
Identifying Drug Targets of Oral Squamous Cell Carcinoma through a Systems Biology Method and Genome-Wide Microarray Data for Drug Discovery by Deep Learning and Drug Design Specifications
Abstract
:1. Introduction

2. Results
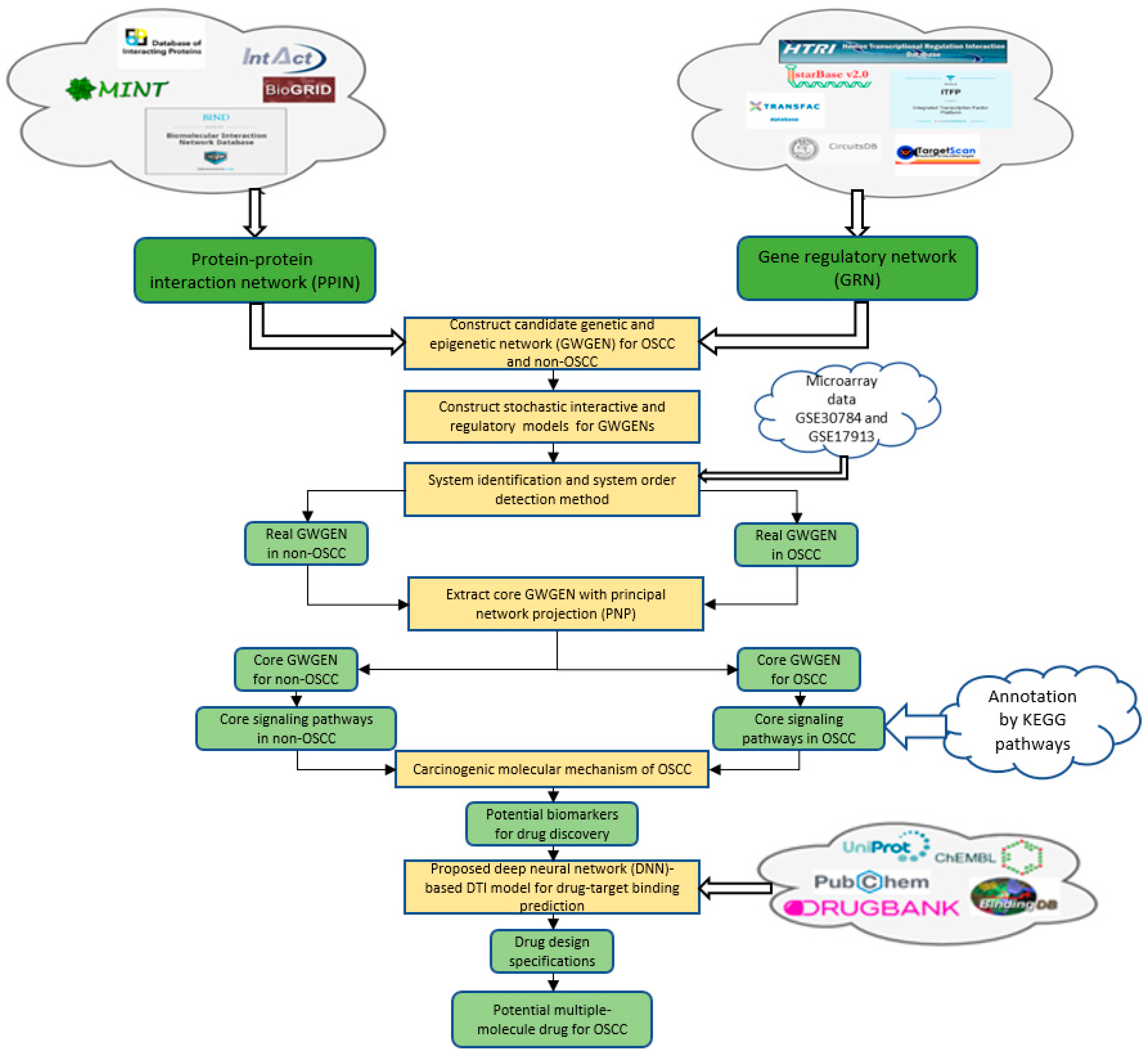
2.1. Overview of the Systems Biology Method and the Systematic Drug Discovery and Design of OSCC
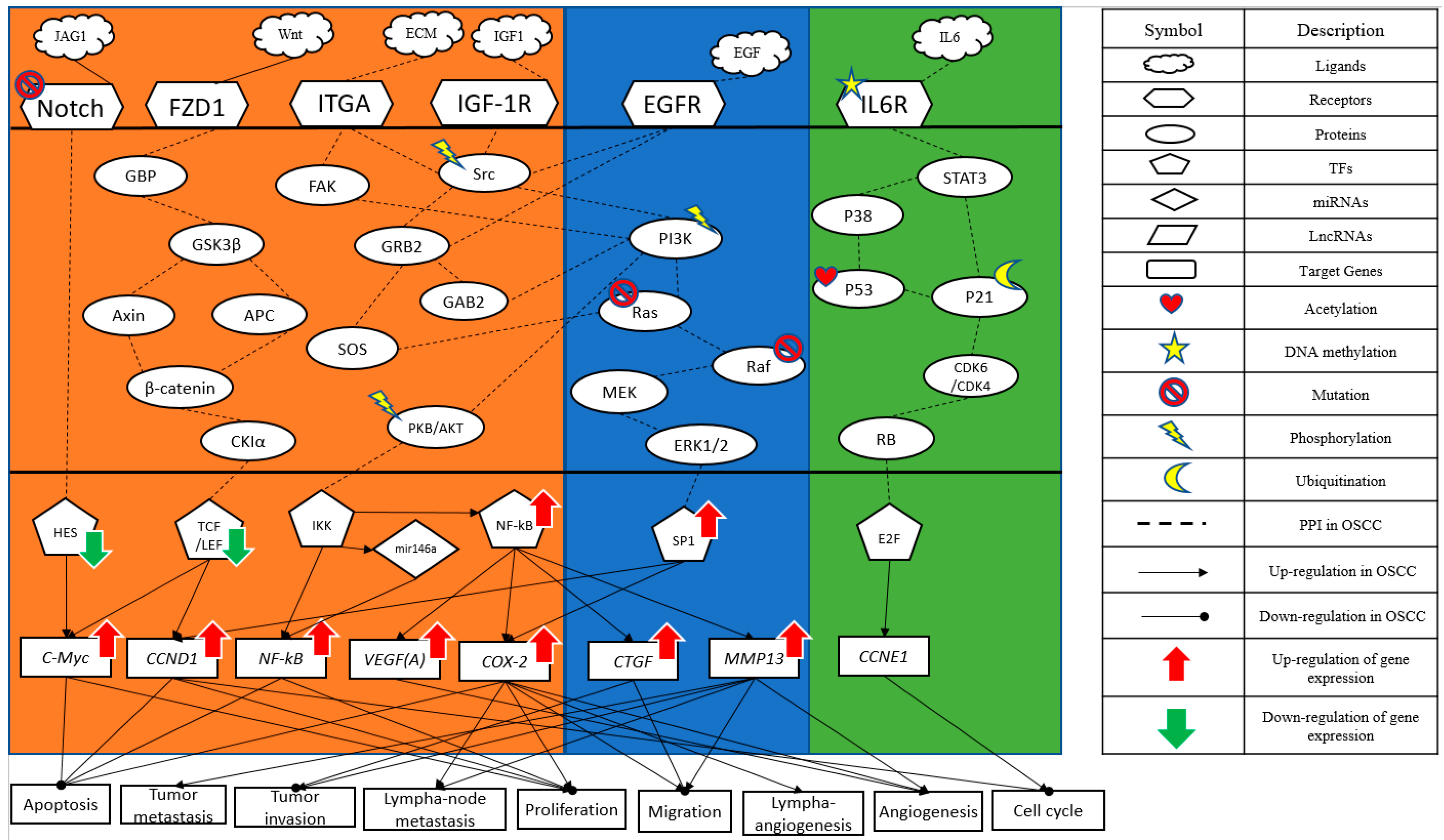
2.2. Investigating the Genetic and Epigenetic Carcinogenic Mechanism of OSCC
- (i)
- Abnormal MAPK signaling pathway in OSCC
- (ii)
- The impact of the Wnt signaling pathway on OSCC
- (iii)
- Notch signaling pathway in OSCC
- (iv)
- Key transcription factor NF-κB on OSCC
2.3. Significant Biomarkers as Drug Targets for the Therapeutic Treatment of OSCC Utilizing the Systematic Drug Discovery Approach


3. Discussion
Potential Multiple-Molecule Drug for the Identified Biomarkers of OSCC
4. Materials and Methods
4.1. A General Review of Constructing Core Genome-Wide Genetic and Epigenetic Networks (GWGENs) of OSCC and Non-OSCC
- (1)
- Constructing the candidate GWGEN: We use big database mining to construct a candidate PPIN and a candidate GRN, including genes, miRNAs and lncRNAs. Note that the candidate GWGEN includes a candidate PPIN and a candidate GRN.
- (2)
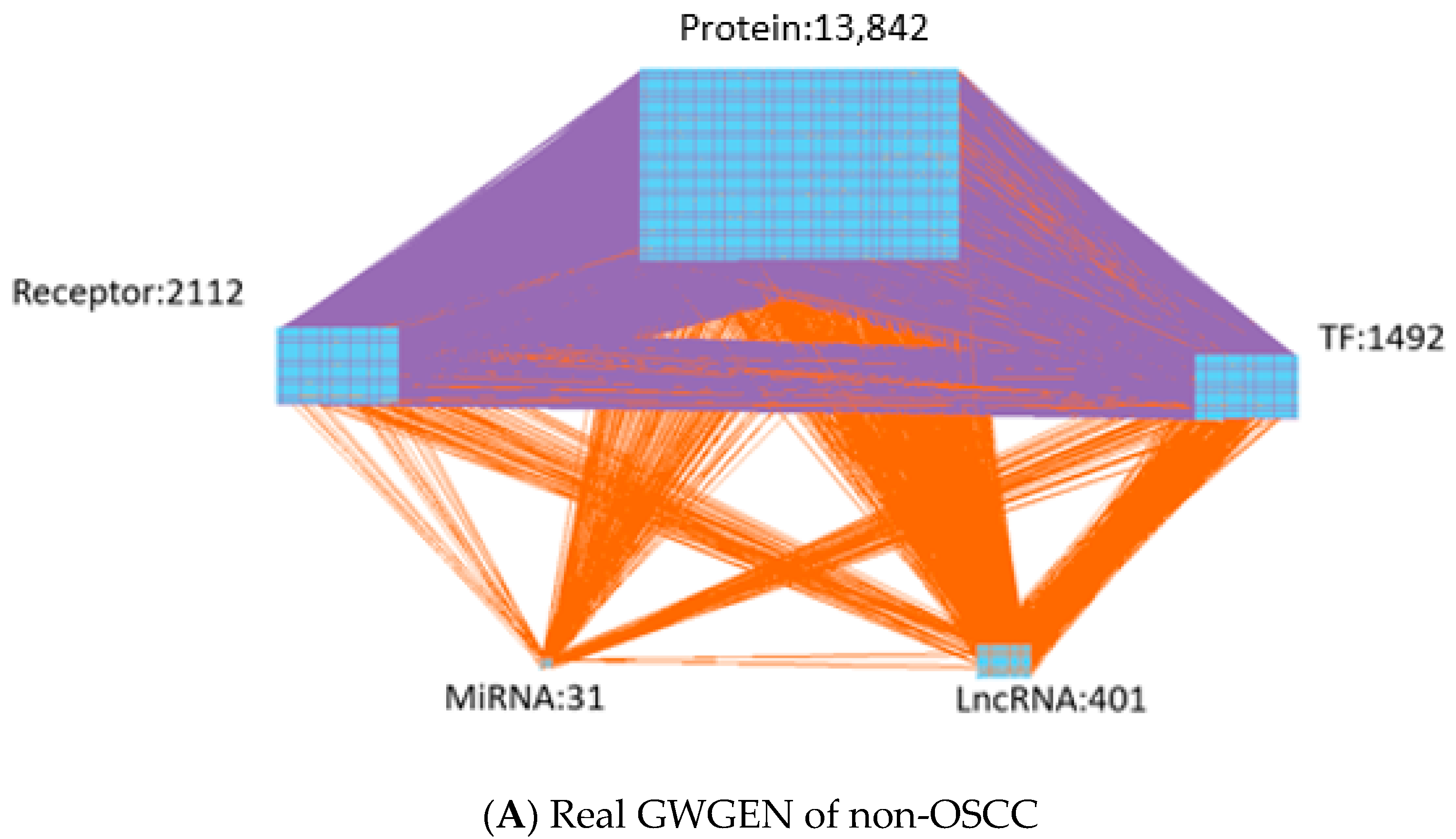
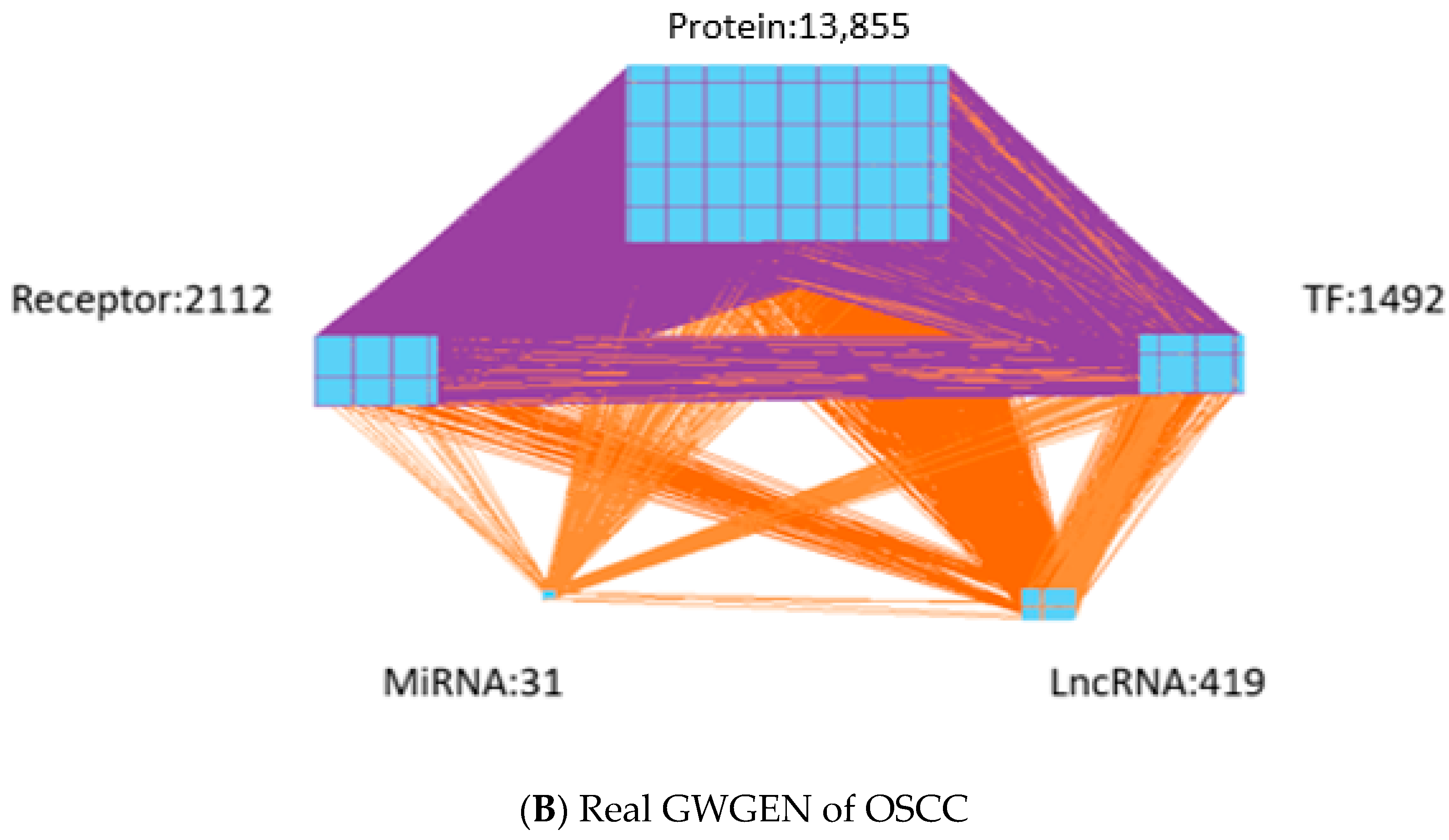
- Identifying real GWGENs: We identify the parameters of PPIN and GRN through the system identification method by solving the corresponding constrained linear least squares estimation problems with the help of the microarray data of OSCC and non-OSCC. After performing system modeling and parameter identification for proteins, genes, miRNAs and lncRNAs in the candidate GWGEN, we used the system order detection method AIC to prune the false positives in the regulation and interactions in the candidate GWGEN to obtain the real GWGENs of OSCC and non-OSCC.
- (3)
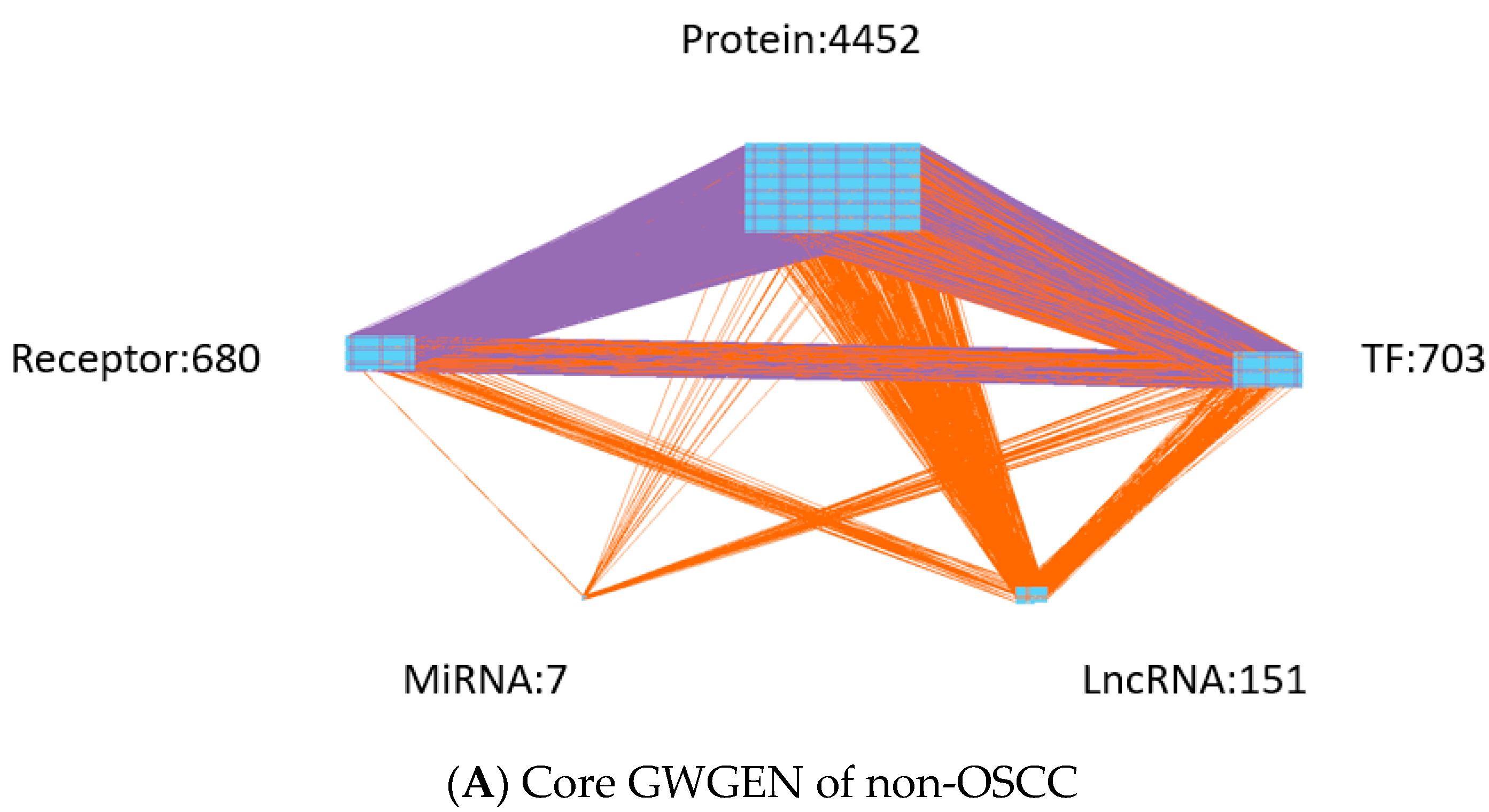
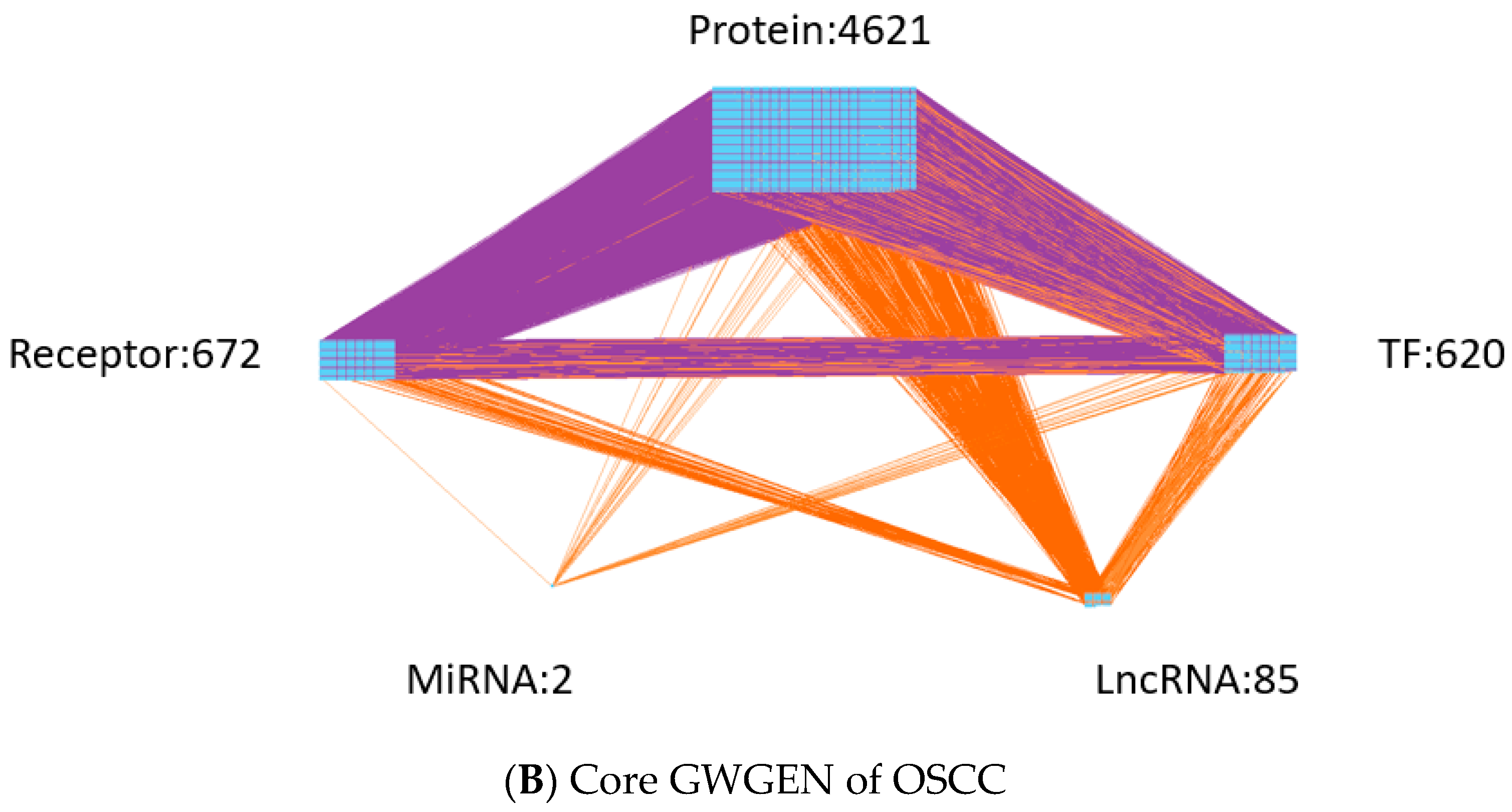
- Extracting the core GWGENs: To extract the core GWGENs of OSCC and non-OSCC, we applied the PNP approach. By doing this, we could compute a projection value for each node in the real GWGENs to 85% significant network structures of real GWGENs. The top 6000 nodes of real GWGENs with the highest projection values have remained as core GWGENs.
- (4)
- Building and comparing the core signaling pathways: The core signaling pathways for cells of OSCC and non-OSCC can be constructed by the annotation of the KEGG pathways of core GWGENs of OSCC and non-OSCC, respectively. We investigated the molecular mechanisms of carcinogenesis of OSCC by comparing the upstream microenvironmental factors, core signaling pathways and their corresponding downstream abnormal cellular functions of OSCC and non-OSCC.
4.2. Data Preprocessing for Constructing the Candidate GWGEN
4.3. System Modeling of the Candidate GWGEN
4.4. The System Identification and System Order Detection Methods for Real GWGENs of OSCC and Non-OSCC from the Candidate GWGEN
4.5. The Principal Network Projection (PNP) Method for the Core GWGENs by Extracting from Real GWGENs
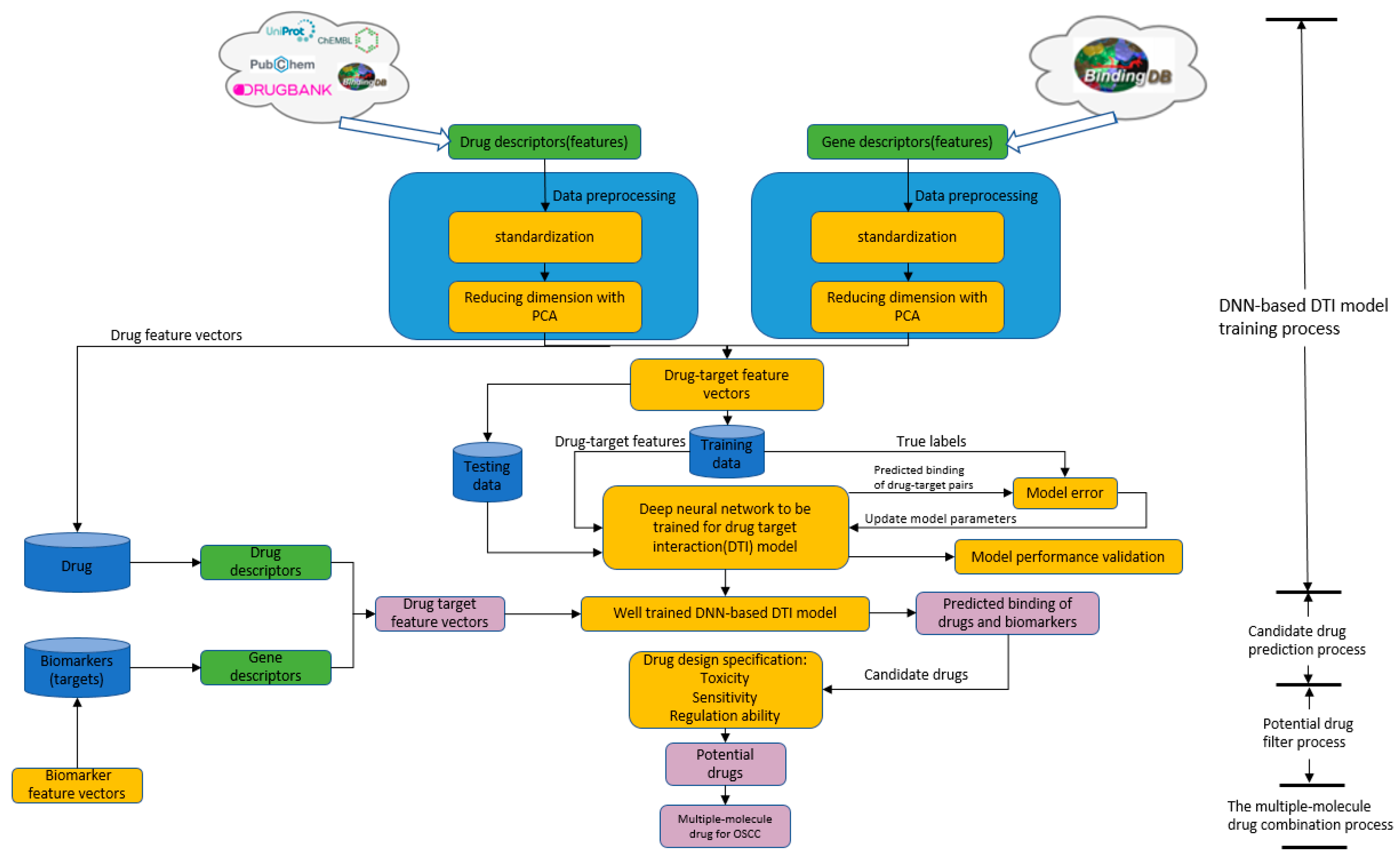
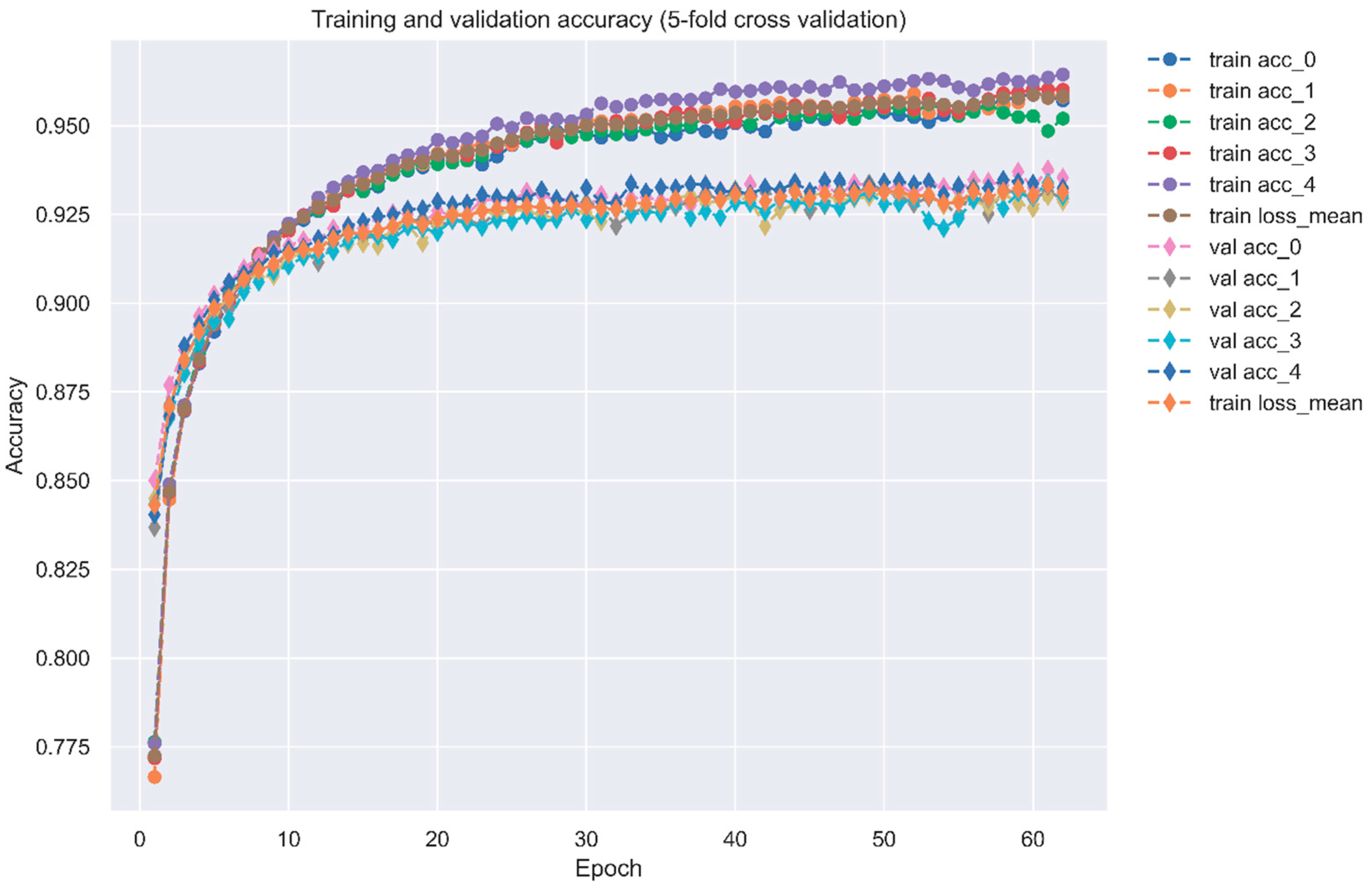
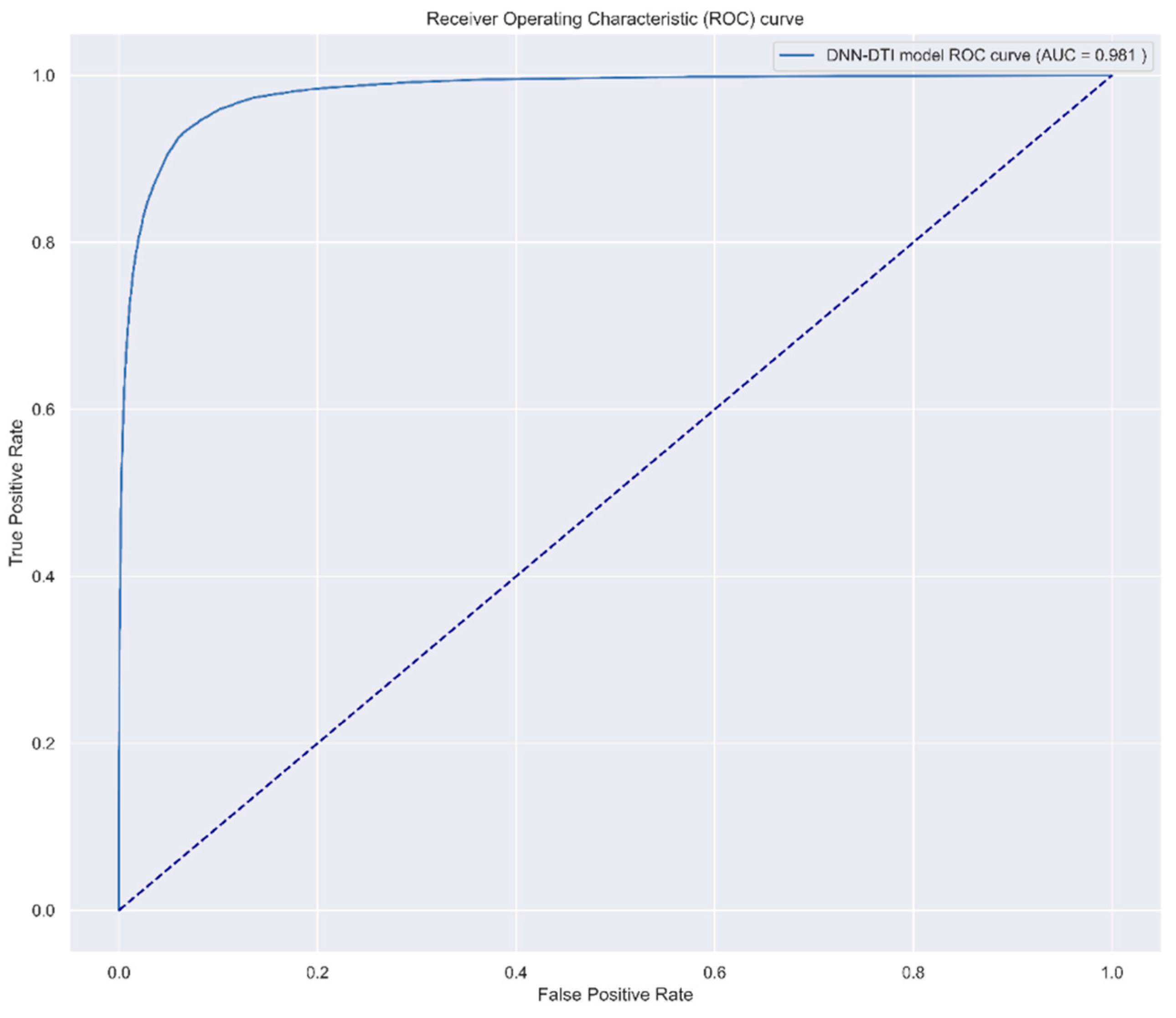
4.6. Systematic Drug Discovery Based on the Drug/Target Interaction Prediction by the DNN-Based DTI Model and Drug Design Specifications for OSCC
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Da Silva, S.D.; Marchi, F.A.; Xu, B.; Bijian, K.; Alobaid, F.; Mlynarek, A.; Rogatto, S.R.; Hier, M.; Kowalski, L.P.; Alaoui-Jamali, M.A. Predominant Rab-GTPase amplicons contributing to oral squamous cell carcinoma progression to metastasis. Oncotarget 2015, 6, 21950. [Google Scholar] [CrossRef] [PubMed]
- Petersen, P.E. Oral cancer prevention and control–the approach of the World Health Organization. Oral Oncol. 2009, 45, 454–460. [Google Scholar] [CrossRef] [PubMed]
- Andisheh-Tadbir, A.; Mehrabani, D.; Heydari, S.T. Epidemiology of squamous cell carcinoma of the oral cavity in Iran. J. Craniofacial Surg. 2008, 19, 1699–1702. [Google Scholar] [CrossRef] [PubMed]
- Dissanayaka, W.L.; Pitiyage, G.; Kumarasiri, P.V.R.; Liyanage, R.L.P.R.; Dias, K.D.; Tilakaratne, W.M. Clinical and histopathologic parameters in survival of oral squamous cell carcinoma. Oral Surg. Oral Med. Oral Pathol. Oral Radiol. 2012, 113, 518–525. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.J.; Chang, J.T.C.; Liao, C.T.; Wang, H.M.; Yen, T.C.; Chiu, C.C.; Lu, Y.C.; Li, H.F.; Cheng, A.J. Head and neck cancer in the betel quid chewing area: Recent advances in molecular carcinogenesis. Cancer Sci. 2008, 99, 1507–1514. [Google Scholar] [CrossRef] [PubMed]
- Otero-Rey, E.M.; Suarez-Alen, F.; Peñamaria-Mallon, M.; Lopez-Lopez, J.; Blanco-Carrion, A. Malignant transformation of oral lichen planus by a chronic inflammatory process. Use of topical corticosteroids to prevent this progression? Acta Odontol. Scand. 2014, 72, 570–577. [Google Scholar] [CrossRef]
- Tang, D.; Tao, D.; Fang, Y.; Deng, C.; Xu, Q.; Zhou, J. TNF-alpha promotes invasion and metastasis via NF-kappa B pathway in oral squamous cell carcinoma. Med. Sci. Monit. Basic Res. 2017, 23, 141. [Google Scholar] [CrossRef]
- Scully, C.; Field, J.; Tanzawa, H. Genetic aberrations in oral or head and neck squamous cell carcinoma (SCCHN): 1. Carcinogen metabolism, DNA repair and cell cycle control. Oral Oncol. 2000, 36, 256–263. [Google Scholar] [CrossRef]
- Feller, L.; Altini, M.; Lemmer, J. Inflammation in the context of oral cancer. Oral Oncol. 2013, 49, 887–892. [Google Scholar] [CrossRef]
- Harada, K.; Ferdous, T.; Itashiki, Y.; Takii, M.; Mano, T.; Mori, Y.; Ueyama, Y. Cepharanthine inhibits angiogenesis and tumorigenicity of human oral squamous cell carcinoma cells by suppressing expression of vascular endothelial growth factor and interleukin-8. Int. J. Oncol. 2009, 35, 1025–1035. [Google Scholar] [CrossRef] [Green Version]
- Chang, S.; Chen, J.-Y.; Chuang, Y.-J.; Chen, B.-S. Systems Approach to Pathogenic Mechanism of Type 2 Diabetes and Drug Discovery Design Based on Deep Learning and Drug Design Specifications. Int. J. Mol. Sci. 2020, 22, 166. [Google Scholar] [CrossRef] [PubMed]
- Ting, C.-T.; Chen, B.-S. Repurposing Multiple-Molecule Drugs for COVID-19-Associated Acute Respiratory Distress Syndrome and Non-Viral Acute Respiratory Distress Syndrome via a Systems Biology Approach and a DNN-DTI Model Based on Five Drug Design Specifications. Int. J. Mol. Sci. 2022, 23, 3649. [Google Scholar] [CrossRef] [PubMed]
- Yeh, S.-J.; Chang, C.-A.; Li, C.-W.; Wang, L.H.-C.; Chen, B.-S. Comparing progression molecular mechanisms between lung adenocarcinoma and lung squamous cell carcinoma based on genetic and epigenetic networks: Big data mining and genome-wide systems identification. Oncotarget 2019, 10, 3760. [Google Scholar] [CrossRef]
- Li, C.-W.; Jheng, B.-R.; Chen, B.-S. Investigating genetic-and-epigenetic networks, and the cellular mechanisms occurring in Epstein–Barr virus-infected human B lymphocytes via big data mining and genome-wide two-sided NGS data identification. PLoS ONE 2018, 13, e0202537. [Google Scholar]
- Hughes, J.P.; Rees, S.; Kalindjian, S.B.; Philpott, K.L. Principles of early drug discovery. Br. J. Pharmacol. 2011, 162, 1239–1249. [Google Scholar] [CrossRef]
- Akhondzadeh, S. The importance of clinical trials in drug development. Avicenna J. Med. Biotechnol. 2016, 8, 151. [Google Scholar]
- Weaver, M.F.; Hopper, J.A.; Gunderson, E.W. Designer drugs 2015: Assessment and management. Addict. Sci. Clin. Pract. 2015, 10, 8. [Google Scholar] [CrossRef] [PubMed]
- Li, A.; Huang, H.-T.; Huang, H.-C.; Juan, H.-F. LncTx: A network-based method to repurpose drugs acting on the survival-related lncRNAs in lung cancer. Comput. Struct. Biotechnol. J. 2021, 19, 3990–4002. [Google Scholar] [CrossRef]
- Cheng, L.-H.; Hsu, T.-C.; Lin, C. Integrating ensemble systems biology feature selection and bimodal deep neural network for breast cancer prognosis prediction. Sci. Rep. 2021, 11, 14914. [Google Scholar]
- Lai, Y.-H.; Chen, W.-N.; Hsu, T.-C.; Lin, C.; Tsao, Y.; Wu, S. Overall survival prediction of non-small cell lung cancer by integrating microarray and clinical data with deep learning. Sci. Rep. 2020, 10, 4679. [Google Scholar]
- Lee, K.-H.; Chang, Y.-C.; Chen, T.-F.; Juan, H.-F.; Tsai, H.-K.; Chen, C.-Y. Connecting MHC-I-binding motifs with HLA alleles via deep learning. Commun. Biol. 2021, 4, 1194. [Google Scholar] [CrossRef] [PubMed]
- Salwinski, L.; Miller, C.S.; Smith, A.J.; Pettit, F.K.; Bowie, J.U.; Eisenberg, D. The database of interacting proteins: 2004 update. Nucleic Acids Res. 2004, 32 (Suppl. 1), D449–D451. [Google Scholar] [CrossRef] [PubMed]
- Orchard, S.; Ammari, M.; Aranda, B.; Breuza, L.; Briganti, L.; Broackes-Carter, F.; Campbell, N.H.; Chavali, G.; Chen, C.; Del-Toro, N. The MIntAct project—IntAct as a common curation platform for 11 molecular interaction databases. Nucleic Acids Res. 2014, 42, D358–D363. [Google Scholar] [CrossRef] [PubMed]
- Stark, C.; Breitkreutz, B.-J.; Reguly, T.; Boucher, L.; Breitkreutz, A.; Tyers, M. BioGRID: A general repository for interaction datasets. Nucleic Acids Res. 2006, 34 (Suppl. 1), D535–D539. [Google Scholar] [CrossRef] [Green Version]
- Zanzoni, A.; Montecchi-Palazzi, L.; Quondam, M.; Ausiello, G.; Helmer-Citterich, M.; Cesareni, G. MINT: A Molecular INTeraction database. FEBS Lett. 2002, 513, 135–140. [Google Scholar] [CrossRef]
- Bovolenta, L.A.; Acencio, M.L.; Lemke, N. HTRIdb: An open-access database for experimentally verified human transcriptional regulation interactions. BMC Genom. 2012, 13, 405. [Google Scholar] [CrossRef]
- Zheng, G.; Tu, K.; Yang, Q.; Xiong, Y.; Wei, C.; Xie, L.; Zhu, Y.; Li, Y. ITFP: An integrated platform of mammalian transcription factors. Bioinformatics 2008, 24, 2416–2417. [Google Scholar] [CrossRef]
- Wingender, E.; Chen, X.; Hehl, R.; Karas, H.; Liebich, I.; Matys, V.; Meinhardt, T.; Prüß, M.; Reuter, I.; Schacherer, F. TRANSFAC: An integrated system for gene expression regulation. Nucleic Acids Res. 2000, 28, 316–319. [Google Scholar] [CrossRef]
- Friard, O.; Re, A.; Taverna, D.; De Bortoli, M.; Corá, D. CircuitsDB: A database of mixed microRNA/transcription factor feed-forward regulatory circuits in human and mouse. BMC Bioinform. 2010, 11, 435. [Google Scholar] [CrossRef]
- Agarwal, V.; Bell, G.W.; Nam, J.-W.; Bartel, D.P. Predicting effective microRNA target sites in mammalian mRNAs. elife 2015, 4, e05005. [Google Scholar] [CrossRef]
- Li, J.-H.; Liu, S.; Zhou, H.; Qu, L.-H.; Yang, J.-H. starBase v2.0: Decoding miRNA-ceRNA, miRNA-ncRNA and protein–RNA interaction networks from large-scale CLIP-Seq data. Nucleic Acids Res. 2014, 42, D92–D97. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.-S.; Wu, C.-C. Systems biology as an integrated platform for bioinformatics, systems synthetic biology, and systems metabolic engineering. Cells 2013, 2, 635–688. [Google Scholar] [CrossRef]
- Li, C.-W.; Chen, B.-S. Investigating core genetic-and-epigenetic cell cycle networks for stemness and carcinogenic mechanisms, and cancer drug design using big database mining and genome-wide next-generation sequencing data. Cell Cycle 2016, 15, 2593–2607. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Pan, C.; Cao, K.; Zhang, J.; Geng, H.; Wu, K.; Wen, J.; Liu, C. Impacts of cigarette smoking on the tumor immune microenvironment in esophageal squamous cell carcinoma. J. Cancer 2022, 13, 413. [Google Scholar] [CrossRef]
- Foy, J.-P.; Bertolus, C.; Michallet, M.-C.; Deneuve, S.; Incitti, R.; Bendriss-Vermare, N.; Albaret, M.-A.; Ortiz-Cuaran, S.; Thomas, E.; Colombe, A. The immune microenvironment of HPV-negative oral squamous cell carcinoma from never-smokers and never-drinkers patients suggests higher clinical benefit of IDO1 and PD1/PD-L1 blockade. Ann. Oncol. 2017, 28, 1934–1941. [Google Scholar] [CrossRef]
- Cheskis, B.J.; Greger, J.; Cooch, N.; McNally, C.; Mclarney, S.; Lam, H.-S.; Rutledge, S.; Mekonnen, B.; Hauze, D.; Nagpal, S. MNAR plays an important role in ERa activation of Src/MAPK and PI3K/Akt signaling pathways. Steroids 2008, 73, 901–905. [Google Scholar] [CrossRef]
- Burotto, M.; Chiou, V.L.; Lee, J.M.; Kohn, E.C. The MAPK pathway across different malignancies: A new perspective. Cancer 2014, 120, 3446–3456. [Google Scholar] [CrossRef]
- Mittal, M.; Kapoor, V.; Mohanti, B.K.; Das, S.N. Functional variants of COX-2 and risk of tobacco-related oral squamous cell carcinoma in high-risk Asian Indians. Oral Oncol. 2010, 46, 622–626. [Google Scholar] [CrossRef]
- Ramos-Garcia, P.; Gil-Montoya, J.; Scully, C.; Ayén, A.; González-Ruiz, L.; Navarro-Triviño, F.; González-Moles, M. An update on the implications of cyclin D1 in oral carcinogenesis. Oral Dis. 2017, 23, 897–912. [Google Scholar] [CrossRef] [PubMed]
- Lakshminarayana, S.; Augustine, D.; Rao, R.S.; Patil, S.; Awan, K.H.; Venkatesiah, S.S.; Haragannavar, V.C.; Nambiar, S.; Prasad, K. Molecular pathways of oral cancer that predict prognosis and survival: A systematic review. J. Carcinog. 2018, 17, 7. [Google Scholar] [CrossRef]
- Freier, K.; Sticht, C.; Hofele, C.; Flechtenmacher, C.; Stange, D.; Puccio, L.; Toedt, G.; Radlwimmer, B.; Lichter, P.; Joos, S. Recurrent coamplification of cytoskeleton-associated genes EMS1 and SHANK2 with CCND1 in oral squamous cell carcinoma. Genes Chromosomes Cancer 2006, 45, 118–125. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Han, J.; Xu, P.; Jian, X.; Huang, X.; Liu, D. Silencing of LncRNA SNHG16 downregulates cyclin D1 (CCND1) to abrogate malignant phenotypes in oral squamous cell carcinoma (OSCC) through upregulating miR-17–5p. Cancer Manag. Res. 2021, 13, 1831. [Google Scholar] [CrossRef] [PubMed]
- Giles, R.H.; Van Es, J.H.; Clevers, H. Caught up in a Wnt storm: Wnt signaling in cancer. Biochim. Biophys. Acta (BBA)-Rev. Cancer 2003, 1653, 1–24. [Google Scholar] [CrossRef]
- Huelsken, J.; Behrens, J. The Wnt signalling pathway. J. Cell Sci. 2002, 115, 3977–3978. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lyou, Y.; Habowski, A.N.; Chen, G.T.; Waterman, M.L. Inhibition of nuclear Wnt signalling: Challenges of an elusive target for cancer therapy. Br. J. Pharmacol. 2017, 174, 4589–4599. [Google Scholar] [CrossRef] [PubMed]
- Xiao, X.; Gu, Y.; Wang, G.; Chen, S. c-Myc, RMRP, and miR-34a-5p form a positive-feedback loop to regulate cell proliferation and apoptosis in multiple myeloma. Int. J. Biol. Macromol. 2019, 122, 526–537. [Google Scholar] [CrossRef]
- Walker, L.; Lynch, M.; Silverman, S.; Fraser, J.; Boulter, J.; Weinmaster, G.; Gasson, J.C. The Notch/Jagged pathway inhibits proliferation of human hematopoietic progenitors in vitro. Stem Cells 1999, 17, 162–171. [Google Scholar] [CrossRef]
- Chai, A.W.Y.; Lim, K.P.; Cheong, S.C. Translational genomics and recent advances in oral squamous cell carcinoma. In Seminars in Cancer Biology; Elsevier: Amsterdam, The Netherlands, 2020; pp. 71–83. [Google Scholar]
- Hijioka, H.; Setoguchi, T.; Miyawaki, A.; Gao, H.; Ishida, T.; Komiya, S.; Nakamura, N. Upregulation of Notch pathway molecules in oral squamous cell carcinoma. Int. J. Oncol. 2010, 36, 817–822. [Google Scholar]
- Sjölund, J.; Manetopoulos, C.; Stockhausen, M.-T.; Axelson, H. The Notch pathway in cancer: Differentiation gone awry. Eur. J. Cancer 2005, 41, 2620–2629. [Google Scholar] [CrossRef]
- Ingram, W.; McCue, K.; Tran, T.; Hallahan, A.; Wainwright, B. Sonic Hedgehog regulates Hes1 through a novel mechanism that is independent of canonical Notch pathway signalling. Oncogene 2008, 27, 1489–1500. [Google Scholar] [CrossRef]
- Subramaniam, D.; Ponnurangam, S.; Ramamoorthy, P.; Standing, D.; Battafarano, R.J.; Anant, S.; Sharma, P. Curcumin induces cell death in esophageal cancer cells through modulating Notch signaling. PLoS ONE 2012, 7, e30590. [Google Scholar] [CrossRef] [PubMed]
- Bi, H.; Ming, L.; Cheng, R.; Luo, H.; Zhang, Y.; Jin, Y. Liver extracellular matrix promotes BM-MSCs hepatic differentiation and reversal of liver fibrosis through activation of integrin pathway. J. Tissue Eng. Regen. Med. 2017, 11, 2685–2698. [Google Scholar] [CrossRef] [PubMed]
- Dayyani, F.; Parikh, N.U.; Varkaris, A.S.; Song, J.H.; Moorthy, S.; Chatterji, T.; Maity, S.N.; Wolfe, A.R.; Carboni, J.M.; Gottardis, M.M. Combined Inhibition of IGF-1R/IR and Src family kinases enhances antitumor effects in prostate cancer by decreasing activated survival pathways. PLoS ONE 2012, 7, e51189. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.; Weinberger, P.M.; Sasaki, C.; Egleston, B.L.; Speier IV, W.F.; Haffty, B.; Kowalski, D.; Camp, R.; Rimm, D.; Vairaktaris, E. Phosphorylation of Akt (Ser473) predicts poor clinical outcome in oropharyngeal squamous cell cancer. Cancer Epidemiol. Biomark. Prev. 2007, 16, 553–558. [Google Scholar] [CrossRef]
- Subarnbhesaj, A.; Miyauchi, M.; Chanbora, C.; Mikuriya, A.; Nguyen, P.T.; Furusho, H.; Ayuningtyas, N.F.; Fujita, M.; Toratani, S.; Takechi, M. Roles of VEGF-Flt-1 signaling in malignant behaviors of oral squamous cell carcinoma. PLoS ONE 2017, 12, e0187092. [Google Scholar] [CrossRef] [Green Version]
- Seçilmiş, D.; Hillerton, T.; Morgan, D.; Tjärnberg, A.; Nelander, S.; Nordling, T.E.; Sonnhammer, E.L. Uncovering cancer gene regulation by accurate regulatory network inference from uninformative data. NPJ Syst. Biol. Appl. 2020, 6, 37. [Google Scholar] [CrossRef]
- Subramanian, A.; Narayan, R.; Corsello, S.M.; Peck, D.D.; Natoli, T.E.; Lu, X.; Gould, J.; Davis, J.F.; Tubelli, A.A.; Asiedu, J.K. A next generation connectivity map: L1000 platform and the first 1,000,000 profiles. Cell 2017, 171, 1437–1452.e17. [Google Scholar] [CrossRef]
- Knox, C.; Law, V.; Jewison, T.; Liu, P.; Ly, S.; Frolkis, A.; Pon, A.; Banco, K.; Mak, C.; Neveu, V. DrugBank 3.0: A comprehensive resource for ‘omics’ research on drugs. Nucleic Acids Res. 2010, 39 (Suppl. 1), D1035–D1041. [Google Scholar] [CrossRef]
- Corsello, S.M.; Nagari, R.T.; Spangler, R.D.; Rossen, J.; Kocak, M.; Bryan, J.G.; Humeidi, R.; Peck, D.; Wu, X.; Tang, A.A. Discovering the anticancer potential of non-oncology drugs by systematic viability profiling. Nat. Cancer 2020, 1, 235–248. [Google Scholar] [CrossRef]
- Li, X.; Guo, S.; Xiong, X.-K.; Peng, B.-Y.; Huang, J.-M.; Chen, M.-F.; Wang, F.-Y.; Wang, J.-N. Combination of quercetin and cisplatin enhances apoptosis in OSCC cells by downregulating xIAP through the NF-κB pathway. J. Cancer 2019, 10, 4509. [Google Scholar] [CrossRef]
- Li, L.; Liu, H.-C.; Wang, C.; Liu, X.; Hu, F.-C.; Xie, N.; Lü, L.; Chen, X.; Huang, H.-Z. Overexpression of β-catenin induces cisplatin resistance in oral squamous cell carcinoma. BioMed Res. Int. 2016, 2016, 5378567. [Google Scholar] [CrossRef] [PubMed]
- Ding, L.; Ren, J.; Zhang, D.; Li, Y.; Huang, X.; Ji, J.; Hu, Q.; Wang, H.; Ni, Y.; Hou, Y. The TLR3 Agonist Inhibit Drug Efflux and Sequentially Consolidates Low-Dose Cisplatin-Based Chemoimmunotherapy while Reducing Side EffectsOrderly Combination poly (I: C)/Low-Dose DDP. Mol. Cancer Ther. 2017, 16, 1068–1079. [Google Scholar] [CrossRef] [PubMed]
- Chou, T.-Y.; Chiu, C.-H.; Li, L.-H.; Hsiao, C.-Y.; Tzen, C.-Y.; Chang, K.-T.; Chen, Y.-M.; Perng, R.-P.; Tsai, S.-F.; Tsai, C.-M. Mutation in the tyrosine kinase domain of epidermal growth factor receptor is a predictive and prognostic factor for gefitinib treatment in patients with non–small cell lung cancer. Clin. Cancer Res. 2005, 11, 3750–3757. [Google Scholar] [CrossRef] [PubMed]
- Taron, M.; Ichinose, Y.; Rosell, R.; Mok, T.; Massuti, B.; Zamora, L.; Mate, J.L.; Manegold, C.; Ono, M.; Queralt, C. Activating mutations in the tyrosine kinase domain of the epidermal growth factor receptor are associated with improved survival in gefitinib-treated chemorefractory lung adenocarcinomas. Clin. Cancer Res. 2005, 11, 5878–5885. [Google Scholar] [CrossRef]
- Moulder, S.L.; Yakes, F.M.; Muthuswamy, S.K.; Bianco, R.; Simpson, J.F.; Arteaga, C.L. Epidermal growth factor receptor (HER1) tyrosine kinase inhibitor ZD1839 (Iressa) inhibits HER2/neu (erb B2)-overexpressing breast cancer cells in vitro and in vivo. Cancer Res. 2001, 61, 8887–8895. [Google Scholar] [PubMed]
- Khan, Z.; Bisen, P.S. Oncoapoptotic signaling and deregulated target genes in cancers: Special reference to oral cancer. Biochim. Biophys. Acta Rev. Cancer 2013, 1836, 123–145. [Google Scholar] [CrossRef]
- Shortt, J.; Johnstone, R.W. Oncogenes in cell survival and cell death. Cold Spring Harb. Perspect. Biol. 2012, 4, a009829. [Google Scholar] [CrossRef]
- Peters, S.; Zimmermann, S.; Adjei, A.A. Oral epidermal growth factor receptor tyrosine kinase inhibitors for the treatment of non-small cell lung cancer: Comparative pharmacokinetics and drug–drug interactions. Cancer Treat. Rev. 2014, 40, 917–926. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Rangel, E.; Inzucchi, S.E. Metformin: Clinical use in type 2 diabetes. Diabetologia 2017, 60, 1586–1593. [Google Scholar] [CrossRef]
- Lv, Z.; Guo, Y. Metformin and its benefits for various diseases. Front. Endocrinol. 2020, 11, 191. [Google Scholar] [CrossRef]
- Chae, Y.K.; Arya, A.; Malecek, M.-K.; Shin, D.S.; Carneiro, B.; Chandra, S.; Kaplan, J.; Kalyan, A.; Altman, J.K.; Platanias, L. Repurposing metformin for cancer treatment: Current clinical studies. Oncotarget 2016, 7, 40767. [Google Scholar] [CrossRef] [PubMed]
- Kasznicki, J.; Sliwinska, A.; Drzewoski, J. Metformin in cancer prevention and therapy. Ann. Transl. Med. 2014, 2, 57. [Google Scholar] [PubMed]
- Li, Y.; Wang, M.; Zhi, P.; You, J.; Gao, J.-Q. Metformin synergistically suppress tumor growth with doxorubicin and reverse drug resistance by inhibiting the expression and function of P-glycoprotein in MCF7/ADR cells and xenograft models. Oncotarget 2018, 9, 2158. [Google Scholar] [CrossRef] [PubMed]
- Shah, R.R. Hyperglycaemia induced by novel anticancer agents: An undesirable complication or a potential therapeutic opportunity? Drug Saf. 2017, 40, 211–228. [Google Scholar] [CrossRef]
- Peng, M.; Darko, K.O.; Tao, T.; Huang, Y.; Su, Q.; He, C.; Yin, T.; Liu, Z.; Yang, X. Combination of metformin with chemotherapeutic drugs via different molecular mechanisms. Cancer Treat. Rev. 2017, 54, 24–33. [Google Scholar] [CrossRef]
- Haslam, E.; Cai, Y. Plant polyphenols (vegetable tannins): Gallic acid metabolism. Nat. Prod. Rep. 1994, 11, 41–66. [Google Scholar] [CrossRef]
- Cai, Y.; Luo, Q.; Sun, M.; Corke, H. Antioxidant activity and phenolic compounds of 112 traditional Chinese medicinal plants associated with anticancer. Life Sci. 2004, 74, 2157–2184. [Google Scholar] [CrossRef]
- Shabani, S.; Rabiei, Z.; Amini-Khoei, H. Exploring the multifaceted neuroprotective actions of gallic acid: A review. Int. J. Food Prop. 2020, 23, 736–752. [Google Scholar] [CrossRef]
- Nikbakht, J.; Hemmati, A.A.; Arzi, A.; Mansouri, M.T.; Rezaie, A.; Ghafourian, M. Protective effect of gallic acid against bleomycin-induced pulmonary fibrosis in rats. Pharmacol. Rep. 2015, 67, 1061–1067. [Google Scholar] [CrossRef]
- Chuang, C.-Y.; Liu, H.-C.; Wu, L.-C.; Chen, C.-Y.; Chang, J.T.; Hsu, S.-L. Gallic acid induces apoptosis of lung fibroblasts via a reactive oxygen species-dependent ataxia telangiectasia mutated-p53 activation pathway. J. Agric. Food Chem. 2010, 58, 2943–2951. [Google Scholar] [CrossRef]
- Bai, J.; Zhang, Y.; Tang, C.; Hou, Y.; Ai, X.; Chen, X.; Zhang, Y.; Wang, X.; Meng, X. Gallic acid: Pharmacological activities and molecular mechanisms involved in inflammation-related diseases. Biomed. Pharmacother. 2021, 133, 110985. [Google Scholar] [CrossRef] [PubMed]
- Yeh, S.-J.; Chen, S.-W.; Chen, B.-S. Investigation of the genome-wide genetic and epigenetic networks for drug discovery based on systems biology approaches in colorectal cancer. Front. Genet. 2020, 11, 117. [Google Scholar] [CrossRef]
- Consortium, U. UniProt: A worldwide hub of protein knowledge. Nucleic Acids Res. 2019, 47, D506–D515. [Google Scholar] [CrossRef]
- Liu, T.; Lin, Y.; Wen, X.; Jorissen, R.N.; Gilson, M.K. BindingDB: A web-accessible database of experimentally determined protein–ligand binding affinities. Nucleic Acids Res. 2007, 35 (Suppl. 1), D198–D201. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Thiessen, P.A.; Bolton, E.E.; Chen, J.; Fu, G.; Gindulyte, A.; Han, L.; He, J.; He, S.; Shoemaker, B.A. PubChem substance and compound databases. Nucleic Acids Res. 2016, 44, D1202–D1213. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Microarray Data | Non-OSCC | OSCC |
|---|---|---|
| GSE30784 and GSE17913 | 102 | 167 |
| Real GWGEN of OSCC | Core GWGEN of OSCC | |
|---|---|---|
| Protein | 13,855 | 4621 |
| Receptor | 2112 | 672 |
| TF | 1492 | 620 |
| miRNA | 31 | 2 |
| LncRNA | 419 | 85 |
| Total nodes | 17,909 | 6000 |
| KEGG Pathway Enrichment Analysis of OSCC Core Signaling Pathways | ||
|---|---|---|
| Pathway | Gene number | p-value |
| Cell cycle | 91 | 8.3 × 10−20 |
| Pathways in cancer | 267 | 3.1 × 10−19 |
| MAPK signaling pathway | 149 | 3.6 × 10−11 |
| Apoptosis | 80 | 3.2 × 10−10 |
| Proteoglycans in cancer | 120 | 1.0 × 10−14 |
| KEGG Pathway Enrichment Analysis of Non-OSCC Core Signaling Pathways | ||
|---|---|---|
| Pathway | Gene number | p-value |
| Cellular senescence | 95 | 2.3 × 10−13 |
| MAPK signaling pathway | 166 | 2.4 × 10−18 |
| Human T-cell leukemia virus 1 infection | 134 | 3.2 × 10−18 |
| Cell cycle | 84 | 4.2 × 10−15 |
| P53 signaling pathway | 46 | 1.6 × 10−7 |
| HES1 (-) | |||
|---|---|---|---|
| Drug | Regulation Ability (L1000) | Sensitivity (PRISM) | Toxicity (LC50, mol/kg) |
| capsaicin | 0.1690 | −0.1217 | 4.202 |
| gabazine | 0.3716 | −0.6103 | 3.079 |
| phenolphthalein | −0.6116 | −0.4833 | 5.297 |
| tetramisole | 0.1036 | 0.1136 | 4.111 |
| gefitinib | 0.2750 | −0.5144 | 5.068 |
| TCF (-) | |||
| Drug | Regulation Ability (L1000) | Sensitivity (PRISM) | Toxicity (LC50, mol/kg) |
| carvedilol | −0.0787 | −0.0906 | 5.014 |
| fipronil | −0.1207 | −0.0939 | 5.534 |
| metformin | 0.0770 | −0.0789 | 2.039 |
| diethylcarbamazine | 0.0501 | −0.0848 | 2.008 |
| dyphylline | 0.1372 | 0.0356 | 2.022 |
| NF-κB (+) | |||
| Drug | Regulation Ability (L1000) | Sensitivity (PRISM) | Toxicity (LC50, mol/kg) |
| sirolimus | −0.0866 | −0.2058 | 3.486 |
| terfenadine | −0.7665 | −0.7406 | 5.437 |
| metformin | −0.2607 | −0.0789 | 2.039 |
| gallic-acid | −1.0620 | 0.6208 | 3.262 |
| gefitinib | −0.3428 | −0.5144 | 5.068 |
| SP1 (+) | |||
| Drug | Regulation Ability (L1000) | Sensitivity (PRISM) | Toxicity (LC50, mol/kg) |
| niridazole | −0.6456 | −0.1400 | 2.746 |
| chlorambucil | −0.0559 | −0.1424 | 3.249 |
| bepridil | 0.7249 | 0.2789 | 5.083 |
| gallic-acid | −0.5239 | 0.6208 | 3.262 |
| disopyramide | −0.3694 | −0.1440 | 3.316 |
| Targets | HES1 | TCF | NF-κB | SP1 | Toxicity (LC50, mol/kg) | Sensitivity (PRISM) | |
|---|---|---|---|---|---|---|---|
| Drugs | |||||||
| metformin | ✓(0.0770) | ✓(−0.2607) | 2.039 | −0.0789 | |||
| gefitinib | ✓(0.2750) | ✓(−0.3428) | 5.068 | −0.5144 | |||
| gallic-acid | ✓(−1.0620) | ✓(−0.5239) | 3.262 | 0.6208 | |||
| metformin | gefitinib | ||||||
 |  | ||||||
| gallic-acid | |||||||
 | |||||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lin, Y.-C.; Chen, B.-S. Identifying Drug Targets of Oral Squamous Cell Carcinoma through a Systems Biology Method and Genome-Wide Microarray Data for Drug Discovery by Deep Learning and Drug Design Specifications. Int. J. Mol. Sci. 2022, 23, 10409. https://doi.org/10.3390/ijms231810409
Lin Y-C, Chen B-S. Identifying Drug Targets of Oral Squamous Cell Carcinoma through a Systems Biology Method and Genome-Wide Microarray Data for Drug Discovery by Deep Learning and Drug Design Specifications. International Journal of Molecular Sciences. 2022; 23(18):10409. https://doi.org/10.3390/ijms231810409
Chicago/Turabian StyleLin, Yi-Chung, and Bor-Sen Chen. 2022. "Identifying Drug Targets of Oral Squamous Cell Carcinoma through a Systems Biology Method and Genome-Wide Microarray Data for Drug Discovery by Deep Learning and Drug Design Specifications" International Journal of Molecular Sciences 23, no. 18: 10409. https://doi.org/10.3390/ijms231810409