activity and neuronal toxicity of Parkinson’s disease-associated LRRK2 is regulated by ArfGAP1. PLoS Genet. 2012, 8, e1002526. [Google Scholar] [CrossRef]
- Couplan, E.; Aiyar, R.S.; Kucharczyk, R.; Kabala, A.; Ezkurdia, N.; Gagneur, J.; St Onge, R.P.; Salin, B.; Soubigou, F.; Le Cann, M.; et al. A yeast-based assay identifies drugs active against human mitochondrial disorders. Proc. Natl. Acad. Sci. USA 2011, 108, 11989–11994. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soczewka, P.; Flis, K.; Tribouillard-Tanvier, D.; di Rago, J.P.; Santos, C.N.; Menezes, R.; Kaminska, J.; Zoladek, T. Flavonoids as Potential Drugs for VPS13-Dependent Rare Neurodegenerative Diseases. Genes 2020, 11, 828. [Google Scholar] [CrossRef] [PubMed]
- Dziurdzik, S.K.; Conibear, E. The Vps13 Family of Lipid Transporters and Its Role at Membrane Contact Sites. Int. J. Mol. Sci. 2021, 22, 2905. [Google Scholar] [CrossRef] [PubMed]
- Ren, J.; Wen, L.; Gao, X.; Jin, C.; Xue, Y.; Yao, X. DOG 1.0: Illustrator of protein domain structures. Cell Res. 2009, 19, 271–273. [Google Scholar] [CrossRef] [PubMed]
- Bankaitis, V.A.; Johnson, L.M.; Emr, S.D. Isolation of yeast mutants defective in protein targeting to the vacuole. Proc. Natl. Acad. Sci. USA 1986, 83, 9075–9079. [Google Scholar] [CrossRef] [Green Version]
- Rothman, J.H.; Howald, I.; Stevens, T.H. Characterization of genes required for protein sorting and vacuolar function in the yeast Saccharomyces cerevisiae. EMBO J. 1989, 8, 2057–2065. [Google Scholar] [CrossRef]
- Brickner, J.H.; Fuller, R.S. SOI1 encodes a novel, conserved protein that promotes TGN-endosomal cycling of Kex2p and other membrane proteins by modulating the function of two TGN localization signals. J. Cell Biol. 1997, 139, 23–36. [Google Scholar] [CrossRef]
- Redding, K.; Brickner, J.H.; Marschall, L.G.; Nichols, J.W.; Fuller, R.S. Allele-specific suppression of a defective trans-Golgi network (TGN) localization signal in Kex2p identifies three genes involved in localization of TGN transmembrane proteins. Mol. Cell Biol. 1996, 16, 6208–6217. [Google Scholar] [CrossRef] [Green Version]
- Rzepnikowska, W.; Flis, K.; Kaminska, J.; Grynberg, M.; Urbanek, A.; Ayscough, K.R.; Zoladek, T. Amino acid substitution equivalent to human chorea-acanthocytosis I2771R in yeast Vps13 protein affects its binding to phosphatidylinositol 3-phosphate. Hum. Mol. Genet. 2017, 26, 1497–1510. [Google Scholar] [CrossRef] [Green Version]
- Dalton, L.E.; Bean, B.D.M.; Davey, M.; Conibear, E. Quantitative high-content imaging identifies novel regulators of Neo1 trafficking at endosomes. Mol. Biol. Cell 2017, 28, 1539–1550. [Google Scholar] [CrossRef] [Green Version]
- De, M.; Oleskie, A.N.; Ayyash, M.; Dutta, S.; Mancour, L.; Abazeed, M.E.; Brace, E.J.; Skiniotis, G.; Fuller, R.S. The Vps13p-Cdc31p complex is directly required for TGN late endosome transport and TGN homotypic fusion. J. Cell Biol. 2017, 216, 425–439. [Google Scholar] [CrossRef] [PubMed]
- Park, J.S.; Thorsness, M.K.; Policastro, R.; McGoldrick, L.L.; Hollingsworth, N.M.; Thorsness, P.E.; Neiman, A.M. Yeast Vps13 promotes mitochondrial function and is localized at membrane contact sites. Mol. Biol. Cell 2016, 27, 2435–2449. [Google Scholar] [CrossRef] [PubMed]
- Galletta, B.J.; Mooren, O.L.; Cooper, J.A. Actin dynamics and endocytosis in yeast and mammals. Curr. Opin. Biotechnol. 2010, 21, 604–610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michelot, A.; Costanzo, M.; Sarkeshik, A.; Boone, C.; Yates, J.R.; Drubin, D.G. Reconstitution and protein composition analysis of endocytic actin patches. Curr. Biol. 2010, 20, 1890–1899. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, J.S.; Neiman, A.M. VPS13 regulates membrane morphogenesis during sporulation in Saccharomyces cerevisiae. J. Cell Sci. 2012, 125, 3004–3011. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakamura, T.S.; Suda, Y.; Muneshige, K.; Fujieda, Y.; Okumura, Y.; Inoue, I.; Tanaka, T.; Takahashi, T.; Nakanishi, H.; Gao, X.D.; et al. Suppression of Vps13 adaptor protein mutants reveals a central role for PI4P in regulating prospore membrane extension. PLoS Genet. 2021, 17, e1009727. [Google Scholar] [CrossRef]
- Scorrano, L.; De Matteis, M.A.; Emr, S.; Giordano, F.; Hajnóczky, G.; Kornmann, B.; Lackner, L.L.; Levine, T.P.; Pellegrini, L.; Reinisch, K.; et al. Coming together to define membrane contact sites. Nat. Communm. 2019, 10, 1287. [Google Scholar] [CrossRef]
- Kumar, N.; Leonzino, M.; Hancock-Cerutti, W.; Horenkamp, F.A.; Li, P.; Lees, J.A.; Wheeler, H.; Reinisch, K.M.; De Camilli, P. VPS13A and VPS13C are lipid transport proteins differentially localized at ER contact sites. J. Cell Biol. 2018, 217, 3625–3639. [Google Scholar] [CrossRef] [Green Version]
- Valverde, D.P.; Yu, S.; Boggavarapu, V.; Kumar, N.; Lees, J.A.; Walz, T.; Reinisch, K.M.; Melia, T.J. ATG2 transports lipids to promote autophagosome biogenesis. J. Cell Biol. 2019, 218, 1787–1798. [Google Scholar] [CrossRef] [Green Version]
- Li, P.; Lees, J.A.; Lusk, C.P.; Reinisch, K.M. Cryo-EM reconstruction of a VPS13 fragment reveals a long groove to channel lipids between membranes. J. Cell Biol. 2020, 219, 1161. [Google Scholar] [CrossRef]
- Prinz, W.A.; Hurley, J.H. A firehose for phospholipids. J. Cell Biol. 2020, 219, 3132. [Google Scholar] [CrossRef] [PubMed]
- Leonzino, M.; Reinisch, K.M.; De Camilli, P. Insights into VPS13 properties and function reveal a new mechanism of eukaryotic lipid transport. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2021, 1866, 159003. [Google Scholar] [CrossRef] [PubMed]
- Lang, A.B.; John Peter, A.T.; Walter, P.; Kornmann, B. ER-mitochondrial junctions can be bypassed by dominant mutations in the endosomal protein Vps13. J. Cell Biol. 2015, 210, 883–890. [Google Scholar] [CrossRef] [Green Version]
- González Montoro, A.; Auffarth, K.; Hönscher, C.; Bohnert, M.; Becker, T.; Warscheid, B.; Reggiori, F.; van der Laan, M.; Fröhlich, F.; Ungermann, C. Vps39 Interacts with Tom40 to Establish One of Two Functionally Distinct Vacuole-Mitochondria Contact Sites. Dev. Cell 2018, 45, 621–636.e627. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bean, B.D.M.; Dziurdzik, S.K.; Kolehmainen, K.L.; Fowler, C.M.S.; Kwong, W.K.; Grad, L.I.; Davey, M.; Schluter, C.; Conibear, E. Competitive organelle-specific adaptors recruit Vps13 to membrane contact sites. J. Cell Biol. 2018, 217, 3593–3607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- John Peter, A.T.; Herrmann, B.; Antunes, D.; Rapaport, D.; Dimmer, K.S.; Kornmann, B. Vps13-Mcp1 interact at vacuole-mitochondria interfaces and bypass ER-mitochondria contact sites. J. Cell Biol. 2017, 216, 3219–3229. [Google Scholar] [CrossRef] [Green Version]
- Park, J.S.; Okumura, Y.; Tachikawa, H.; Neiman, A.M. SPO71 encodes a developmental stage-specific partner for Vps13 in Saccharomyces cerevisiae. Eukaryot. Cell 2013, 12, 1530–1537. [Google Scholar] [CrossRef] [Green Version]
- Kolakowski, D.; Rzepnikowska, W.; Kaniak-Golik, A.; Zoladek, T.; Kaminska, J. The GTPase Arf1 Is a Determinant of Yeast Vps13 Localization to the Golgi Apparatus. Int. J. Mol. Sci. 2021, 22, 2274. [Google Scholar] [CrossRef]
- Kolakowski, D.; Kaminska, J.; Zoladek, T. The binding of the APT1 domains to phosphoinositides is regulated by metal ions in vitro. Biochim. Biophys. Acta Biomembr. 2020, 1862, 183349. [Google Scholar] [CrossRef]
- Velayos-Baeza, A.; Vettori, A.; Copley, R.R.; Dobson-Stone, C.; Monaco, A.P. Analysis of the human VPS13 gene family. Genomics 2004, 84, 536–549. [Google Scholar] [CrossRef]
- Ueno, S.; Maruki, Y.; Nakamura, M.; Tomemori, Y.; Kamae, K.; Tanabe, H.; Yamashita, Y.; Matsuda, S.; Kaneko, S.; Sano, A. The gene encoding a newly discovered protein, chorein, is mutated in chorea-acanthocytosis. Nat. Genet. 2001, 28, 121–122. [Google Scholar] [CrossRef] [PubMed]
- Rampoldi, L.; Dobson-Stone, C.; Rubio, J.P.; Danek, A.; Chalmers, R.M.; Wood, N.W.; Verellen, C.; Ferrer, X.; Malandrini, A.; Fabrizi, G.M.; et al. A conserved sorting-associated protein is mutant in chorea-acanthocytosis. Nat. Genet. 2001, 28, 119–120. [Google Scholar] [CrossRef] [PubMed]
- Rubio, J.P.; Danek, A.; Stone, C.; Chalmers, R.; Wood, N.; Verellen, C.; Ferrer, X.; Malandrini, A.; Fabrizi, G.M.; Manfredi, M.; et al. Chorea-acanthocytosis: Genetic linkage to chromosome 9q21. Am. J. Hum. Genet. 1997, 61, 899–908. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peikert, K.; Danek, A.; Hermann, A. Current state of knowledge in Chorea-Acanthocytosis as core Neuroacanthocytosis syndrome. Eur. J. Med. Genet. 2018, 61, 699–705. [Google Scholar] [CrossRef]
- Dobson-Stone, C.; Danek, A.; Rampoldi, L.; Hardie, R.J.; Chalmers, R.M.; Wood, N.W.; Bohlega, S.; Dotti, M.T.; Federico, A.; Shizuka, M.; et al. Mutational spectrum of the CHAC gene in patients with chorea-acanthocytosis. Eur. J. Hum. Genet. 2002, 10, 773–781. [Google Scholar] [CrossRef] [Green Version]
- Saiki, S.; Sakai, K.; Murata, K.Y.; Saiki, M.; Nakanishi, M.; Kitagawa, Y.; Kaito, M.; Gondo, Y.; Kumamoto, T.; Matsui, M.; et al. Primary skeletal muscle involvement in chorea-acanthocytosis. Mov. Disord. 2007, 22, 848–852. [Google Scholar] [CrossRef]
- Nishida, Y.; Nakamura, M.; Urata, Y.; Kasamo, K.; Hiwatashi, H.; Yokoyama, I.; Mizobuchi, M.; Sakurai, K.; Osaki, Y.; Morita, Y.; et al. Novel pathogenic VPS13A gene mutations in Japanese patients with chorea-acanthocytosis. Neurol. Genet. 2019, 5, e332. [Google Scholar] [CrossRef] [Green Version]
- Dobson-Stone, C.; Rampoldi, L.; Bader, B.; Walker, R.H.; Danek, A.; Monaco, A.P. Chorea-Acanthocytosis; University of Washington: Seattle, WA, USA, 2019. [Google Scholar]
- Gold, M.M.; Shifteh, K.; Bello, J.A.; Lipton, M.; Kaufman, D.M.; Brown, A.D. Chorea-acanthocytosis: A mimicker of Huntington disease case report and review of the literature. Neurologist 2006, 12, 327–329. [Google Scholar] [CrossRef]
- Lu, X.; Wang, W.; Liu, Y.; Song, N.; Li, M.; Mu, X.; Zhang, N.; Chen, Q.; Jiang, L.; Kong, X.; et al. Establishment and characterization of human induced pluripotent stem cell line from a Parkinson’s disease patient harboring VPS13A gene mutation. Stem. Cell Res. 2022, 60, 102685. [Google Scholar] [CrossRef]
- Bayreuther, C.; Borg, M.; Ferrero-Vacher, C.; Chaussenot, A.; Lebrun, C. [Chorea-acanthocytosis without acanthocytes]. Rev. Neurol. 2010, 166, 100–103. [Google Scholar] [CrossRef]
- Yeshaw, W.M.; van der Zwaag, M.; Pinto, F.; Lahaye, L.L.; Faber, A.I.; Gómez-Sánchez, R.; Dolga, A.M.; Poland, C.; Monaco, A.P.; van IJzendoorn, S.C.; et al. Human VPS13A is associated with multiple organelles and influences mitochondrial morphology and lipid droplet motility. eLife 2019, 8, e43561. [Google Scholar] [CrossRef] [PubMed]
- Muñoz-Braceras, S.; Tornero-Écija, A.R.; Vincent, O.; Escalante, R. VPS13A is closely associated with mitochondria and is required for efficient lysosomal degradation. Dis. Models Mech. 2019, 12, 6681. [Google Scholar] [CrossRef] [Green Version]
- Muñoz-Braceras, S.; Calvo, R.; Escalante, R. TipC and the chorea-acanthocytosis protein VPS13A regulate autophagy in Dictyostelium and human HeLa cells. Autophagy 2015, 11, 918–927. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmidt, E.M.; Schmid, E.; Münzer, P.; Hermann, A.; Eyrich, A.K.; Russo, A.; Walker, B.; Gu, S.; vom Hagen, J.M.; Faggio, C.; et al. Chorein sensitivity of cytoskeletal organization and degranulation of platelets. FASEB J. 2013, 27, 2799–2806. [Google Scholar] [CrossRef] [PubMed]
- Föller, M.; Hermann, A.; Gu, S.; Alesutan, I.; Qadri, S.M.; Borst, O.; Schmidt, E.M.; Schiele, F.; vom Hagen, J.M.; Saft, C.; et al. Chorein-sensitive polymerization of cortical actin and suicidal cell death in chorea-acanthocytosis. FASEB J. 2012, 26, 1526–1534. [Google Scholar] [CrossRef]
- Shiokawa, N.; Nakamura, M.; Sameshima, M.; Deguchi, A.; Hayashi, T.; Sasaki, N.; Sano, A. Chorein, the protein responsible for chorea-acanthocytosis, interacts with β-adducin and β-actin. Biochem. Biophys. Res. Commun. 2013, 441, 96–101. [Google Scholar] [CrossRef]
- Porro, F.; Rosato-Siri, M.; Leone, E.; Costessi, L.; Iaconcig, A.; Tongiorgi, E.; Muro, A.F. beta-adducin (Add2) KO mice show synaptic plasticity, motor coordination and behavioral deficits accompanied by changes in the expression and phosphorylation levels of the alpha- and gamma-adducin subunits. Genes Brain Behav. 2010, 9, 84–96. [Google Scholar] [CrossRef]
- Franco, T.; Low, P.S. Erythrocyte adducin: A structural regulator of the red blood cell membrane. Transfus. Clin. Biol. 2010, 17, 87–94. [Google Scholar] [CrossRef] [Green Version]
- Stanslowsky, N.; Reinhardt, P.; Glass, H.; Kalmbach, N.; Naujock, M.; Hensel, N.; Lübben, V.; Pal, A.; Venneri, A.; Lupo, F.; et al. Neuronal Dysfunction in iPSC-Derived Medium Spiny Neurons from Chorea-Acanthocytosis Patients Is Reversed by Src Kinase Inhibition and F-Actin Stabilization. J. Neurosci. 2016, 36, 12027–12043. [Google Scholar] [CrossRef] [Green Version]
- De Franceschi, L.; Tomelleri, C.; Matte, A.; Brunati, A.M.; Bovee-Geurts, P.H.; Bertoldi, M.; Lasonder, E.; Tibaldi, E.; Danek, A.; Walker, R.H.; et al. Erythrocyte membrane changes of chorea-acanthocytosis are the result of altered Lyn kinase activity. Blood 2011, 118, 5652–5663. [Google Scholar] [CrossRef] [Green Version]
- Park, J.S.; Neiman, A.M. XK is a partner for VPS13A: A molecular link between Chorea-Acanthocytosis and McLeod Syndrome. Mol. Biol. Cell 2020, 31, 2425–2436. [Google Scholar] [CrossRef] [PubMed]
- Ryoden, Y.; Segawa, K.; Nagata, S. Requirement of Xk and Vps13a for the P2X7-mediated phospholipid scrambling and cell lysis in mouse T cells. Proc. Natl. Acad. Sci. USA 2022, 119, e2119286119. [Google Scholar] [CrossRef] [PubMed]
- Urata, Y.; Nakamura, M.; Sasaki, N.; Shiokawa, N.; Nishida, Y.; Arai, K.; Hiwatashi, H.; Yokoyama, I.; Narumi, S.; Terayama, Y.; et al. Novel pathogenic XK mutations in McLeod syndrome and interaction between XK protein and chorein. Neurol. Genet. 2019, 5, e328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peikert, K.; Hermann, A.; Danek, A. XK-Associated McLeod Syndrome: Nonhematological Manifestations and Relation to VPS13A Disease. Transfus. Med. Hemother. 2022, 49, 4–12. [Google Scholar] [CrossRef] [PubMed]
- Kolehmainen, J.; Black, G.C.; Saarinen, A.; Chandler, K.; Clayton-Smith, J.; Träskelin, A.L.; Perveen, R.; Kivitie-Kallio, S.; Norio, R.; Warburg, M.; et al. Cohen syndrome is caused by mutations in a novel gene, COH1, encoding a transmembrane protein with a presumed role in vesicle-mediated sorting and intracellular protein transport. Am. J. Hum. Genet. 2003, 72, 1359–1369. [Google Scholar] [CrossRef] [Green Version]
- Rodrigues, J.M.; Fernandes, H.D.; Caruthers, C.; Braddock, S.R.; Knutsen, A.P. Cohen Syndrome: Review of the Literature. Cureus 2018, 10, e3330. [Google Scholar] [CrossRef] [Green Version]
- Seifert, W.; Holder-Espinasse, M.; Kühnisch, J.; Kahrizi, K.; Tzschach, A.; Garshasbi, M.; Najmabadi, H.; Walter Kuss, A.; Kress, W.; Laureys, G.; et al. Expanded mutational spectrum in Cohen syndrome, tissue expression, and transcript variants of COH1. Hum. Mutat. 2009, 30, E404–E420. [Google Scholar] [CrossRef]
- Seifert, W.; Kühnisch, J.; Maritzen, T.; Horn, D.; Haucke, V.; Hennies, H.C. Cohen syndrome-associated protein, COH1, is a novel, giant Golgi matrix protein required for Golgi integrity. J. Biol. Chem. 2011, 286, 37665–37675. [Google Scholar] [CrossRef] [Green Version]
- Seifert, W.; Kühnisch, J.; Maritzen, T.; Lommatzsch, S.; Hennies, H.C.; Bachmann, S.; Horn, D.; Haucke, V. Cohen syndrome-associated protein COH1 physically and functionally interacts with the small GTPase RAB6 at the Golgi complex and directs neurite outgrowth. J. Biol. Chem. 2015, 290, 3349–3358. [Google Scholar] [CrossRef] [Green Version]
- Goud, B.; Yang, C.; Roa, M.; Martinez, O.; Slepnev, V. Study of Rab6, a ras-like GTP-binding protein associated with the Golgi complex. Ann. N. Y. Acad. Sci. 1994, 733, 340–343. [Google Scholar] [CrossRef]
- Martinez, O.; Antony, C.; Pehau-Arnaudet, G.; Berger, E.G.; Salamero, J.; Goud, B. GTP-bound forms of rab6 induce the redistribution of Golgi proteins into the endoplasmic reticulum. Proc. Natl. Acad. Sci. USA 1997, 94, 1828–1833. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grigoriev, I.; Splinter, D.; Keijzer, N.; Wulf, P.S.; Demmers, J.; Ohtsuka, T.; Modesti, M.; Maly, I.V.; Grosveld, F.; Hoogenraad, C.C.; et al. Rab6 regulates transport and targeting of exocytotic carriers. Dev. Cell 2007, 13, 305–314. [Google Scholar] [CrossRef] [Green Version]
- Duplomb, L.; Duvet, S.; Picot, D.; Jego, G.; El Chehadeh-Djebbar, S.; Marle, N.; Gigot, N.; Aral, B.; Carmignac, V.; Thevenon, J.; et al. Cohen syndrome is associated with major glycosylation defects. Hum. Mol. Genet. 2014, 23, 2391–2399. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.K.; Lee, S.K.; Choi, S.; Huh, Y.H.; Kwak, J.H.; Lee, Y.S.; Jang, D.J.; Lee, J.H.; Lee, K.; Kaang, B.K.; et al. Autophagy pathway upregulation in a human iPSC-derived neuronal model of Cohen syndrome with VPS13B missense mutations. Mol. Brain 2020, 13, 69. [Google Scholar] [CrossRef] [PubMed]
- Jansen, I.E.; Ye, H.; Heetveld, S.; Lechler, M.C.; Michels, H.; Seinstra, R.I.; Lubbe, S.J.; Drouet, V.; Lesage, S.; Majounie, E.; et al. Discovery and functional prioritization of Parkinson’s disease candidate genes from large-scale whole exome sequencing. Genome Biol. 2017, 18, 22. [Google Scholar] [CrossRef] [Green Version]
- Smolders, S.; Philtjens, S.; Crosiers, D.; Sieben, A.; Hens, E.; Heeman, B.; Van Mossevelde, S.; Pals, P.; Asselbergh, B.; Dos Santos Dias, R.; et al. Contribution of rare homozygous and compound heterozygous VPS13C missense mutations to dementia with Lewy bodies and Parkinson’s disease. Acta Neuropathol. Commun. 2021, 9, 25. [Google Scholar] [CrossRef] [PubMed]
- Schormair, B.; Kemlink, D.; Mollenhauer, B.; Fiala, O.; Machetanz, G.; Roth, J.; Berutti, R.; Strom, T.M.; Haslinger, B.; Trenkwalder, C.; et al. Diagnostic exome sequencing in early-onset Parkinson’s disease confirms VPS13C as a rare cause of autosomal-recessive Parkinson’s disease. Clin. Genet. 2018, 93, 603–612. [Google Scholar] [CrossRef] [Green Version]
- Saxena, R.; Hivert, M.F.; Langenberg, C.; Tanaka, T.; Pankow, J.S.; Vollenweider, P.; Lyssenko, V.; Bouatia-Naji, N.; Dupuis, J.; Jackson, A.U.; et al. Genetic variation in GIPR influences the glucose and insulin responses to an oral glucose challenge. Nat. Genet. 2010, 42, 142–148. [Google Scholar] [CrossRef]
- Strawbridge, R.J.; Dupuis, J.; Prokopenko, I.; Barker, A.; Ahlqvist, E.; Rybin, D.; Petrie, J.R.; Travers, M.E.; Bouatia-Naji, N.; Dimas, A.S.; et al. Genome-wide association identifies nine common variants associated with fasting proinsulin levels and provides new insights into the pathophysiology of type 2 diabetes. Diabetes 2011, 60, 2624–2634. [Google Scholar] [CrossRef] [Green Version]
- Ingelsson, E.; Langenberg, C.; Hivert, M.F.; Prokopenko, I.; Lyssenko, V.; Dupuis, J.; Mägi, R.; Sharp, S.; Jackson, A.U.; Assimes, T.L.; et al. Detailed physiologic characterization reveals diverse mechanisms for novel genetic Loci regulating glucose and insulin metabolism in humans. Diabetes 2010, 59, 1266–1275. [Google Scholar] [CrossRef] [Green Version]
- Grarup, N.; Overvad, M.; Sparsø, T.; Witte, D.R.; Pisinger, C.; Jørgensen, T.; Yamauchi, T.; Hara, K.; Maeda, S.; Kadowaki, T.; et al. The diabetogenic VPS13C/C2CD4A/C2CD4B rs7172432 variant impairs glucose-stimulated insulin response in 5,722 non-diabetic Danish individuals. Diabetologia 2011, 54, 789–794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lesage, S.; Drouet, V.; Majounie, E.; Deramecourt, V.; Jacoupy, M.; Nicolas, A.; Cormier-Dequaire, F.; Hassoun, S.M.; Pujol, C.; Ciura, S.; et al. Loss of VPS13C Function in Autosomal-Recessive Parkinsonism Causes Mitochondrial Dysfunction and Increases PINK1/Parkin-Dependent Mitophagy. Am. J. Hum. Genet. 2016, 98, 500–513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, R.Y.; Xue, H.; Yu, L.; Velayos-Baeza, A.; Monaco, A.P.; Liu, F.T. Identification of VPS13C as a Galectin-12-Binding Protein That Regulates Galectin-12 Protein Stability and Adipogenesis. PLoS ONE 2016, 11, e0153534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, R.Y.; Yu, L.; Graham, J.L.; Hsu, D.K.; Lloyd, K.C.; Havel, P.J.; Liu, F.T. Ablation of a galectin preferentially expressed in adipocytes increases lipolysis, reduces adiposity, and improves insulin sensitivity in mice. Proc. Natl. Acad. Sci. USA 2011, 108, 18696–18701. [Google Scholar] [CrossRef] [Green Version]
- Yang, R.Y.; Hsu, D.K.; Yu, L.; Chen, H.Y.; Liu, F.T. Galectin-12 is required for adipogenic signaling and adipocyte differentiation. J. Biol. Chem. 2004, 279, 29761–29766. [Google Scholar] [CrossRef] [Green Version]
- Gauthier, J.; Meijer, I.A.; Lessel, D.; Mencacci, N.E.; Krainc, D.; Hempel, M.; Tsiakas, K.; Prokisch, H.; Rossignol, E.; Helm, M.H.; et al. Recessive mutations in >VPS13D cause childhood onset movement disorders. Ann. Neurol. 2018, 83, 1089–1095. [Google Scholar] [CrossRef]
- Seong, E.; Insolera, R.; Dulovic, M.; Kamsteeg, E.J.; Trinh, J.; Brüggemann, N.; Sandford, E.; Li, S.; Ozel, A.B.; Li, J.Z.; et al. Mutations in VPS13D lead to a new recessive ataxia with spasticity and mitochondrial defects. Ann. Neurol. 2018, 83, 1075–1088. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Xu, Y.Y.; Gao, Y.; Li, J.M.; Liu, X.W.; Wang, M.Q.; Deng, H.; Xiao, L.L.; Ren, H.B.; Xiong, B.T.; et al. Deep brain stimulation for chorea-acanthocytosis: A systematic review. Neurosurg. Rev. 2022; online ahead of print. [Google Scholar] [CrossRef]
- Anding, A.L.; Wang, C.; Chang, T.K.; Sliter, D.A.; Powers, C.M.; Hofmann, K.; Youle, R.J.; Baehrecke, E.H. Vps13D Encodes a Ubiquitin-Binding Protein that Is Required for the Regulation of Mitochondrial Size and Clearance. Curr. Biol. 2018, 28, 287–295.e286. [Google Scholar] [CrossRef] [Green Version]
- Guillén-Samander, A.; Leonzino, M.; Hanna, M.G.; Tang, N.; Shen, H.; De Camilli, P. VPS13D bridges the ER to mitochondria and peroxisomes via Miro. J. Cell Biol. 2021, 220, e202010004. [Google Scholar] [CrossRef]
- Baldwin, H.A.; Wang, C.; Kanfer, G.; Shah, H.V.; Velayos-Baeza, A.; Dulovic-Mahlow, M.; Brüggemann, N.; Anding, A.; Baehrecke, E.H.; Maric, D.; et al. VPS13D promotes peroxisome biogenesis. J. Cell Biol. 2021, 220, 202001188. [Google Scholar] [CrossRef]
- Wang, J.; Fang, N.; Xiong, J.; Du, Y.; Cao, Y.; Ji, W.K. An ESCRT-dependent step in fatty acid transfer from lipid droplets to mitochondria through VPS13D-TSG101 interactions. Nat. Commun. 2021, 12, 1252. [Google Scholar] [CrossRef] [PubMed]
- Peikert, K.; Glaß, H.; Federti, E.; Matte, A.; Pelzl, L.; Akgün, K.; Ziemssen, T.; Ordemann, R.; Lang, F.; Patients, T.N.F.T.; et al. Targeting Lyn Kinase in Chorea-Acanthocytosis: A Translational Treatment Approach in a Rare Disease. J. Pers. Med. 2021, 11, 392. [Google Scholar] [CrossRef] [PubMed]
- Federti, E.; Matte, A.; Riccardi, V.; Peikert, K.; Alper, S.L.; Danek, A.; Walker, R.H.; Siciliano, A.; Iatcenko, I.; Hermann, A.; et al. Adaptative Up-Regulation of PRX2 and PRX5 Expression Characterizes Brain from a Mouse Model of Chorea-Acanthocytosis. Antioxidants 2021, 11, 76. [Google Scholar] [CrossRef] [PubMed]
- Peikert, K.; Federti, E.; Matte, A.; Constantin, G.; Pietronigro, E.C.; Fabene, P.F.; Defilippi, P.; Turco, E.; Del Gallo, F.; Pucci, P.; et al. Therapeutic targeting of Lyn kinase to treat chorea-acanthocytosis. Acta Neuropathol. Commun. 2021, 9, 81. [Google Scholar] [CrossRef] [PubMed]
- Soczewka, P.; Kolakowski, D.; Smaczynska-de Rooij, I.; Rzepnikowska, W.; Ayscough, K.R.; Kaminska, J.; Zoladek, T. Yeast-model-based study identified myosin- and calcium-dependent calmodulin signalling as a potential target for drug intervention in chorea-acanthocytosis. Dis. Models Mech. 2019, 12, dmm036830. [Google Scholar] [CrossRef] [Green Version]
- Goodson, H.V.; Anderson, B.L.; Warrick, H.M.; Pon, L.A.; Spudich, J.A. Synthetic lethality screen identifies a novel yeast myosin I gene (MYO5): Myosin I proteins are required for polarization of the actin cytoskeleton. J. Cell Biol. 1996, 133, 1277–1291. [Google Scholar] [CrossRef] [Green Version]
- Geli, M.I.; Riezman, H. Role of type I myosins in receptor-mediated endocytosis in yeast. Science 1996, 272, 533–535. [Google Scholar] [CrossRef]
- Cyert, M.S. Calcineurin signaling in Saccharomyces cerevisiae: How yeast go crazy in response to stress. Biochem. Biophys. Res. Commun. 2003, 311, 1143–1150. [Google Scholar] [CrossRef]
- Cyert, M.S.; Kunisawa, R.; Kaim, D.; Thorner, J. Yeast has homologs (CNA1 and CNA2 gene products) of mammalian calcineurin, a calmodulin-regulated phosphoprotein phosphatase. Proc. Natl. Acad. Sci. USA 1991, 88, 7376–7380. [Google Scholar] [CrossRef] [Green Version]
- Cyert, M.S.; Thorner, J. Regulatory subunit (CNB1 gene product) of yeast Ca2+/calmodulin-dependent phosphoprotein phosphatases is required for adaptation to pheromone. Mol. Cell Biol. 1992, 12, 3460–3469. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Ishii, S.; Tokai, M.; Tsutsumi, H.; Ohki, O.; Akada, R.; Tanaka, K.; Tsuchiya, E.; Fukui, S.; Miyakawa, T. The Saccharomyces cerevisiae genes (CMP1 and CMP2) encoding calmodulin-binding proteins homologous to the catalytic subunit of mammalian protein phosphatase 2B. Mol. Gen. Genet. 1991, 227, 52–59. [Google Scholar] [CrossRef] [PubMed]
- Stathopoulos, A.M.; Cyert, M.S. Calcineurin acts through the CRZ1/TCN1-encoded transcription factor to regulate gene expression in yeast. Genes Dev. 1997, 11, 3432–3444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stathopoulos-Gerontides, A.; Guo, J.J.; Cyert, M.S. Yeast calcineurin regulates nuclear localization of the Crz1p transcription factor through dephosphorylation. Genes Dev. 1999, 13, 798–803. [Google Scholar] [CrossRef] [Green Version]
- Caraveo, G.; Soste, M.; Cappelleti, V.; Fanning, S.; van Rossum, D.B.; Whitesell, L.; Huang, Y.; Chung, C.Y.; Baru, V.; Zaichick, S.; et al. FKBP12 contributes to α-synuclein toxicity by regulating the calcineurin-dependent phosphoproteome. Proc. Natl. Acad. Sci. USA 2017, 114, E11313–E11322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wardaszka, P.; Soczewka, P.; Sienko, M.; Zoladek, T.; Kaminska, J. Partial Inhibition of Calcineurin Activity by Rcn2 as a Potential Remedy for Vps13 Deficiency. Int. J. Mol. Sci. 2021, 22, 1193. [Google Scholar] [CrossRef]
- Mehta, S.; Li, H.; Hogan, P.G.; Cunningham, K.W. Domain architecture of the regulators of calcineurin (RCANs) and identification of a divergent RCAN in yeast. Mol. Cell Biol. 2009, 29, 2777–2793. [Google Scholar] [CrossRef] [Green Version]
- Kingsbury, T.J.; Cunningham, K.W. A conserved family of calcineurin regulators. Genes Dev. 2000, 14, 1595–1604. [Google Scholar] [CrossRef]
- Francis, C.E.; Bai, Y. Differential expression of cyclosporine A-Induced calcineurin isoform-specific matrix metalloproteinase 9 (MMP-9) in renal fibroblasts. Biochem. Biophys. Res. Commun. 2018, 503, 2549–2554. [Google Scholar] [CrossRef]
- Usuda, N.; Arai, H.; Sasaki, H.; Hanai, T.; Nagata, T.; Muramatsu, T.; Kincaid, R.L.; Higuchi, S. Differential subcellular localization of neural isoforms of the catalytic subunit of calmodulin-dependent protein phosphatase (calcineurin) in central nervous system neurons: Immunohistochemistry on formalin-fixed paraffin sections employing antigen retrieval by microwave irradiation. J. Histochem. Cytochem. 1996, 44, 13–18. [Google Scholar] [CrossRef]
- Eastwood, S.L.; Salih, T.; Harrison, P.J. Differential expression of calcineurin A subunit mRNA isoforms during rat hippocampal and cerebellar development. Eur. J. Neurosci. 2005, 22, 3017–3024. [Google Scholar] [CrossRef]
- Zhang, Y.; Liu, R.B.; Cao, Q.; Fan, K.Q.; Huang, L.J.; Yu, J.S.; Gao, Z.J.; Huang, T.; Zhong, J.Y.; Mao, X.T.; et al. USP16-mediated deubiquitination of calcineurin A controls peripheral T cell maintenance. J. Clin. Investig. 2019, 129, 2856–2871. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Cheng, N.; Wang, P.; Li, J.; Jia, A.; Li, W.; Zhang, N.; Yin, Y.; Tong, L.; Wei, Q.; et al. A novel peptide exerts potent immunosuppression by blocking the two-site interaction of NFAT with calcineurin. J. Biol. Chem. 2020, 295, 2760–2770. [Google Scholar] [CrossRef] [PubMed]
- Noguchi, H.; Sugimoto, K.; Miyagi-Shiohira, C.; Nakashima, Y.; Kobayashi, N.; Saitoh, I.; Watanabe, M.; Noguchi, Y. RCAN-11R peptide provides immunosuppression for fully mismatched islet allografts in mice. Sci. Rep. 2017, 7, 3043. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kipanyula, M.J.; Kimaro, W.H.; Seke Etet, P.F. The Emerging Roles of the Calcineurin-Nuclear Factor of Activated T-Lymphocytes Pathway in Nervous System Functions and Diseases. J. Aging Res. 2016, 2016, 5081021. [Google Scholar] [CrossRef] [Green Version]
- Mizuguchi, T.; Nakashima, M.; Kato, M.; Okamoto, N.; Kurahashi, H.; Ekhilevitch, N.; Shiina, M.; Nishimura, G.; Shibata, T.; Matsuo, M.; et al. Loss-of-function and gain-of-function mutations in PPP3CA cause two distinct disorders. Hum. Mol. Genet. 2018, 27, 1421–1433. [Google Scholar] [CrossRef]
- Caraveo, G.; Auluck, P.K.; Whitesell, L.; Chung, C.Y.; Baru, V.; Mosharov, E.V.; Yan, X.; Ben-Johny, M.; Soste, M.; Picotti, P.; et al. Calcineurin determines toxic versus beneficial responses to α-synuclein. Proc. Natl. Acad. Sci. USA 2014, 111, E3544–E3552. [Google Scholar] [CrossRef] [Green Version]
- Prakriya, M.; Lewis, R.S. Store-Operated Calcium Channels. Physiol. Rev. 2015, 95, 1383–1436. [Google Scholar] [CrossRef] [Green Version]
- Wegierski, T.; Kuznicki, J. Neuronal calcium signaling via store-operated channels in health and disease. Cell Calcium 2018, 74, 102–111. [Google Scholar] [CrossRef]
- Ureshino, R.P.; Erustes, A.G.; Bassani, T.B.; Wachilewski, P.; Guarache, G.C.; Nascimento, A.C.; Costa, A.J.; Smaili, S.S.; Pereira, G.J.D.S. The Interplay between Ca2+ Signaling Pathways and Neurodegeneration. Int. J. Mol. Sci. 2019, 20, 6004. [Google Scholar] [CrossRef] [Green Version]
- Huang, D.X.; Yu, X.; Yu, W.J.; Zhang, X.M.; Liu, C.; Liu, H.P.; Sun, Y.; Jiang, Z.P. Calcium Signaling Regulated by Cellular Membrane Systems and Calcium Homeostasis Perturbed in Alzheimer’s Disease. Front. Cell Dev. Biol. 2022, 10, 834962. [Google Scholar] [CrossRef]
- Catoni, C.; Calì, T.; Brini, M. Calcium, Dopamine and Neuronal Calcium Sensor 1: Their Contribution to Parkinson’s Disease. Front. Mol. Neurosci. 2019, 12, 55. [Google Scholar] [CrossRef] [PubMed]
- Pchitskaya, E.; Popugaeva, E.; Bezprozvanny, I. Calcium signaling and molecular mechanisms underlying neurodegenerative diseases. Cell Calcium 2018, 70, 87–94. [Google Scholar] [CrossRef] [PubMed]
- Marambaud, P.; Dreses-Werringloer, U.; Vingtdeux, V. Calcium signaling in neurodegeneration. Mol. Neurodegener. 2009, 4, 20. [Google Scholar] [CrossRef] [PubMed]
- Pelzl, L.; Elsir, B.; Sahu, I.; Bissinger, R.; Singh, Y.; Sukkar, B.; Honisch, S.; Schoels, L.; Jemaà, M.; Lang, E.; et al. Lithium Sensitivity of Store Operated Ca2+ Entry and Survival of Fibroblasts Isolated from Chorea-Acanthocytosis Patients. Cell Physiol. Biochem. 2017, 42, 2066–2077. [Google Scholar] [CrossRef] [Green Version]
- Pelzl, L.; Hauser, S.; Elsir, B.; Sukkar, B.; Sahu, I.; Singh, Y.; Höflinger, P.; Bissinger, R.; Jemaà, M.; Stournaras, C.; et al. Lithium Sensitive ORAI1 Expression, Store Operated Ca. Sci. Rep. 2017, 7, 6457. [Google Scholar] [CrossRef]
- Bezprozvanny, I. Calcium signaling and neurodegenerative diseases. Trends Mol. Med. 2009, 15, 89–100. [Google Scholar] [CrossRef] [Green Version]
- Belaidi, A.A.; Bush, A.I. Iron neurochemistry in Alzheimer’s disease and Parkinson’s disease: Targets for therapeutics. J. Neurochem. 2016, 139 (Suppl. 1), 179–197. [Google Scholar] [CrossRef] [Green Version]
- Ejaz, H.W.; Wang, W.; Lang, M. Copper Toxicity Links to Pathogenesis of Alzheimer’s Disease and Therapeutics Approaches. Int. J. Mol. Sci. 2020, 21, 7660. [Google Scholar] [CrossRef]
- Gromadzka, G.; Tarnacka, B.; Flaga, A.; Adamczyk, A. Copper Dyshomeostasis in Neurodegenerative Diseases-Therapeutic Implications. Int. J. Mol. Sci. 2020, 21, 9259. [Google Scholar] [CrossRef]
- Behl, T.; Madaan, P.; Sehgal, A.; Singh, S.; Anwer, M.K.; Makeen, H.A.; Albratty, M.; Mohan, S.; Bungau, S. Mechanistic Insights Expatiating the Redox-Active-Metal-Mediated Neuronal Degeneration in Parkinson’s Disease. Int. J. Mol. Sci. 2022, 23, 678. [Google Scholar] [CrossRef]
- Ackerman, C.M.; Chang, C.J. Copper signaling in the brain and beyond. J. Biol. Chem. 2018, 293, 4628–4635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fox, J.H.; Kama, J.A.; Lieberman, G.; Chopra, R.; Dorsey, K.; Chopra, V.; Volitakis, I.; Cherny, R.A.; Bush, A.I.; Hersch, S. Mechanisms of copper ion mediated Huntington’s disease progression. PLoS ONE 2007, 2, e334. [Google Scholar] [CrossRef] [PubMed]
- Aaseth, J.; Skalny, A.V.; Roos, P.M.; Alexander, J.; Aschner, M.; Tinkov, A.A. Copper, Iron, Selenium and Lipo-Glycemic Dysmetabolism in Alzheimer’s Disease. Int. J. Mol. Sci. 2021, 22, 9461. [Google Scholar] [CrossRef] [PubMed]
- Schrag, M.; Mueller, C.; Oyoyo, U.; Smith, M.A.; Kirsch, W.M. Iron, zinc and copper in the Alzheimer’s disease brain: A quantitative meta-analysis. Some insight on the influence of citation bias on scientific opinion. Prog. Neurobiol. 2011, 94, 296–306. [Google Scholar] [CrossRef] [Green Version]
- Squitti, R.; Faller, P.; Hureau, C.; Granzotto, A.; White, A.R.; Kepp, K.P. Copper Imbalance in Alzheimer’s Disease and Its Link with the Amyloid Hypothesis: Towards a Combined Clinical, Chemical, and Genetic Etiology. J. Alzheimers Dis. 2021, 83, 23–41. [Google Scholar] [CrossRef]
- Li, D.D.; Zhang, W.; Wang, Z.Y.; Zhao, P. Serum Copper, Zinc, and Iron Levels in Patients with Alzheimer’s Disease: A Meta-Analysis of Case-Control Studies. Front. Aging Neurosci. 2017, 9, 300. [Google Scholar] [CrossRef] [Green Version]
- Guan, C.; Dang, R.; Cui, Y.; Liu, L.; Chen, X.; Wang, X.; Zhu, J.; Li, D.; Li, J.; Wang, D. Characterization of plasma metal profiles in Alzheimer’s disease using multivariate statistical analysis. PLoS ONE 2017, 12, e0178271. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.H.; Lee, S.M.; Baik, S.K. Demonstration of striatopallidal iron deposition in chorea-acanthocytosis by susceptibility-weighted imaging. J. Neurol. 2011, 258, 321–322. [Google Scholar] [CrossRef]
- Soczewka, P.; Tribouillard-Tanvier, D.; di Rago, J.P.; Zoladek, T.; Kaminska, J. Targeting Copper Homeostasis Improves Functioning of vps13Δ Yeast Mutant Cells, a Model of VPS13-Related Diseases. Int. J. Mol. Sci. 2021, 22, 2248. [Google Scholar] [CrossRef]
- de Andrade Teles, R.B.; Diniz, T.C.; Costa Pinto, T.C.; de Oliveira Júnior, R.G.; Gama E Silva, M.; de Lavor, É.; Fernandes, A.W.C.; de Oliveira, A.P.; de Almeida Ribeiro, F.P.R.; da Silva, A.A.M.; et al. Flavonoids as Therapeutic Agents in Alzheimer’s and Parkinson’s Diseases: A Systematic Review of Preclinical Evidences. Oxid. Med. Cell. Longev. 2018, 2018, 7043213. [Google Scholar] [CrossRef]
- Kumar, S.; Pandey, A.K. Chemistry and biological activities of flavonoids: An overview. Sci. World J. 2013, 2013, 162750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodríguez-García, C.; Sánchez-Quesada, C.; Toledo, E.; Delgado-Rodríguez, M.; Gaforio, J.J. Naturally Lignan-Rich Foods: A Dietary Tool for Health Promotion? Molecules 2019, 24, 917. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nabavi, S.F.; Braidy, N.; Habtemariam, S.; Orhan, I.E.; Daglia, M.; Manayi, A.; Gortzi, O.; Nabavi, S.M. Neuroprotective effects of chrysin: From chemistry to medicine. Neurochem. Int. 2015, 90, 224–231. [Google Scholar] [CrossRef] [PubMed]
- Panche, A.N.; Diwan, A.D.; Chandra, S.R. Flavonoids: An overview. J. Nutr. Sci. 2016, 5, e47. [Google Scholar] [CrossRef] [Green Version]
- Antonini, A.; Abbruzzese, G.; Barone, P.; Bonuccelli, U.; Lopiano, L.; Onofrj, M.; Zappia, M.; Quattrone, A. COMT inhibition with tolcapone in the treatment algorithm of patients with Parkinson’s disease (PD): Relevance for motor and non-motor features. Neuropsychiatr. Dis. Treat. 2008, 4, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Willman, C.; Tadi, P. StatPearls. 2022. Available online: https://www.ncbi.nlm.nih.gov/books/NBK560593/ (accessed on 29 April 2022).
- Bors, W.; Heller, W.; Michel, C.; Saran, M. Flavonoids as antioxidants: Determination of radical-scavenging efficiencies. Methods Enzymol. 1990, 186, 343–355. [Google Scholar] [CrossRef]
- Fernandez, M.T.; Mira, M.L.; Florêncio, M.H.; Jennings, K.R. Iron and copper chelation by flavonoids: An electrospray mass spectrometry study. J. Inorg. Biochem. 2002, 92, 105–111. [Google Scholar] [CrossRef]
- Rice-Evans, C.A.; Miller, N.J.; Paganga, G. Structure-antioxidant activity relationships of flavonoids and phenolic acids. Free Radic Biol. Med. 1996, 20, 933–956. [Google Scholar] [CrossRef]
- Ruotolo, R.; Marchini, G.; Ottonello, S. Membrane transporters and protein traffic networks differentially affecting metal tolerance: A genomic phenotyping study in yeast. Genome Biol. 2008, 9, R67. [Google Scholar] [CrossRef] [Green Version]
- Jiang, L.; Cao, C.; Zhang, L.; Lin, W.; Xia, J.; Xu, H.; Zhang, Y. Cadmium-induced activation of high osmolarity glycerol pathway through its Sln1 branch is dependent on the MAP kinase kinase kinase Ssk2, but not its paralog Ssk22, in budding yeast. FEMS Yeast Res. 2014, 14, 1263–1272. [Google Scholar] [CrossRef]
- Dix, D.R.; Bridgham, J.T.; Broderius, M.A.; Byersdorfer, C.A.; Eide, D.J. The FET4 gene encodes the low affinity Fe(II) transport protein of Saccharomyces cerevisiae. J. Biol. Chem. 1994, 269, 26092–26099. [Google Scholar] [CrossRef]
- Hechtberger, P.; Zinser, E.; Saf, R.; Hummel, K.; Paltauf, F.; Daum, G. Characterization, quantification and subcellular localization of inositol-containing sphingolipids of the yeast, Saccharomyces cerevisiae. Eur. J. Biochem. 1994, 225, 641–649. [Google Scholar] [CrossRef] [PubMed]
- Kraft, M.L. Sphingolipid Organization in the Plasma Membrane and the Mechanisms That Influence It. Front. Cell Dev. Biol. 2016, 4, 154. [Google Scholar] [CrossRef] [PubMed]
- Goins, L.; Spassieva, S. Sphingoid bases and their involvement in neurodegenerative diseases. Adv. Biol. Regul. 2018, 70, 65–73. [Google Scholar] [CrossRef]
- López-García, B.; Gandía, M.; Muñoz, A.; Carmona, L.; Marcos, J.F. A genomic approach highlights common and diverse effects and determinants of susceptibility on the yeast Saccharomyces cerevisiae exposed to distinct antimicrobial peptides. BMC Microbiol. 2010, 10, 289. [Google Scholar] [CrossRef] [Green Version]
- Kaniak-Golik, A.; Skoneczna, A. Mitochondria-nucleus network for genome stability. Free Radic Biol. Med. 2015, 82, 73–104. [Google Scholar] [CrossRef] [Green Version]
- Abdel Hadi, L.; Di Vito, C.; Marfia, G.; Ferraretto, A.; Tringali, C.; Viani, P.; Riboni, L. Sphingosine Kinase 2 and Ceramide Transport as Key Targets of the Natural Flavonoid Luteolin to Induce Apoptosis in Colon Cancer Cells. PLoS ONE 2015, 10, e0143384. [Google Scholar] [CrossRef] [Green Version]
- Pena, M.M.; Puig, S.; Thiele, D.J. Characterization of the Saccharomyces cerevisiae high affinity copper transporter Ctr3. J. Biol. Chem. 2000, 275, 33244–33251. [Google Scholar] [CrossRef] [Green Version]
- Dancis, A.; Yuan, D.S.; Haile, D.; Askwith, C.; Eide, D.; Moehle, C.; Kaplan, J.; Klausner, R.D. Molecular characterization of a copper transport protein in S. cerevisiae: An unexpected role for copper in iron transport. Cell 1994, 76, 393–402. [Google Scholar] [CrossRef]
- Dancis, A.; Haile, D.; Yuan, D.S.; Klausner, R.D. The Saccharomyces cerevisiae copper transport protein (Ctr1p). Biochemical characterization, regulation by copper, and physiologic role in copper uptake. J. Biol. Chem. 1994, 269, 25660–25667. [Google Scholar] [CrossRef]
- Li, H.; Wang, J.; Wu, C.; Wang, L.; Chen, Z.S.; Cui, W. The combination of disulfiram and copper for cancer treatment. Drug Discov. Today 2020, 25, 1099–1108. [Google Scholar] [CrossRef] [PubMed]
- Kirshner, J.R.; He, S.; Balasubramanyam, V.; Kepros, J.; Yang, C.Y.; Zhang, M.; Du, Z.; Barsoum, J.; Bertin, J. Elesclomol induces cancer cell apoptosis through oxidative stress. Mol. Cancer Ther. 2008, 7, 2319–2327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wangpaichitr, M.; Wu, C.; You, M.; Maher, J.C.; Dinh, V.; Feun, L.G.; Savaraj, N. N′,N′-Dimethyl-N′,N′-bis(phenylcarbonothioyl) Propanedihydrazide (Elesclomol) Selectively Kills Cisplatin Resistant Lung Cancer Cells through Reactive Oxygen Species (ROS). Cancers 2009, 1, 23–38. [Google Scholar] [CrossRef] [PubMed]
- Reeder, N.L.; Kaplan, J.; Xu, J.; Youngquist, R.S.; Wallace, J.; Hu, P.; Juhlin, K.D.; Schwartz, J.R.; Grant, R.A.; Fieno, A.; et al. Zinc pyrithione inhibits yeast growth through copper influx and inactivation of iron-sulfur proteins. Antimicrob. Agents Chemother. 2011, 55, 5753–5760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, D.S.; Dancis, A.; Klausner, R.D. Restriction of copper export in Saccharomyces cerevisiae to a late Golgi or post-Golgi compartment in the secretory pathway. J. Biol. Chem. 1997, 272, 25787–25793. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, D.S.; Stearman, R.; Dancis, A.; Dunn, T.; Beeler, T.; Klausner, R.D. The Menkes/Wilson disease gene homologue in yeast provides copper to a ceruloplasmin-like oxidase required for iron uptake. Proc. Natl. Acad. Sci. USA 1995, 92, 2632–2636. [Google Scholar] [CrossRef] [Green Version]
- Stearman, R.; Yuan, D.S.; Yamaguchi-Iwai, Y.; Klausner, R.D.; Dancis, A. A permease-oxidase complex involved in high-affinity iron uptake in yeast. Science 1996, 271, 1552–1557. [Google Scholar] [CrossRef]
- Askwith, C.; Eide, D.; Van Ho, A.; Bernard, P.S.; Li, L.; Davis-Kaplan, S.; Sipe, D.M.; Kaplan, J. The FET3 gene of S. cerevisiae encodes a multicopper oxidase required for ferrous iron uptake. Cell 1994, 76, 403–410. [Google Scholar] [CrossRef]
- Gaxiola, R.A.; Yuan, D.S.; Klausner, R.D.; Fink, G.R. The yeast CLC chloride channel functions in cation homeostasis. Proc. Natl. Acad. Sci. USA 1998, 95, 4046–4050. [Google Scholar] [CrossRef] [Green Version]
- Soma, S.; Latimer, A.J.; Chun, H.; Vicary, A.C.; Timbalia, S.A.; Boulet, A.; Rahn, J.J.; Chan, S.S.L.; Leary, S.C.; Kim, B.E.; et al. Elesclomol restores mitochondrial function in genetic models of copper deficiency. Proc. Natl. Acad. Sci. USA 2018, 115, 8161–8166. [Google Scholar] [CrossRef] [Green Version]
- Guthrie, L.M.; Soma, S.; Yuan, S.; Silva, A.; Zulkifli, M.; Snavely, T.C.; Greene, H.F.; Nunez, E.; Lynch, B.; De Ville, C.; et al. Elesclomol alleviates Menkes pathology and mortality by escorting Cu to cuproenzymes in mice. Science 2020, 368, 620–625. [Google Scholar] [CrossRef] [PubMed]
- Schlecht, U.; Suresh, S.; Xu, W.; Aparicio, A.M.; Chu, A.; Proctor, M.J.; Davis, R.W.; Scharfe, C.; St Onge, R.P. A functional screen for copper homeostasis genes identifies a pharmacologically tractable cellular system. BMC Genom. 2014, 15, 263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blackman, R.K.; Cheung-Ong, K.; Gebbia, M.; Proia, D.A.; He, S.; Kepros, J.; Jonneaux, A.; Marchetti, P.; Kluza, J.; Rao, P.E.; et al. Mitochondrial electron transport is the cellular target of the oncology drug elesclomol. PLoS ONE 2012, 7, e29798. [Google Scholar] [CrossRef] [PubMed]
- Modica-Napolitano, J.S.; Bharath, L.P.; Hanlon, A.J.; Hurley, L.D. The Anticancer Agent Elesclomol Has Direct Effects on Mitochondrial Bioenergetic Function in Isolated Mammalian Mitochondria. Biomolecules 2019, 9, 298. [Google Scholar] [CrossRef] [Green Version]
- Beeler, T.J.; Fu, D.; Rivera, J.; Monaghan, E.; Gable, K.; Dunn, T.M. SUR1 (CSG1/BCL21), a gene necessary for growth of Saccharomyces cerevisiae in the presence of high Ca2+ concentrations at 37 degrees C, is required for mannosylation of inositolphosphorylceramide. Mol. Gen. Genet. 1997, 255, 570–579. [Google Scholar] [CrossRef]
- Megyeri, M.; Riezman, H.; Schuldiner, M.; Futerman, A.H. Making Sense of the Yeast Sphingolipid Pathway. J. Mol. Biol. 2016, 428, 4765–4775. [Google Scholar] [CrossRef]
- Descazeaud, V.; Mestre, E.; Marquet, P.; Essig, M. Calcineurin regulation of cytoskeleton organization: A new paradigm to analyse the effects of calcineurin inhibitors on the kidney. J. Cell. Mol. Med. 2012, 16, 218–227. [Google Scholar] [CrossRef]
- Xiong, T.Q.; Chen, L.M.; Tan, B.H.; Guo, C.Y.; Li, Y.N.; Zhang, Y.F.; Li, S.L.; Zhao, H.; Li, Y.C. The effects of calcineurin inhibitor FK506 on actin cytoskeleton, neuronal survival and glial reactions after pilocarpine-induced status epilepticus in mice. Epilepsy Res. 2018, 140, 138–147. [Google Scholar] [CrossRef]
- Glaß, H.; Neumann, P.; Pal, A.; Reinhardt, P.; Storch, A.; Sterneckert, J.; Hermann, A. Combined Dendritic and Axonal Deterioration Are Responsible for Motoneuronopathy in Patient-Derived Neuronal Cell Models of Chorea-Acanthocytosis. Int. J. Mol. Sci. 2020, 21, 1797. [Google Scholar] [CrossRef] [PubMed] [Green Version]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kaminska, J.; Soczewka, P.; Rzepnikowska, W.; Zoladek, T. Yeast as a Model to Find New Drugs and Drug Targets for VPS13-Dependent Neurodegenerative Diseases. Int. J. Mol. Sci. 2022, 23, 5106. https://doi.org/10.3390/ijms23095106
Kaminska J, Soczewka P, Rzepnikowska W, Zoladek T. Yeast as a Model to Find New Drugs and Drug Targets for VPS13-Dependent Neurodegenerative Diseases. International Journal of Molecular Sciences. 2022; 23(9):5106. https://doi.org/10.3390/ijms23095106
Chicago/Turabian StyleKaminska, Joanna, Piotr Soczewka, Weronika Rzepnikowska, and Teresa Zoladek. 2022. "Yeast as a Model to Find New Drugs and Drug Targets for VPS13-Dependent Neurodegenerative Diseases" International Journal of Molecular Sciences 23, no. 9: 5106. https://doi.org/10.3390/ijms23095106