
A Mouse Model of Glycogen Storage Disease Type IX-Beta: A Role for Phkb in Glycogenolysis
 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
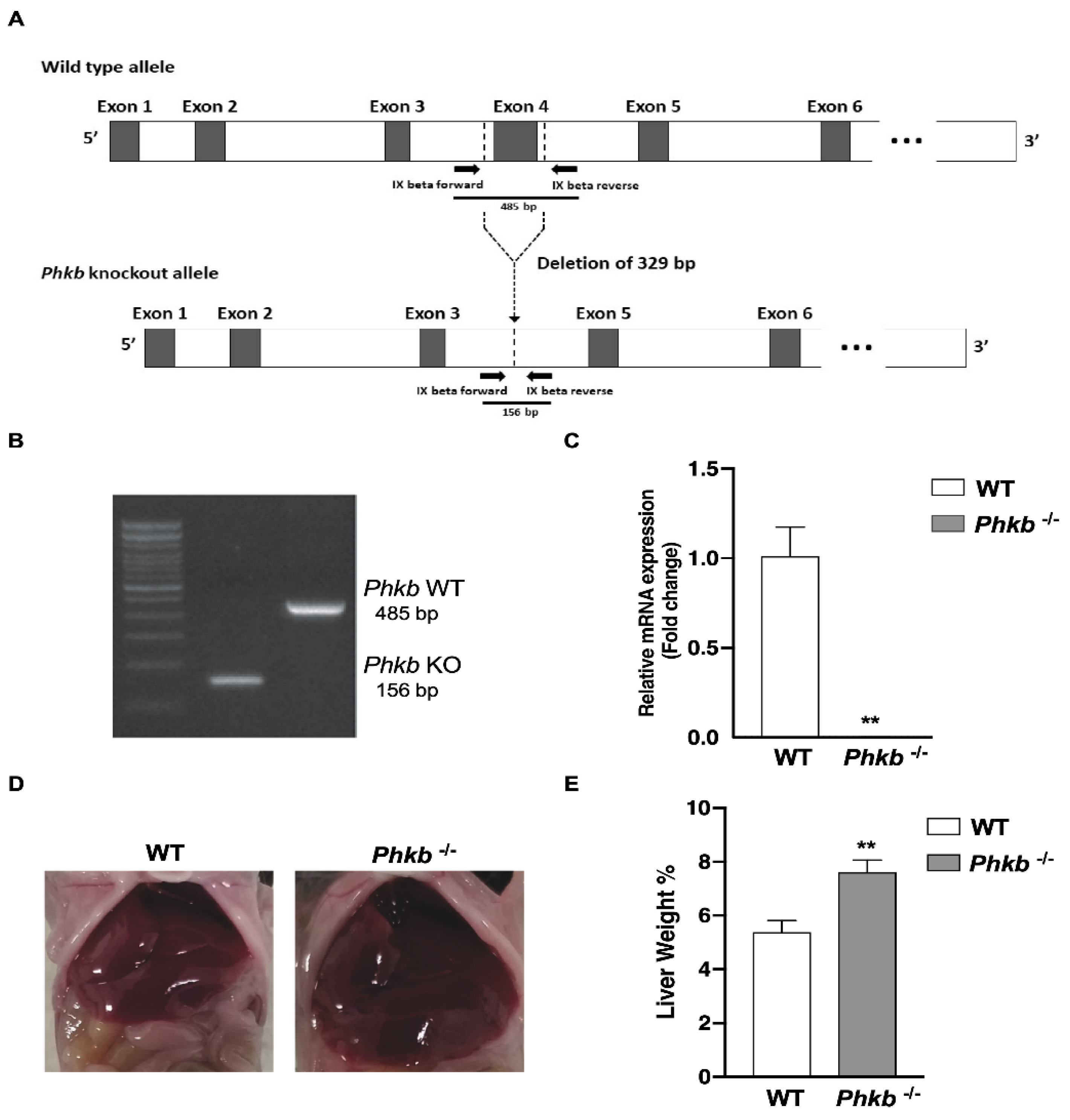
2.1. Characterization of Phosphorylase Kinase Beta Deficiency: A Model for GSD-IX-Beta
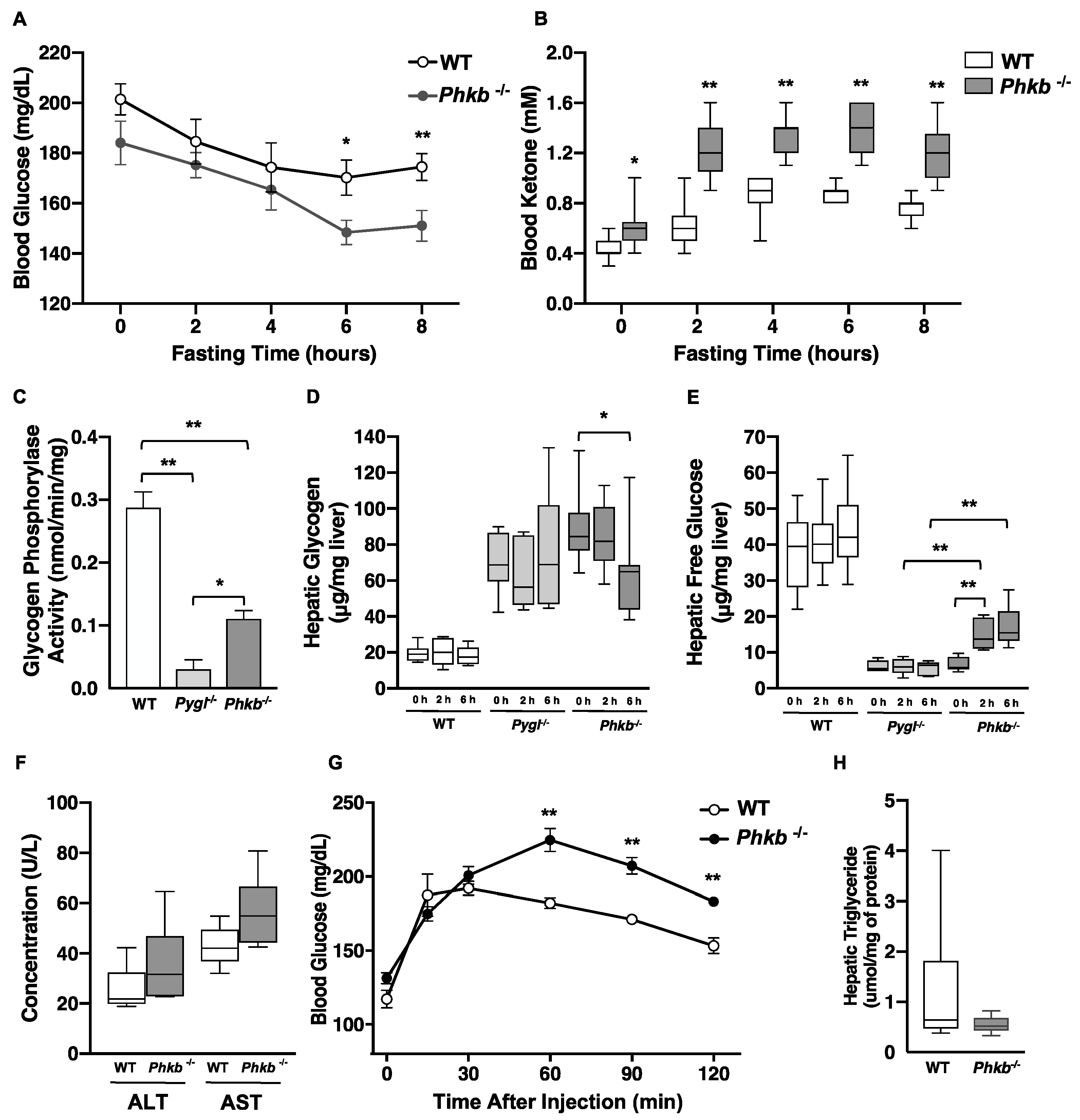
2.2. Phkb−/− Mice Showed Partial Glycogenolysis and Endogenous Glucose Production
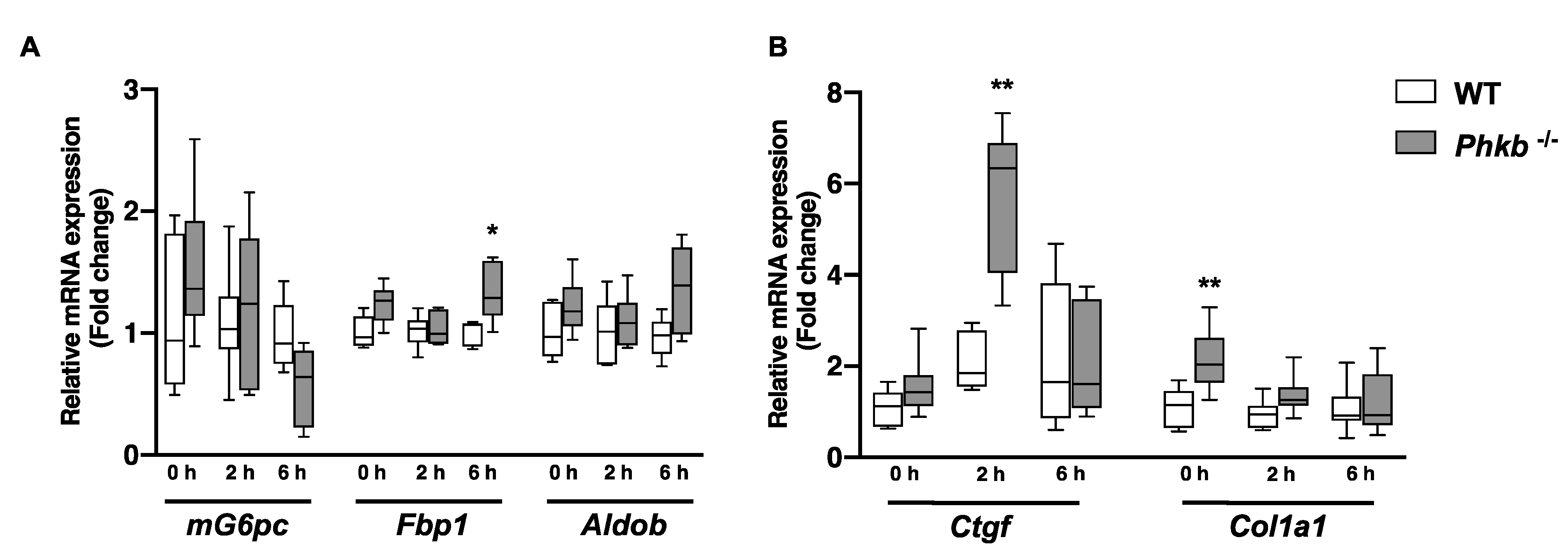
2.3. Alternatively Upregulated Pathways: Gluconeogenesis and Fatty Acid Metabolism
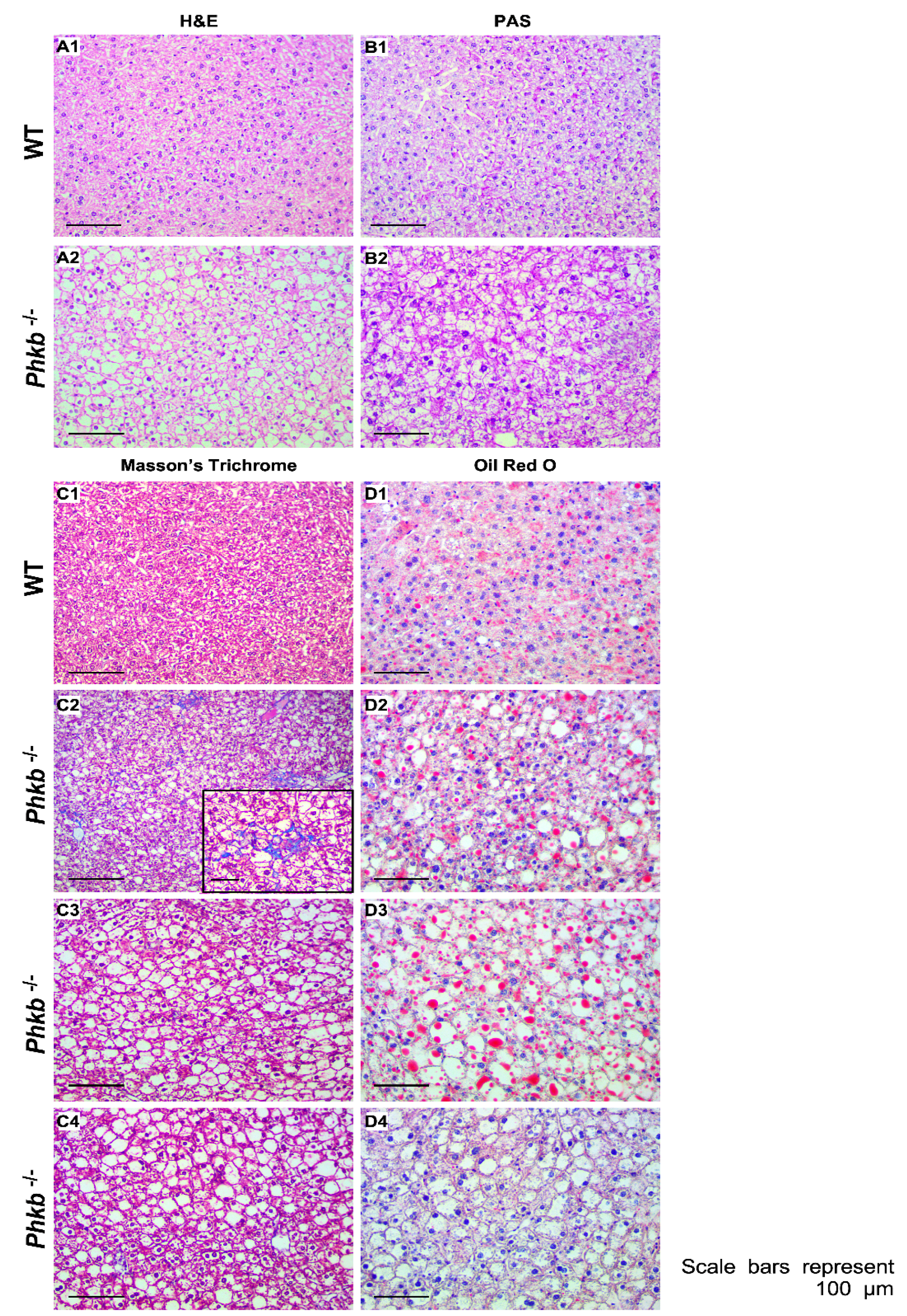
2.4. Phkb−/− Exhibit Excessive Glycogen Accumulation with a Mild Profibrogenic Phenotype
3. Discussion
4. Materials and Methods
4.1. Animal Studies
4.2. Genotyping
4.3. Serum Biochemistry
4.4. Fasting Glucose and Ketones
4.5. Histopathology of Phkb−/− Mice
4.6. Liver Glycogen, Free Glucose, and Triglycerides Contents Determination
4.7. Liver Glycogen Phosphorylase Activity Assay
4.8. Gene Expression Analysis
4.9. Insulin Tolerance Test and Pyruvate Tolerance Test
4.10. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Roscher, A.; Patel, J.; Hewson, S.; Nagy, L.; Feigenbaum, A.; Kronick, J.; Raiman, J.; Schulze, A.; Siriwardena, K.; Mercimek-Mahmutoglu, S. The natural history of glycogen storage disease types VI and IX: Long-term outcome from the largest metabolic center in Canada. Mol. Genet. Metab. 2014, 113, 171–176. [Google Scholar] [CrossRef] [PubMed]
- Newgard, C.B.; Hwang, P.K.; Fletterick, R.J. The family of glycogen phosphorylases: Structure and function. Crit. Rev. Biochem. Mol. Biol. 1989, 24, 69–99. [Google Scholar] [CrossRef] [PubMed]
- Llavero, F.; Sastre, A.A.; Montoro, M.L.; Gálvez, P.; Lacerda, H.M.; Parada, L.A.; Zugaza, J.L. McArdle Disease: New Insights into Its Underlying Molecular Mechanisms. Int. J. Mol. Sci. 2019, 20, 5919. [Google Scholar] [CrossRef] [PubMed]
- Barford, D.; Johnson, L.N. The allosteric transition of glycogen phosphorylase. Nature 1989, 340, 609–616. [Google Scholar] [CrossRef] [PubMed]
- Sutherland, E.W., Jr.; Wosilait, W.D. Inactivation and activation of liver phosphorylase. Nature 1955, 175, 169–170. [Google Scholar] [CrossRef]
- Herbert, M.; Goldstein, J.L.; Rehder, C.; Austin, S.; Kishnani, P.S.; Bali, D.S. Phosphorylase Kinase Deficiency. In GeneReviews®; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Gripp, K.W., Mirzaa, G.M., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Willems, P.J.; Gerver, W.J.; Berger, R.; Fernandes, J. The natural history of liver glycogenosis due to phosphorylase kinase deficiency: A longitudinal study of 41 patients. Eur. J. Pediatr. 1990, 149, 268–271. [Google Scholar] [CrossRef]
- Kishnani, P.S.; Austin, S.L.; Abdenur, J.E.; Arn, P.; Bali, D.S.; Boney, A.; Chung, W.K.; Dagli, A.I.; Dale, D.; Koeberl, D.; et al. Diagnosis and management of glycogen storage disease type I: A practice guideline of the American College of Medical Genetics and Genomics. Genet. Med. 2014, 16, e1. [Google Scholar] [CrossRef]
- Wolfsdorf, J.I.; Holm, I.A.; Weinstein, D.A. Glycogen storage diseases. Phenotypic, genetic, and biochemical characteristics, and therapy. Endocrinol. Metab. Clin. N. Am. 1999, 28, 801–823. [Google Scholar] [CrossRef]
- Fernandes, S.A.; Cooper, G.E.; Gibson, R.A.; Kishnani, P.S. Benign or not benign? Deep phenotyping of liver Glycogen Storage Disease IX. Mol. Genet. Metab. 2020, 131, 299–305. [Google Scholar] [CrossRef]
- Li, C.; Huang, L.; Tian, L.; Chen, J.; Li, S.; Yang, Z. PHKG2 mutation spectrum in glycogen storage disease type IXc: A case report and review of the literature. J. Pediatr. Endocrinol. Metab. 2018, 31, 331–338. [Google Scholar] [CrossRef]
- Sovik, O.; deBarsy, T.; Maehle, B. Phosphorylase kinase deficiency: Severe glycogen storage disease with evidence of autosomal recessive mode of inheritance. Eur. J. Pediatr. 1982, 139, 210. [Google Scholar] [CrossRef] [PubMed]
- Beauchamp, N.J.; Dalton, A.; Ramaswami, U.; Niinikoski, H.; Mention, K.; Kenny, P.; Kolho, K.L.; Raiman, J.; Walter, J.; Treacy, E.; et al. Glycogen storage disease type IX: High variability in clinical phenotype. Mol. Genet. Metab. 2007, 92, 88–99. [Google Scholar] [CrossRef] [PubMed]
- Kishnani, P.S.; Goldstein, J.; Austin, S.L.; Arn, P.; Bachrach, B.; Bali, D.S.; Chung, W.K.; El-Gharbawy, A.; Brown, L.M.; Kahler, S.; et al. Diagnosis and management of glycogen storage diseases type VI and IX: A clinical practice resource of the American College of Medical Genetics and Genomics (ACMG). Genet. Med. 2019, 21, 772–789. [Google Scholar] [CrossRef] [PubMed]
- Kilimann, M.W. Molecular genetics of phosphorylase kinase: cDNA cloning, chromosomal mapping and isoform structure. J. Inherit. Metab. Dis. 1990, 13, 435–441. [Google Scholar] [CrossRef] [PubMed]
- Bali, D.S.; Goldstein, J.L.; Fredrickson, K.; Rehder, C.; Boney, A.; Austin, S.; Weinstein, D.A.; Lutz, R.; Boneh, A.; Kishnani, P.S. Variability of disease spectrum in children with liver phosphorylase kinase deficiency caused by mutations in the PHKG2 gene. Mol. Genet. Metab. 2014, 111, 309–313. [Google Scholar] [CrossRef] [PubMed]
- Maichele, A.J.; Burwinkel, B.; Maire, I.; Søvik, O.; Kilimann, M.W. Mutations in the testis/liver isoform of the phosphorylase kinase γ subunit (PHKG2) cause autosomal liver glycogenosis in the gsd rat and in humans. Nat. Genet. 1996, 14, 337–340. [Google Scholar] [CrossRef] [PubMed]
- Lei, K.J.; Chen, H.; Pan, C.J.; Ward, J.M.; Mosinger, B., Jr.; Lee, E.J.; Westphal, H.; Mansfield, B.C.; Chou, J.Y. Glucose-6-phosphatase dependent substrate transport in the glycogen storage disease type-1a mouse. Nat. Genet. 1996, 13, 203–209. [Google Scholar] [CrossRef]
- Chen, L.Y.; Shieh, J.J.; Lin, B.; Pan, C.J.; Gao, J.L.; Murphy, P.M.; Roe, T.F.; Moses, S.; Ward, J.M.; Lee, E.J.; et al. Impaired glucose homeostasis, neutrophil trafficking and function in mice lacking the glucose-6-phosphate transporter. Hum. Mol. Genet. 2003, 12, 2547–2558. [Google Scholar] [CrossRef]
- Liu, K.M.; Wu, J.Y.; Chen, Y.T. Mouse model of glycogen storage disease type III. Mol. Genet. Metab. 2014, 111, 467–476. [Google Scholar] [CrossRef]
- Gibson, R.A.; Lim, J.A.; Choi, S.J.; Flores, L.; Clinton, L.; Bali, D.; Young, S.; Asokan, A.; Sun, B.; Kishnani, P.S. Characterization of liver GSD IX γ2 pathophysiology in a novel Phkg2−/− mouse model. Mol. Genet. Metab. 2021, 133, 269–276. [Google Scholar] [CrossRef] [PubMed]
- Kido, J.; Mitsubuchi, H.; Watanabe, T.; Sugawara, K.; Sasai, H.; Fukao, T.; Nakamura, K. A female patient with GSD IXc developing multiple and recurrent hepatocellular carcinoma: A case report and literature review. Hum. Genome Var. 2021, 8, 45. [Google Scholar] [CrossRef] [PubMed]
- Bashan, N.; Iancu, T.C.; Lerner, A.; Fraser, D.; Potashnik, R.; Moses, S.W. Glycogenosis due to liver and muscle phosphorylase kinase deficiency. Pediatr. Res. 1981, 15, 299–303. [Google Scholar] [CrossRef] [PubMed]
- Burwinkel, B.; Maichele, A.J.; Aagenaes, O.; Bakker, H.D.; Lerner, A.; Shin, Y.S.; Strachan, J.A.; Kilimann, M.W. Autosomal glycogenosis of liver and muscle due to phosphorylase kinase deficiency is caused by mutations in the phosphorylase kinase β subunit (PHKB). Hum. Mol. Genet. 1997, 6, 1109–1115. [Google Scholar] [CrossRef] [PubMed]
- Wilson, L.H.; Cho, J.H.; Estrella, A.; Smyth, J.A.; Wu, R.; Chengsupanimit, T.; Brown, L.M.; Weinstein, D.A.; Lee, Y.M. Liver Glycogen Phosphorylase Deficiency Leads to Profibrogenic Phenotype in a Murine Model of Glycogen Storage Disease Type VI. Hepatol. Commun. 2019, 3, 1544–1555. [Google Scholar] [CrossRef]
- Taub, R. Liver regeneration: From myth to mechanism. Nat. Rev. Mol. Cell Biol. 2004, 5, 836–847. [Google Scholar] [CrossRef]
- Geisler, C.E.; Hepler, C.; Higgins, M.R.; Renquist, B.J. Hepatic adaptations to maintain metabolic homeostasis in response to fasting and refeeding in mice. Nutr. Metab. 2016, 13, 62. [Google Scholar] [CrossRef]
- Mallette, L.E.; Exton, J.H.; Park, C.R. Control of Gluconeogenesis from Amino Acids in the Perfused Rat Liver. J. Biol. Chem. 1969, 244, 5713–5723. [Google Scholar] [CrossRef]
- Legouis, D.; Faivre, A.; Cippà, P.E.; De Seigneux, S. Renal gluconeogenesis: An underestimated role of the kidney in systemic glucose metabolism. Nephrol. Dial. Transplant. 2022, 37, 1417–1425. [Google Scholar] [CrossRef]
- Newman, J.C.; Verdin, E. β-hydroxybutyrate: Much more than a metabolite. Diabetes Res. Clin. Pr. 2014, 106, 173–181. [Google Scholar] [CrossRef]
- van den Berg, I.E.; van Beurden, E.A.; de Klerk, J.B.; van Diggelen, O.P.; Malingre, H.E.; Boer, M.M.; Berger, R. Autosomal recessive phosphorylase kinase deficiency in liver, caused by mutations in the gene encoding the β subunit (PHKB). Am. J. Hum. Genet. 1997, 61, 539–546. [Google Scholar] [CrossRef] [Green Version]
- Guénet, J.-L. Animal models of human genetic diseases: Do they need to be faithful to be useful? Mol. Genet. Genom. 2011, 286, 1–20. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Arends, C.J.; Wilson, L.H.; Estrella, A.; Kwon, O.S.; Weinstein, D.A.; Lee, Y.M. A Mouse Model of Glycogen Storage Disease Type IX-Beta: A Role for Phkb in Glycogenolysis. Int. J. Mol. Sci. 2022, 23, 9944. https://doi.org/10.3390/ijms23179944
Arends CJ, Wilson LH, Estrella A, Kwon OS, Weinstein DA, Lee YM. A Mouse Model of Glycogen Storage Disease Type IX-Beta: A Role for Phkb in Glycogenolysis. International Journal of Molecular Sciences. 2022; 23(17):9944. https://doi.org/10.3390/ijms23179944
Chicago/Turabian StyleArends, Charles J., Lane H. Wilson, Ana Estrella, Oh Sung Kwon, David A. Weinstein, and Young Mok Lee. 2022. "A Mouse Model of Glycogen Storage Disease Type IX-Beta: A Role for Phkb in Glycogenolysis" International Journal of Molecular Sciences 23, no. 17: 9944. https://doi.org/10.3390/ijms23179944