
Staphylococcus aureus Exfoliative Toxin E, Oligomeric State and Flip of P186: Implications for Its Action Mechanism
 , , , , , , , and
, , , , , , , and
Abstract
:
1. Introduction
2. Results
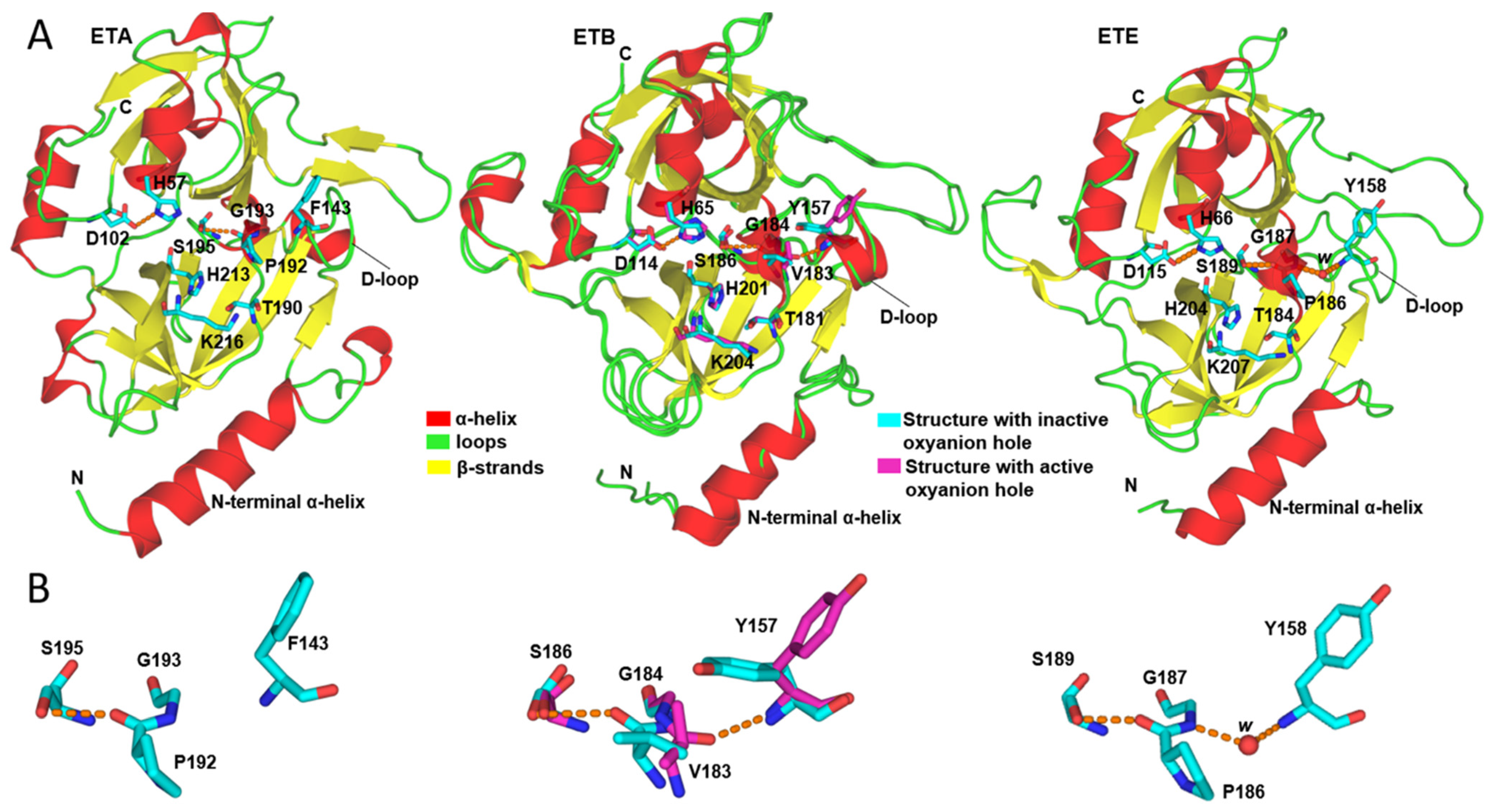
2.1. Refinement of the ETE Crystal Structure
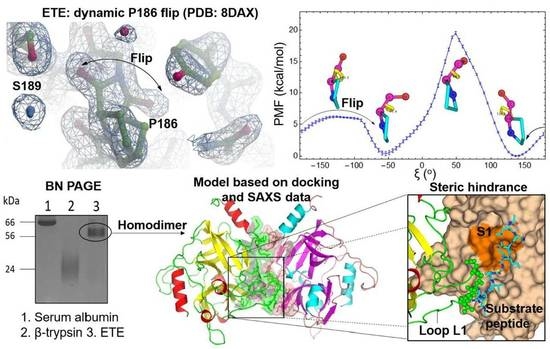
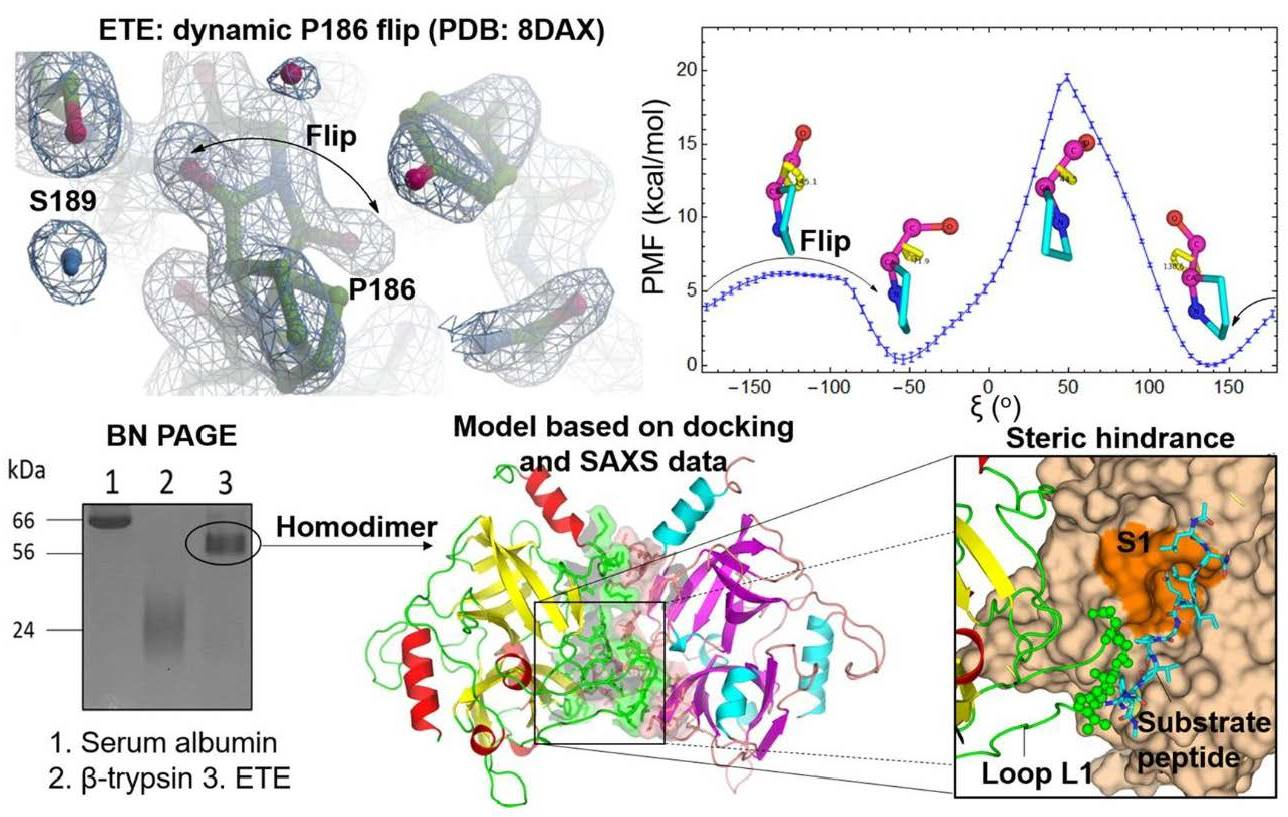
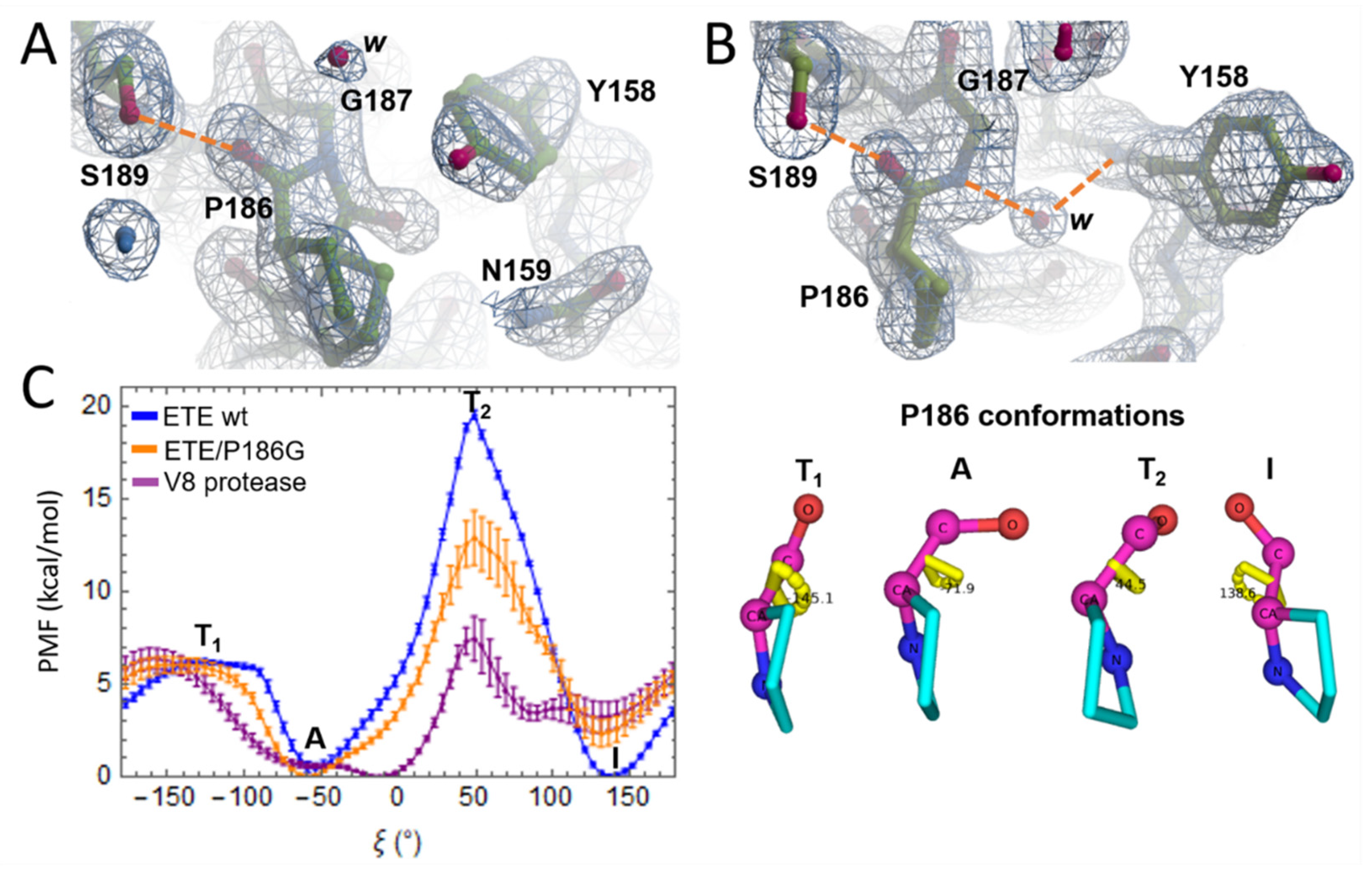
2.2. Oxyanion Hole and P186 Flip
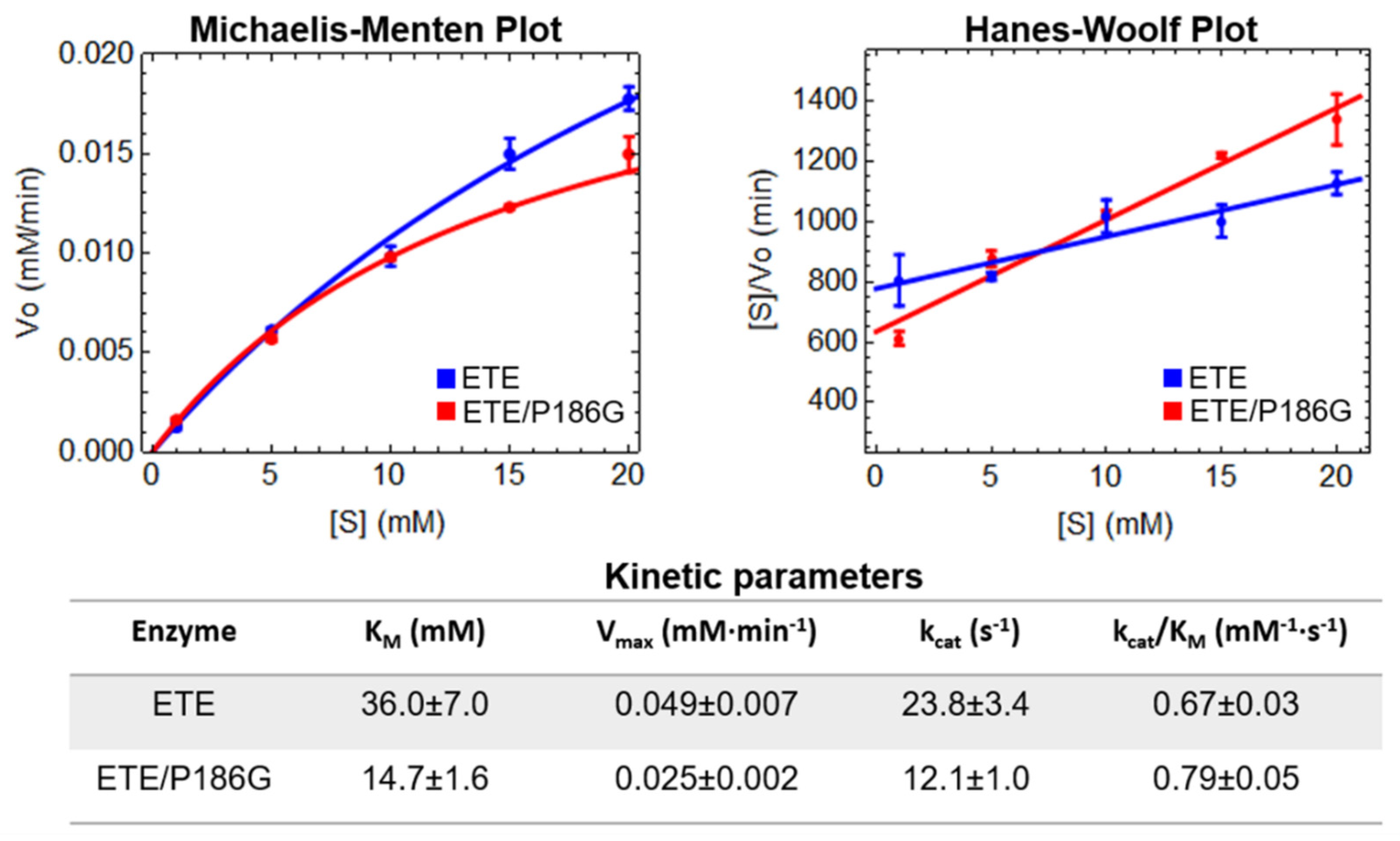
2.3. Esterolytic Activity of ETE Wild-Type and ETE/P186G Mutant
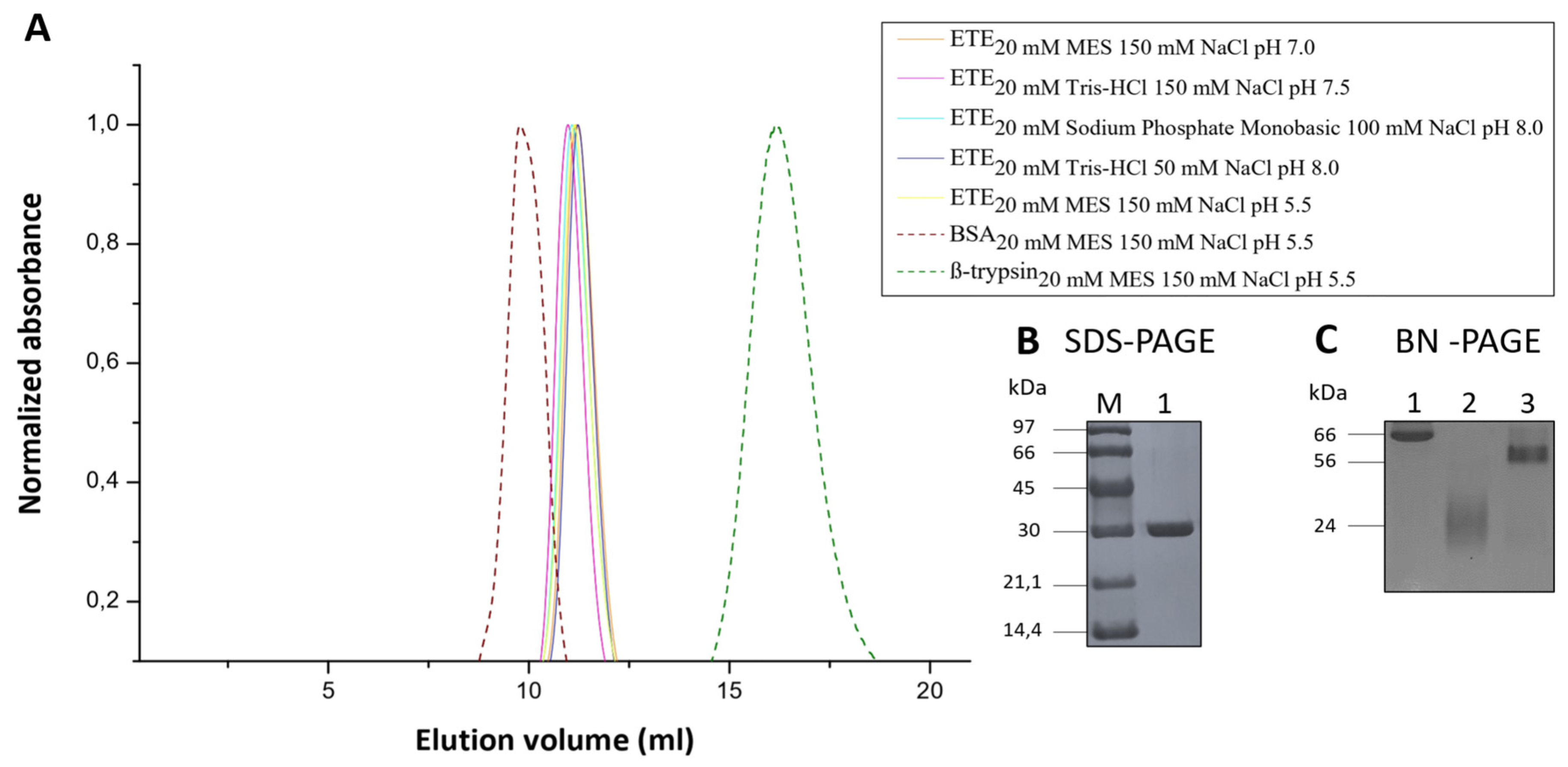
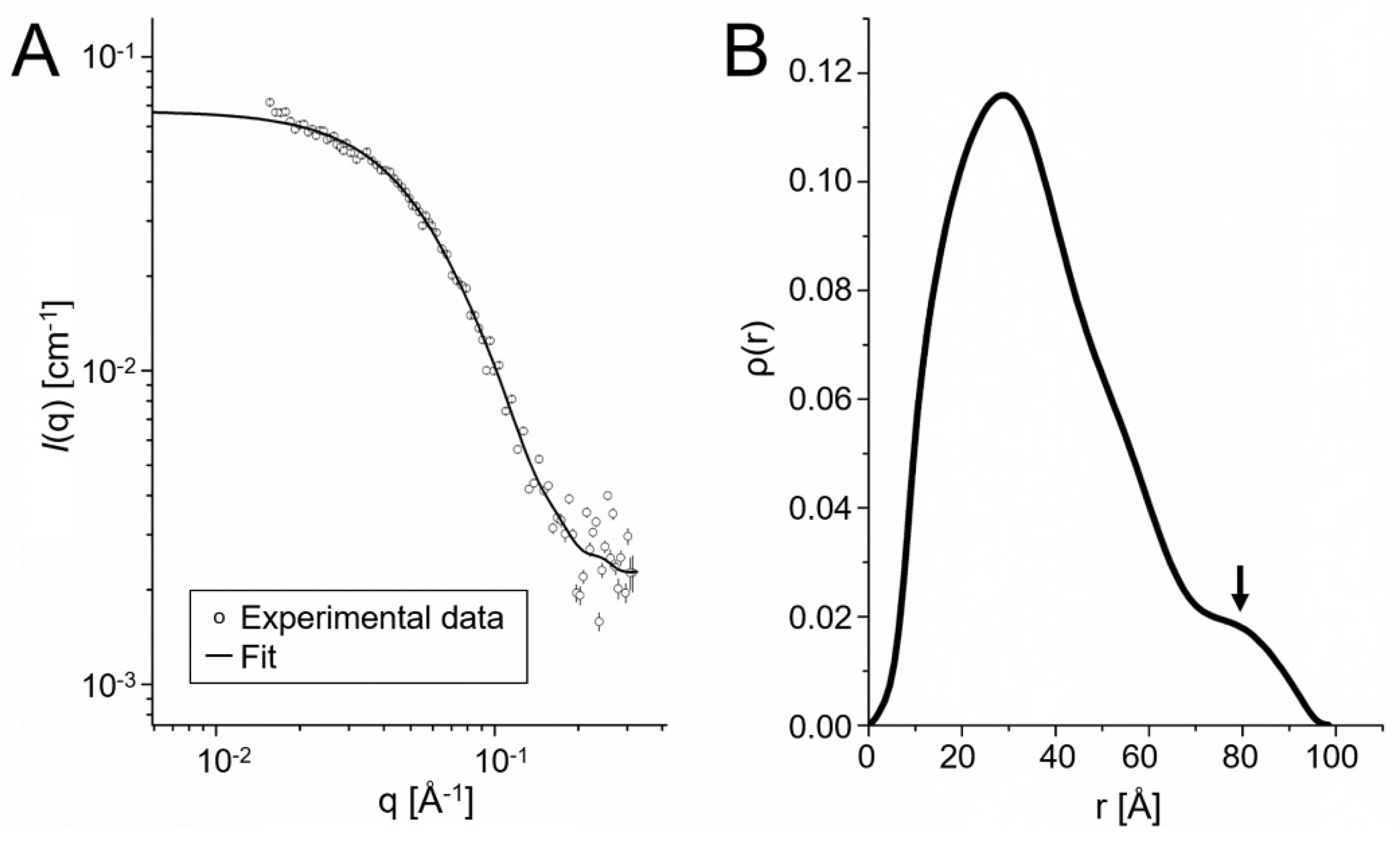
2.4. Oligomeric State of ETE in Solution
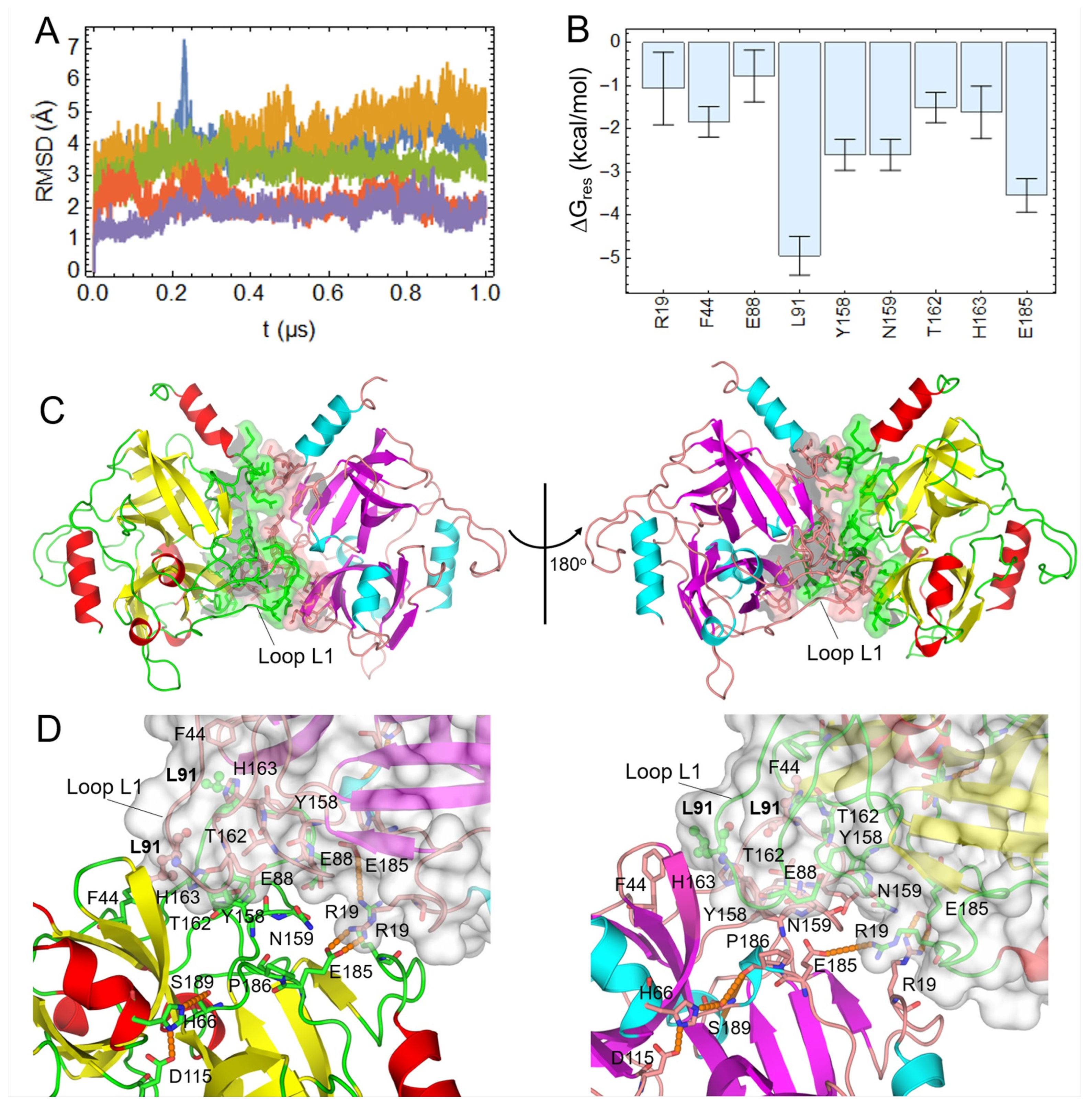
2.5. Prediction of the ETE Homodimer Structure in Solution
3. Discussion
4. Materials and Methods
4.1. Protein Expression and Purification
4.2. SDS-PAGE and BN-PAGE 15%
4.3. Enzymatic Assays
4.4. Crystallization, Data Collection, Processing and Structure Determination
4.5. Small-Angle X-ray Scattering Experiments
4.6. Computational Approaches to Predict the Conformation of ETE Homodimers
4.7. Molecular Dynamics Simulations
4.8. MM-GBSA Free Energy Calculations
4.9. Umbrella Sampling Free Energy Calculations
4.10. Trajectory Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bukowski, M.; Wladyka, B.; Dubin, G. Exfoliative toxins of Staphylococcus aureus. Toxins 2010, 2, 1148–1165. [Google Scholar] [CrossRef] [PubMed]
- Mariutti, R.B.; Tartaglia, N.R.; Le Loir, Y.; Nishifuji, K. Exfoliative toxins of Staphylococcus aureus. In The Rise of Virulence and Resistance in Staphylococcus aureus; Enany, S., Crotty Alexander, L.E., Eds.; IntechOpen: London, UK, 2017; pp. 127–143. [Google Scholar]
- Amagai, M.; Yamaguchi, T.; Hanakawa, Y.; Nishifuji, K.; Sugai, M.; Stanley, J.R. Staphylococcal exfoliative toxin B specifically cleaves desmoglein 1. J. Investig. Dermatol. 2002, 118, 845–850. [Google Scholar] [CrossRef] [PubMed]
- Hanakawa, Y.; Schechter, N.M.; Lin, C.; Nishifuji, K.; Amagai, M.; Stanley, J.R. Enzymatic and molecular characteristics of the efficiency and specificity of exfoliative toxin cleavage of desmoglein 1. J. Biol. Chem. 2004, 279, 5268–5277. [Google Scholar] [CrossRef] [PubMed]
- Hanakawa, Y.; Selwood, T.; Woo, D.; Lin, C.; Schechter, N.M.; Stanley, J.R. Calcium-dependent conformation of desmoglein 1 is required for its cleavage by exfoliative toxin. J. Investig. Dermatol. 2003, 121, 383–389. [Google Scholar] [CrossRef] [PubMed]
- O’Toole, P.W.; Foster, T.J. Nucleotide sequence of the epidermolytic toxin A gene of Staphylococcus aureus. J. Bacteriol. 1987, 169, 3910–3915. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.Y.; Schmidt, J.J.; Johnson-Winegar, A.D.; Spero, L.; Iandolo, J.J. Sequence determination and comparison of the exfoliative toxin A and toxin B genes from Staphylococcus aureus. J. Bacteriol. 1987, 169, 3904–3909. [Google Scholar] [CrossRef]
- Yamaguchi, T.; Nishifuji, K.; Sasaki, M.; Fudaba, Y.; Aepfelbacher, M.; Takata, T.; Ohara, M.; Komatsuzawa, H.; Amagai, M.; Sugai, M. Identification of the Staphylococcus aureus etd pathogenicity island which encodes a novel exfoliative toxin, ETD, and EDIN-B. Infect. Immun. 2002, 70, 5835–5845. [Google Scholar] [CrossRef]
- Le Marechal, C.; Jardin, J.; Jan, G.; Even, S.; Pulido, C.; Guibert, J.M.; Hernandez, D.; Francois, P.; Schrenzel, J.; Demon, D.; et al. Staphylococcus aureus seroproteomes discriminate ruminant isolates causing mild or severe mastitis. Vet. Res. 2011, 42, 35. [Google Scholar] [CrossRef]
- Le Marechal, C.; Seyffert, N.; Jardin, J.; Hernandez, D.; Jan, G.; Rault, L.; Azevedo, V.; Francois, P.; Schrenzel, J.; van de Guchte, M.; et al. Molecular basis of virulence in Staphylococcus aureus mastitis. PLoS ONE 2011, 6, e27354. [Google Scholar] [CrossRef]
- Kapral, F.A.; Miller, M.M. Product of Staphylococcus aureus responsible for the scalded-skin syndrome. Infect. Immun. 1971, 4, 541–545. [Google Scholar] [CrossRef] [Green Version]
- Arbuthnott, J.P.; Kent, J.; Lyell, A.; Gemmell, C.G. Toxic epidermal necrolysis produced by an extracellular product of Staphylococcus aureus. Br. J. Derm. 1971, 85, 145–149. [Google Scholar] [CrossRef] [PubMed]
- Melish, M.E.; Glasgow, L.A. The staphylococcal scalded-skin syndrome. N. Engl. J. Med. 1970, 282, 1114–1119. [Google Scholar] [CrossRef] [PubMed]
- Imanishi, I.; Nicolas, A.; Caetano, A.B.; Castro, T.L.P.; Tartaglia, N.R.; Mariutti, R.; Guedon, E.; Even, S.; Berkova, N.; Arni, R.K.; et al. Exfoliative toxin E, a new Staphylococcus aureus virulence factor with host-specific activity. Sci. Rep. 2019, 9, 16336. [Google Scholar] [CrossRef] [PubMed]
- Wegener, H.C.; Andresen, L.O.; Bille-Hansen, V. Staphylococcus hyicus virulence in relation to exudative epidermitis in pigs. Can. J. Vet. Res. 1993, 57, 119–125. [Google Scholar]
- Chen, S.; Wang, Y.; Chen, F.; Yang, H.; Gan, M.; Zheng, S.J. A highly pathogenic strain of Staphylococcus sciuri caused fatal exudative epidermitis in piglets. PLoS ONE 2007, 2, e147. [Google Scholar]
- Li, H.; Wang, Y.; Ding, L.; Zheng, S.J. Staphylococcus sciuri exfoliative toxin C (ExhC) is a necrosis-inducer for mammalian cells. PLoS ONE 2011, 6, e23145. [Google Scholar] [CrossRef]
- Demidyuk, I.V.; Chukhontseva, K.N.; Kostrov, S.V. Glutamyl Endopeptidases: The Puzzle of Substrate Specificity. Acta Nat. 2017, 9, 17–33. [Google Scholar] [CrossRef]
- Cavarelli, J.; Prevost, G.; Bourguet, W.; Moulinier, L.; Chevrier, B.; Delagoutte, B.; Bilwes, A.; Mourey, L.; Rifai, S.; Piemont, Y.; et al. The structure of Staphylococcus aureus epidermolytic toxin A, an atypic serine protease, at 1.7 A resolution. Structure 1997, 5, 813–824. [Google Scholar] [CrossRef]
- Vath, G.M.; Earhart, C.A.; Rago, J.V.; Kim, M.H.; Bohach, G.A.; Schlievert, P.M.; Ohlendorf, D.H. The structure of the superantigen exfoliative toxin A suggests a novel regulation as a serine protease. Biochemistry 1997, 36, 1559–1566. [Google Scholar] [CrossRef]
- Papageorgiou, A.C.; Plano, L.R.; Collins, C.M.; Acharya, K.R. Structural similarities and differences in Staphylococcus aureus exfoliative toxins A and B as revealed by their crystal structures. Protein Sci. 2000, 9, 610–618. [Google Scholar] [CrossRef]
- Iyori, K.; Futagawa-Saito, K.; Hisatsune, J.; Yamamoto, M.; Sekiguchi, M.; Ide, K.; Son, W.G.; Olivry, T.; Sugai, M.; Fukuyasu, T.; et al. Staphylococcus pseudintermedius exfoliative toxin EXI selectively digests canine desmoglein 1 and causes subcorneal clefts in canine epidermis. Vet. Derm. 2011, 22, 319–326. [Google Scholar]
- Bailey, C.J.; Redpath, M.B. The esterolytic activity of epidermolytic toxins. Biochem. J. 1992, 284 Pt 1, 177–180. [Google Scholar] [PubMed]
- Rago, J.V.; Vath, G.M.; Tripp, T.J.; Bohach, G.A.; Ohlendorf, D.H.; Schlievert, P.M. Staphylococcal exfoliative toxins cleave alpha- and beta-melanocyte-stimulating hormones. Infect. Immun. 2000, 68, 2366–2368. [Google Scholar] [PubMed] [Green Version]
- Mariutti, R.B.; Souza, T.A.; Ullah, A.; Caruso, I.P.; de Moraes, F.R.; Zanphorlin, L.M.; Tartaglia, N.R.; Seyffert, N.; Azevedo, V.A.; Le Loir, Y.; et al. Crystal structure of Staphylococcus aureus exfoliative toxin D-like protein: Structural basis for the high specificity of exfoliative toxins. Biochem Biophys Res Commun 2015, 467, 171–177. [Google Scholar]
- Krissinel, E.; Henrick, K. Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 2007, 372, 774–797. [Google Scholar]
- Jimenez-Garcia, B.; Elez, K.; Koukos, P.I.; Bonvin, A.M.; Vangone, A. PRODIGY-crystal: A web-tool for classification of biological interfaces in protein complexes. Bioinformatics 2019, 35, 4821–4823. [Google Scholar]
- Shibukawa, M.; Miyake, A.; Eda, S.; Saito, S. Determination of the cis-trans isomerization barriers of L-alanyl-L-proline in aqueous solutions and at water/hydrophobic interfaces by on-line temperature-jump relaxation HPLC and dynamic on-column reaction HPLC. Anal. Chem. 2015, 87, 9280–9287. [Google Scholar]
- Rago, J.V.; Vath, G.M.; Bohach, G.A.; Ohlendorf, D.H.; Schlievert, P.M. Mutational analysis of the superantigen staphylococcal exfoliative toxin A (ETA). J. Immunol. 2000, 164, 2207–2213. [Google Scholar]
- Kawalec, M.; Potempa, J.; Moon, J.L.; Travis, J.; Murray, B.E. Molecular diversity of a putative virulence factor: Purification and characterization of isoforms of an extracellular serine glutamyl endopeptidase of Enterococcus faecalis with different enzymatic activities. J. Bacteriol. 2005, 187, 266–275. [Google Scholar]
- Meijers, R.; Blagova, E.V.; Levdikov, V.M.; Rudenskaya, G.N.; Chestukhina, G.G.; Akimkina, T.V.; Kostrov, S.V.; Lamzin, V.S.; Kuranova, I.P. The crystal structure of glutamyl endopeptidase from Bacillus intermedius reveals a structural link between zymogen activation and charge compensation. Biochemistry 2004, 43, 2784–2791. [Google Scholar]
- DeLano, W.L. PyMOL, 2.1.0; DeLano Scientific: San Carlos, CA, USA, 2002. [Google Scholar]
- Hanes, C.S. Studies on plant amylases: The effect of starch concentration upon the velocity of hydrolysis by the amylase of germinated barley. Biochem. J. 1932, 26, 1406–1421. [Google Scholar] [PubMed]
- Li, H.; Li, X.; Lu, Y.; Wang, X.; Zheng, S.J. Staphylococcus sciuri exfoliative toxin C is a dimer that modulates macrophage functions. Can. J. Microbiol. 2011, 57, 722–729. [Google Scholar] [PubMed]
- Oliveira, C.L.P. Investigating macromolecular complexes in solution by small angle X-Ray scattering. In Current Trends in X-Ray Crystallography; Chandasekaran, A., Ed.; IntechOpen: London, UK, 2011; pp. 367–392. [Google Scholar]
- Kozakov, D.; Hall, D.R.; Xia, B.; Porter, K.A.; Padhorny, D.; Yueh, C.; Beglov, D.; Vajda, S. The ClusPro web server for protein-protein docking. Nat. Protoc. 2017, 12, 255–278. [Google Scholar]
- Porollo, A.; Meller, J. Prediction-based fingerprints of protein-protein interactions. Proteins 2007, 66, 630–645. [Google Scholar]
- Gaber, A.; Pavsic, M. Modeling and structure determination of homo-oligomeric proteins: An overview of challenges and current approaches. Int. J. Mol. Sci. 2021, 22, 9081. [Google Scholar] [PubMed]
- Peters, K.; Braun, H.P. Comparative analyses of protein complexes by blue native DIGE. In Difference Gel Electrophoresis (DIGE) Methods and Protocols; Cramer, R., Westermeier, R., Eds.; Humana Press: New York, NY, USA, 2012; pp. 145–154. [Google Scholar]
- Kabsch, W. XDSS. Acta Cryst. D Biol Cryst. 2010, 66, 125–132. [Google Scholar]
- Murshudov, G.N.; Skubak, P.; Lebedev, A.A.; Pannu, N.S.; Steiner, R.A.; Nicholls, R.A.; Winn, M.D.; Long, F.; Vagin, A.A. REFMAC5 for the refinement of macromolecular crystal structures. Acta Cryst. D Biol. Cryst. 2011, 67, 355–367. [Google Scholar]
- Emsley, P.; Cowtan, K. Coot: Model-building tools for molecular graphics. Acta Cryst. D Biol. Cryst. 2004, 60, 2126–2132. [Google Scholar]
- Chen, V.B.; Arendall, W.B., 3rd; Headd, J.J.; Keedy, D.A.; Immormino, R.M.; Kapral, G.J.; Murray, L.W.; Richardson, J.S.; Richardson, D.C. MolProbity: All-atom structure validation for macromolecular crystallography. Acta Cryst. D Biol. Cryst. 2010, 66, 12–21. [Google Scholar]
- Hammersley, A.P. FIT2D: A multi-purpose data reduction, analysis and visualization program. J. Appl. Cryst. 2016, 49, 646–652. [Google Scholar]
- Oliveira, C.L.P.; Vorup-Jensen, T.; Andersen, C.B.F.; Andersen, G.R.; Pedersen, J.S. Discovering new features of protein complexes structures by small-angle X-ray scattering. In Applications of Synchrotron Light to Scattering and Diffraction in Materials and Life Sciences; Gomez, M., Nogales, A., Garcia-Gutierrez, M.C., Ezquerra, T.A., Eds.; Springer Berlin: Heidelberg, Germany, 2009; pp. 231–244. [Google Scholar]
- Petoukhov, M.V.; Franke, D.; Shkumatov, A.V.; Tria, G.; Kikhney, A.G.; Gajda, M.; Gorba, C.; Mertens, H.D.; Konarev, P.V.; Svergun, D.I. New developments in the ATSAS program package for small-angle scattering data analysis. J. Appl. Cryst. 2012, 45, 342–350. [Google Scholar]
- Yan, Y.; Tao, H.; He, J.; Huang, S.Y. The HDOCK server for integrated protein-protein docking. Nat. Protoc. 2020, 15, 1829–1852. [Google Scholar] [PubMed]
- Dominguez, C.; Boelens, R.; Bonvin, A.M. HADDOCK: A protein-protein docking approach based on biochemical or biophysical information. J. Am. Chem. Soc. 2003, 125, 1731–1737. [Google Scholar] [CrossRef]
- Park, T.; Baek, M.; Lee, H.; Seok, C. GalaxyTongDock: Symmetric and asymmetric ab initio protein-protein docking web server with improved energy parameters. J. Comput. Chem. 2019, 40, 2413–2417. [Google Scholar] [PubMed]
- Ko, J.; Park, H.; Heo, L.; Seok, C. GalaxyWEB server for protein structure prediction and refinement. Nucleic. Acids Res. 2012, 40, W294–W297. [Google Scholar] [CrossRef]
- Christoffer, C.; Bharadwaj, V.; Luu, R.; Kihara, D. LZerD protein-protein docking webserver enhanced with de novo structure prediction. Front. Mol. Biosci. 2021, 8, 724947. [Google Scholar]
- de Vries, S.J.; Bonvin, A.M. CPORT: A consensus interface predictor and its performance in prediction-driven docking with HADDOCK. PLoS ONE 2011, 6, e17695. [Google Scholar]
- Park, T.; Won, J.; Baek, M.; Seok, C. GalaxyHeteromer: Protein heterodimer structure prediction by template-based and ab initio docking. Nucleic Acids Res. 2021, 49, W237–W241. [Google Scholar]
- Evans, R.; O’Neill, M.; Pritzel, A.; Antropova, N.; Senior, A.; Green, T. Protein complex prediction with AlphaFold-Multimer. Biorxiv [Preprint] 2021. [Google Scholar] [CrossRef]
- Ghani, U.; Desta, I.; Jindal, A.; Khan, O.; Jones, G.; Kotelnikov, S.; Padhorny, D.; Vajda, S.; Kozakov, D. Improved docking of protein models by a combination of alphafold2 and cluspro. Biorxiv [Preprint] 2021. [Google Scholar] [CrossRef]
- Mirdita, M.; Schutze, K.; Moriwaki, Y.; Heo, L.; Ovchinnikov, S.; Steinegger, M. ColabFold: Making protein folding accessible to all. Nat. Methods 2022, 19, 679–682. [Google Scholar] [CrossRef] [PubMed]
- Gordon, J.C.; Myers, J.B.; Folta, T.; Shoja, V.; Heath, L.S.; Onufriev, A. H++: A server for estimating pKas and adding missing hydrogens to macromolecules. Nucleic Acids Res. 2005, 33, W368–W371. [Google Scholar] [CrossRef] [PubMed]
- Case, D.A.; Belfon, K.; Ben-Shalom, I.Y.; Brozell, S.R.; Cerutti, D.S.; Cheatham, T.E., III; Cruzeiro, V.W.D.; Darden, T.A.; Duke, R.E.; Giambasu, G.; et al. Amber 2020; University of California: San Francisco, CA, USA, 2020. [Google Scholar]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the accuracy of protein side chain and backbone parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef] [PubMed]
- Salomon-Ferrer, R.; Gotz, A.W.; Poole, D.; Le Grand, S.; Walker, R.C. Routine microsecond molecular dynamics simulations with AMBER on GPUs. 2. Explicit solvent particle mesh ewald. J. Chem. Theory Comput. 2013, 9, 3878–3888. [Google Scholar] [CrossRef] [PubMed]
- Hernandez Gonzalez, J.E.; Hernandez Alvarez, L.; Pascutti, P.G.; Valiente, P.A. Predicting binding modes of reversible peptide-based inhibitors of falcipain-2 consistent with structure-activity relationships. Proteins 2017, 85, 1666–1683. [Google Scholar] [CrossRef]
- Hopkins, C.W.; Le Grand, S.; Walker, R.C.; Roitberg, A.E. Long-time-step molecular dynamics through hydrogen mass repartitioning. J. Chem. Theory Comput. 2015, 11, 1864–1874. [Google Scholar] [CrossRef]
- Miller, B.R., 3rd; McGee, T.D., Jr.; Swails, J.M.; Homeyer, N.; Gohlke, H.; Roitberg, A.E. MMPBSA.py: An efficient program for end-state free energy calculations. J. Chem. Theory Comput. 2012, 8, 3314–3321. [Google Scholar] [CrossRef]
- Onufriev, A.; Bashford, D.; Case, D.A. Exploring protein native states and large-scale conformational changes with a modified generalized born model. Proteins 2004, 55, 383–394. [Google Scholar] [CrossRef]
- Kästner, J. Umbrella sampling. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2011, 1, 932–942. [Google Scholar] [CrossRef]
- Heinzelmann, G.; Henriksen, N.M.; Gilson, M.K. Attach-pull-release calculations of ligand binding and conformational changes on the first BRD4 bromodomain. J. Chem. Theory Comput. 2017, 13, 3260–3275. [Google Scholar] [CrossRef]
- Kumar, S.; Rosenberg, J.M.; Bouzida, D.; Swendsen, R.H.; Kollman, P.A. The weighted histogram analysis method for free-energy calculations on biomolecules. I. The method. J. Comput. Chem. 1992, 13, 1011–1021. [Google Scholar] [CrossRef]
- Shao, J.; Tanner, S.W.; Thompson, N.; Cheatham, T.E. Clustering molecular dynamics trajectories: 1. Characterizing the performance of different clustering algorithms. J. Chem. Theory Comput. 2007, 3, 2312–2334. [Google Scholar] [CrossRef] [PubMed]
- Hong, P.; Koza, S.; Bouvier, E.S. Size-Exclusion Chromatography for the Analysis of Protein Biotherapeutics and their Aggregates. J. Liq. Chromatogr. Relat. Technol. 2012, 35, 2923–2950. [Google Scholar] [CrossRef] [PubMed]
- Madhusudhan, M.S.; Webb, B.M.; Marti-Renom, M.A.; Eswar, N.; Sali, A. Alignment of multiple protein structures based on sequence and structure features. Protein Eng. Des. Sel. 2009, 22, 569–574. [Google Scholar] [CrossRef] [PubMed]
- Larkin, M.A.; Blackshields, G.; Brown, N.P.; Chenna, R.; McGettigan, P.A.; McWilliam, H.; Valentin, F.; Wallace, I.M.; Wilm, A.; Lopez, R.; et al. Clustal W and Clustal X version 2.0. Bioinformatics 2007, 23, 2947–2948. [Google Scholar] [CrossRef]
- Robin, S.; Zhu, J.; Galons, H.; Chuong, P.-H.; Claude, J.R.; Tomas, A.; Viossat, B. A convenient asymmetric synthesis of thalidomide. Tetrahedron Asymmetry 1995, 6, 1249–1252. [Google Scholar] [CrossRef]
- Doudou, S.; Burton, N.A.; Henchman, R.H. Standard Free Energy of Binding from a One-Dimensional Potential of Mean Force. J Chem Theory Comput 2009, 5, 909–918. [Google Scholar] [CrossRef]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1, 19–25. [Google Scholar] [CrossRef]
- Hernandez Gonzalez, J.E.; Hernandez Alvarez, L.; Pascutti, P.G.; Leite, V.B.P. Prediction of Noncompetitive Inhibitor Binding Mode Reveals Promising Site for Allosteric Modulation of Falcipain-2. J. Phys. Chem. B 2019, 123, 7327–7342. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Data Collection | ETE |
|---|---|
| Wavelength (Å) | 1.459 |
| Resolution range (Å) | 43.71–1.61 (1.67–1.61) |
| Space symmetry group | P 4(3) 2(1) 2 |
| Cell dimensions: | |
| a, b, c (Å) | 97.74, 97.74, 116.65 |
| α, β, γ (º) | 90.00, 90.00, 90.00 |
| Total reflections | 1,847,482 (175,190) |
| Unique reflections | 72,995 (7181) |
| Multiplicity | 25.3 (24.4) |
| Completeness range (%) | 99.94 (99.85) |
| Mean I/σI | 24.66 (1.07) |
| R-merge | 0.076 (2.829) |
| Rmeas | 0.077 (2.889) |
| Correlation coefficient ½ | 1 (0.52) |
| R(work)/R(free) | 0.2206/0.2523 |
| Number of protein chains in AU | 2 |
| Solvent water molecules | 145 |
| R.M.S: | |
| Bond lengths (Å) | 0.015 |
| Bond angles (º) | 2.00 |
| Ramachandran analysis: | |
| Favored (%) | 95.33% |
| Allowed (%) | 4.27% |
| Disallowed (%) | 0.41% |
| Average B-factor (Å2) | 36.50 |
| PDB accession code | 8DAX |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gismene, C.; Hernández González, J.E.; Santisteban, A.R.N.; Ziem Nascimento, A.F.; dos Santos Cunha, L.; de Moraes, F.R.; de Oliveira, C.L.P.; Oliveira, C.C.; Jocelan Scarin Provazzi, P.; Pascutti, P.G.; et al. Staphylococcus aureus Exfoliative Toxin E, Oligomeric State and Flip of P186: Implications for Its Action Mechanism. Int. J. Mol. Sci. 2022, 23, 9857. https://doi.org/10.3390/ijms23179857
Gismene C, Hernández González JE, Santisteban ARN, Ziem Nascimento AF, dos Santos Cunha L, de Moraes FR, de Oliveira CLP, Oliveira CC, Jocelan Scarin Provazzi P, Pascutti PG, et al. Staphylococcus aureus Exfoliative Toxin E, Oligomeric State and Flip of P186: Implications for Its Action Mechanism. International Journal of Molecular Sciences. 2022; 23(17):9857. https://doi.org/10.3390/ijms23179857
Chicago/Turabian StyleGismene, Carolina, Jorge Enrique Hernández González, Angela Rocio Niño Santisteban, Andrey Fabricio Ziem Nascimento, Lucas dos Santos Cunha, Fábio Rogério de Moraes, Cristiano Luis Pinto de Oliveira, Caio C. Oliveira, Paola Jocelan Scarin Provazzi, Pedro Geraldo Pascutti, and et al. 2022. "Staphylococcus aureus Exfoliative Toxin E, Oligomeric State and Flip of P186: Implications for Its Action Mechanism" International Journal of Molecular Sciences 23, no. 17: 9857. https://doi.org/10.3390/ijms23179857