
The Effect of Uridine on the State of Skeletal Muscles and the Functioning of Mitochondria in Duchenne Dystrophy

 ,
,  ,
, 
Abstract
:1. Introduction
2. Results
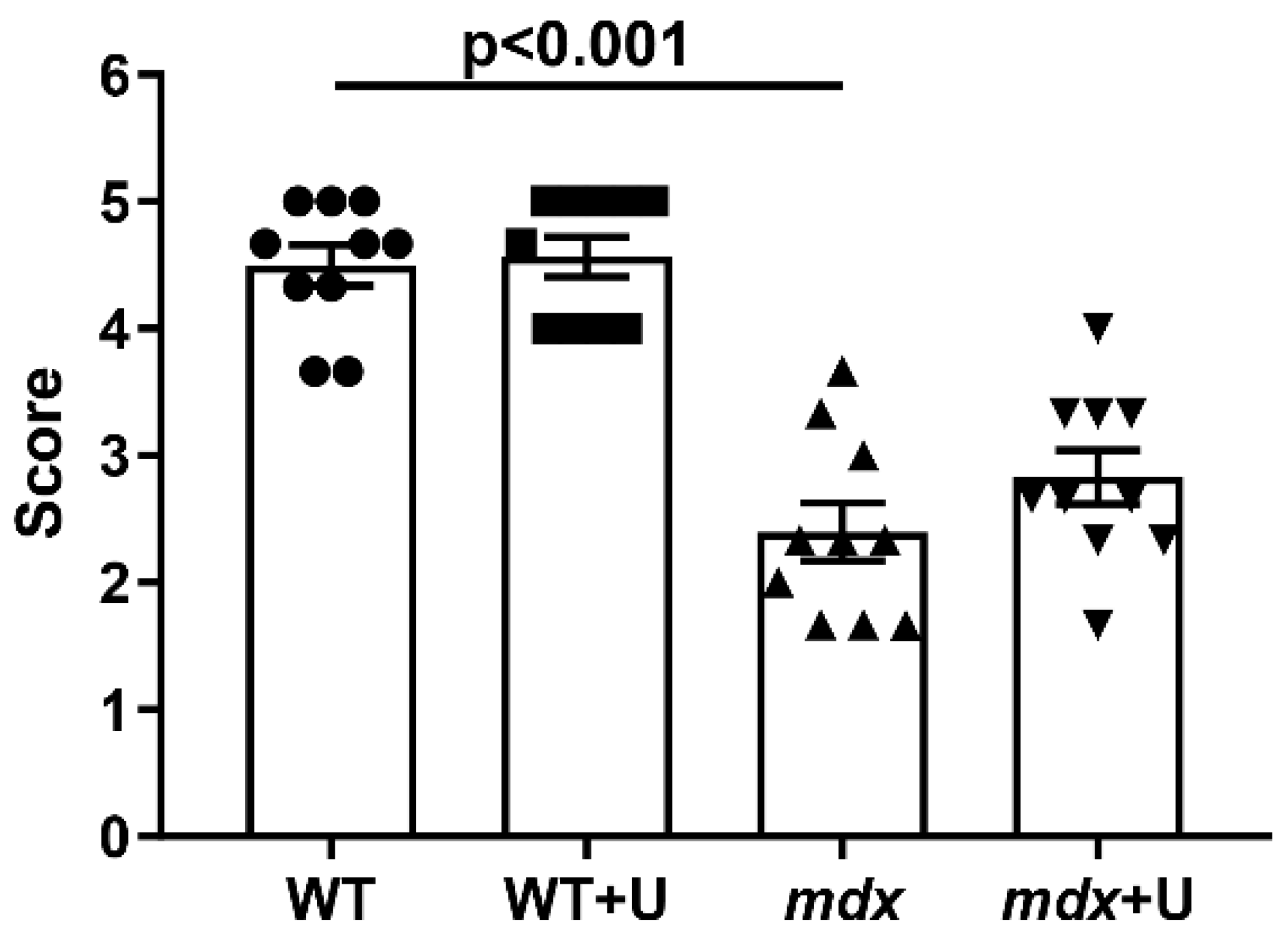
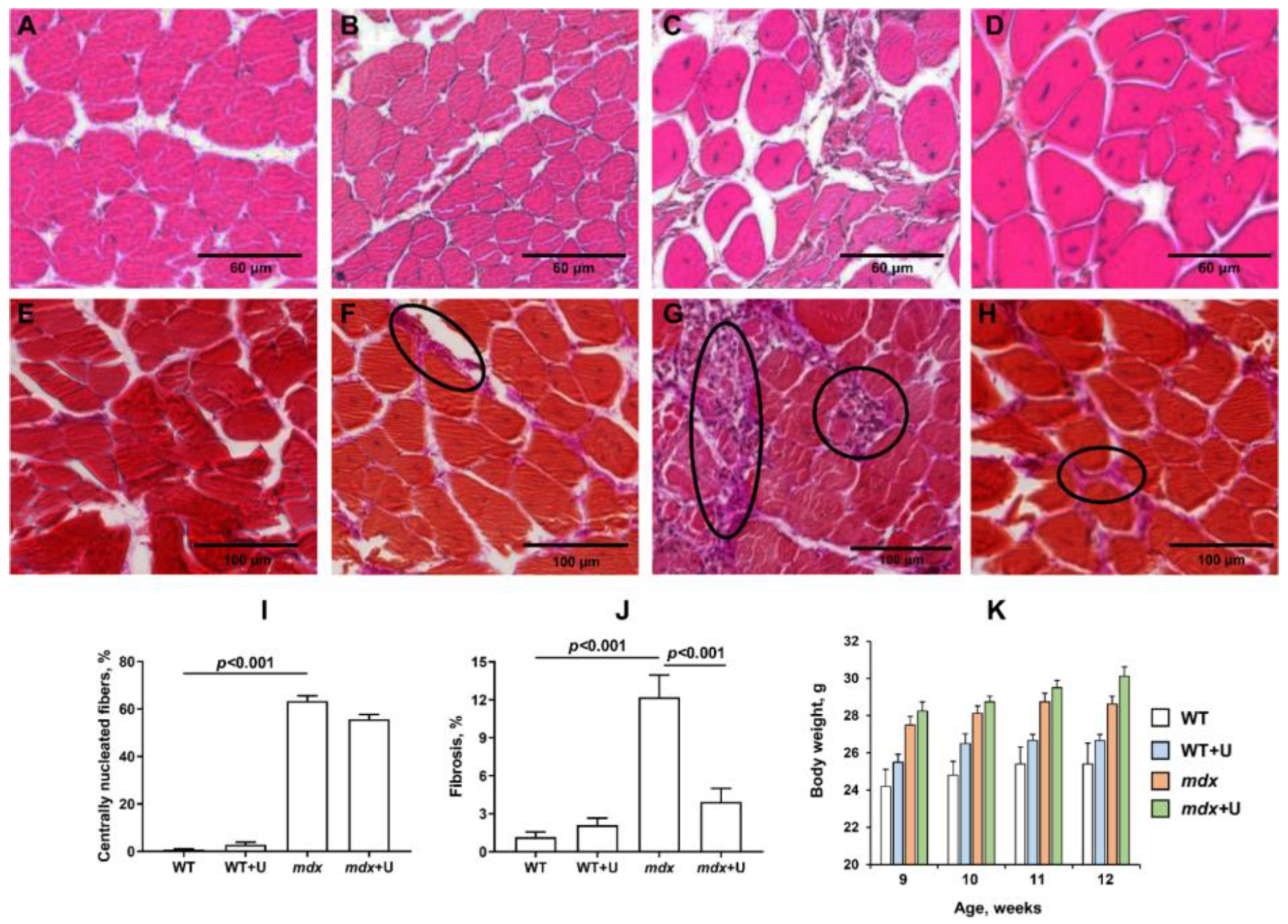
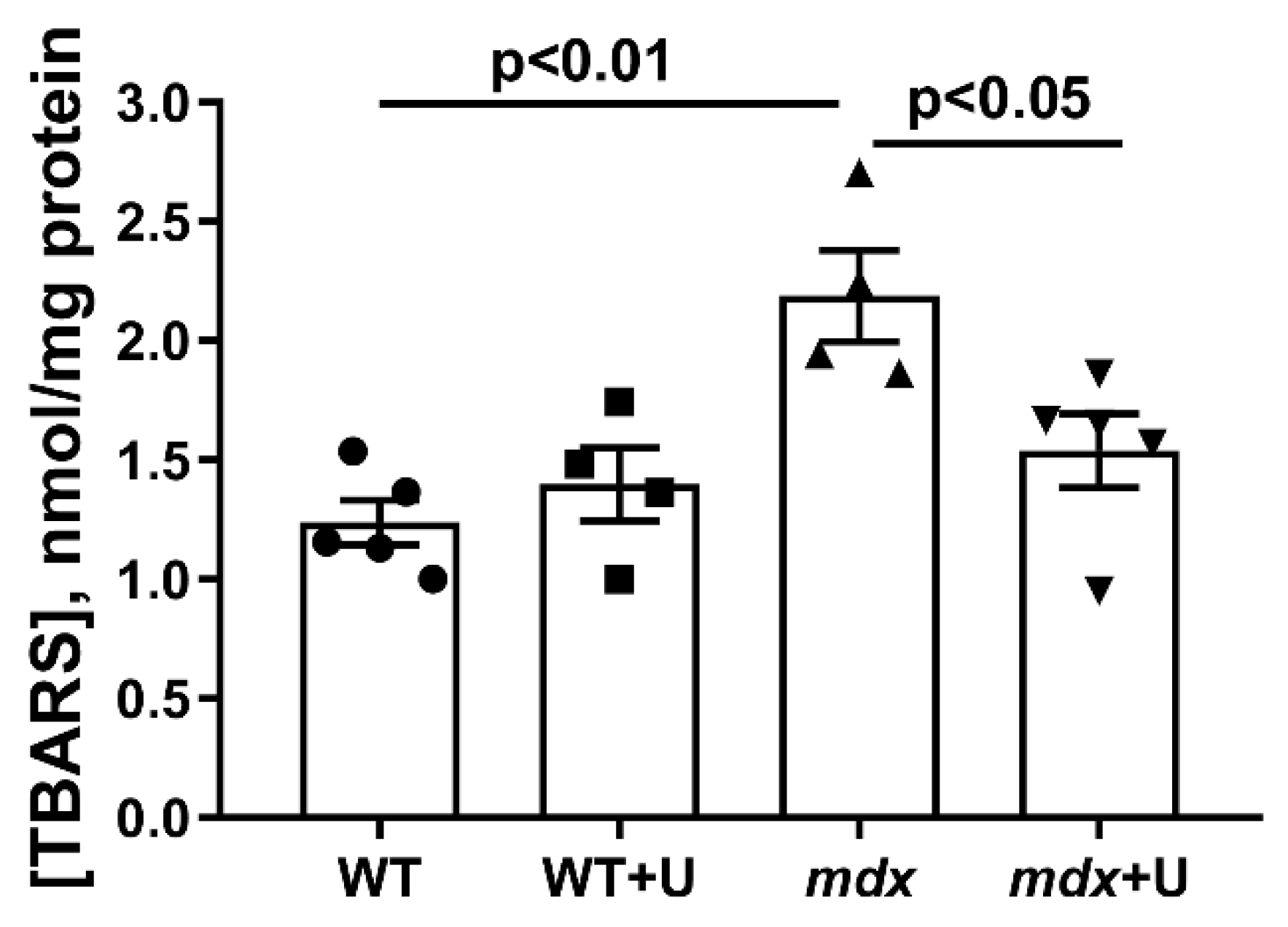
2.1. Effect of Uridine Treatment on the Somatic and Biochemical Characteristics of Mice and Skeletal Muscle Health
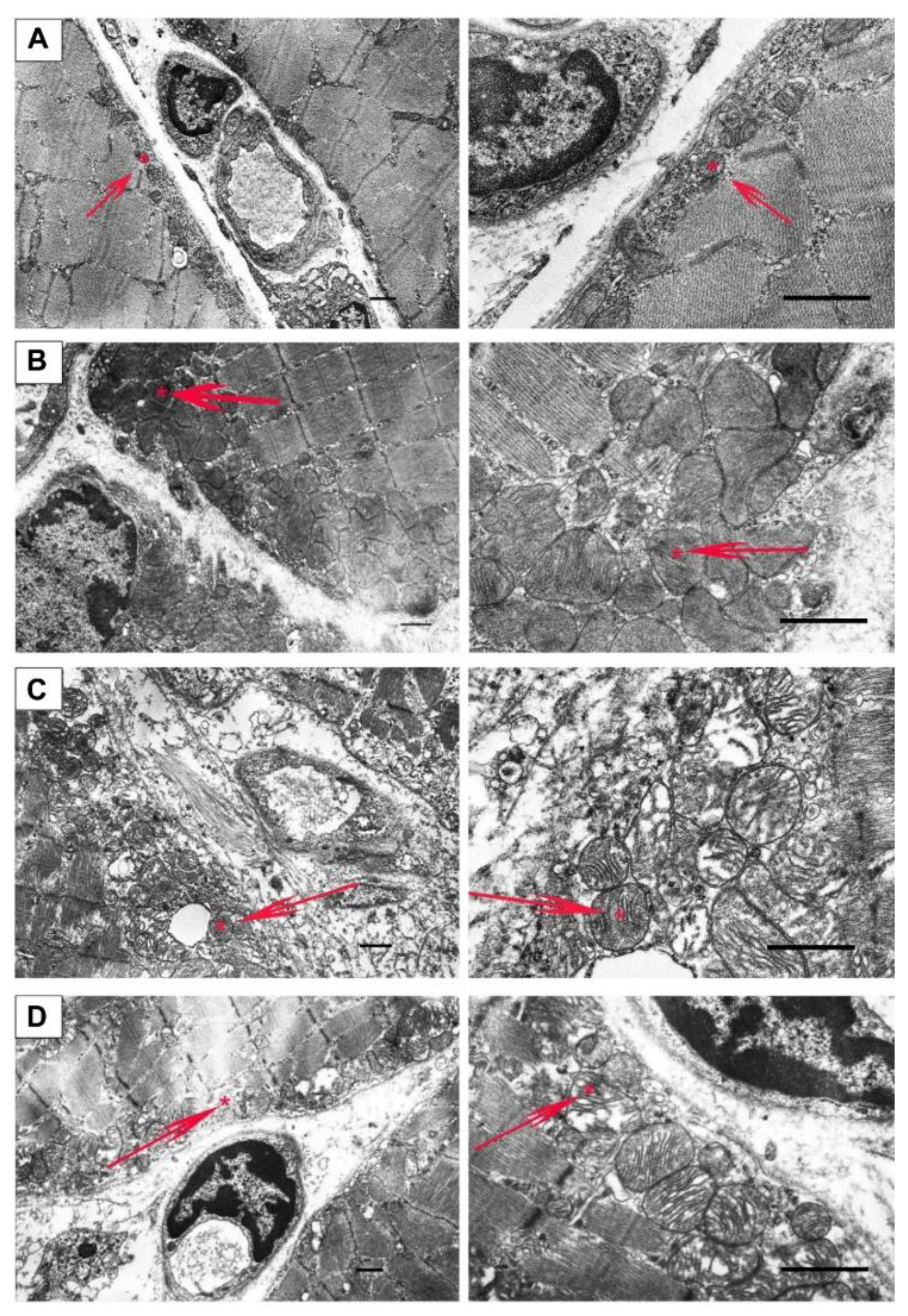
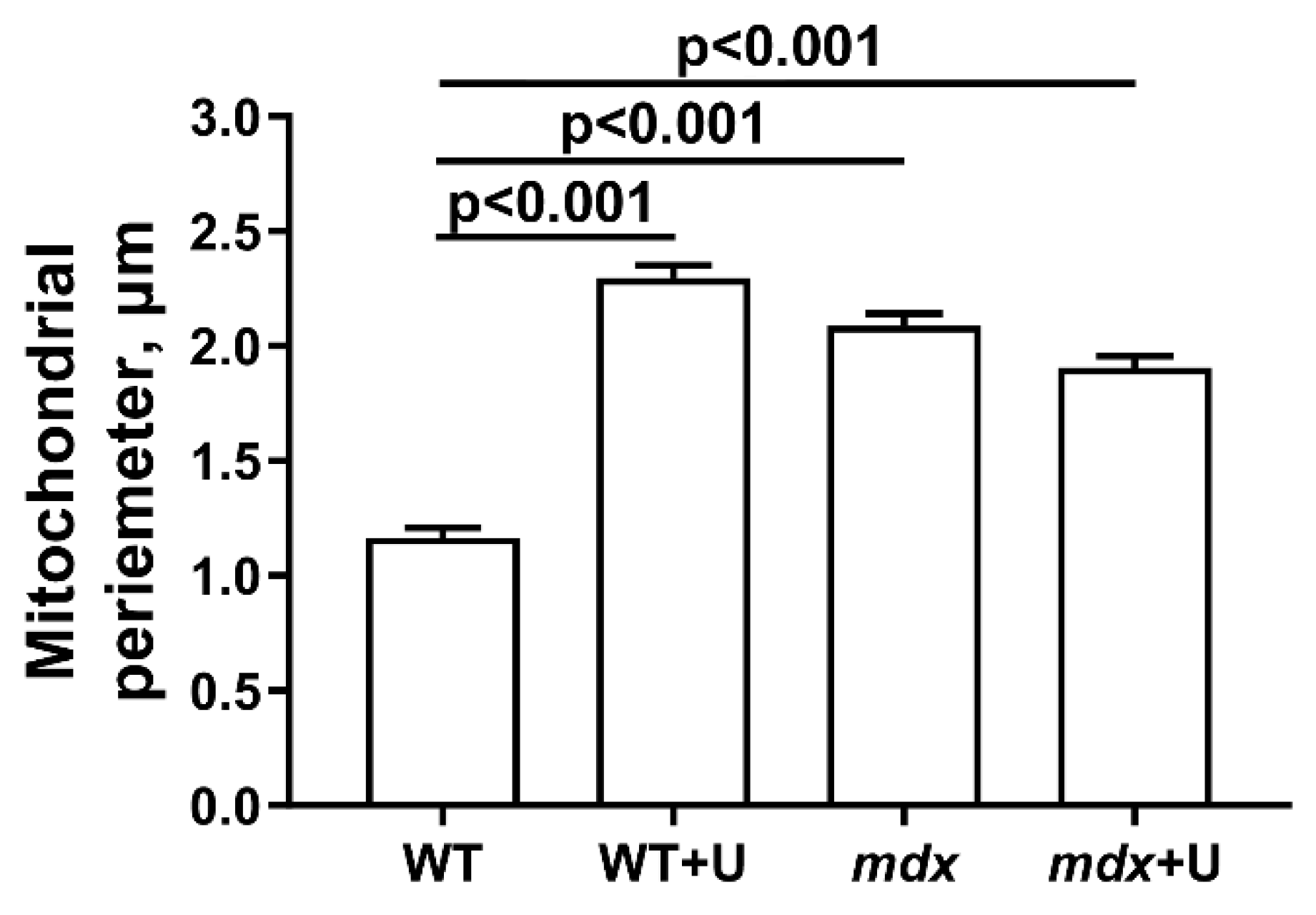
2.2. Effect of Uridine on the Ultrastructure of Skeletal Muscle Mitochondria
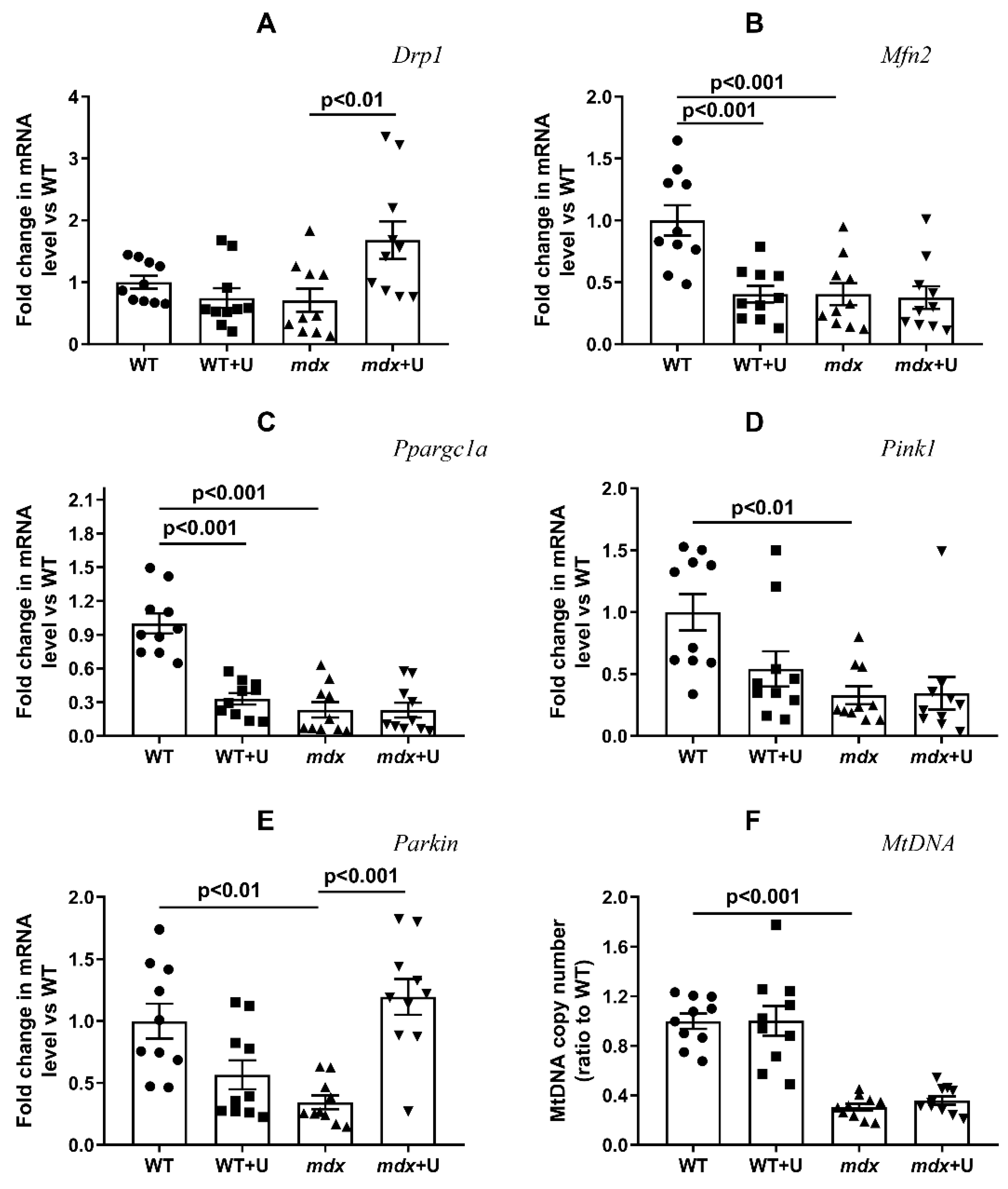
2.3. Effect of Uridine on the mRNA Expression of Proteins Responsible for Mitochondrial Dynamics, Biogenesis, and Mitophagy
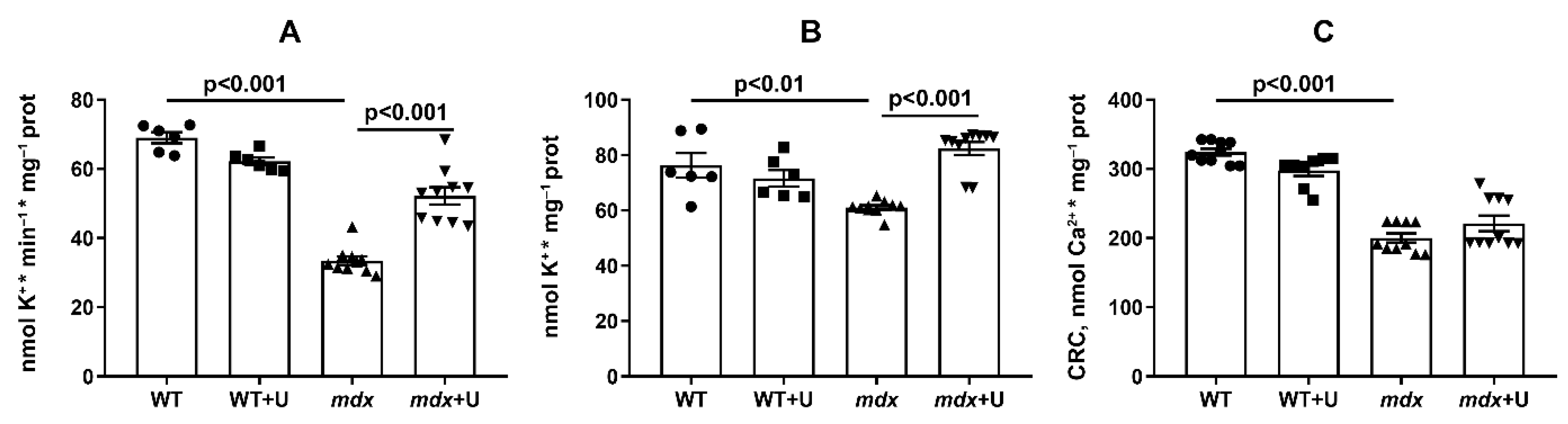
2.4. The Effect of Uridine on the Functioning of Skeletal Muscle Mitochondria in Dystrophin Deficient Mice
3. Discussion
4. Materials and Methods
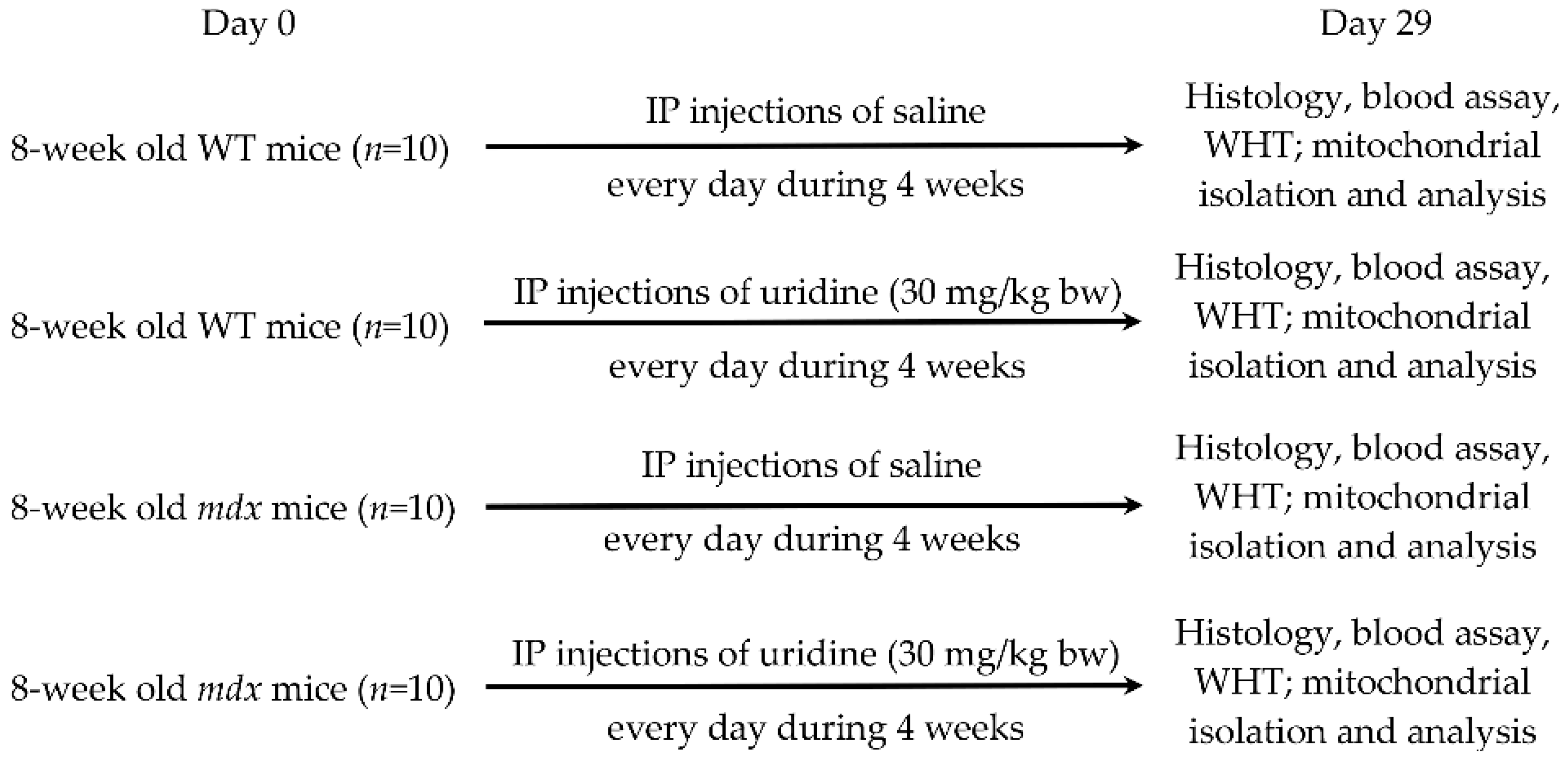
4.1. Experimental Animals
4.2. Uridine Administration
4.3. Blood Analysis
4.4. Histological Examination of Skeletal Muscle Tissues
4.5. Wire-Hanging Test
4.6. Transmission Electron Microscopy
4.7. RNA Extraction, Reverse Transcription, and Quantitative Real-Time PCR
4.8. Quantification of Mitochondrial DNA
4.9. Skeletal Muscle Mitochondria Isolation and Determination of Functional Parameters
4.10. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Emery, A.E. Population frequencies of inherited neuromuscular diseases—A world survey. Neuromuscul. Disord. 1991, 1, 19–29. [Google Scholar] [CrossRef]
- Wahlgren, L.; Kroksmark, A.K.; Tulinius, M.; Sofou, K. One in five patients with Duchenne muscular dystrophy dies from other causes than cardiac or respiratory failure. Eur. J. Epidemiol. 2022, 37, 147–156. [Google Scholar] [CrossRef] [PubMed]
- Timpani, C.A.; Hayes, A.; Rybalka, E. Revisiting the dystrophin-ATP connection: How half a century of research still implicates mitochondrial dysfunction in duchenne muscular dystrophy aetiology. Med. Hypotheses 2015, 85, 1021–1033. [Google Scholar] [CrossRef]
- Rybalka, E.; Timpani, C.A.; Stathis, C.G.; Hayes, A.; Cooke, M.B. Metabogenic and Nutriceutical Approaches to Address Energy Dysregulation and Skeletal Muscle Wasting in Duchenne Muscular Dystrophy. Nutrients 2015, 7, 9734–9767. [Google Scholar] [CrossRef] [PubMed]
- Hess, J. Phosphorylase activity and glycogen, glucose-6-phosphate, and lactic acid content of human skeletal muscle in various myopathies. J. Lab. Clin. Med. 1965, 66, 452–463. [Google Scholar] [PubMed]
- Chinet, A.; Even, P.; Decrouy, A. Dystrophin-dependent efficiency of metabolic pathways in mouse skeletal muscles. Experientia 1994, 50, 602–605. [Google Scholar] [CrossRef]
- Lindsay, A.; Chamberlain, C.M.; Witthuhn, B.A.; Lowe, D.A.l.; Ervast, J.M. Dystrophinopathy-associated dysfunction of Krebs cycle metabolism. Hum. Mol. Genet. 2019, 28, 942–951. [Google Scholar] [CrossRef]
- Rybalka, E.; Timpani, C.A.; Cooke, M.B.; Williams, A.D.; Hayes, A. Defects in mitochondrial ATP synthesis in dystrophindeficient Mdx skeletal muscles may be caused by complex I insufficiency. PLoS ONE 2014, 9, e115763. [Google Scholar] [CrossRef]
- Hughes, M.C.; Ramos, S.V.; Turnbull, P.C.; Rebalka, I.A.; Cao, A.; Monaco, C.M.F.; Varah, N.E.; Edgett, B.A.; Huber, J.S.; Tadi, P.; et al. Early myopathy in Duchenne muscular dystrophy is associated with elevated mitochondrial H2O2 emission during impaired oxidative phosphorylation. J. Cachexia Sarcopenia Muscle 2019, 10, 643–661. [Google Scholar] [CrossRef]
- Gaglianone, R.B.; Santos, A.T.; Bloise, F.F.; Ortiga-Carvalho, T.M.; Costa, M.L.; Quirico-Santos, T.; da Silva, W.S.; Mermelstein, C. Reduced mitochondrial respiration and increased calcium deposits in the EDL muscle, but not in soleus, from 12-week-old dystrophic mdx mice. Sci. Rep. 2019, 9, 1986. [Google Scholar] [CrossRef]
- Dubinin, M.V.; Talanov, E.Y.; Tenkov, K.S.; Starinets, V.S.; Mikheeva, I.B.; Sharapov, M.G.; Belosludtsev, K.N. Duchenne muscular dystrophy is associated with the inhibition of calcium uniport in mitochondria and an increased sensitivity of the organelles to the calcium-induced permeability transition. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165674. [Google Scholar] [CrossRef] [PubMed]
- Dubinin, M.V.; Talanov, E.Y.; Tenkov, K.S.; Starinets, V.S.; Belosludtseva, N.V.; Belosludtsev, K.N. The Effect of Deflazacort Treatment on the Functioning of Skeletal Muscle Mitochondria in Duchenne Muscular Dystrophy. Int. J. Mol. Sci. 2020, 21, 8763. [Google Scholar] [CrossRef] [PubMed]
- Angelini, G.; Mura, G.; Messina, G. Therapeutic approaches to preserve the musculature in Duchenne muscular dystrophy: The importance of the secondary therapies. Exp. Cell Res. 2022, 410, 112968. [Google Scholar] [CrossRef]
- Zhang, Y.; Guo, S.; Xie, C.; Fang, J. Uridine Metabolism and Its Role in Glucose, Lipid, and Amino Acid Homeostasis. Biomed Res. Int. 2020, 2020, 7091718. [Google Scholar] [CrossRef]
- Connolly, G.P.; Duley, J.A. Uridine and its nucleotides: Biological actions, therapeutic potentials. Trends Pharmacol. Sci. 1999, 20, 218–225. [Google Scholar] [CrossRef]
- Yamamoto, T.; Koyama, H.; Kurajoh, M.; Shoji, T.; Tsutsumi, Z.; Moriwaki, Y. Biochemistry of uridine in plasma. Clin. Chim. Acta 2011, 412, 1712–1724. [Google Scholar] [CrossRef]
- Le, T.T.; Ziemba, A.; Urasaki, Y.; Hayes, E.; Brotman, S.; Pizzorno, G. Disruption of uridine homeostasis links liver pyrimidine metabolism to lipid accumulation. J. Lipid. Res. 2013, 54, 1044–1057. [Google Scholar] [CrossRef]
- Thomson, W.; Guest, K.E. A trial of therapy by nucleosides and nucleotides in muscular dystrophy. J. Neurol. Neurosurg. Psychiatry 1963, 26, 111. [Google Scholar] [CrossRef]
- Bulon, V.V.; Krylova, I.B.; Selina, E.N.; Rodionova, O.M.; Evdokimova, N.R.; Sapronov, N.S.; Mironova, G.D. Antiarrhythmic effect of uridine and uridine-5′-monophosphate in acute myocardial ischemia. Bull. Exp. Biol. Med. 2014, 157, 728–731. [Google Scholar] [CrossRef]
- Krylova, I.B.; Bulion, V.V.; Selina, E.N.; Mironova, G.D.; Sapronov, N.S. Effect of uridine on energy metabolism, LPO, and antioxidant system in the myocardium under conditions of acute coronary insufficiency. Bull. Exp. Biol. Med. 2012, 153, 644–646. [Google Scholar] [CrossRef]
- Krylova, I.B.; Kachaeva, E.V.; Rodionova, O.M.; Negoda, A.E.; Evdokimova, N.R.; Balina, M.I.; Sapronov, N.S.; Mironova, G.D. The cardioprotective effect of uridine and uridine-5′-monophosphate: The role of the mitochondrial ATP-dependent potassium channel. Exp. Gerontol. 2006, 41, 697–703. [Google Scholar] [CrossRef] [PubMed]
- Krylova, I.B.; Selina, E.N.; Bulion, V.V.; Rodionova, O.M.; Evdokimova, N.R.; Belosludtseva, N.V.; Shigaeva, M.I.; Mironova, G.D. Uridine treatment prevents myocardial injury in rat models of acute ischemia and ischemia/reperfusion by activating the mitochondrial ATP-dependent potassium channel. Sci. Rep. 2021, 11, 16999. [Google Scholar] [CrossRef] [PubMed]
- Urasaki, Y.; Pizzorno, G.; Le, T.T. Chronic Uridine Administration Induces Fatty Liver and Pre-Diabetic Conditions in Mice. PLoS ONE 2016, 11, e0146994. [Google Scholar] [CrossRef] [PubMed]
- Salera, S.; Menni, F.; Moggio, M.; Guez, S.; Sciacco, M.; Esposito, S. Nutritional Challenges in Duchenne Muscular Dystrophy. Nutrients 2017, 9, 594. [Google Scholar] [CrossRef]
- Pant, M.; Sopariwala, D.H.; Bal, N.C.; Lowe, J.; Delfín, D.A.; Rafael-Fortney, J.; Periasamy, M. Metabolic dysfunction and altered mitochondrial dynamics in the utrophin-dystrophin deficient mouse model of Duchenne muscular dystrophy. PLoS ONE 2015, 10, e0123875. [Google Scholar] [CrossRef]
- Dubinin, M.V.; Starinets, V.S.; Talanov, E.Y.; Mikheeva, I.B.; Belosludtseva, N.V.; Belosludtsev, K.N. Alisporivir Improves Mitochondrial Function in Skeletal Muscle of mdx Mice but Suppresses Mitochondrial Dynamics and Biogenesis. Int. J. Mol. Sci. 2021, 22, 9780. [Google Scholar] [CrossRef]
- Giovarelli, M.; Zecchini, S.; Catarinella, G.; Moscheni, C.; Sartori, P.; Barbieri, C.; Roux-Biejat, P.; Napoli, A.; Vantaggiato, C.; Cervia, D.; et al. Givinostat as metabolic enhancer reverting mitochondrial biogenesis deficit in Duchenne muscular dystrophy. Pharmacol. Res. 2021, 170, 105751. [Google Scholar] [CrossRef]
- Mironova, G.D.; Negoda, A.E.; Marinov, B.S.; Paucek, P.; Costa, A.D.; Grigoriev, S.M.; Skarga, Y.Y.; Garlid, K.D. Functional distinctions between the mitochondrial ATP-dependent K+ channel (mitoKATP) and its inward rectifier subunit (mitoKIR). J. Biol. Chem. 2004, 279, 32562–32568. [Google Scholar] [CrossRef]
- Baranova, O.V.; Skarga, Y.Y.; Negoda, A.E.; Mironova, G.D. Inhibition of 2,4-dinitrophenol-induced potassium efflux by adenine nucleotides in mitochondria. Biochemistry 2000, 65, 218–222. [Google Scholar]
- Kharraz, Y.; Guerra, J.; Pessina, P.; Serrano, A.L.; Muñoz-Cánoves, P. Understanding the process of fibrosis in Duchenne muscular dystrophy. Biomed Res. Int. 2014, 2014, 965631. [Google Scholar] [CrossRef]
- Kypson, J.; Hait, G. Effects of uridine and inosine on glucose metabolism in skeletal muscle and activated lipolysis in adipose tissue. J. Pharmacol. Exp. Ther. 1976, 19, 565–574. [Google Scholar]
- Urasaki, Y.; Pizzorno, G.; Le, T.T. Uridine affects liver protein glycosylation, insulin signaling, and heme biosynthesis. PLoS ONE 2014, 9, e99728. [Google Scholar] [CrossRef]
- Paucek, P.; Mironova, G.; Mahdi, F.; Beavis, A.D.; Woldegiorgis, G.; Garlid, K.D. Reconstitution and partial purification of the glibenclamide-sensitive, ATP-dependent K+ channel from rat liver and beef heart mitochondria. J. Biol. Chem. 1992, 267, 26062–26069. [Google Scholar] [CrossRef]
- Paggio, A.; Checchetto, V.; Campo, A.; Menabò, R.; Di Marco, G.; Di Lisa, F.; Szabo, I.; Rizzuto, R.; De Stefani, D. Identification of an ATP-sensitive potassium channel in mitochondria. Nature 2019, 572, 609–613. [Google Scholar] [CrossRef] [PubMed]
- Graciotti, L.; Becker, J.; Granata, A.L.; Procopio, A.D.; Tessarollo, L.; Fulgenzi, G. Dystrophin is required for the normal function of the cardio-protective K(ATP) channel in cardiomyocytes. PLoS ONE 2011, 6, e27034. [Google Scholar] [CrossRef]
- Belosludtsev, K.N.; Dubinin, M.; Belosludtseva, N.; Mironova, G.D. Mitochondrial Ca2+ Transport: Mechanisms, Molecular Structures, and Role in Cells. Biochemistry 2019, 84, 593–607. [Google Scholar] [CrossRef] [PubMed]
- Bonora, M.; Patergnani, S.; Ramaccini, D.; Morciano, G.; Pedriali, G.; Kahsay, A.E.; Bouhamida, E.; Giorgi, C.; Wieckowski, M.R.; Pinton, P. Physiopathology of the permeability transition pore: Molecular mechanisms in human pathology. Biomolecules 2020, 10, 998. [Google Scholar] [CrossRef]
- Schiavone, M.; Zulian, A.; Menazza, S.; Petronilli, V.; Argenton, F.; Merlini, L.; Sabatelli, P.; Bernardi, P. Alisporivir rescues defective mitochondrial respiration in Duchenne muscular dystrophy. Pharmacol. Res. 2017, 125, 122–131. [Google Scholar] [CrossRef]
- Deacon, R.M. Measuring motor coordination in mice. J. Vis. Exp. 2013, 75, e2609. [Google Scholar] [CrossRef]
- Ye, J.; Coulouris, G.; Zaretskaya, I.; Cutcutache, I.; Rozen, S.; Madden, T.L. Primer-BLAST: A tool to design target-specific primers for polymerase chain reaction. BMC Bioinform. 2012, 13, 134. [Google Scholar] [CrossRef]
- Schmittgen, T.D.; Livak, K.J. Analyzing real-time PCR data by the comparative CT method. Nat. Protoc. 2008, 3, 1101–1108. [Google Scholar] [CrossRef] [PubMed]
- Quiros, P.M.; Goyal, A.; Jha, P.; Auwerx, J. Analysis of mtDNA/nDNA ratio in mice. Curr. Protoc. Mouse Biol. 2017, 7, 47–54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belosludtsev, K.N.; Belosludtseva, N.V.; Kosareva, E.A.; Talanov, E.Y.; Gudkov, S.V.; Dubinin, M.V. Itaconic acid impairs the mitochondrial function by the inhibition of complexes II and IV and induction of the permeability transition pore opening in rat liver mitochondria. Biochimie 2020, 176, 150–157. [Google Scholar] [CrossRef] [PubMed]
- Belosludtseva, N.V.; Starinets, V.S.; Mikheeva, I.B.; Serov, D.A.; Astashev, M.E.; Belosludtsev, M.N.; Dubinin, M.V.; Belosludtsev, K.N. Effect of the MPT Pore Inhibitor Alisporivir on the Development of Mitochondrial Dysfunction in the Heart Tissue of Diabetic Mice. Biology 2021, 10, 839. [Google Scholar] [CrossRef] [PubMed]
- Dubinin, M.V.; Talanov, E.Y.; Tenkov, K.S.; Starinets, V.S.; Mikheeva, I.B.; Belosludtsev, K.N. Transport of Ca2+ and Ca2+-dependent permeability transition in heart mitochondria in the early stages of Duchenne muscular dystrophy. Biochim. Biophys. Acta Bioenerg. 2020, 1861, 148250. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Animals | Creatine Kinase | AST | LDH |
|---|---|---|---|
| U/L | |||
| WT (n = 10) | 339.5 ± 57.6 | 26.7 ± 3.0 | 325.9 ± 25.4 |
| WT+U (n = 10) | 449.3 ± 59.4 | 28.7 ± 5.1 | 372.7 ± 32.5 |
| mdx (n = 10) | 854.3 ± 69.2 ** | 97.2 ± 9.6 ** | 1027.6 ± 103.7 ** |
| mdx+U (n = 10) | 823.6 ± 101.8 * | 120.7 ± 12.6 ** | 1116.9 ± 145.4 ** |
| Group | V Respiration, nmol O2 * min−1 * mg−1 Protein | RCR | ADP/O | |||
|---|---|---|---|---|---|---|
| State 2 | State 3 | State 4 | State 3UDNP | |||
| WT | 24.1 ± 1.9 | 201.2 ± 6.7 | 45.1 ± 3.6 | 221.3 ± 9.1 | 4.6 ± 0.3 | 1.2 ± 0.1 |
| WT+U | 25.9 ± 2.3 | 156.2 ± 7.4 * | 38.3 ± 1.9 | 175.8 ± 6.3 * | 4.2 ± 0.3 | 1.3 ± 0.1 |
| mdx | 20.2 ± 1.4 | 171.4 ± 5.8 * | 56.3 ± 8.3 | 187.3 ± 7.1 * | 3.7 ± 0.5 | 1.1 ± 0.1 |
| mdx+U | 27.3 ± 2.7 | 154.7 ± 5.5 * | 34.7 ± 2.8 # | 174.0 ± 6.6 * | 4.5 ± 0.3 | 1.3 ± 0.1 |
| Gene | Forward (5’→3’) | Reverse (5’→3’) |
|---|---|---|
| Pink1 | TTGCCCCACACCCTAACATC | GCAGGGTACAGGGGTAGTTCT |
| Parkin | AGCCAGAGGTCCAGCAGTTA | GAGGGTTGCTTGTTTGCAGG |
| Drp1 | TTACAGCACACAGGAATTGT | TTGTCACGGGCAACCTTTTA |
| Mfn2 | CACGCTGATGCAGACGGAGAA | ATCCCAGCGGTTGTTCAGG |
| Ppargc1a | CTGCCATTGTTAAGACCGAG | GTGTGAGGAGGGTCATCGTT |
| Rplp2 | CGGCTCAACAAGGTCATCAGTGA | AGCAGAAACAGCCACAGCCCCAC |
| Nd4 | ATTATTATTACCCGATGAGGGAACC | ATTAAGATGAGGGCAATTAGCAGT |
| Gapdh | GTGAGGGAGATGCYCAGTGT | CTGGCATTGCTCTCAATGAC |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dubinin, M.V.; Starinets, V.S.; Belosludtseva, N.V.; Mikheeva, I.B.; Chelyadnikova, Y.A.; Penkina, D.K.; Vedernikov, A.A.; Belosludtsev, K.N. The Effect of Uridine on the State of Skeletal Muscles and the Functioning of Mitochondria in Duchenne Dystrophy. Int. J. Mol. Sci. 2022, 23, 10660. https://doi.org/10.3390/ijms231810660
Dubinin MV, Starinets VS, Belosludtseva NV, Mikheeva IB, Chelyadnikova YA, Penkina DK, Vedernikov AA, Belosludtsev KN. The Effect of Uridine on the State of Skeletal Muscles and the Functioning of Mitochondria in Duchenne Dystrophy. International Journal of Molecular Sciences. 2022; 23(18):10660. https://doi.org/10.3390/ijms231810660
Chicago/Turabian StyleDubinin, Mikhail V., Vlada S. Starinets, Natalia V. Belosludtseva, Irina B. Mikheeva, Yuliya A. Chelyadnikova, Daria K. Penkina, Alexander A. Vedernikov, and Konstantin N. Belosludtsev. 2022. "The Effect of Uridine on the State of Skeletal Muscles and the Functioning of Mitochondria in Duchenne Dystrophy" International Journal of Molecular Sciences 23, no. 18: 10660. https://doi.org/10.3390/ijms231810660