
Two Important Anticancer Mechanisms of Natural and Synthetic Chalcones


1
Department of Chemistry, Faculty of Pharmacy, Iuliu Hatieganu University, 400012 Cluj-Napoca, Romania
2
Advanced Materials and Applied Technologies Laboratory, Institute of Research-Development-Innovation in Applied Natural Sciences, “Babes-Bolyai” University, Fantanele Str. 30, 400294 Cluj-Napoca, Romania
*
Author to whom correspondence should be addressed.
Int. J. Mol. Sci. 2022, 23(19), 11595; https://doi.org/10.3390/ijms231911595
Submission received: 24 August 2022
/
Revised: 25 September 2022
/
Accepted: 27 September 2022
/
Published: 30 September 2022
(This article belongs to the Special Issue Novel Biological Molecules for Cancer Treatments)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:ATP-binding cassette subfamily G and tubulin pharmacological mechanisms decrease the effectiveness of anticancer drugs by modulating drug absorption and by creating tubulin assembly through polymerization. A series of natural and synthetic chalcones have been reported to have very good anticancer activity, with a half-maximal inhibitory concentration lower than 1 µM. By modulation, it is observed in case of the first mechanism that methoxy substituents on the aromatic cycle of acetophenone residue and substitution of phenyl nucleus by a heterocycle and by methoxy or hydroxyl groups have a positive impact. To inhibit tubulin, compounds bind to colchicine binding site. Presence of methoxy groups, amino groups or heterocyclic substituents increase activity.
1. Introduction
Cancer is characterized by uncontrolled, progressive, rapid and pathological cell proliferation and is one of the most aggressive diseases in the world [1,2]. This condition is a correlation of pathologies characterized by uncontrolled and abnormal cell growth, which are based on gene mutations [3,4]. The altered activity of some transcription factors determines the appearance of many types of cancers [5]. Heterogeneity of cancer cells targets is associated eith genomic instability, gene variation in tumor formation and ability of tumor cells to have excessive phenotypic variations [6]. This has important clinical consequences, as tumor formations with complex subclonal structures can be aggressive and have the ability to initiate resistance to therapy and form metastases [7,8]. The formation of metastases to distant organs is often incurable and is a determining factor in the death of cancer patients, which in these cases, is frequently associated with dysfunctions of vital organs [9,10]. Cancer cells have aberrant redox homeostasis, reactive oxygen (ROS) species by being pro-tumorigenic and, at high levels of ROS, have cytotoxic activity [11,12]. Due to aberrant redox homeostasis in tumor cells and their microenvironment, these reactive species are insoluble in carcinogenesis, but also in anticancer therapy. Numerous studies have shown that the role of ROS in malignant cells, positive or negative, depends on factors such as tumor type, cancer stage, therapeutic strategies, duration of cell exposure to ROS, specificity and ROS level [13,14]. The increased number and clinical importance of molecular biomarkers in usual practice allow anticancer therapies to have an increased specificity on a particular genetic complex in tumor formation. The disadvantages of these biomarkers are related to the expected duration of analyses and the need to take samples from tissues [15]. There are four major types of approaches available for cancer treatment (radiotherapy, surgery, immunotherapy and drug therapy) [16,17,18,19,20]. Less than a quarter of patients with common cancers receive appropriate therapy [21]. Early diagnosis of cancer provides the opportunity to choose the appropriate therapeutic intervention. Among methods used for early diagnosis are tumor markers and imaging techniques, such as computed tomography, magnetic resonance imaging, position emission tomography and ultrasound scanning endoscopy (cytogenetic and cell genetics screening) and so forth [22]. Standard treatment for most cancers includes chemotherapeutic agents. The response to this type of therapy varies for each patient, especially in case of poorly characterized cancers. Tumor heterogeneity, the presence of cancer stem cells and the plasticity of these cells indicate the need to use combination therapies and/or targeted therapies [23]. Platinum compounds are a major component of chemotherapy for various types of cancer. Three platinum compounds used in anticancer therapy-cisplatin, carboplatin and oxaliplatin-have the ability to distort the normal functioning of DNA (deoxyribonucleic acid) by generating monoadducts [24]. Unlike chemotherapy, targeted therapy is considered more effective and safer for the treatment of cancer [25]. In recent years, immunotherapy has become a major alternative to conventional anticancer therapy, which aims to increase the quality of life of patients and prolong life [26]. Immunotherapy, ideal for fighting cancer, acts on the immune system to eradicate malignant cells [27]. There are two therapeutic directions of immunotherapy: blocking immune checkpoints by antibodies that block immune system inhibitory receptors in tumors, and adoptive cell therapy by which T cells are designed to express chimeric antigenic receptors [28,29,30,31]. It is also known that most recently approved targeted anticancer therapies are specific for coding oncoproteins for mutant somatic genes. These agents include proteins essential for the maintenance of specific cell descendants or proteins that inhibit immune responses, such as protein 1 of programmed cell death (PD-1) [32]. Immunotherapy, based on immunostimulatory monoclonal antibodies, explains the involvement of an immune response in anticancer therapy. PD1/PDL1 blockade demonstrates the importance of targeted immune mechanisms in tumor formation, by which T cell response in tumor formation is rehabilitated [33]. The most common problem with antitumor therapy is resistance to therapy [34,35,36,37,38,39,40,41]. Some cancer cells initiate resistance to many chemotherapeutic agents, others acquire this characteristic through mutations in the process of carcinogenesis [42]. This is correlated with cassette-binding ATP (ABC) transporters, which are P-glycoprotein (P-gP/ABCB1), multidrug resistance protein 1 (MRP1/ABCC1) and breast cancer resistance protein (BCRP/ABCG2). One method of treatment is the elimination of these resistant cancer cells, using multidrug ABC transporters [43,44,45,46,47,48,49,50,51].
Diet is known to play an important role in the prevention, onset and progression of cancer [52]. Due to remarkable chemical diversity of natural products, many biologically active compounds have been isolated from plants, microbes and other living organisms, which have been shown to have anticancer activity [53].
Numerous studies on cell cultures and animal models show that different natural compounds with various chemical structures have the ability to act as chemopreventive and chemotherapeutic agents. These biologically active molecules come from structural classes such as polyphenols, alkaloids, terpenoids and organosulfur compounds and so forth [17].
2. Flavonoids
Flavonoids are the largest class of low-molecular-weight polyphenolic secondary metabolites with a benzopyron residue (C6-C3-C6) in the molecule. They consist of two aromatic rings joined by a bridge of three carbon atoms that form an oxygenated heterocycle [54,55,56,57,58,59]. Over 10,000 flavonoids have been described, many of which are present in large quantities in fruits and vegetables [60]. Flavonoids are biosynthesized by phenylpropanoid, starting from phenylalanine [61]. Chalcone synthetase is the first enzyme in flavonoid biosynthesis and catalyzes the formation of chalcones from one molecule of p-coumaroyl-CoA and three molecules of malonyl-CoA [62]. Depending on the degree of oxidation of the central nucleus, flavonoids are classified into flavones, flavanones, flavonols (proanthocyanidins), dihydroflavonols, isoflavones, isoflavanones, chalcones, aurones, anthocyanidins and tannins (Supplementary Figure S1) [63,64,65,66].
Flavonoid compounds are associated with a number of biological activities, their antioxidant, anticancer, anti-inflammatory, antidiabetic, antimicrobial, antidyslipidemic, neuroprotective, antiosteoporotic, cytoprotective, hepatoprotective, vasoprotective properties being much studied [67,68,69,70,71,72,73,74,75]. Flavonoids are known to decrease cardiovascular risk and cancer-associated mortality [76]. Experimental studies also show that dietary flavonoids interact with rhodopsin and modulate the functions of visual pigments [77]. The structural diversity of flavonoids is associated with chemical changes such as hydroxylation, methylation, acylation, glycosylation, etc. In plants, glycosylation is the most common modification of flavonoids, being responsible for increasing solubility and stability, maintaining bioactivity, regulating transport and accumulating and/or decreasing toxicity of these compounds [78]. By methylation of hydroxy groups of flavonoids, compounds with superior stability, a longer metabolism time, a favorable transport rate and, implicitly, a better absorption in target tissues are obtained [79]. For example, hesperidin (a natural flavanone, Tables S1 and S2, Compound 1) has low water solubility and has low absorption in the small intestine. By methylation of hesperidin in an alkaline medium, hesperidin methyl chalcone (Tables S1 and S2, Compound 2) is formed, a compound with superior water solubility and favorable intestinal absorption, bioavailability and tissue distribution [80]. In general, metallic complex combinations of flavonoids have superior cytotoxic, anti-inflammatory and redox properties [81].
Flavonoid compounds are associated with a number of biological activities, their antioxidant, anticancer, anti-inflammatory, antidiabetic, antimicrobial, antidyslipidemic, neuroprotective, antiosteoporotic, cytoprotective, hepatoprotective, vasoprotective properties being much studied [67,68,69,70,71,72,73,74,75]. Flavonoids are known to decrease cardiovascular risk and cancer-associated mortality [76]. Experimental studies also show that dietary flavonoids interact with rhodopsin and modulate the functions of visual pigments [77]. The structural diversity of flavonoids is associated with chemical changes such as hydroxylation, methylation, acylation, glycosylation, etc. In plants, glycosylation is the most common modification of flavonoids, being responsible for increasing solubility and stability, maintaining bioactivity, regulating transport and accumulating and/or decreasing toxicity of these compounds [78]. By methylation of hydroxy groups of flavonoids, compounds with superior stability, a longer metabolism time, a favorable transport rate and, implicitly, a better absorption in target tissues are obtained [79]. For example, hesperidin (a natural flavanone, Tables S1 and S2, Compound 1) has low water solubility and has low absorption in the small intestine. By methylation of hesperidin in an alkaline medium, hesperidin methyl chalcone (Tables S1 and S2, Compound 2) is formed, a compound with superior water solubility and favorable intestinal absorption, bioavailability and tissue distribution [80]. In general, metallic complex combinations of flavonoids have superior cytotoxic, anti-inflammatory and redox properties [81].
2.1. Chalcones
Compounds with an enone system in molecules continue to be of particular interest due to their simple chemistry, the ease with which they are obtained by chemical synthesis, their biological importance and the possibility of being precursors in organic chemistry [82]. Chalcones (chalconoids, 1,3-diaril-2-propen-1-ones) are natural and synthetic compounds in which aromatic residues are joined by an α, β-unsaturated electrophiles chain of three carbon atoms (Supplementary Figure S1) [66,83,84,85,86,87,88]. Reactive hydrogens from the basic structure of chalcones determine the possibility of these compounds being structurally modified in order to obtain a large number of derivatives [89]. Chalcones are precursors for many heterocyclic compounds such as cyanopyridines, 2-pyrazolines, pyrazoles, isoxazoles, pyrimidine-2-ones, flavanones and flavones and so forth [90,91]. Data from literature indicate that molecules with a 1,3-diaryl-2-propen-1-one residue have numerous biological activities and, for this reason, are of particular pharmacological importance [92,93]. Among the most important biological activities of chalcones are antitumor, antibacterial, anti-inflammatory, antifungal, cytotoxic, antiviral, antimalarial, antituberculous, antiparasitic, antiulcer, analgesic, antipyretic, antihepatotoxic, etc., properties [94,95,96,97,98,99,100,101,102,103,104,105,106,107,108,109,110,111,112,113,114,115,116,117,118,119,120]. Chalcones have an extended conjugate system, which facilitates their binding to bioactive molecules, such as enzymes and DNA [121]. For example, chalcones are active on some enzymes such as acetylcholinesterase and carbonic anhydrase, being active in glaucoma, obesity, osteoporosis, epilepsy, Parkinson’s and Alzheimer’s [122]. The presence of α, β-unsaturated carbonyl group, which acts as a Michael acceptor, facilitates the interaction of chalcones with sulfhydryl groups in cysteine residues or with thiol groups. This interaction is considered to be responsible for the numerous biological properties of these compounds [123,124]. The biological activity of chalcones is also strongly influenced by their chemical structure, especially by substituents on two aromatic residues. It has advantages related to its ability to regulate molecular pathways related to cancer, favorably influence apoptosis, metastases and the response to cellular stress. Numerous methoxychalcones with antitumor properties have been identified, the presence of the methoxy group being favorable for this activity [125,126,127,128]. Since chalcones are active in vitro and in vivo in cancers susceptible to therapy, as well as in case of resistant ones, these compounds represent an important skeleton for the identification of new anticancer agents. In addition, 1,3-diaryl-2-propen-1-ones represent an important class of natural small molecules with anticancer chemotherapeutic action [129]. For example, chalcones are important pharmacophores for many natural products, such as flavokawaina, millepachine and xanthohumol (XN) (Tables S1 and S2, Compounds 3–7) [130]. XN (Tables S1 and S2, Compound 7), a prenylated chalcone from female hop inflorescences, inhibits the growth of MCF-7 (human breast adenocarcinoma cancer cells) and induces their apoptosis. Prenylchalcone is also effective in hepatocellular carcinoma and prostate cancer [131]. Mechanisms of the anticancer activity of XN include inhibition of initiation and progression of carcinogenesis, induction of apoptosis, and inhibition of angiogenesis [132,133]. Natural compounds with 1,3-diaryl-2-propen-1-one residue in molecules can be readily converted to various by-products by exposure to UV light. Natural chalcones can also be metabolized rapidly by drug metabolism enzymes, which causes a short half-life in vivo. For these reasons, numerous series’ of stable chalcone-like compounds have been obtained whose biological properties have been evaluated [134]. The most widely used method of obtaining chalcones is Claisen-Schmidt condensation (Supplementary Figure S2) [66]. During the reaction, the base (catalyst) attacks α-hydrogen of acetophenone and forms an enolate anion (carbanion). The carbanion attacks the carbonyl group of benzaldehyde and forms a β-hydroxycarbonyl intermediate, from which chalcone is obtained [135,136,137,138,139,140,141].
Synthetic chalcones can also be obtained by Suzuki reaction, Friedel-Crafts acylation, Wittig reaction and photo-Fries rearrangement of phenyl cinnamate [142]. Among synthetic chalcones, hybrid molecules with indole in the molecule are widely used in anticancer therapy. For example, Yan J et al. synthesized a series of 29 indole chalcones in order to evaluate their antiproliferative activity. The most active compound in the series, (E)-3-(6-Methoxy-1H-indol-3-yl)-2-methyl-1-(3,4,5-trimethoxyphenyl)propen-2-en-1-one (Tables S1–S3, Compound 8), has IC50 values = 3–9 nM on six cancer cell lines. The new tubulin polymerization inhibitor binds to the colchicine binding site. Studies of cellular mechanisms show that derivative blocks the cell cycle in the G2/M phase and induces apoptosis consecutively with decreasing mitochondrial membrane potential. The indole compound has low cytotoxicity compared to normal human cells and has a similar potency to therapy-resistant cells [143].
Given the need to identify new anticancer therapies and the growing interest in identifying the biological properties of chalcones, we evaluated the antitumor mechanisms of these compounds.
2.1.1. Anticancer Activity of Chalcones
The anticancer potential of chalcones is correlated with their ability to act on various molecular targets such as ABCG2 (ATP binding cassette subfamily G), tubulin, activated nuclear B cell growth (NF-κB), vascular endothelial growth factor (VEGF), tyrosine kinase receptor (EGFR), mesenchymal-epithelial transition factor (MET), 5-α reductase, ACP-reductase, histone deacetylase, p53, CDC25B (protein tyrosine phosphatase), retinoic acid receptors, estrogenic topoisomerase receptors and MDM2 [66,144,145,146,147]. In order to obtain chalcones with superior anticancer properties, three methods of modulation of natural chalcones were used: (1) modulation of substituents at the level of the two aromatic residues (aldehyde and acetophenone), (2) replacement of aromatic residues with heterocycles and (3) obtaining hybrid molecules by conjugating chalcones with other molecules with antitumor properties [66]. Since studies performed to highlight the anticancer activity of chalcones are numerous, and mechanisms by which they exert this property are multiple, we wanted to conduct a study on two important antitumor mechanisms of these compounds.
2.1.2. ABCG2
ABCG2 (also called BCRP, breast cancer resistance protein) is a 655 amino acid protein that has a molecular weight of 72 kDa and consists of an N-terminal nucleotide-binding domain and a six-segment C-terminal transmembrane domain, as well as a hydrophobic and an extracellular loop between TM5 and TM6 [148,149]. Having a membrane localization, protein is part of the ATP-binding cassette (ABC) drug transporter family and has the ability to use the energy formed by hydrolysis of ATP [150]. It contains a transmembrane binding domain and a nucleotide-binding domain and is activated by homodimerization. Some high-performance 3D structures of ABCG2 protein binding to different substrates and inhibitors indicate the molecular mechanisms of ABCG2 substrate selection, binding and transport [151]. Many ABCG2 substrates are additional substrates for other ABC transporters, P-glycoprotein (ABCB1), so the net effect of the availability of a dual-substrate drug (ABCB1/ABCG2) is attributed to combined action of two transporters [152]. ABCG2 actively recognizes and transports distinct molecules, often hydrophobic, many of which have a polycyclic structure or a relatively flat shape [153]. BCRP is an efflux transporter with an important role in detoxification by removing endo- and xenobiotics from many cell types (e.g., uric acid, steroid metabolites), in modulating drug absorption and having an important role in various pharmacokinetic stages. Similarly, it determines resistance to anticancer therapy [154,155,156,157,158,159]. In addition, a transporter reduces the transfer of therapeutic substances to tumor cells. ABCG2 also has an important role in the protection of stem cells. It has been shown that the number of tumorigenic stem cells is directly proportional to the progression of cancers, these cells being involved in the initiation of tumorigenesis, tumor angiogenesis, metastases, drug resistance and recurrences. Stem cells are characterized by increased drug and chemotherapeutic resistance due to their expression in the efflux pump of ABCG2 [160]. It is located on the interface of the blood-brain barrier (efflux capacity of the protein protects the central nervous system from compounds with endogenous toxicity), placenta, liver, kidneys, colon and small intestine [161,162,163,164,165,166,167]. The transporter is involved in decreasing the oral bioavailability, tissue distribution and effectiveness of many therapeutic agents [168]. ABCG2 transporters are known to be responsible for limited exposure of the brain to anticancer drugs, the effectiveness of which reduces it, especially in case of brain metastases [169]. The clinical significance of BCRP expression is correlated with the response to therapy and the prognosis of gastrointestinal, lung, breast, head and neck tumors, ovarian, prostate, glioblastomas and leukemias and so forth [164]. BCRP has important functions in excluding antitumor drugs from various cancer cells, being a significant therapeutic barrier. Since it has subspecies polyspecificity and is present in many tissues, protein is an important factor in resistance to therapy [170,171,172]. In some tumor formations, ABCG2 is strongly overexpressed, which is correlated with unfavorable clinical outcomes for these tumors [173]. Because the carrier is crucial for the pharmacokinetics of certain compounds, the US Food and Drug Administration and European Medicines Agency have indicated that pharmacokinetic studies and drug–drug interactions have been performed for it [174]. Functional characterization indicates that ABCG2 carries a large number of different substances, including methotrexate, mitoxantrone, SN-38 and various tyrosine kinase modulators (e.g., imatinib, nilotinib), which act as both substrates and inhibitors of ABCG2. Multidrug resistance proteins 1 and 2, which are encoded by ABCG2 genes, play an important role in the excretion of tyrosine kinase inhibitors. In addition, ABCG2 recognizes a wide range of positively or negatively modified substances and is resistant to most topoisomerases I or II, such as topotecan or doxorubicin, which is the reason for therapeutic failure [175,176,177,178,179]. ABCG2 polymorphism explains the low accumulation of anticancer agents (doxorubicin, tyrosine kinase inhibitors, adriamycin, platinum compounds, sorafenib and mitoxantrone) in cells and their altered chemotherapeutic response [180]. Previous studies have indicated the architecture of BCRP and the structural basis of the inhibition of this protein by small molecules and antibodies [82]. The inhibition of these transporters has a favorable impact on anticancer therapy by co-administering them with chemotherapeutic agents. There is a growing need to identify selective inhibitors that optimize drug absorption and prevent possible side effects. Currently, there are a limited number of ABCG2 inhibitors, which are used as pharmacological tools in studies to highlight pathological/physiological role of this transporter, to cross the blood-brain barrier of these biologically active molecules and to reduce resistance to polytherapy [181].
2.1.3. Chalcones with Activity on ABCG2
Licochalcone A (LCA, Tables S1–S3, Compound 9, Figure 1), the most studied natural licochalcone, is present in roots of Glycyrrhiza inflata, has known pro-apoptotic and antiproliferative properties on different cell lines. Wu et al. determined the effect of LCA on multidrug-resistant ABCG2-overexpressing cancer cells. LCA has been shown to significantly alter the chemosensitivity of R482-HEK293 (ABCG2-transfected HEK293 human cells), S1-M1-80 (human colon cancer) and H460-MX20 (human non-small-cell lung cancer NSCLC) cells. For two known substrates of ABCG2 (mitoxantrone and topotecan), the activity was directly correlated with concentration [182,183].
Fan X et al. evaluated the inhibitory effects of some flavonoids on resistant proteins in breast cancer. The chalcones included in the study are LCA and licochalcone B, echinatin, isoliquiritin and isoliquiritigenin (Tables S1 and S2, Compounds 9–13, Figure 2). A computational model (CDOCKER) was used to investigate molecular models of BCRP binding. The spatial conformation of BCRP-docked chalcones is different from that of mitoxantrone substrate. Mitoxantrone has a Pi-Alkyl potential with Val442 and two conventional bonds with Thr435 and Gln398. Analyzed chalcones have presumed potential Π-Π interactions with Phe439 and/or potential Pi-Alkyl interactions with Val546, which are essential for the strong inhibition of BCRP by flavonoids. Results show that the inhibitory potential of LCA and its analogue, isoliquiritigenin, is not correlated with conventional hydrogen bonds. Studies to identify essential pharmacophores responsible for inhibition and biological activity of chalcones on BCRP show that the basic elements are aromatic nuclei, hydrophobic groups and acceptors of hydrogen bonds [184].
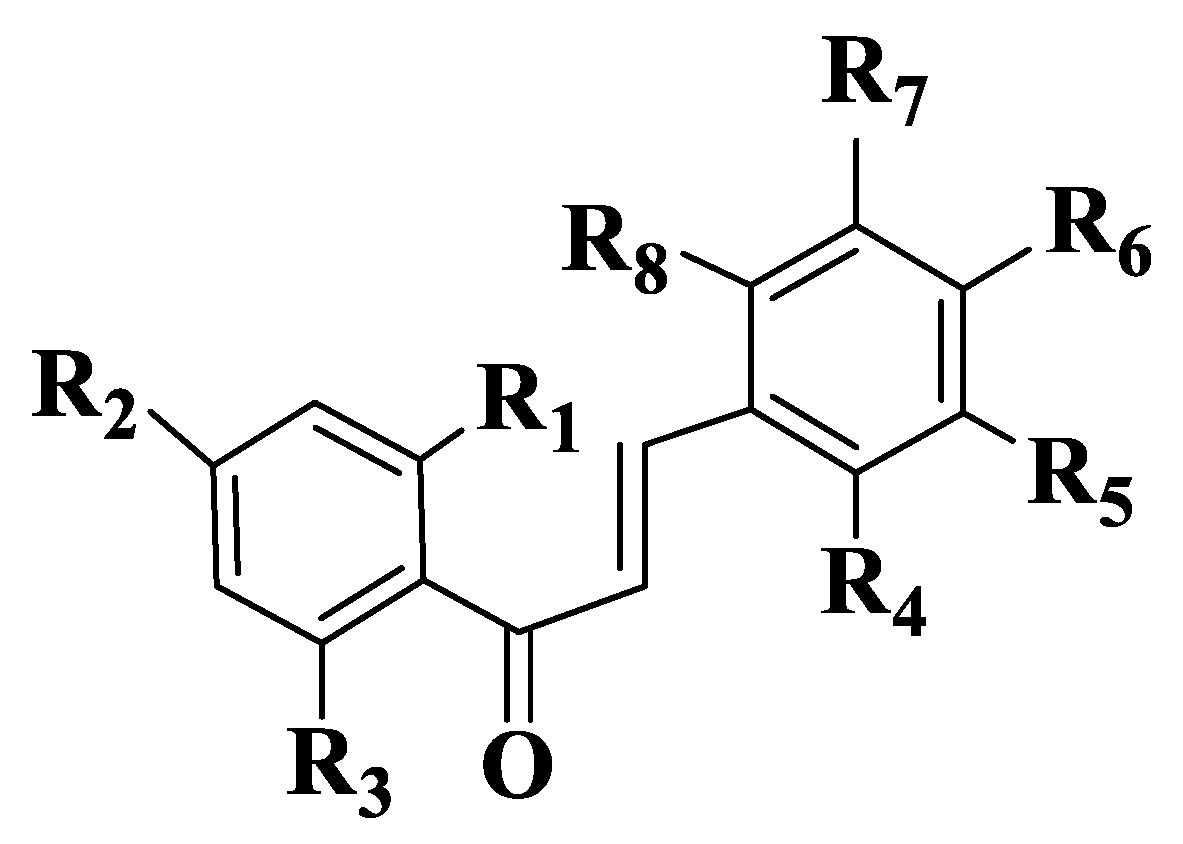
A series of 44 chalcones and their analogues (Tables S1–S3, Compounds 14–57) were synthesized to evaluate the inhibitory potential and selectivity of ABCG2 by evaluating the effect on the transporter of mitoxantrone, a known substrate of ABCG2. Cell lines determined were ABCG2-transfected human fibroblast HEK293 (immortalized human embryonic kidney cells). Substituents on the two subunits (aldehyde and acetophenone) are the groups frequently present in natural compounds (methoxy and hydroxy groups), and their positions are 2,4 and 6 on acetophenone and 2, 3, 4, 5 and 6 on aldehyde. From the preliminary analysis, it was identified that -OR substituents are the most favorable for inhibitory activity. The substitution of aldehyde with chlorine in positions 2 or 3 also increases the inhibition of mitoxantrone efflux. In contrast, chlorine substitution at position 4 has a negative impact on ABCG2-mediated efflux inhibition [185]. The inhibitory activity of ABCG2 for the 44 chalcones was expressed as a percentage for the concentrations of 2 μM and 10 μΜ. For non-heterocyclic chalcones (Tables S1–S3, Compounds 14–47, Figure 3), the best inhibitory capacity is for compounds that are substituted on acetophenone with two methoxy groups at positions 2 and 4 and a hydroxy group at position 6 (Tables S1–S3, Compounds 40–47). On aldehyde, the position and number of methoxy groups are important. A substitution with -OMe in positions 2, 6 and 3, 5 (Tables S1–S3, Compounds 18, 21, 22, 24, 26, 27, 31, 34, 36, 37, 41, 45, 46) is the most favorable for inhibitory activity. A substitution with a single methoxy group at position 2 or 3 (Tables S1 and S2, Compounds 15, 16) is more favorable than the unsubstituted compound (Tables S1 and S2, Compound 14). The activity of indole analogs is very different for the 1-indolyl and 3-indolyl series (Tables S1–S3, Compounds 48–57), which is explained by the impact of the electronic distribution in molecules on inhibitory activity. In a series of 1-indolylchalcones (Tables S1–S3, Compounds 48–52), heterocycle has a positive impact, the activity of these compounds being similar to that of the chalcones substituted on acetophenone with two methoxy groups in positions 2 and 4 and a hydroxy group in position 6 (Tables S1–S3, Compounds 40–47). Even though the basic structure is the same for all synthesized and analyzed compounds, there is a strong link between the structure of the compound and its activity. The positive contribution of the hydroxy group at position 6 of acetophenone is correlated with the electrostatic properties of these compounds (Tables S1–S3, Compounds 40, 41, 42). Similarly, the favorable contribution of the methoxy group on 5 position of aldehyde is explained (Tables S1–S3, Compound 41). On the other hand, the positive contribution of the methoxy group on 2 position of aldehyde (Tables S1–S3, Compounds 40, 50, 51) and the methoxy group at position 2 of acetophenone (Tables S1 and S2, Compounds 40, 41, 44) is due to steric effects. The negative effect of the methoxy group on 4 position (Compound 45 versus Compound 40 and Compound 46 versus Compound 41) is due to electrostatic properties. The remaining N-methyl-1-indolyl does not show particular interactions with favorable impact, indicating that hydrophobic interactions are essential for the binding capacity of compounds. In conclusion, it can be stated about chalcones and indolylphenylpropenones analyzed that they are potential inhibitors of ABCG2. From the 44 compounds, two representatives (Tables S1–S3, Compounds 51, 40), which have a methoxy group on position 4 of aldehyde, are potential candidates for preclinical studies [185].
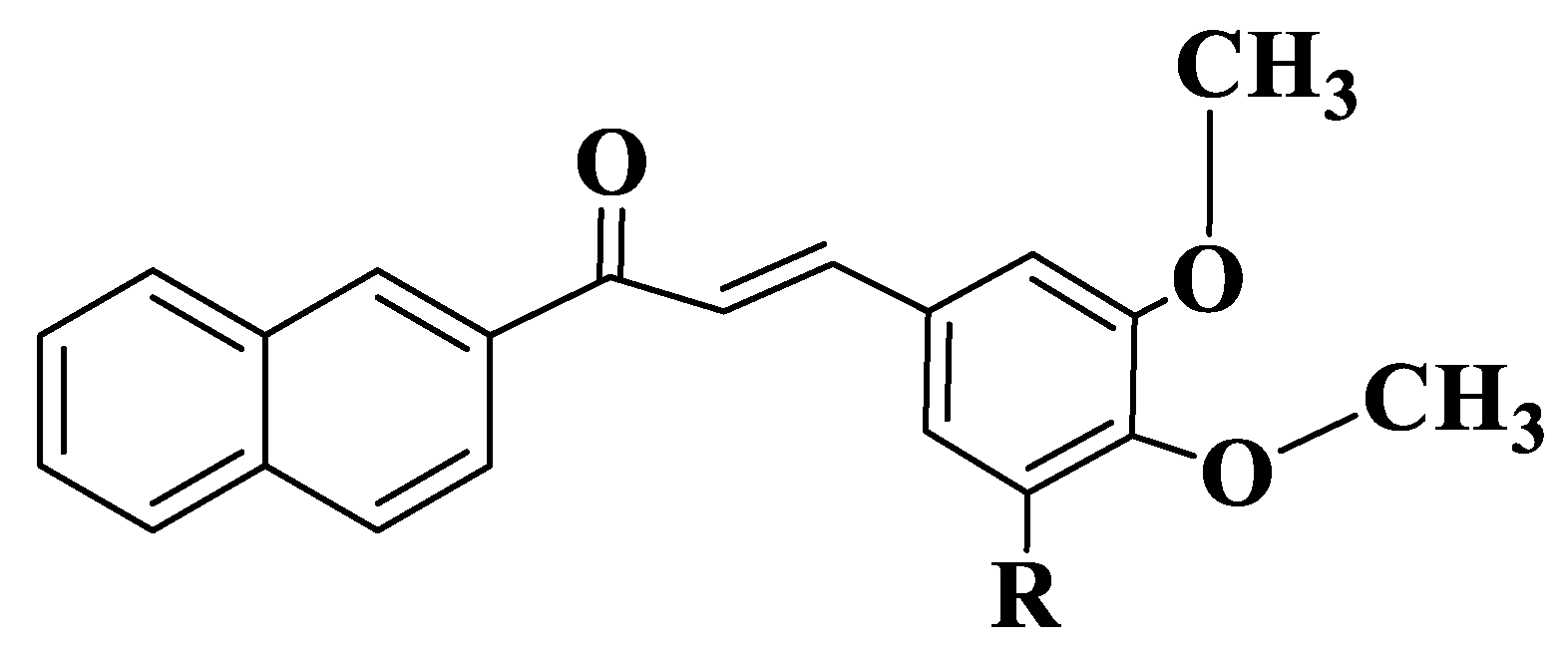
The fact that methoxy groups are favorable for the action of chalcones on ABCG2 was also performed for four naphthochalcones (Tables S1–S3, Compounds 58–61, Figure 4), which were synthesized to evaluate their effect on cancer cell growth on five different entities, using MTT analysis (2,5-diphenyl-2H-tetrazolium bromide). Compounds exhibit dose-dependent inhibitory activity. 3-Halophenyl chalcones (Compounds 58–60) have, at micromolar concentrations, an inhibitory capacity that is twice lower than the trimethoxylated derivative (Compound 61). Since chalcones substituted with two methoxy groups at positions 3, 4 inhibit BCRP-type ABC efflux transporters, it is normal for MCF-7/Topo cell lines to be approximately 10 times more sensitive to chalcone action compared to other cell lines. Halophenyl chalcones have IC50 values between 5 and 9 μM (Compounds 58–60), and the trimethoxylated derivative is 2–3 times more active (Compound 61). Trimethoxynaphthochalcone (Compound 61) inhibits both BCRP and P-gp transporters. Chalcone induces apoptosis through intrinsic pathways, and a Michael-type electrophilic system is essential for this activity [186].
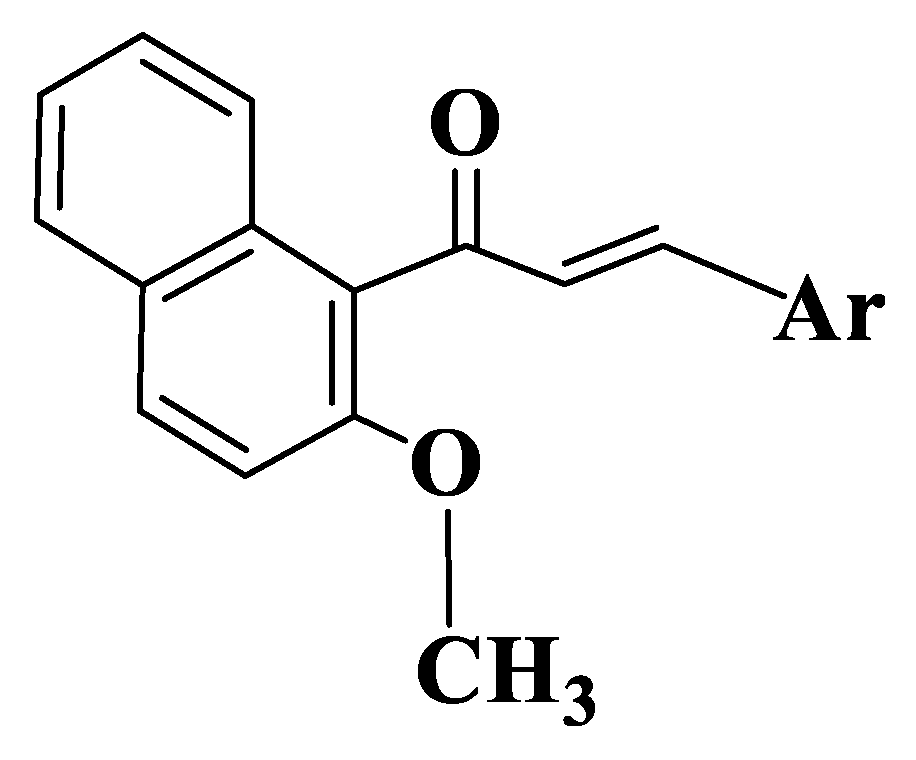
Since the presence of a quinazoline residue and methoxy groups is known to be correlated with a strong inhibition of ABCG [187], they synthesized a series of 22 quinazoline chalcones (Tables S1–S3, Compounds 62–83) containing methoxy groups in the molecule. Initial results indicated the importance of substituents on the aromatic nucleus of aldehyde. Depending on acetophenone substituents, the unsubstituted aldehyde compounds are inactive. Among variants analyzed, the substitution of aldehyde with two methoxy groups in positions 3 and 4 (Compounds 64, 66, 69, 74, 76, 80) is favorable for ABCG2 inhibitory activity. The introduction of a quinazoline nucleus into molecules causes an increase in activity. Compounds from a series that have a meta-acryloyl phenyl residue on acetophenone (Compounds 62–65) show an inhibitory effect on ABCG2 with IC50 values between 0.2 and 2 μΜ. Chalcone with three methoxy groups in positions 6 and 7 of quinazoline and in position 4 of aldehyde (Compound 65) is the most active (IC50 = 0.32 μM). The substitution of acetophenone with two methoxy groups at positions 3 and 4 (Compounds 62, 64) causes a decrease in activity. In case of quinazolines substituted on 2 position of phenyl (Compounds 66–68, Figure 5), the results obtained are different. Chalcone substituted with two methoxy groups in positions 3 and 4 of aldehyde (Compound 66) is the most active, followed by the chalcone substituted on meta position with methoxy (Compound 68). An analogue with methoxy group on para position (Compound 67) is the least active. By introducing an additional methoxy group on 2 phenyl residue of compounds, (Compounds 69–71) are obtained derivatives with similar activities to unsubstituted quinazoline chalcones (Compounds 62–65).
In case of compounds with an acetophenone para-acryloylphenyl residue (Tables S1–S3, Compounds 72–83), the most active compounds are methoxy-substituted monosubstituted derivatives. The substituted derivatives in positions 6 and 7 of quinazoline with methoxy groups and monosubstituted ones are the least active (Compounds 72–75). Compared to compounds from the first series (with a meta-acryloylphenyl residue), the para-substituted compounds have a lower activity, substitution on 2 position of phenyl on the heterocycle level being a favorable variant (Compound 62-IC50 = 1.30 μM versus Compound 72-IC50 = 0.82 μM and Compound 64-IC50 = 1.71 μM versus Compound 74-IC50 = 1.23 μM). Two derivatives substituted with methoxy groups at positions 6 and 7 of quinazoline and with a single methoxy group on aldehyde (Compounds 65, 75) have the best activity of the eight unsubstituted derivatives on 2 position of quinazoline. For quinazoline chalcones with a 2-phenyl residue (Compounds 76–78), a similar effect to that of derivatives 66–68 was observed. From these, compound 76 has the best activity (IC50 = 0.29 μM), and the least active is the compound with a methoxy group on 4 position of aldehyde (Compound 78, IC50 = 3.55 μM). Similar to meta-acryloyl compounds, the substitution of phenyl residue on quinazoline with two methoxy groups on 3 and 4 results in an increase in the activity of para-acryloyl derivatives as well. Compound 80 is the most active in the series (IC50 = 0.19 μM). Compound 81 (IC50 = 0.36 μM) is also a potent inhibitor of Ko143 (the most potent BCRP inhibitor known) and is the most active inhibitor of ABCG2 [188].
In another series of heterocyclic chalcones, in which inhibitory activity on ABCG2 was tested and obtained by Winter et al., 12 new compounds with a quinoxaline residue were synthesized (Tables S1–S3, Compounds 84–95). Depending on the t number and position of existing methoxy groups on the phenyl residue of acetophenone, chalcones have significant inhibitory effects on ABCG2. The best inhibitory properties have compounds substituted with two or three methoxy groups on acetophenone (Compounds 85, 86, 87, 88, 90, 91, 93, 95). Chalcone having a hydroxy group on 4 position of acetophenone has a much lower ability to inhibit ABCG2 (Compound 89). Compared to chalcones in which the aldehyde subunit has a 3,4-methylenedioxyphenyl or 2-naphthyl residue, chalcones with a quinoxaline residue are much more active. Following evaluations, acetophenone substituents were found to have a significant influence on the ABCG2 inhibitory activity of chalcones. Substitution with chlorine, bromine, trifluoromethyl, nitro, cyano and hydroxy is not favorable for this activity [189].
Another study included 35 chalcones substituted in positions 2, 3 and 4 of acetophenone with a benzamide residue and in positions 3, 4 of aldehyde residue (especially with methoxy groups) (Tables S1–S3, Compounds 96–131), which were synthesized to evaluate their inhibitory potential on ABCG2 by Pheophorbide A and Hoechst 33342 methods, using MDCK II BCRP cell lines. Depending on the position of amide on the rest of acetophenone (ortho, meta or para), the series of compounds was divided into three groups. The first group includes compounds (Compounds 96–98) in which amide is located on the meta position of acetophenone and is substituted with a phenyl, 4-nitrophenyl or 3-bromophenyl residues. The best activity has a phenyl-substituted amide compound (Compound 96, IC50 = 2.18 μM in pheophorbid A). The 4-Nitrophenyl-substituted compound (Compound 97) is not active. The second group has compounds (Compounds 99–115) in which amide is located on the para position, and it is substituted with halogens, methoxy, cyano or nitro. With the exception of chlorine-substituted compounds (Compounds 105–107), meta-substituted compounds are more active than para-substituted or disubstituted compounds. Of the para-benzamide compounds tested, the compound in which phenyl is not substituted on amide is the most active (Compound 99, IC50 = 1.30 μM in pheophorbid A), and the least active compound is the one substituted with fluorine in 2 position of phenyl from amide (Compound 108). Compounds with nitro or cyano groups on benzyl phenyl (Compounds 100–102, 109, 110) have inhibitory activities on ABCG2 similar to halogen-substituted chalcones (Compounds 103–108). The least active is a compound substituted with trifluoromethyl (Compound 110, IC50 = 16.1 μM in pheophorbid A). In case of methoxylated derivatives of phenyl benzamide, the contribution of methoxy groups is additive. The most active are compounds with a methoxy group on 2 or 3 positions of phenyl (Compounds 111, 112), and the least active is the compound substituted in position 4 (Compound 113). The third group of compounds contains an amide group on ortho position of acetophenone (Compounds 116–131). Compared to derivatives from the para-substituted series, ortho-substituted chalcones are less active (except for compounds with nitro groups). The substitution of phenyl from the amide group with a chlorine causes a slight increase in their activity, this being the least active compound in the group (Compound 128, IC50 = 0.77 μM in pheophorbid A). The 3-Methoxylated derivative (Compound 122) has an activity similar to unsubstituted derivative. The replacement of phenyl with a 2-thienyl residue (Compound 127) is favorable, as it is the most active compound. The derivative with a 3-quinolinyl residue (Compound 123) is twice as potent as the one with a 3-pyridyl residue (Compound 125). The replacement of 3,4-dimethoxy groups on aldehyde with chlorine or 3-methoxy-4-fluoro (Compounds 130, 131) resulted in a significant decrease in inhibitory capacity [190].
Solórzano et al. also synthesized six tariquidar-chalcones (Tables S1–S3, Compounds 132–137, Figure 6) in order to evaluate their cytotoxic potential, starting from the idea that tranquilizer analogues are selective inhibitors of ABCG2 susceptible to hydrolysis (which limits their use in biochemical and biological studies). Synthesized chalcones were investigated by Hoechst 33342 microplate analysis using MCF-7/Topo cells with overexpressed ABCG2. Standards used were fumitremorgin C and tariquidar. Chalcones have a maximum activity between 72 and 111%. Compounds with a methyl group on 4 position of aminochalcone (Compounds 135–137) have a higher potency and a maximum inhibitory effect compared to unsubstituted compounds (Compounds 132–134), indicating the importance of the substitution on 4 position of acetophenone. Their maximum effect is between 86 and 111%. A compound substituted with three methyl residues-a group on 4 position of acetophenone and two groups on tetrahydroisoquinoline (Compound 135) has an inhibitory capacity similar to standard (fumitremorgin). The introduction of an ethylene or triethylene glycol residue (Compounds 133–134, 136–137) on tetraisoquinoline residue does not influence IC50 [191].
2.1.4. Tubulin
Microtubules are cellular structures with a fundamental role in many essential biological processes, such as cytoskeletal architecture, intracellular transport, motility, chromosomal segregation and mitosis [92,192,193,194,195,196,197]. In the interface, microtubules are organized on cytoplasm in the form of a matrix-type network [198]. These are non-covalent mesoscopic polymers composed of α,β-tubulin protein heterodimers, which associate with and form protofilaments, which subsequently bind laterally and form microtubules. These dynamic structures are constantly elongated or shortened in all phases of cell cycle by adding or removing tubulin heterodimers from the ends of microtubules [199,200,201,202]. The assembly of protofilaments to microtubules is done spontaneously, with a number of protofilaments between 9 and 16, resulting in microtubules with different diameters [203]. It is known that the organization of microtubules changes significantly during the cell cycle. On the interface, microtubules form matrices in the cytoplasm and are relatively stable, and during mitosis, they form a bipolar mitotic spindle and become dynamic. When proliferative cells are exposed to microtubule inhibitors, bipolar spindle formation and the attachment of microtubules to kinetochores is inhibited, thus, activating the control point of division spindle assembly [204]. The stabilization of microtubules by acetylation is involved in cell migration [205]. Tubulin, in a depolymerized form, has the ability to influence cell physiology in various forms. For example, many microtubule-associated proteins have numerous sites of interaction with tubulin and microtubules. Thus, proteins containing the domain of overexpressed tumor genes use such multivalences to produce microtubule assembly. Mainly, tubulin, through its ability to influence the activity of proteins associated with microtubules, forms strong loops that connect the dynamics of microtubules to their basic matrix [206]. The dynamics of microtubules are strongly correlated with the regulation of cellular functions, such as intracellular transport and pathological processes [207]. This is essential for the optimal functioning of microtubules, in particular for the formation of dividing spindle in mitosis process. The disruption of this dynamic causes changes in cellular functions, influences the replication process and induces apoptosis. For these reasons, microtubules are one of the most studied targets of anticancer therapy. They actively participate in the formation of the centrosome, a formation characteristic of the G2/M phase of the cell cycle [208,209,210,211,212]. The unique feature of microtubule-binding agents, which is not present in other classes of anticancer agents, is their complexity and structural diversity, which determine many possibilities for optimization and modulation [213]. Compounds that interfere with cell division as a result of binding to α,β-dimers, oligomers or polymers have been extensively studied recently. Antimitotic agents, including different natural, synthetic or semi-synthetic products, have various chemical structures. Agents that interact with tubulin in the same binding region also belong to this category. Antimitotic agents include inhibitors of microtubule assembly (binds to Vinca domain or have sulfhydryl groups that alkylate tubulin) and microtubule-stabilizing agents (compounds that have a very high binding affinity, e.g., paclitaxel, docetaxyl, epothilone) [214,215,216,217]. Agents that target the field of colchicine (colchicine, podophyllotoxin, combrestatin A4) or those that bind to the domain of Vinca alkaloids (vincristine, vinblastine, vinorelbine) are defined as inhibitors of tubulin assembly as destabilizing agents [218,219,220,221].
Colchicine has a particular interest due to its antimitotic properties. The therapeutic potential of colchicine binding site has been investigated for its applications in chemotherapy. Colchicine binds strongly to the β subunit of tubulin. Cys241 residue forms hydrogen bonds with the trimethoxyphenyl residue of colchicine, and tubulin residues Thr179 and Val181 form hydrogen bonds with the troponelone residue of colchicine [218,222]. It is an increased need to identify new agents that bind to colchicine binding site. It is known that the intensity of the bond is dependent on the interaction of trimethoxyphenyl residue of colchicine with the binding site, and inhibition is achieved by interactions between oxygen atoms of the topolone network [223].
2.1.5. Antitubulin Chalcones
Malik et al. evaluated the potential for the inhibition of microtubule polymerization by semisynthetic chalcones (Tables S1–S3, Compound 138, (E)-1-(2-(allyloxy)-4-methoxyphenyl)-3-(4-methoxyphenyl)prop-2-en-1-one, Figure 7). Assays were performed in vitro on HCT116 (human colorectal carcinoma) and MV-4-11 (human acute myeloid leukemia) cell lines. The derivative was observed to be a robust inducer of histone H3 to S10 phosphorylation after 8 h of treatment with 25 μM of the compound. The treatment of HCT116 or MV-4-11 cells with different concentrations of chalcone at 8 h resulted in a significant increase in histone H3 phosphorylation at S10, even at concentrations of 0.78 μM. Chalcone-induced cell cycle blockade was also confirmed by a FACS analysis of MV-4-11 cells. Through biochemical tests performed, it was observed that the derivative inhibits polymerization of microtubules depending on concentration (2–25 μM). Chalcone docking studies indicate its binding to a colchicine binding site, one of the aromatic residues being placed on trimethoxyphenyl test region of colchicine. The compound forms two hydrogen bonds with tubulin on Leu255 and Cys241 residues. The phenyl residue adjacent to the carbonyl group forms Π-σ interactions with Leu248, and the other aromatic residue interacts with residues Ala180 and Lys254 [224].
Millepachine (Tables S1 and S2, Compound 6), a chalcone extracted from seeds of Milletia pachycarpa, has in vitro anticancer properties. It is known that flavonoids act by mechanisms such as cell cycle blocking by inhibiting cyclin-dependent kinase and by inhibiting tubulin polymerization. Hybrid molecule of millepachine with indole (Tables S1 and S2, Compound 139) also inhibits the polymerization of tubulin on hepatocellular cancer cell lines 3.6 times stronger than natural chalcone. An increase in activity can be attributed to the introduction of indole residue into molecules, this being a promoter for the binding of the compound on the tubulin cavity. Similarly, millepachine, indole derivative distorts dynamics of the intracellular tubulin-microtubule system and, implicitly, blocks cell cycle by blocking their G2/M phase. Two compounds (Tables S1–S3, Compounds 140, 141) were used to evaluate the class of antitubulin agents (stabilizers or destabilizers of tubulin) to identify in which chalcones belong. The amount of each compound used for microtubule dynamics methods is 15 μM with paclitaxel and combrestatin A-4 as controls. Paclitaxel (10 μM) stimulates tubulin polymerization, and combrestatin inhibits it. For the chalcones analyzed, a marked inhibition of polymerization was observed, with data showing that these compounds have the ability to effectively inhibit tubulin polymerization and act as destabilizing agents. To determine the possible binding of derivatives, docking studies were performed to demonstrate that these compounds have the same binding site as colchicine and 4-hydroxymillepachine. Binding patterns were generated for four compounds (Compounds 140, 141, colchicine and 4-hydroxymillepachine) on the colchicine binding site. The surflex docking score is 12.53 for compound 140 (€-2-(2-methoxy-5-(3-(5-methoxy-2,2-dimethyl-2H-chromen-8-yl)-3-oxoprop-1-en-1-yl)phenoxy)-N-(2-methoxyphenyl)acetamide) and 12.31 for compound 141 ((E)-N-(2-chlorophenyl)-2-(2-methoxy-5-(3-oxo-3-(3,4,5-trimethoxyphenyl)prop-1-en-1-yl)phenoxy)acetamide); a higher score indicating a higher affinity for the mode of interaction of compound 141. In the binding site, compound 140 is surrounded by Cys241, Leu248, Lys352 and Leu352. In particular, compound 140 forms three hydrogen bonds with polar amino acid Asn101, indicating a possible strong electrostatic interaction with protein. In addition, the hydrophobic residue of compound 140 is embedded in a pocket that interacts with several hydrophobic residues that facilitate strong tubulin binding. Compound 141 binds similarly to colchicine. In this case, docking studies showed that the 3,4,5-trimethoxyphenyl residue of compound 141, similar to colchicine, is compatible with a hydrophobic site and adopts an energetically stable conformation. In addition, the methoxy group and carbonyl oxygen of t 3,4,5-trimethoxyphenyl residue and the oxygen atom of amide from compound 141 act as acceptors and establish three hydrogen bonds with Ser178, Tyr224 and Asn249, which are consistent with the observation that tubulin heterodimer is stabilized by colchicine and confirms that this subunit is essential for binding. Similarly, an interaction with Ser178 or Tyr224 in the 4-hydroxymillepachine can be observed. Essential electrostatic interactions between methoxy groups of 3,4,5-trimethoxyphenyl residue and Ser178, Lys254 and Asn101 residues of the neighboring α subunit are observed on a binding pocket, demonstrating a plausible competitive mechanism of action on the colchicine binding site [130].
Another study was developed by Wang G et al., who synthesized a series of naphthalenchalcones (Tables S1–S3, Compounds 142–161, Figure 8) in order to evaluate their ability to inhibit tubulin polymerization. To evaluate the possibility that the representatives of this class have an action on the microtubule system, compound (E)-3-(3-hydroxy-4-methoxyphenyl)-1-(2-methoxynaphthalen-1-yl)prop-2-en-1-one (Compound 142), one of the most active compounds from the series, has been investigated for its ability to inhibit tubulin polymerization in a direct correlation with its concentration. This is a precise indicator of its action by interfering with the polymerization of microtubules. Treatment with 3, 6, 12.5 and 25 μM of the compound inhibited tubulin polymerization by 21, 41, 60 and 82%. Compound 142 is more active than the control compound, colchicine (IC50 value of compound 142 = 8.4 μM, IC50 value of colchicine = 10.6 μM). These results show that derivative 142 is an inhibitor of tubulin polymerization, which binds to tubulin and influences polymerization of microtubules. The determination of tubulin polymerization inhibitory activity for compound 142 was followed by an investigation of the cellular mechanism of action on MCF-7 cancer cells, using cytometric analysis. To elucidate the molecular mechanism of the derivative, the effect on cell cycle progression in MCF-7 cells was initially studied. The control group has typical cell cycle characteristics in G1, S and G2/M phases. In contrast, after treatment of MCF-7 cells with 2 μM of compound 142, the accumulation of cancer cells was detected in the G2/M phase with an intensity 5.5 times higher than in the control group (84.55% for the treated group and 15.19% for the control group). The results indicate the potential of chalcone to block the cell cycle in the G2/M phase and to stop cell mitosis, thus, inhibiting the proliferation of MCF-7 cells. To explain how to bind this class of tubulin compounds, a docking study was performed for chalcone 142 which is based on the binding pocket of tubulin colchicine. The results estimate a binding energy of −8.8 kcal/mol. The compound adopts “L-shaped” conformation on tubulin pocket. The 2-Methoxynaphtyl group of the compound is located in the hydrophobic pocket, is surrounded by residues Cys241, Leu248, Ala250, Leu252 and Leu255 and forms a strong hydrophobic bond. The detailed analysis shows that phenyl residue from the center of compound 142 forms a cation-Π-type interaction with Lys254 residue. Gln11, Leu248 and Leu 255 residues form interactions through three hydrogen bonds with compound 142 [225].
A new series of chalcone derivatives with a diaryl ether residue (Tables S1–S3, Compounds 162–177, Figure 9) were modulated and synthesized in order to evaluate their activity on tubulin polymerization. Of the compounds obtained, chalcone substituted with a methoxy group on an aromatic ring of aldehyde (compound 163, (E)-3-(4-methoxyphenyl)-1-(3-(3,4,5-trimethoxyphenoxy)phenyl)prop-2-en-1-one) is the most active on MCF-7, HepG 2 (hepatoblastoma-derived cell line) and HTC116 cancer lines. Tubulin inhibition activity for chalcone 163 was evaluated in vitro, using colchicine as a standard. Tubulin protein was mixed with different concentrations of compound 163 (0.8 μM, 1.5 μM, 3.0 μM, 6.0 μM, 12.5 μM and 25 μM). In case of the inhibition of tubulin protein by derivative 163, a tendency to decrease the intensity of fluorescence similar to colchicine was observed. IC50 values for compound 163 and colchicine are 20 μM and 10.6 μΜ. The cytometric method using MCF-7 cells was used to examine the cytotoxic effect of chalcone (2.5 μM and 5.0 μM). Cells from the control group have 59.61% of cells in phase G1, 18.11% in phase G2 and 22.27% on phase S of the cell cycle. Treatment with 2.5 μM of the compound did not significantly influence the cell cycle. The use of 5.0 μΜ of the compound greatly increased the percentage in G2/M phase cells (50.64%). This result shows that the derivative has increased antiproliferative activity on MCF-7 cells by increasing the percentage of cells in the G2/M phase. Docking studies suggest that derivative interacts and binds to the binding site of colchicine at tubulin level [226].
Du et al. analyzed a new indole chalcone, (3-6-methoxy-2-methyl-1H-indol-3-yl)-1-pyridyl-2-propen-1-one) (Tables S1 and S2, Compound 178), in order to identify the mechanism by which it induces cell death and to assess whether it crosses the blood-brain barrier. The analyzed chalcone acts as a destabilizing agent for microtubules and induces mitosis suppression and programmed cell death depending on the activity of caspases on glioblastoma and on various cancer cell lines. The activity of compound 178 is correlated with its ability to bind to the binding site of β-tubulin colchicine [227]. Other indole-chalcone derivatives were analyzed from the point of view of anti-tumor activity, related to tubulin and TrxR [228].
Another study by Canela et al. comprises a series of chalcones with a dioxolane residue in acetophenone (Tables S1–S3, Compounds 179–187, Figure 10), given that this substitution is frequently present in natural colchicine binding ligands. Using impedance cell growth monitoring, a time- and dose-dependent antimitotic effect was identified on MDA-MB-231 (human breast adenocarcinoma) cancer cells treated with (E)-3-(3“-amino-4”-methoxyphenyl)-1-(5’-Methoxy-3’,4’-methylenedioxyphenyl)-2-methylpropen-2-en-1-one (Compound 185), the most active compound from the series. At concentrations greater than 5 nM, the compound causes a rapid and continuous decrease in cell index, indicating a reduction in cell adhesion and/or toxicity. Using low concentrations of the compound, a decrease in the cellular index was observed at the first 14 h and a restoration occurred at 24–48 h, with the properties being similar to those of compounds that act on tubulin. Flow cytometry indicates an accumulation of cells in the G2/M phase 24 h after treatment with 10 nM compound. The induction of apoptosis was confirmed by measuring the translocation of phosphatidylserine from the cytoplasmic medium to the extracellular medium and by the activation of caspase 3 by the compound. The evaluation of the antitubulin activity of compound 185 on MDA-MB-231 cell cultures (human breast adenocarcinoma) shows the derivative blocks mitosis. Using flow cytometry, an accumulation of cells in the G2/M phase after 24 h of treatment with 10 nM of the compound was observed. Chalcone also causes a dose-dependent increase in subG1 phase cells and a sub-diploid DNA content, which is characteristic of apoptotic cells (45% ± 9 and 19% ± 8 to 10 and 1 nM of the compound). The induction of apoptosis was confirmed by measuring the translocation of phosphatidylserine from the cytoplasm into the extracellular environment of the cytoplasmic membrane and by activating caspase 3 by the compound [229].
Lindamulage et al. synthesized 24 quinoline chalcones to evaluate their anticancer activity. Two chalcones of series, (E)-3-(3-(2-methoxyphenyl)-3-(oxoprop-1-enyl)quinolin-2(1H)-one (Tables S1–S3, Compound 188) and (E)-6-methoxy-3(3-(2-methoxyphenyl)-3-oxoprop-1-enyl)quinolin-2(1H)-one (Tables S1–S3, Compound 189) show increased efficiency and selectivity of the cells. Both chalcones bind to the colchicine binding site and cause a prolongation of mitosis by acting on the dividing spindle, leading to cell death. Compounds destroy tumor cells with overexpressed MDR1 and MRP1, which are resistant to colchicine, ABT and paclitaxel. Chalcones 188 and 189 have a strong synergistic effect on tumor cells, including pluritherapy-resistant tumors. Since the analyzed quinazoline derivatives block the functioning of microtubules, the activity of polymerizing microtubules in the absence and presence of chalcones was examined. In vitro studies have shown that the activity of chalcones on microtubules is similar to the activity of nocodozole, but different from that of paclitaxel. This indicates the link between the action of chalcones and the polymerization of microtubules. Studies of intrinsic fluorescence of tryptophan, a method frequently used to determine the binding affinity of compounds to tubulin heterodimers, indicate the possibility of compounds to bind to the binding site of colchicine [230].
A series of 25 heterocyclic chalcones (Tables S1–S3, Compounds 190–214, Figure 11) were synthesized to evaluate their cytotoxic potential in vitro. After evaluating the interaction capacity between synthesized compounds and microtubules, it was shown that compound 190 had the capacity to inhibit tubulin polymerization in vitro. Chalcone interrupts cell cycle in the G2/M phase, an indicator of tubulin polymerization. The ability of the compound to inhibit tubulin polymerization was determined by combrestatin A4 and paclitaxel. Experimental data show that the hybrid molecule is a potent inhibitor of tubulin polymerization, having IC50 values = 9.66 ± 0.06 μM [231].
Qiu et al. also synthesized a series of the shikonium-derived chalcones (Tables S1–S3, Compounds 215–232, Figure 12) to evaluate their potential to inhibit tubulin polymerization by in vitro assays. IC50 values are similar for inhibiting tubulin polymerization and for cellular antiproliferation testing. Among the synthesized compounds, compound 226 has the best anti-tubulin activities, having IC50 values = 2.98 ± 0.53 μM. Studies of mechanisms of action show that derivative 226 can induce apoptosis of MCF-7 cells, reduce the potential of mitochondrial membranes and cause an accumulation of cells in the G2/M phase of the cell cycle, and the effect is to disrupt the microtubule system similarly to standard compound, colchicine. Of the compounds analyzed, derivative 226 has the best energy, the value of this compound being −64.6074 kcal/mol. The compound binds to the binding site of colchicine on tubulin through three hydrogen bonds with Ser178, Tyr202 and Lys254 residues. Data from in silico analysis show that derivative 224 is a potent inhibitor of tubulin polymerization [232].
Starting from the anticancer activity of natural and synthetic chalcones, Yan W et al. synthesized a 1,2,3-triazole hybrid of chalcones (Tables S1–S3, Compound 233) and tested the anticancer activity of compounds on liver cells lines. (E)-1-(4-((1-allyl-1H-1,2,3-triazol-4-yl)methoxyphenyl)-3-(2,4-dichlorophenyl)propen-2-en-1-one (Compound 233) shows inhibitory values IC50 = 2.34 μM against tubulin polymerization, a value that indicates the potential of the compound to be a new candidate for novel antitubulin agents [233].
3. Conclusions
ABCG2 and tubulin are mechanisms by which cancer cells resist treatment. ABCG2 (also called BCRP) decreases the transfer of drugs to cancer cells. It is also sought to find substances that inhibit the formation of tubulin assembly under the influence of tumor cells, through the binding of the compounds to the colchicine binding site. Compound 41 and compound 80 have very good anticancer activity against ABCG2, having an IC50 of 0.17 µM and 0.19 µM (Figure 13), respectively, due to the methoxy substituents on the aromatic nucleus of the acetophenone residue, and the substitution of the phenyl residue by a heterocycle (quinazoline) with two methoxy groups. For the second mechanism, compounds 8 and 185 have an IC50 of 0.003 µM and 0.0039 µM (Figure 13), respectively. This strong activity is explained by the substitution of methoxy groups at the acetophenonic residue but also by the presence of a heterocycle (indole) in the molecule of compound 8. In compound 185, the activity is determined by the condensation of dioxolane with the phenyl of the acetophenonic group and by the existence of methoxy and amino substituents in the molecule.
4. Methods
The articles were selected from the PubMed and Google Scholar databases, taking into account the most representative articles about cancer and flavonoids, and about natural, semi-synthetic and synthetic chalcones with anticancer activity, with an emphasis on the mechanisms of ATP binding cassette subfamily G and tubulin. The inclusion criterion was that the articles were from the period 2010–2022, and the exclusion criterion was other mechanisms of anticancer action, different from ABCG2 and tubulin.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/ijms231911595/s1, Figure S1: Basic structure of flavonoids. Figure S2: Claisen-Schmidt reaction. Table S1: Chemical structures of flavonoids. Table S2: The name and chemical formula of compounds. Table S3: Half-maximal inhibitory concentration (IC50) of the most effective chalcones compounds.
Author Contributions
All authors contributed equally to the preparation of this manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
| ABCG2 | ATP Binding Cassette Subfamily G Member 2 |
| BCRP | breast cancer resistant protein |
| DNA | deoxyribonucleic acid |
| EGFR | tyrosine kinase receptor |
| H460-MX20 | human non-small cell lung cancer NSCLC |
| HCT116 | human colorectal carcinoma cell line |
| Heg 2 | hepatoblastoma-derived cell line |
| HEK293 | immortalized human embryonic kidney cells |
| Ko143 | the most potent BCRP inhibitor known |
| LCA | licochalcone A |
| MCF-7 | human breast adenocarcinoma |
| MDA-MB-231 | human breast adenocarcinoma |
| MET | mesenchymal epithelial transition factor |
| MTT | 2,5-diphenyl-2H-tetrazolium bromide |
| MV-4-11 | human acute myeloid leukemia cell line |
| NF-ĸB | activated nuclear B cell growth |
| R482-HEK293 | ABCG2 transfected HEX293 human cells |
| ROS | reactive oxygen species |
| S1-M1-80 | human colon cancer |
| VEGF | vascular endothelial growth factor |
| XN | xanthohumol |
References
- Li, P.; Jiang, H.; Zhang, W.; Li, Y.; Zhao, M.; Zhou, W. Synthesis of carbazole derivatives containing chalcone analogs as non-intercalative topoisomerase II catalytic inhibitors and apoptosis inducers. Eur. J. Med. Chem. 2018, 145, 498–510. [Google Scholar] [CrossRef]
- Woodman, C.; Vundu, G.; George, A.; Wilson, C. Applications and strategies in nanodiagnosis and nanotherapy in lung cancer. Semin. Cancer Biol. 2021, 69, 349–364. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Jiménez, F.; Muiños, F.; Sentís, I.; Deu-Pons, J.; Reyes-Salazar, I.; Arnedo-Pac, C.; Mularoni, L.; Pich, O.; Bonet, J.; Kranas, H.; et al. A compendium of mutational cancer A driver genes 1. Nat. Rev. Cancer 2020, 20, 555–572. [Google Scholar] [CrossRef] [PubMed]
- Williams, N.; Lee, J.; Mitchell, E.; Moore, L.; Baxter, E.J.; Hewinson, J.; Dawson, K.J.; Menzies, A.; Godfrey, A.L.; Green, A.R.; et al. Life histories of myeloproliferative neoplasms inferred from phylogenies. Nature 2022, 602, 162–168. [Google Scholar] [CrossRef]
- Goodall, G.; Wickramasinghe, V. RNA in cancer. Nat. Rev. Cancer 2021, 21, 22–36. [Google Scholar] [CrossRef]
- Ganesh, K.; Massagué, J. Targeting metastatic cancer. Nat. Med. 2021, 27, 34–44. [Google Scholar] [CrossRef] [PubMed]
- Tarabichi, M.; Salcedo, A.; Deshwar, A.; Leathlobhair, M.N.; Wintersinger, J.; Wedge, D.; Van Loo, P.; Morris, Q.; Boutros, P. A practical guide to cancer subclonal reconstruction from DNA sequencing. Nat. Methods 2021, 18, 144–155. [Google Scholar] [CrossRef] [PubMed]
- Kwan, E.M.; Wyatt, A.W. Androgen receptor genomic alterations and treatment resistance in metastatic prostate cancer. Prostate 2022, 82 (Suppl. S1), S25–S36. [Google Scholar] [CrossRef]
- Bergers, G.; Fendt, S.M. The metabolism of cancer cells during metastasis. Nat. Rev. Cancer 2021, 21, 162–180. [Google Scholar] [CrossRef]
- Riggio, A.; Varley, K.; Welm, A. The lingering mysteries of metastatic recurrence in breast cancer. Br. J. Cancer 2021, 124, 13–16. [Google Scholar] [CrossRef]
- Hayes, J.D.; Dinkova-Kostova, A.T.; Tew, K.D. Oxidative Stress in Cancer. Cancer Cell 2020, 38, 167–197. [Google Scholar] [CrossRef]
- Kong, N.; Zhang, H.; Feng, C.; Liu, C.; Xiao, Y.; Zhang, X. Arsenene-mediated multiple independently targeted reactive oxygen species burst for cancer therapy. Nat. Commun. 2021, 12, 4777. [Google Scholar] [CrossRef]
- Zhang, J.; Duan, D.; Song, Z.L.; Liu, T.; Hou, Y.; Fang, J. Small molecules regulating reactive oxygen species homeostasis for cancer therapy. Med. Res. Rev. 2021, 41, 342–394. [Google Scholar] [CrossRef]
- Cheung, E.C.; Vousden, K.H. The role of ROS in tumour development and progression. Nat. Rev. Cancer 2022, 22, 280–297. [Google Scholar] [CrossRef]
- Echle, A.; Rindtorff, N.T.; Brinker, T.J.; Luedde, T.; Pearson, A.T.; Kather, J.N. Deep learning in cancer pathology: A new generation of clinical biomarkers. Br. J. Cancer 2021, 124, 686–696. [Google Scholar] [CrossRef]
- Paidakula, S.; Nerella, S.; Vadde, R.; Kamal, A. Design and synthesis of 4β-Acetamidobenzofuranone-podophyllotoxin hybrids and their anti-cancer evaluation. Bioorg. Med. Chem. Lett. 2019, 29, 2153–21566. [Google Scholar] [CrossRef]
- Kashyap, D.; Tuli, H.S.; Yerer, M.B.; Sharma, A.; Sak, K.; Srivastava, S.; Pandey, A.; Gang, V.K.; Sethi, G.; Bishayee, A. Natural product-based nanoformulation for cancer therapy: Opportunities and challenges. Semin. Cancer Biol. 2021, 69, 5–23. [Google Scholar] [CrossRef]
- Li, Y.; Zheng, X.; Chu, Q. Bio-based nanomaterials for cancer therapy. Nano Today 2021, 38, 101134. [Google Scholar] [CrossRef]
- Chargari, C.; Peignaux, K.; Escande, A.; Renard, S.; Lafond, C.; Petit, A.; Hannoun-Lévi, J.M.; Durdux, C.; Haie-Méder, C. Radiotherapy for endometrial cancer. Cancer Radiother. 2022, 26, 309–314. [Google Scholar] [CrossRef]
- Stine, Z.E.; Schug, Z.T.; Salvino, J.M.; Dang, C.V. Targeting cancer metabolism in the era of precision oncology. Nat. Rev. Drug Discov. 2022, 21, 141–162. [Google Scholar] [CrossRef]
- Boehm, J.; Garnett, M.; Adams, D.; Francies, H.; Golub, T.; Hahn, W.; Iorio, F.; McFarland, J.; Parts, L.; Vazques, F. Cancer research needs a better map. Nature 2021, 589, 514–516. [Google Scholar] [CrossRef] [PubMed]
- Raj, S.; Khurana, S.; Choudhari, R.; Kesari, K.K.; Kamal, M.A.; Garg, N.; Ruokolainen, J.; Das, B.; Kumar, D. Specific targeting cancer cells with nanoparticles and drug delivery in cancer therapy. Semin. Cancer Biol. 2021, 69, 166–177. [Google Scholar] [CrossRef] [PubMed]
- Augustine, R.; Kalva, N.S.; Ahmad, R.; Zahid, A.A.; Hasan, S.; Nayeem, A.; McClements, L.; Hasan, A. 3D Bioprinted cancer models: Revolutionizing personalized cancer therapy. Transl. Oncol. 2021, 14, 101015. [Google Scholar] [CrossRef] [PubMed]
- Rottenberg, S.; Disler, C.; Perego, P. The rediscovery of platinum-based cancer therapy. Nat. Rev. Cancer 2021, 21, 37–50. [Google Scholar] [CrossRef]
- Chen, H.; Lin, Z.; Arnst, K.; Miller, D.; Li, W. Tubulin Inhibitor-Based Antibody-Drug Conjugates for Cancer Therapy. Molecules 2017, 22, 1281. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wang, M.; Wu, H.X.; Xu, R.H. Advancing to the era of cancer immunotherapy. Cancer Commun. 2021, 41, 803–829. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Jiang, Q.; Zhao, X.; Zhao, R.; Wang, Y.; Wang, Y.; Liu, J.; Shang, Y.; Zhao, S.; Wu, T.; et al. A DNA nanodevice-based vaccine forcancer immunotherapy. Nat. Mater. 2021, 20, 421–430. [Google Scholar] [CrossRef]
- Tay, R.E.; Richardson, E.; Toh, H.C. Revisiting the role of CD4+ T cells in cancer immunotherapy—New insights into old paradigms. Cancer Gene Ther. 2021, 28, 5–17. [Google Scholar] [CrossRef]
- Zeng, Z.; Zhang, C.; Li, J.; Cui, D.; Jiang, Y.; Pu, K. Activatable Polymer Nanoenzymes for Photodynamic Immunometabolic Cancer Therapy. Adv. Mater. 2021, 33, 2007247. [Google Scholar] [CrossRef]
- He, J.; Xiong, X.; Yang, H.; Li, D.; Liu, X.; Li, S.; Liao, S.; Chen, S.; Wen, X.; Yu, K.; et al. Defined tumor antigen-specific T cells potentiate personalized TCR-T cell therapy and prediction of immunotherapy response. Cell Res. 2022, 32, 530–542. [Google Scholar] [CrossRef]
- Luoma, A.M.; Suo, S.; Wang, Y.; Gunasti, L.; Porter, C.; Nabilsi, N.; Tadros, J.; Ferretti, A.P.; Liao, S.; Gurer, C.; et al. Tissue-resident memory and circulating T cells are early responders to pre-surgical cancer immunotherapy. Cell 2022, 185, 2918–2935.e29. [Google Scholar] [CrossRef]
- Hahn, W.; Bader, J.; Braun, T.; Califano, A.; Clemons, P.; Druker, B.; Edward, A.; Fu, H.; Jagu, S.; Kemp, C.; et al. An expanded universe of cancer targets. Cell 2021, 184, 1142–1155. [Google Scholar] [CrossRef]
- Sun, J.; Lu, Q.; Sanmanmed, M.; Wang, J. Singlec-15 as an emerging target for next–generation cancer immunotherapy. Clin. Cancer Res. 2021, 27, 680–688. [Google Scholar] [CrossRef]
- Angelis, M.L.; Francescangeli, F.; Torre, F.; Zeuner, A. Stem Cell Plasticity and Dormancy in the Development of Cancer Therapy Resistance. Front. Oncol. 2019, 9, 626. [Google Scholar] [CrossRef]
- Sabnis, A.J.; Bivona, T.G. Principles of Resistance to Targeted Cancer Therapy: Lessons from Basic and Translational Cancer Biology. Trends Mol. Med. 2019, 25, 185–197. [Google Scholar] [CrossRef]
- Reggiani, F.; Gobbi, G.; Ciarrocchi, A.; Ambrosetti, C.; Sancisi, V. Mutiple roles and context-specific mechanisms underlying YAP and TAZ-mediated resistance to anticancer therapy. Rev. Cancer 2020, 1873, 188341. [Google Scholar]
- Sachs, J.; Döhl, K.; Weber, A.; Bonus, M.; Ehlers, F.; Fleischer, E.; Klinger, A.; Gohlke, H.; Pietruszka, J.; Schmitt, L. Novel 3,4-Dihydroisocoumarins inhibit Human P-gp and BCRP in Multidrug Resistant Tumors and Demonstrate Substrate Inhibiton of Yeast Pdr5. Front. Pharmacol. 2019, 10, 400. [Google Scholar] [CrossRef]
- Ihmaid, S.; Ahmed, H.; Zayed, M. The Design and Development of Potent Small Molecules as Anticancer Agents Targeting EGFR TK and Tubulin Polymerization. Int. J. Mol. Sci. 2018, 19, 408. [Google Scholar] [CrossRef]
- Vesely, M.D.; Zhang, T.; Chen, L. Resistance Mechanisms to Anti-PD Cancer Immunotherapy. Annu. Rev. Immunol. 2022, 40, 45–74. [Google Scholar] [CrossRef]
- Passaro, A.; Brahmer, J.; Antonia, S.; Mok, T.; Peters, S. Managing Resistance to Immune Checkpoint Inhibitors in Lung Cancer: Treatment and Novel Strategies. J. Clin. Oncol. 2022, 40, 598–610. [Google Scholar] [CrossRef]
- Zhang, C.; Liu, X.; Jin, S.; Chen, Y.; Guo, R. Ferroptosis in cancer therapy: A novel approach to reversing drug resistance. Mol. Cancer 2022, 21, 47. [Google Scholar] [CrossRef]
- Majidinia, M.; Mirza-Aghazadeh-Attari, M.; Rahimi, M.; Mihanfar, A.; Karimian, A.; Safa, A.; Yousefi, B. Overcoming multidrug resistance in cancer: Recent progress in nanotechnology and new horizons. IUBMB Life 2020, 72, 855–871. [Google Scholar] [CrossRef]
- Kaya, S.; Gökce, H.; Arslan, T.; Alpaslan, G. Synthesis, spectroscopic characterization, DFT computations, nonlinear optical profile and molecular docking study of a novel chalcone derivative. J. Mol. Struct. 2019, 1202, 127270. [Google Scholar] [CrossRef]
- Yin, H.; Dong, J.; Cai, Y.; Shi, X.; Wang, H.; Liu, G.; Tang, Y.; Liu, J.; Ma, L. Design, synthesis and biological evaluation of chalcones as reversers of P-glycoprotein-mediated multidrug resistance. Eur. J. Med. Chem. 2019, 180, 350–366. [Google Scholar] [CrossRef]
- Bhardwaj, B.; Baidya, A.T.K.; Amin, S.A.; Adhikari, N.; Jha, T.; Gayen, S. Insight into structural features of phenyltetrazole derivatives as ABCG2 inhibitors for the treatment of multidrug resistance in cancer. SAR QSAR Environ. Res. 2019, 30, 457–475. [Google Scholar] [CrossRef]
- Köhler, S.C.; Silbermann, K.; Wiese, M. Phenyltetrazolyl-phenylamides: Substituent impact on modulation capability and selectivity toward the efflux protein ABCG2 and investigation of interaction with the transporter. Eur. J. Med. Chem. 2016, 124, 881–895. [Google Scholar] [CrossRef]
- Pérès, B.; Nasr, R.; Zarioh, M.; Lecerf-Schmidt, F.; Di Pietro, A.; Baubichon-Cortay, H.; Boumendjel, A. Ferrocene-embedded flavonoids targeting the Achilles hell of multidrug-resistant cancer cells through collateral sensitivity. Eur. J. Med. Chem. 2017, 130, 346–353. [Google Scholar] [CrossRef]
- Ding, R.; Jin, S.; Pabon, K.; Scotto, K. A role for ABCG2 beyond drug transport: Regulation of autophagy. Autophagy 2016, 12, 737–751. [Google Scholar] [CrossRef]
- Khunweeraphong, N.; Stockner, T.; Kuchler, K. The structure of the human ABC transporter ABCG2 reveals a novel mechanism for drug extrusion. Sci. Rep. 2017, 7, 13767. [Google Scholar] [CrossRef]
- Modi, A.; Roy, D.; Sharma, S.; Vishnoi, J.R.; Pareek, P.; Elhence, P.; Sharma, P.; Purohit, P. ABC transporters in breast cancer: Their roles in multidrug resistance and beyond. J. Drug Target. 2022, 1–21. [Google Scholar] [CrossRef]
- Yuan, T.; Hu, J.; Zhu, X.; Yin, H.; Yin, J. Oxidative stress-mediated up-regulation of ABC transporters in lung cancer cells. J. Biochem. Mol. Toxicol. 2022, 36, e23095. [Google Scholar] [CrossRef]
- Tuli, H.S.; Mittal, S.; Aggarwal, D.; Parashar, G.; Parashar, N.C.; Upadhyay, S.K.; Barwal, T.S.; Jain, A.; Kaur, G.; Savla, R.; et al. Path of Silibinin from diet to medicine: A dietary polyphenolic flavonoid having potential anti-cancer therapeutic significance. Semin. Cancer Biol. 2021, 73, 196–218. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.; Jian, J.J.; Ding, J. Natural Products in Cancer Therapy: Past, Present and Future. Nat. Prod. Bioprospect. 2021, 11, 5–13. [Google Scholar] [CrossRef] [PubMed]
- Obaid, R.; Mughal, E.U.; Naeem, N.; Sadiq, A.; Alsantali, R.; Jasses, R.; Moussa, Z.; Ahmed, S. Natural and synthetic flavonoid derivatives as new potential tyrosinase inhibitors: A systematic review. RCS Adv. 2021, 11, 22159. [Google Scholar] [CrossRef] [PubMed]
- Böttner, L.; Grabe, V.; Gablenz, S.; Böhme, N.; Appenroth, K.; Gershenzon, J.; Hubert, M. Differential localization of flavonoid glucosides in an aquatic plant implicates different functions under abiotic stress. Plant Cell Environ. 2021, 44, 900–911. [Google Scholar] [CrossRef]
- Tow, W.K.; Goh, A.P.; Sundralingam, U.; Palanisamy, U.D.; Sivasothy, Y. Flavonoid Composition and Pharmacological Properties of Elaeis guineensis Jacq. Leaf Extracts: A Systematic Review. Pharmaceuticals 2021, 14, 961. [Google Scholar] [CrossRef]
- Du, T.; Fan, Y.; Cao, H.; Song, Z.; Dong, B.; Liu, T.; Yang, W.; Wang, M.; Niu, L.; Yang, Q.; et al. Transcriptome analysis revealed key genes involved in flavonoid metabolism in response to jasmonic acid in pigeon pea (Cajanus cajan (L.) Millsp.). Plant Physiol. Biochem. 2021, 168, 410–422. [Google Scholar] [CrossRef]
- Yen, S.C.; Chen, L.C.; Huang, H.L.; Ngo, S.T.; Wu, Y.W.; Lin, T.E.; Sung, T.Y.; Lien, S.T.; Tseng, H.J.; Pan, S.L.; et al. Investigation of Selected Flavonoid Derivatives as Potent FLT3 Inhibitors for the Potential Treatment of Acute Myeloid Leukemia. J. Nat. Prod. 2021, 84, 1–10. [Google Scholar] [CrossRef]
- Kubra, G.; Khan, M.; Munir, F.; Gul, A.; Shah, T.; Hussain, A.; Caparrós-Ruiz, D.; Amir, R. Expression Characterization of Flavonoid Biosynthetic Pathway Genes and Transcription Factors in Peanut under Water Deficit Conditions. Front. Plant Sci. 2021, 12, 680368. [Google Scholar] [CrossRef]
- Kim, J.Y.; Lee, S.I.; Kim, J.; Muthusamy, M.; Jeong, M.J. Specific audible sound waves improve flavonoid contents and antioxidative properties of sprouts. Sci. Hortic. 2021, 276, 109746. [Google Scholar] [CrossRef]
- Ančić, N.; Patelou, E.; Papanikolaou, A.; Kanioura, A.; Valdesturli, C.; Arapitsas, P.; Skorić, M.; Dragićević, M.; Gašic, U.; Koukounaras, A.; et al. Comparative Metabolite and Gene Expression Analyses in Combination with Gene Characterization Revealed the Patterns of Flavonoid Accumulation During Cistus creticus subsp. creticus Fruit Development. Front. Plant Sci. 2021, 12, 619634. [Google Scholar] [CrossRef]
- Park, S.I.; Park, H.L.; Bhoo, S.H.; Lee, S.W.; Cho, M.H. Biochemical and Molecular Characterization of the Rice Chalcone Isomerase Family. Plants 2021, 10, 2064. [Google Scholar] [CrossRef]
- Boulebd, H. Structure-activity relationship of antioxidant prenylated (iso)flavonoid-type compounds: Quantum chemistry and molecular docking studies. J. Biomol. Struct. Dyn. 2021, 1–10. [Google Scholar] [CrossRef]
- do Rosario, V.A.; Schoenaker, D.; Kent, K.; Weston-Green, K.; Charlton, K. Association between flavonoid intake and risk of hypertension in two cohorts of Australian women: A longitudinal study. Eur. J. Nutr. 2021, 60, 2507–2519. [Google Scholar] [CrossRef]
- del Río, J.C.; Rencoret, J.; Gutiérrez, A.; Lan, W.; Kim, H.; Ralph, J. Lignin Monomers Derived from the Flavonoid and Hydroxystilbene Biosynthetic Pathways Lignin Monomers Derived from the Monolignol Biosynthetic Pathway. Res. Adv. Polyphen. Res. 2021, 7, 177–205. [Google Scholar]
- Constantinescu, T.; Lungu, C.N. Anticancer Activity of Natural and Synthetic Chalcones. Int. J. Mol. Sci. 2021, 22, 11306. [Google Scholar] [CrossRef]
- Yang, J.; Wang, X.; Zhang, C.; Ma, L.; Wei, T.; Zhao, Y.; Peng, X. Comparative study of inhibition mechanisms of structurally different flavonoid compounds on α-glucosidase and synergistic effect with acarbose. Food Chem. 2021, 347, 129056. [Google Scholar] [CrossRef]
- Duan, Y.; Ying, Z.; He, F.; Ying, X.; Jia, L.; Yang, G. A new skeleton flavonoid and a new lignan from Portulaca oleracea L. and their activities. Fitoterapia 2021, 153, 104993. [Google Scholar] [CrossRef]
- Liu, Z.; Silva, J.; Shao, A.; Liang, J.; Wallner, M.; Shao, X.; Li, M.; Olsen, R. Flavonoid compounds isolated from Tibetan herbs, binding to GABAA receptor with anxiolytic property. J. Ethnopharmacol. 2021, 267, 113630. [Google Scholar] [CrossRef]
- Acevedo-Fani, A.; Ochoa-Grimaldo, A.; Loveday, S.; Singh, H. Digestive dynamics of yoghurt structure impacting the release and bioaccessibility of the flavonoid rutin. Food Hydrocoll. 2021, 111, 106215. [Google Scholar] [CrossRef]
- Bellavia, D.; Caradonna, F.; Dimarco, E.; Costa, V.; Carina, V.; De Luca, A.; Raimondi, L.; Fini, M.; Gentile, C.; Giavaresi, G. Non-flavonoid polyphenols in osteoporosis: Preclinical evidence. Trends Endocrinol. Metab. 2021, 32, 515–529. [Google Scholar] [CrossRef]
- Bencheikh, N.; Bouhrim, M.; Merrouni, I.A.; Boutahiri, S.; Kharchoufa, L.; Addi, M.; Tungmunnithum, D.; Hano, C.; Eto, B.; Legssyer, A.; et al. Antihyperlipidemic and Antioxidant Activities of Flavonoid-Rich Extract of Ziziplus lotus (L.) Lam. Fruits. Appl. Sci. 2021, 11, 7788. [Google Scholar] [CrossRef]
- Birhanie, Z.M.; Xiao, A.; Yang, D.; Huang, S.; Zhang, C.; Zhao, L.; Liu, L.; Li, J.; Chen, A.; Tang, J.; et al. Polysaccharides, Total Phenolic, and Flavonoid Content from Different Kenaf (Hibiscus cannabinus L.) Genotypes and Their Antioxidants and Antibacterial Properties. Plants 2021, 10, 1900. [Google Scholar] [CrossRef]
- Cirmi, S.; Maugeri, A.; Lombardo, G.E.; Russo, C.; Musumeci, L.; Gangemi, S.; Calapai, G.; Barreca, D.; Navvara, M. A Flavonoid-Rich Extract of Mandarin Juice Counteracts 6-OHDA-Induced Oxidative Stress in SH-SY5Y Cells and Modulates Parkinson-Related Genes. Antioxidants 2021, 10, 539. [Google Scholar] [CrossRef]
- Zhu, J.; Zhao, W.; Li, R.; Guo, D.; Li, H.; Wang, Y.; Mei, W.; Peng, S. Identification and Characterization of Chalcone Isomerase Genes Involved in Flavonoid Production in Dracaena cambodiana. Front. Plant Sci. 2021, 12, 616396. [Google Scholar] [CrossRef]
- Goris, T.; Cuadrat, R.; Braune, A. Flavonoid-Modifying Capabilities of the Human Gut Microbiome—An In Silico Study. Nutrients 2021, 13, 2688. [Google Scholar] [CrossRef]
- Davinelli, S.; Ali, S.; Scapagnini, G.; Costagliola, C. Effects of Flavonoid Supplementation on Common Eye Disorders: A Systematic Review and Meta-Analysis of Clinical Trials. Front. Nutr. 2021, 8, 651441. [Google Scholar] [CrossRef]
- Feng, C.Y.; Li, S.S.; Taguchi, G.; Wu, Q.; Yin, D.D.; Gu, Z.Y.; Wu, J.; Xu, W.Z.; Li, C.; Wang, L.S. Enzymatic basis for stepwise C-glycosylation in the formation of flavonoid di-C-glycosides in sacred lotus (Nelumbo nucifera Gaertn.). Plant J. 2021, 106, 351–365. [Google Scholar] [CrossRef]
- Attari, F.; Keighobadi, F.; Abdollahi, M.; Arefian, E.; Lotfizadeh, R.; Sepehi, H.; Farimani, M.M. Inhibitory effect of flavonoid xanthomicrol on triple-negative breast tumor via regulation of cancer-associated microRNAs. Phytother. Res. 2021, 35, 1967–1982. [Google Scholar] [CrossRef]
- Guazelli, C.; Fattori, V.; Ferraz, C.; Borghi, S.; Baracat, M.; Verri Jr, W. Antioxidant and anti-inflammatory effects of hesperidin methyl chalcone in experimental ulcerative colits. Chem. Biol. Interact. 2021, 333, 109315. [Google Scholar] [CrossRef]
- Vančo, J.; Trávníček, Z.; Hošek, J.; Malina, T. Copper (II) Complexes Containing Natural Flavonoid Pomiferin Show Considerable In Vitro Cytotoxicity and Anti-Inflammatory Effects. Int. J. Mol. Sci. 2021, 22, 7626. [Google Scholar] [CrossRef]
- Manolaridis, I.; Jackson, S.; Taylor, N.; Kowal, J.; Stahlberg, H.; Locher, K. Cryo-EM structures of human ABCG2 mutant trapped in ATP-bound and substrate-bound states. Nature 2018, 563, 426–430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, D.; Park, J.C.; Lee, H.N.; Moon, J.H.; Ahn, H.; Park, K.; Hong, J. In vitro osteogenic differentiation and antibacterial potentials of chalcone. Mol. Pharm. 2018, 15, 3197–3204. [Google Scholar] [CrossRef] [PubMed]
- Kostopoulou, I.; Detsi, A. Recent Developments on Tyrosinase Inhibitors based on the Chalcone and Aurone Scaffolds. Curr. Enzym. Inhib. 2018, 14, 3–17. [Google Scholar] [CrossRef]
- Zhuang, C.; Zhang, W.; Sheng, C.; Zhang, W.; Xing, C.; Miao, Z. Chalcone: A Privileged Structure in Medicinal Chemistry. Chem. Rev. 2017, 117, 7762–7810. [Google Scholar] [CrossRef]
- Rashid, H.; Xu, Y.; Ahmad, N.; Muhammad, Y.; Wang, L. Promising anti-inflammatory effects of chalcones via inhibition of cyclooxygenase, prostaglandin E2, inducible NO synthase and nuclear factor ĸB activities. Bioorg. Chem. 2019, 87, 335–365. [Google Scholar] [CrossRef]
- Zhang, Y.Q.; Wen, Z.H.; Wan, K.; Yuan, D.; Zeng, X.; Liang, G.; Zhu, J.; Xu, B.; Luo, H. A novel synthesized 3′,5′-diprenylated chalcone mediates the proliferation of human leukemia cells by regulating apoptosis and autophagy pathways. Biomed. Pharmacother. 2018, 106, 794–804. [Google Scholar] [CrossRef]
- Kamal, A.; Reddy, M.K.; Viswanath, A. The design and development of imidazothiazole-chalcone derivatives as potential anticancer drugs. Expert Opin. Drug. Discov. 2013, 8, 289–304. [Google Scholar] [CrossRef]
- Kahssay, S.W.; Hailu, G.S.; Desta, K.T. Design, Synthesis, Characterization and In Vivo Antidiabetic Activity Evaluation of Some Chalcone Derivatives. Drug Des. Dev. Ther. 2021, 15, 3119–3129. [Google Scholar] [CrossRef]
- Annath, H.; Manayil, J.; Thompson, J.; Marr, A.; Raja, R. Contrasting structure-property relationships in amorphous, hierarchical and microporous aluminophosphate catalysts for Claisen-Schmidt condensation reactions. Appl. Catal. A Gen. 2021, 627, 118376. [Google Scholar] [CrossRef]
- Xue, K.; Sun, G.; Zhang, Y.; Chen, X.; Zhou, Y.; Hou, J.; Long, H.; Zhang, Z.; Lei, M.; Wu, W. A new method for the synthesis of chalcone derivatives promoted by PPh3/I2 under non-alkaline conditions. Synth. Commun. 2020, 51, 625–634. [Google Scholar] [CrossRef]
- Arnst, K.; Banerjee, S.; Chen, H.; Deng, S.; Hwang, D.J.; Li, W.; Miller, D. Current advances of tubulin inhibitors as dual acting small molecules for cancer therapy. Med. Res. Rev. 2019, 39, 1398–1426. [Google Scholar] [CrossRef]
- Yin, H.; Shi, X.; Wang, H.; Liu, G.; Ma, L. VB1 Promoted Green Synthesis of Chalcones and Its Neuroprotection Potency Evaluation. Processes 2019, 7, 236. [Google Scholar] [CrossRef]
- Madhavi, S.; Sreenivasulu, R.; Yazala, J.P.; Raju, R.R. Synthesis of chalcone incorporated quinazoline derivatives as anticancer agents. Saudi Pharm. J. 2017, 25, 275–279. [Google Scholar] [CrossRef]
- Yadav, P.; Lal, K.; Kumar, A.; Kumar, S.; Jaglan, S.; Bhushan, S. Green synthesis and anticancer potential of chalcone linked-1,2,3-triazoles. Eur. J. Med. Chem. 2017, 126, 944–953. [Google Scholar] [CrossRef]
- Singh, A.; Gut, J.; Rosenthal, P.J.; Kumar, V. 4-Aminoquinoline-ferrocenyl-chalcone conjugates: Synthesis and anti-plasmodial evaluation. Eur. J. Med. Chem. 2017, 125, 269–277. [Google Scholar] [CrossRef]
- Gomes, M.N.; Braga, R.C.; Grzelak, E.M.; Neves, B.J.; Muratov, E.; Ma, R.; Klein, L.; Cho, S.; Oliveira, G.; Franzlau, S.; et al. QSAR-driven design, synthesis and discovery of potent chalcone derivatives with antitubercular activity. Eur. J. Med. Chem. 2017, 137, 126–138. [Google Scholar] [CrossRef]
- Singh, A.; Rani, A.; Gut, J.; Rosenthal, P.; Kumar, V. Piperazine-linked 4-aminoquinoline-chalcone/ferrocenyl-chalcone conjugates: Synthesis and antiplasmodial evaluation. Chem. Biol. Drug Des. 2017, 90, 590–595. [Google Scholar] [CrossRef]
- Ortalli, M.; Ilari, A.; Colotti, G.; De Ionna, I.; Battista, T.; Bisi, A.; Gobbi, S.; Rampa, A.; Di Martino, R.; Gentilomi, G.; et al. Identification of chalcone-based antileishmanial agents targeting trypanothione reductase. Eur. J. Med. Chem. 2018, 152, 527–541. [Google Scholar] [CrossRef]
- Kant, R.; Kumar, D.; Agarwal, D.; Devi, R.; Tilak, R.; Awasthi, S.K.; Agarwal, A. Synthesis of newer 1,2,3-triazole linked chalcone and flavone hybrid compounds and evaluation of their antimicrobial and cytotoxic activities. Eur. J. Med. Chem. 2016, 113, 34–49. [Google Scholar] [CrossRef]
- Mazzone, G.; Galano, A.; Alvarez-Idaboy, J.R.; Russo, N. Coumarin−Chalcone Hybrids as Peroxyl Radical Scavengers: Kinetics and Mechanisms. J. Chem. Inf. Model. 2016, 56, 662–670. [Google Scholar] [CrossRef]
- Cheng, P.; Huang, X.; Wang, X.; Gong, M. Chalcone hybrids and their antimalarial activity. Arch. Pharm. Chem. Life Sci. 2020, 353, e1900350. [Google Scholar] [CrossRef] [PubMed]
- Singh, G.; Arora, A.; Kalra, P.; Kumar, I.; Espinosa, C.; Estebanc, M.A.; Sinha, S.; Goyal, K.; Sehgal, R. A strategic approach to the synthesis of ferrocene appended chalcone linked triazole allied organosilatranes: Antibacterial, antifungal, antiparasitic and antioxidant studies. Bioorg. Med. Chem. 2019, 27, 188–195. [Google Scholar] [CrossRef] [PubMed]
- Polo, E.; Ibarra-Arellano, N.; Prent-Peñaloza, L.; Morales-Bayuelo, A.; Henao, J.; Galdámez, A.; Gutierrez, M. Ultrasound-assisted synthesis of novel chalcone, heterochalcone and bis-chalcone derivatives and the evaluation of their antioxidant properties and as acetylcholinesterase inhibitors. Bioorg. Chem. 2019, 90, 103034. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, C.; Ko, P.; Chang, Y.; Kapoor, M.; Liang, Y.; Chu, H.; Lin, H.; Horng, J.; Hsu, M. Design and Synthesis of Benzimidazole-Chalcone Derivatives as Potential Anticancer Agents. Molecules 2019, 24, 3259. [Google Scholar] [CrossRef]
- Kurt, B.Z.; Kandas, N.O.; Dag, A.; Sonmez, F.; Kucukislamogh, M. Synthesis and biological evaluation of novel coumarin-chalcone derivatives containing urea moiety as potential anticancer agents. Arab. J. Chem. 2020, 13, 1120–1129. [Google Scholar] [CrossRef]
- Constantinescu, T.; Lungu, C.N.; Lung, I. Lipophilicity as a Central Component of Drug-like Properties of Chalcones and Flavonoid Derivatives. Molecules 2019, 24, 1505. [Google Scholar] [CrossRef]
- Zenger, K.; Dutta, S.; Wolff, H.; Genton, M.G.; Kraus, B. In vitro structure-toxicity relationship of chalcones in human hepatic stellate cells. Toxicology 2015, 336, 26–36. [Google Scholar] [CrossRef]
- Yadav, P.; Lal, K.; Kumar, L.; Kumar, A.; Kumar, A.; Paul, A.K.; Kumar, R. Synthesis, crystal structure and antimicrobial potential of some fluorinated chalcone-1,2,3-triazole conjugates. Eur. J. Med. Chem. 2018, 155, 263–274. [Google Scholar] [CrossRef]
- Mohamed, M.; Shaykoon, M.; Abdelrahman, M.; Aboraia, A.S.; Abuo-Rahma, G. Design, synthesis, docking studies and biological evaluation of novel chalcone derivatives as potential histone deacetylase inhibitors. Bioorg. Chem. 2017, 72, 32–41. [Google Scholar] [CrossRef]
- Ahmad, Z.; Kumar, S.; Rao, S.; Sharma, S.; Mahajan, G.; Behl, A.; Kumar, A.; Sharma, P.; Kamal, A.; Bhushan, S.; et al. A novel quinazolinone chalcone derivative induces mitochondrial dependent apoptosis and inhibits PI3K/Akt/mTOR signaling pathway in human colon cancer HCT-116 cells. Food Chem. Toxicol. 2016, 87, 1–11. [Google Scholar]
- Zhou, Y.; Zhong, B.; Min, X.; Hou, Y.; Lin, L.; Wu, Q.; Shi, J.; Chen, X. Therapeutic potential of isobavachalcone, a natural flavonoid, in murine experimental colitis by inhibiting NF-κB p65. Phytother. Res. 2021, 35, 5861–5870. [Google Scholar] [CrossRef]
- Sahoo, S.K.; Rani, B.; Gaikwad, N.B.; Ahmad, M.N.; Kaul, G.; Shukla, M.; Nanduri, S.; Dasgupta, A.; Chopra, S.; Yaddanapudi, V.M. Synthesis and structure-activity relationship of new chalcone linked 5-phenyl-3-isoxazolecarboxylic acid methyl esters potentially active against drug resistant Mycobacterium tuberculosis. Eur. J. Med. Chem. 2021, 222, 113580. [Google Scholar] [CrossRef]
- Wang, X.; Zhou, L.Y.; Tan, R.X.; Liang, G.P.; Fang, S.X.; Li, W.; Xie, M.; Wen, Y.H.; Wu, J.Q.; Chen, Y.P. Design, Synthesis, and Evaluation of Chalcone Derivatives as Multifunctional Agents against Alzheimer´s Disease. Chem. Biodivers. 2021, 18, e2100341. [Google Scholar] [CrossRef]
- Emam, S.; Sonousi, A.; Osman, E.; Hwang, D.; Kim, G.D.; Hassan, R. Design and synthesis of methoxyphenyl- and coumarin-based chalcone derivatives as anti-inflammatory agents by inhibition of NO production and down-regulation of NF-κB in LPS-induced RAW264.7 macrophage cells. Bioorg. Chem. 2021, 107, 104630. [Google Scholar] [CrossRef]
- Constantinescu, T. Natural and Synthetic Chalcones as Privileged Molecules with Antioxidant Activities. In Natural and Synthetic Chalcones as Privileged Molecules with Antioxidant Activities; BP International: London, UK, 2022; pp. 1–91. [Google Scholar] [CrossRef]
- Elkanzi, N.; Hrichi, H.; Alolayan, R.A.; Derafa, W.; Zahou, F.M.; Bakr, R.B. Synthesis of Chalcones Derivatives and Their Biological Activities: A Review. ACS Omega 2022, 7, 27769–27786. [Google Scholar] [CrossRef]
- Möller, G.; Temml, V.; Cala Peralta, A.; Gruet, O.; Richomme, P.; Séraphin, D.; Viault, G.; Kraus, L.; Huber-Cantonati, P.; Schopfhauser, E.; et al. Analogues of Natural Chalcones as Efficient Inhibitors of AKR1C3. Metabolites 2022, 12, 99. [Google Scholar] [CrossRef]
- Bukhari, S.N.A. Synthesis and evaluation of new chalcones and oximes as anticancer agents. RSC Adv. 2022, 12, 10307–10320. [Google Scholar] [CrossRef]
- Tan, Q.W.; He, L.Y.; Zhang, S.S.; He, Z.W.; Liu, W.H.; Zhang, L.; Guan, L.P.; Wang, S.H. Design, Synthesis, and Biological Activity of Chalcone Analogs Containing 4-Phenylquinolin and Benzohydrazide. Chem. Biodivers. 2022, 19, e202100610. [Google Scholar] [CrossRef]
- Hu, Y.; Hu, C.; Pan, G.; Yu, C.; Asari, M.F.; Yadav Bheemanaboina, R.; Cheng, Y.; Zhou, C.; Zhang, J. Novel chalcone-conjugated, multi-flexible end-group coumarin thiazole hybrids as potential antibacterial repressors against methicillin-resistant Staphylococcus aureus. Eur. J. Med. Chem. 2021, 222, 113628. [Google Scholar] [CrossRef]
- Arslan, T. Design, synthesis of novel peripherally tetra-chalcone substituted phthalocyanines and their inhibitory effects on acetylcholinesterase and carbonic anhydrases (hCA I and II). J. Organomet. Chem. 2021, 951, 122021. [Google Scholar] [CrossRef]
- Sharma, A.; Chakravarti, B.; Prasad, M.; Siddiqui, J.; Konwar, R.; Tripathi, R. Synthesis and anti breast cancer activity of biphenyl based chalcones. Bioorg. Med. Chem. 2010, 18, 4711–4720. [Google Scholar] [CrossRef]
- Thapa, P.; Upadhyay, S.; Suo, W.; Singh, V.; Gurung, P.; Lee, S.E.; Sharma, R.; Sharma, M. Chalcone and its analogs: Therapeutic and diagnostic applications in Alzheimer’s disease. Bioorg. Chem. 2021, 108, 104681. [Google Scholar] [CrossRef]
- Komoto, T.T.; Bernardes, M.; Mesquitam, T.B.; Bortolotto, L.; Silva, G.; Bitencourt, T.A.; Baek, S.J.; Marins, M.; Fachin, A.L. Chalcones Repressed the AURKA and MDR Proteins Involved in Metastasis and Multiple Drug Resistance in Breast Cancer Cell Lines. Molecules 2018, 23, 2018. [Google Scholar] [CrossRef]
- Mielcke, T.; Muradás, T.; Filippi-Chiela, E.; Amaral, M.E.; Kist, L.; Bogo, M.; Mascarello, A.; Neuenfeldt, P.; Nunes, R.; Campos, M. Mechanisms underlying the antiproliferative effects of a series of quinoxaline-derived chalcones. Sci. Rep. 2017, 7, 15850. [Google Scholar] [CrossRef]
- Nigam, S.; Pande Narayan, A.; Reddy, N.; Rao, V. Investigating the potential of tetrahydropyridinyl chalcones as useful agents against breast carcinoma: An in vitro and in vivo study. Res. Chem. Intermed. 2017, 44, 901–924. [Google Scholar] [CrossRef]
- Hawash, M.; Kahraman, D.C.; Eren, F.; Atalay, R.C.; Baytas, S.N. Synthesis and biological evaluation of novel pyrazolic chalcone derivatives as novel hepatocellular carcinoma therapeutics. Eur. J. Med. Chem. 2017, 129, 12–26. [Google Scholar] [CrossRef]
- Singh, N.; Chandra, R. Probing the binding interaction of ortho-vanillin derived chalcone with lysozyme: A biophysical studies aided by in silico calculations. J. Mol. Liq. 2020, 321, 114490. [Google Scholar] [CrossRef]
- Huang, X.; Huang, R.; Li, L.; Gou, S.; Wang, H. Synthesis and biological evaluation of novel chalcone derivatives as a new class of microtubule destabilizing agents. Eur. J. Med. Chem. 2017, 132, 11–25. [Google Scholar] [CrossRef]
- Harish, V.; Haque, E.; Śmiech, M.; Taniguchi, H.; Jamieson, S.; Tewari, D.; Bishayee, A. Xanthohumol for Human Malignancies: Chemistry, Pharmacokinetics and Molecular Targets. Int. J. Mol. Sci. 2021, 22, 4478. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Yin, H.; Qian, X.; Dong, J.; Qian, Z.; Miao, J. Xanthohumol, a Prenylated Chalcone from Hops, Inhibits the Viability and Stemness of Doxorubicin-Resistant MCF-7/ADR Cells. Molecules 2017, 22, 36. [Google Scholar] [CrossRef] [PubMed]
- Seitz, T.; Hackl, C.; Freese, K.; Dietrich, P.; Mahli, A.; Thasler, R.M.; Thasler, W.E.; Lang, S.A.; Bosserhoff, A.K.; Hellebrand, C. Xanthohumol, a Prenylated Chalcone Derived from Hops, Inhibits Growth and Metastasis of Melanoma Cells. Cancers 2021, 13, 511. [Google Scholar] [CrossRef] [PubMed]
- Huo, P.C.; Hu, Q.; Shu, S.; Zhou, Q.H.; He, R.J.; Hou, J.; Guan, X.Q.; Tu, D.Z.; Hou, X.D.; Liu, P.; et al. Design, synthesis and biological evaluation of novel chalcone-like compounds as potent and reversible pancreatic lipase inhibitors. Bioorg. Med. Chem. 2021, 29, 115853. [Google Scholar] [CrossRef] [PubMed]
- Wijawanti, L.W.; Swasono, R.T.; Le, W.; Jumina, J. Synthesis and Evaluation of Chalcone Derivatives as Novel Sunscreen Agent. Molecules 2021, 26, 2698. [Google Scholar] [CrossRef] [PubMed]
- Jioty; Gaur, R.; Kumar, Y.; Cheema, H.S.; Kapkoti, S.D.; Darokar, M.; Khan, F.; Bhakuni, R.S. Synthesis, molecular modelling studies of indolyl chalcone derivatives and their antimalarial activity evaluation. Nat. Prod. Res. 2021, 35, 3261–3268. [Google Scholar]
- Hu, S.; Zhao, T.; Xu, K.; Ji, S.; Cao, L.; Teng, B. Growth and characterization of a chalcone derivative DMMC with strong SHG efficiency for NLO applications. Optik 2021, 231, 166410. [Google Scholar] [CrossRef]
- Vlasiou, M.; Al Hatahta, A. Spectroscopic evaluation of chalcone derivatives and their zinc metal complexes: A combined experimental and computational approach studying the interactions of the complexes with the serum albumin. J. Mol. Struct. 2021, 1232, 130052. [Google Scholar] [CrossRef]
- Rai, P.; Chettri, P.; Kar, S.; Nagar, M.A.; Srisastava, S.; Golakoti, N.R. Synthesis, characterization and structure–activity relationship of non-linear optical response of chalcone derivatives with in silico insights. Chem. Pap. 2021, 75, 2603–2615. [Google Scholar] [CrossRef]
- Urbonavičius, A.; Fortunato, G.; Ambrazaitytė, E.; Plytninkienė, E.; Bieliauskas, A.; Milišiūnaitė, V.; Luisi, R.; Arbačiauskienė, E.; Krikštolaitytė, S.; Šačkus, A. Synthesis and Characterization of Novel Heterocyclic Chalcones from 1-Phenyl-1H-pyrazol-3-ol. Molecules 2022, 27, 3752. [Google Scholar] [CrossRef]
- Bala, D.; Jinga, L.I.; Popa, M.; Hanganu, A.; Voicescu, M.; Bleotu, C.; Tarko, L.; Nica, S. Design, Synthesis, and Biological Evaluation of New Azulene-Containing Chalcones. Materials 2022, 15, 1629. [Google Scholar] [CrossRef]
- Jain, S.; Kumar, S.; Lamba, B.Y.; Patra, J.; Mahindroo, N. Nanocatalysts: Applications in synthesis of chalcones—A review. Synth. Commun. 2020, 51, 1–12. [Google Scholar] [CrossRef]
- Yan, J.; Chen, J.; Zhang, S.; Hu, J.; Huang, L.; Li, X. Synthesis, Evaluation, and Mechanism Study of Novel Indole-Chalcone Derivatives Exerting Effective Antitumor Activity Through Microtubule Destabilization In Vitro and In Vivo. J. Med. Chem. 2016, 59, 5264–5283. [Google Scholar] [CrossRef]
- Stanojković, T.; Marković, V.; Matić, Z.; Mladenović, M.; Petrović, N.; Krivokuća, A.; Petrović, M.; Joksović, M. Highly selective anthraquinone-chalcone hybrids as potential antileukemia agents. Bioorg. Med. Chem. Lett. 2018, 28, 2593–2598. [Google Scholar] [CrossRef]
- Zhou, B.; Xing, C. Diverse Molecular Targets for Chalcones with Varied Bioactivities. Med. Chem. 2015, 5, 388. [Google Scholar] [CrossRef]
- Desai, V.; Desai, S.; Naik, S.; Palyekar, U.; Joshi, S.; Dixit, S. Novel quinoxalinyl chalcone hybrid scaffolds as enoyl ACP reductase inhibitors: Synthesis, molecular docking and biological evaluation. Bioorg. Med. Chem. Lett. 2017, 27, 2174–2180. [Google Scholar] [CrossRef]
- Liu, W.; He, M.; Li, Y.; Peng, Z.; Wang, G. A review on synthetic chalcone derivatives as tubulin polymerisation inhibitors. J. Enzyme Inhib. Med. Chem. 2022, 37, 9–38. [Google Scholar] [CrossRef]
- Basseville, A.; Hall, M.D.; Chau, C.H.; Robey, R.W.; Gottesman, M.; Figg, W.D.; Bates, S. The ABCG2 Multidrug Transporter. In ABC Transporters; Springer: Cham, Switzerland, 2016; pp. 195–226. [Google Scholar]
- Cleophas, M.C.; Joosten, L.A.; Stamp, L.K.; Dalberth, N.; Woodward, O.M.; Merriman, T.R. ABCG2 polymorphisms in gout: Insights into disease susceptibility and treatment approaches. Pharmacogenet. Pers. Med. 2017, 10, 129–142. [Google Scholar] [CrossRef]
- Wu, C.P.; Li, Y.Q.; Hung, T.H.; Chang, Y.T.; Huang, Y.H.; Wu, Y.S. Sophora flavanone G Resensitizes ABCG2-Overexpressing Multidrug-Resistant Non-Small-Cell Lung Cancer Cells to Chemotherapeutic Drugs. J. Nat. Prod. 2021, 84, 2544–2553. [Google Scholar] [CrossRef]
- Eckenstaler, R.; Benndorf, R. The Role of ABCG2 in the Pathogenesis of Primary Hyperuricemia and Gout—An Update. Int. J. Mol. Sci. 2021, 22, 6678. [Google Scholar] [CrossRef]
- El Biali, M.; Karch, R.; Philippe, C.; Haslacher, H.; Tournier, N.; Hacker, M.; Zeitlinger, M.; Schmidl, D.; Langer, O.; Bauer, M. ABCB1 and ABCG2 Together Limit the Distribution of ABCB1/ABCG2 Substrates to the Human Retina and the ABCG2 Single Nucleotide Polymorphism Q141K (c.421C>A) May Lead to Increased Drug Exposure. Front. Pharmacol. 2021, 12, 698966. [Google Scholar] [CrossRef]
- Kowal, J.; Ni, D.; Jackson, S.; Manolaridis, I.; Stahlberg, H.; Locher, K. Structural Basis of Drug Recognition by the Multidrug Transporter ABCG2. J. Mol. Biol. 2021, 433, 166980. [Google Scholar] [CrossRef]
- Liu, L.; Zhao, T.; Shan, L.; Cao, L.; Zhu, X.; Xue, Y. Estradiol regulates intestinal ABCG2 to promote urate excretion via the PI3K/Akt pathway. Nutr. Metab. 2021, 18, 63. [Google Scholar] [CrossRef]
- Blanco-Paniagua, E.; García-Lino, A.; García-Mateos, D.; Alvarez, A.; Merino, G. Role of the ABCG2 transporter in plasma levels and tissue accumulation of the anti-inflammatory tolfenamic acid in mice. Chem. Biol. Interact. 2021, 345, 109537. [Google Scholar] [CrossRef]
- Yang, H.J.; Liu, M.; Kim, M.J.; Park, S. The haplotype of SLC2A9_rs3733591, the hyperuricaemia risk and alcohol, chicken and processed meat intakes and smoking interact with its risk. Int. J. Food Sci. Nutr. 2021, 72, 391–401. [Google Scholar] [CrossRef]
- Ibrahim, M.; Badr, E.; Abdelrahman, A.; Almansour, N.M.; Shawky, A.; Mekhemer, G.; Alrumaihi, F.; Moustafa, M.; Atia, M. Prospective Drug Candidates as Human Multidrug Transporter ABCG2 Inhibitors: An In Silico Drug Discovery Study. Cell Biochem. Biophys. 2021, 79, 189–200. [Google Scholar] [CrossRef]
- Takei, R.; Cadzow, M.; Markie, D.; Bixley, M.; Phipps-Green, A.; Major, T.; Li, C.; Choi, H.; Li, Z.; Hu, H.; et al. Trans-ancestral dissection of urate- and gout-associated major loci SLC2A9 and ABCG2 reveals primate-specific regulatory effects. J. Hum. Gen. 2021, 66, 225. [Google Scholar] [CrossRef]
- Garcia-Lino, A.; Gomez-Gomez, A.; Garcia-Mateos, D.; de la Fuente, A.; Alvarez, A.; Pozo, O.; Merino, G. Analysis of the interaction between tryptophan-related compounds and ATP-binding cassette transporter G2 (ABCG2) using targeted metabolomics. Food Chem. 2021, 344, 128665. [Google Scholar] [CrossRef]
- Xu, H.; Liu, T.; Li, W.; Yao, Q. SMAR1 attenuates the stemness of osteosarcoma cells via through suppressing ABCG2 transcriptional activity. Environ. Toxicol. 2021, 36, 1090–1098. [Google Scholar] [CrossRef]
- Mózner, O.; Bartos, Z.; Zámbó, B.; Homolya, L.; Hegedűs, T.; Sarkadi, B. Cellular Processing of the ABCG2 Transporter—Potential Effects on Gout and Drug Metabolism. Cells 2019, 8, 1215. [Google Scholar] [CrossRef]
- Jackson, S.M.; Manolaridis, I.; Kowal, J.; Zechner, M.; Taylor, N.; Bause, M.; Bauer, S.; Bartholomaeurs, R.; Bernhardt, G.; Koenig, B.; et al. Structural basis of small-molecule inhibition of human multidrug transporter ABCG2. Nat. Struct. Mol. Biol. 2018, 25, 333–340. [Google Scholar] [CrossRef]
- Xu, L.; Huang, T.; Hu, H.; Wang, M.; Shi, S.; Yang, Q.; Lin, F.; Qiang, Y.Y.; Mei, Y.; Lang, Y.H.; et al. The development transcription factor IRF6 attenuates ABCG2 gene expression and distinctively reverses stemness phenotype in nasoparyngeal carcinoma. Cancer Lett. 2018, 431, 230–243. [Google Scholar] [CrossRef]
- Wang, Y.; Sparidans, R.; Potters, S.; Lebre, M.; Beijnen, J.; Schinkel, A. ABCB1 and ABCG2, but not CYP3A4 limit oral availability and brain accumulation of the RET inhibitor pralsetinib. Pharmacol. Res. 2021, 172, 105850. [Google Scholar] [CrossRef] [PubMed]
- Toyoda, Y.; Pavecová, K.; Bohatá, J.; Ješina, P.; Kubota, Y.; Suzuki, H.; Takada, T.; Stiburkova, B. Identification of Two Dysfunctional Variants in the ABCG2 Urate Transporter Associated with Pediatric-Onset of Familial Hyperuricemia and Early-Onset Gout. Int. J. Mol. Sci. 2021, 22, 1935. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.C.; Park, Y.H.; Won, J.H.; Kim, M.; Kwon, B.; Lee, S.J.; Kim, D.H.; Shin, J.G.; Seo, S.K. Repression of PPAR-γ reduces the ABCG2-mediated efflux activity of M2 macrophages. Int. J. Biochem. Cell Biol. 2021, 130, 105895. [Google Scholar] [CrossRef] [PubMed]
- Pavlič, R.; Vidic, S.; Anko, M.; Knific, T.; Büdefeld, T.; Marton, K.; Sinreih, M.; Poschner, S.; Jäger, W.; Frković-Grazio, S.; et al. Altered Profile of E1-S Transporters in Endometrial Cancer: Lower Protein Levels of ABCG2 and OSTβ and Up-Regulation of SLCO1B3 Expression. Int. J. Mol. Sci. 2021, 22, 3819. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.P.; Hung, C.Y.; Lusvarghi, S.; Chang, Y.F.; Hsiao, S.H.; Hunag, Y.H.; Hunag, T.H.; Yu, J.S.; Ambudkar, S. Overexpression of Human ABCB1 and ABCG2 Reduces the Susceptibility of Cancer Cells to the Histone Deacetylase 6-Specific Inhibitor Citarinostat. Int. J. Mol. Sci. 2021, 22, 2592. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Wu, Z.X.; Wang, J.Q.; Teng, Q.X.; Lei, Z.N.; Lusvarghi, S.; Ambudkar, S.; Chen, Z.S.; Yang, D.H. OTS964, a TOPK Inhibitor, Is Susceptible to ABCG2-Mediated Drug Resistance. Front. Pharmacol. 2021, 12, 620874. [Google Scholar] [CrossRef] [PubMed]
- Taylor, N.; Manolaridis, I.; Jackson, S.; Kowal, J.; Stahlberg, H.; Kaspar, P. Structure of the human multidrug transporter ABCG2. Nature 2017, 546, 504–509. [Google Scholar] [CrossRef]
- Bartos, Z.; Homolya, L. Identification of Specific Trafficking Defects of Naturally Occurring Variants of the Human ABCG2 Transporter. Front. Cell Dev. Biol. 2021, 9, 615729. [Google Scholar] [CrossRef]
- Liu, C.; Xing, W.; Yu, H.; Zhang, W.; Si, T. ABCB1 and ABCG2 restricts the efficacy of gedatolisib (PF-05212384), a PI3K inhibitor in colorectal cancer cells. Cancer Cell Int. 2021, 21, 108. [Google Scholar] [CrossRef]
- Homoloya, L. Medically Important Alterations in Transport Function and Trafficking of ABCG2. Int. J. Mol. Sci. 2021, 22, 2786. [Google Scholar] [CrossRef]
- Nagy, T.; Tóth, Á.; Telbisz, Á.; Sarkadi, B.; Tordai, H.; Tordai, A. The transport pathway in the ABCG2 protein and its regulation revealed by molecular dynamics simulations. Cell. Mol. Life Sci. 2021, 78, 2329–2339. [Google Scholar] [CrossRef]
- Peña-Solórzano, D.; Stark, S.A.; König, B.; Sierra, C.A.; Ochoa-Puentes, C. ABCG2/BCRP: Specific and Nonspecific Modulators. Med. Res. Rev. 2016, 37, 987–1050. [Google Scholar] [CrossRef]
- Toyoda, Y.; Takada, T.; Suzuki, H. Inhibitors of Human ABCG2: From Technical Background to Recent Updates with Clinical Implications. Front. Pharmacol. 2019, 10, 208. [Google Scholar] [CrossRef]
- Yin, W.; Xiang, D.; Wang, T.; Zhang, Y.; Pham, C.; Zhou, S.; Jiang, G.; Hou, Y.; Zhu, Y.; Han, Y.; et al. The inhibition of ABCB1/MDR1 or ABCG2/BCRP enables doxorubicin to eliminate liver cancer stem cells. Sci. Rep. 2021, 11, 10791. [Google Scholar] [CrossRef]
- Loscocco, F.; Visani, G.; Ruzzo, A.; Bagaloni, I.; Fuligni, F.; Galimberti, S.; Di Paolo, A.; Stagno, F.; Pregno, P.; Annuziata, M.; et al. Clinical Relevance of ABCB1, Polymorphisms in Chronic Myeloid Leukemia Patients Treated with Nilotinib. Front. Oncol. 2021, 11, 672287. [Google Scholar] [CrossRef]
- Buks, R.; Brusson, M.; Cochet, S.; Galochkina, T.; Cassinat, B.; Nemazanyy, I.; Peyrard, T.; Kiladjian, J.J.; de Brevern, A.G.; Azouzi, S.; et al. ABCG2 Is Overexpressed on Red Blood Cells in Ph-Negative Myeloproliferative Neoplasms and Potentiates Ruxolitinib-Induced Apoptosis. Int. J. Mol. Sci. 2021, 22, 3530. [Google Scholar] [CrossRef]
- Turiján-Espinoza, E.; Ruíz-Rodríguez, V.M.; Uresti-Rivera, E.E.; Martínez-Leija, E.; Zermeño-Nava, J.; Guel-Pañola, A.; Romano-Moreno, S.; Vargas-Morales, J.M.; Portales-Pérez, D.P. Clinical utility of ABCB1 and ABCG2 genotyping for assessing the clinical and pathological response to FAC therapy in Mexican breast cancer patients. Cancer Chemoth. Pharmacol. 2021, 87, 843–853. [Google Scholar] [CrossRef]
- Antoni, F.; Wifling, D.; Bernhardt, G.J. Water-Soluble Inhibitors of ABCG2 (BCRP)-A Fragment-Based and Computational Approach. Eur. J. Med. Chem. 2020, 210, 112958. [Google Scholar] [CrossRef]
- Wu, C.P.; Lusvarghi, S.; Hsiao, S.H.; Liu, T.C.; Li, Y.Q.; Huang, Y.H.; Hung, T.; Ambudkar, S. Licochalcone A Selectively Resensitizes ABCG2-Overexpressing Multidrug-Resistant Cancer Cells to Chemotherapeutic Drugs. J. Nat. Prod. 2020, 83, 1461–1472. [Google Scholar] [CrossRef]
- Michalkova, R.; Mirossay, L.; Gazdova, M.; Kello, M.; Mojzis, J. Molecular Mechanisms of Antiproliferative Effects of Natural Chalcones. Cancers 2021, 13, 2730. [Google Scholar] [CrossRef]
- Fan, X.; Bai, J.; Zhao, S.; Hu, M.; Sun, Y.; Wang, B.; Ji, M.; Jin, J.; Wang, X.; Hu, J.; et al. Evaluation of inhibitory effects of flavonoids on breast cancer resistance protein (BCRP): From library screening to biological evaluation to structure-activity relationship. Toxicol. Vitro 2019, 61, 104642. [Google Scholar] [CrossRef]
- Valdameri, G.; Gauthier, C.; Terreux, R.; Day, B.; Winnischofer, S.; Rocha, M.; Frachet, V.; Ronot, X.; Di Pietro, A.; Boumendjel, A. Investigation of Chalcones as Selective Inhibitors of the Breast Cancer Resistance Protein: Critical Role of Methoxylation in both Inhibition Potency and Cytotoxicity. J. Med. Chem. 2012, 55, 3193–3200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmitt, F.; Draut, H.; Biersack, B.; Schobert, R. Halogenated naphthochalcones and structurally related naphthopyrazolines with antitumor activity. Bioorg. Med. Chem. Lett. 2016, 26, 5168–5171. [Google Scholar] [CrossRef]
- Krapf, M.; Gallus, J.; Wiese, M. Synthesis and biological investigation of 2,4-substituted quinazolines as highly potent inhibitors of breast cancer resistance protein (ABCG2). Eur. J. Med. Chem. 2017, 139, 587–611. [Google Scholar] [CrossRef]
- Kraege, S.; Stefan, K.; Juvale, K.; Ross, T.; Willmes, T.; Wiese, M. The combination of quinazoline and chalcone moieties leads to novel potent heterodimeric modulators of Breast Cancer Resistance Protein (BCRP/ABCG2). Eur. J. Med. Chem. 2016, 117, 212–229. [Google Scholar] [CrossRef]
- Winter, E.; Gozzi, G.J.; Daflon-Yunes, N.; Terreux, R.; Gauthier, C.; Mascarello, A.; Leal, P.C.; Cadena, S.; Yunes, R.A.; Nunes, R.J.; et al. Quinoxaline-substituted chalcones as new inhibitors of breast cancer resistance protein ABCG2: Polyspecificity at B-ring position. Drug Des. Dev. Ther. 2014, 8, 609–619. [Google Scholar]
- Kraege, S.; Köhler, S.; Wiese, M. Acryloylphenylcarboxamides: A New Class of Breast Cancer Resistance Protein (ABCG2) Modulators. Chem. Med. Chem. 2016, 11, 2422–2435. [Google Scholar] [CrossRef]
- Solórzano, D.P.; Scholler, M.; Bernhardt, G.; Buschauer, A.; König, B.; Puentes, C.O. Tariquidar-related chalcones and ketones as ABCG2 modulators. ACS Med. Chem. Lett. 2018, 9, 854–859. [Google Scholar] [CrossRef]
- Sultana, F.; Reddy Bonam, S.; Reddy, V.G.; Nayak, V.L.; Akunuri, R.; Routhu Rani, S.; Alarifi, A.; Halmuthur, K.S.; Kamal, A. Synthesis of benzo[d]imidazo[2,1-b]thiazole-chalcone conjugates as microtubule targeting and apoptosis inducing agents. Bioorg. Chem. 2018, 76, 1–12. [Google Scholar] [CrossRef]
- Wang, G.; Peng, Z.; Zhang, J.; Qiu, J.; Xie, Z.; Gong, Z. Synthesis, biological evaluation and molecular docking studies of aminochalcone derivatives as potential anticancer agents by targeting tubulin colchicine binding site. Bioorg. Chem. 2018, 78, 332–340. [Google Scholar] [CrossRef]
- Li, L.; Quan, D.; Chen, J.; Ding, J.; Zhao, J.; Lv, L.; Chen, J. Design, synthesis, and biological evaluation of 1-substitute-2-aryl imidazoles targeting tubulin polymerization as potential anticancer agents. Eur. J. Med. Chem. 2019, 184, 111732. [Google Scholar] [CrossRef] [PubMed]
- Fu, D.; Fu, L.; Liu, Y.; Wang, J.; Wang, Y.; Han, B.; Li, X.; Zhang, C.; Li, F.; Song, J.; et al. Structure-Activity Relationship Studies of β-Lactam-azide Analogues as Orally Active Antitumor Agents Targeting the Tubulin Colchicine Site. Sci. Rep. 2017, 7, 12788. [Google Scholar] [CrossRef] [PubMed]
- Mirzaei, H.; Shokrzadeh, M.; Modanloo, M.; Ziar, A.; Hossein, G.; Emami, S. New indole-based chalconoids as tubulin-targeting antiproliferative agents. Bioorg. Chem. 2017, 75, 86–98. [Google Scholar] [CrossRef]
- Hagita, A.; Wada-Kakuda, S.; Nobuhara, M.; Kakuda, N.; Miyasaka, T. Biochemical and Biophysical Research Communications Quantitative fractionation of tissue microtubules with distinct biochemical properties reflecting their stability and lability. Biochem. Biophys. Res. Commun. 2021, 560, 186–191. [Google Scholar] [CrossRef]
- Minagawa, M.; Shirato, M.; Toya, M.; Sato, M. Dual Impact of a Benzimidazole Resistant β-Tubulin on Microtubule Behavior in Fission Yeast. Cells 2021, 10, 1042. [Google Scholar] [CrossRef]
- Alswah, M.; Bayoumi, A.; Elgamal, K.; Elmorsy, A.; Ihmaid, S.; Ahmed, H. Design, Synthesis and Cytotoxic Evaluation of Novel Chalcone Derivatives Bearing Thiazole [4,3-a]-quinoxaline Moieties as Potent Anticancer Agents with Dual EGFR Kinase and Tubulin Polymerization. Molecules 2018, 23, 48. [Google Scholar] [CrossRef]
- Parker, A.; Teo, W.S.; Mc Carroll, J.; Kavallaris, M. An Emerging Role for Tubulin Isotypes in Modulating Cancer Biology and Chemotherapy Resistance. Int. J. Mol. Sci. 2017, 18, 1434. [Google Scholar] [CrossRef]
- Fong, A.; Durkin, A.; Lee, H. The Potential of Combining Tubulin-Targeting Anticancer Therapeutics and Immune Therapy. Int. J. Mol. Sci. 2019, 20, 586. [Google Scholar] [CrossRef]
- Chen, J.; Kholina, E.; Szyk, A.; Fedorov, V.; Kovalenko, I.; Gudimchuk, N.; Roll Mecak, A. α-tubulin tail modifications regulate microtubule stability through selective effector recruitment, not changes in intrinsic polymer dynamics. Dev. Cell 2021, 56, 2016–2028. [Google Scholar] [CrossRef]
- Böhler, A.; Vermeulen, B.; Würtz, M.; Zupa, E.; Pfeffer, S.; Schiebel, E. The gamma-tubulin ring complex: Deciphering the molecular organization and assembly mechanism of a major vertebrate microtubule nucleator. Probl. Paradig. 2021, 43, 2100114. [Google Scholar] [CrossRef]
- Morishita, J.; Nurse, P. Identification of novel microtubule inhibitors effective in fission yeast and human cells and their effects on breast cancer cell lines. R. Soc. Publ. 2021, 11, 210161. [Google Scholar] [CrossRef]
- Yoshimoto, S.; Morita, H.; Okamura, K.; Hiraki, A.; Hashimoto, S. αTAT1-induced tubulin acetylation promotes ameloblastoma migration and invasion. Lab. Investig. 2021, 102, 80–89. [Google Scholar] [CrossRef]
- Ohi, R.; Strothman, C.; Zanic, M. Impact of the ‘tubulin economy’ on the formation and function of the microtubule cytoskeleton. Curr. Opin. Cell Biol. 2021, 68, 81–89. [Google Scholar] [CrossRef]
- Oh, S.; You, E.; Ko, P.; Jeong, J.; Keum, S.; Rhee, S. Communications Genetic disruption of tubulin acetyltransferase, a TAT1, inhibits proliferation and invasion of colon cancer cells through decreases in Wnt1/β-catenin signaling. Biochem. Biophy. Res. Commun. 2017, 482, 8–14. [Google Scholar] [CrossRef]
- Bonandi, E.; Foschi, F.; Marucci, C.; Dapiaggi, F.; Sironi, M.; Pieraccini, S.; Chistodoulou, M.; Balauger, F.; Diaz, F.; Zidar, N.; et al. Synthesis of Thicolchicine-Based Conjugates: Investigation towards Bivalent Tubulin/Microtubules Binders. ChemPlusChem 2019, 84, 98–102. [Google Scholar] [CrossRef]
- Hura, N.; Naaz, A.; Prassanawar, S.S.; Guchhait, S.K.; Panda, D. Drug-Clinical Agent Molecular Hybrid: Synthesis of Diaryl(trifluoromethyl)pyrazoles as Tubulin Targeting Anticancer Agents. ABS Omega 2018, 3, 1955–1969. [Google Scholar] [CrossRef]
- Sun, W.X.; Ji, Y.J.; Wan, Y.; Han, H.W.; Lin, H.Y.; Lu, G.H.; Qi, J.L.; Wang, X.M.; Yang, Y.H. Design and synthesis of piperazine acetate podophyllotoxin ester derivatives targeting tubulin depolymerization as new anticancer agents. Bioorg. Med. Chem. Lett. 2017, 27, 4066–4074. [Google Scholar] [CrossRef]
- Wang, Z.Z.; Sun, W.X.; Wang, X.; Zhang, Y.H.; Qiu, H.Y.; Qi, J.L.; Pan, Y.J.; Lu, G.H.; Wang, X.M.; Yu, F.G.; et al. Design, synthesis, biological evaluation, and 3D-QSAR analysis of podophyllotoxin–dioxazole combination as tubulin targeting anticancer agents. Chem. Biol. Drug Des. 2017, 90, 236–243. [Google Scholar] [CrossRef]
- López-López, E.; Cerda-García-Rojas, C.; Medina-Franco, J. Tubulin Inhibitors: A Chemoinformatic Analysis Using Cell-Based Data. Molecules 2021, 26, 2483. [Google Scholar] [CrossRef]
- Khan, I.; Gaikapati, K.R.; Saik, A.B.; Makani, V.K.K.; Rahim, A.; Shareef, M.A.; Reddy, G.; Pal-Bhadra, M.; Kamal, A.; Kumar, G. Design, synthesis and biological evaluation of 1,4-dihydro indeno[1,2-c]pyrazole linked oxindole analogues as potential anticancer agents targeting tubulin and inducing p53 dependent apoptosis. Eur. J. Med. Chem. 2018, 144, 104–115. [Google Scholar] [CrossRef]
- La Regina, G.; Coluccia, A.; Naccarato, V.; Silvestri, R. Towards modern anticancer agents that interact with tubulin. Eur. J. Pharm. Sci. 2019, 131, 58–68. [Google Scholar] [CrossRef] [PubMed]
- Sakchaisri, K.; Kim, S.; Hwang, J.; Soung, N.K.; Lee, H.; Choi, T.W.; Lee, Y.; Shwetha, B.; Srinivasrao, G.; Pham, T.; et al. Anticancer activity of a novel small molecule tubulin inhibitor STK899704. PLoS ONE 2017, 12, e0173311. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.N.; Wang, J.J.; Ji, Y.T.; Zhao, G.D.; Tang, L.Q.; Zhang, C.M.; Guo, X.L.; Liu, Z.P. Design, Synthesis and Biological Evaluation of 1-Methyl-1,4-dihydroindeno [1,2-c]pyrazole Analogues as Potential Anticancer Agents Targeting Tubulin Colchicine Binding Site. J. Med. Chem. 2016, 59, 5341–5355. [Google Scholar] [CrossRef] [PubMed]
- Erem, E.; Watts, N.; Sackett, D.; Wingfield, P. Conformational changes in tubulin upon binding cryptophycin-52 reveal its mechanism of action. J. Biol. Chem. 2021, 297, 101138. [Google Scholar] [CrossRef]
- Majcher, U.; Klejborowska, G.; Kaik, M.; Maj, E.; Wietrzyk, J.; Moshari, M.; Preto, J.; Tuszynski, J.; Hunczyński, A. Synthesis and Biological Evaluation of Novel Triple-Modified Colchicine Derivatives as Potent Tubulin-Targeting Anticancer Agents. Cell 2018, 7, 216. [Google Scholar] [CrossRef]
- Bai, L.F.; Qian, H.H.; Jiang, D.W.; Wang, X.M.; Wang, R.L. Design, Synthesis and Anti-tubulin Activity of Novel Dinitro Diphenyl Ether Derivatives as Potent Anticancer Agent. Med. Chem. 2017, 7, 393–397. [Google Scholar]
- Gaspari, R.; Prota, A.; Bargsten, K.; Andrea, E.; Bargsten, K.; Cavalli, A.; Steinmetz, M. Structural Basis of cis- and trans-Combretastatin Binding to Tubulin. Chem 2017, 2, 102–113. [Google Scholar] [CrossRef]
- Li, W.; Shuai, W.; Xu, F.; Sun, H.; Xu, S.; Yao, H.; Liu, J.; Yao, H.; Zhu, Z.; Xu, Y. Discovery of Novel 4-Arylisochromenes as Anticancer Agents Inhibiting Tubulin Polymerization. ACS Med. Chem. Lett. 2018, 9, 974–979. [Google Scholar]
- Binarova, P.; Tuszynski, J. Tubulin: Structure, Functions and Roles in Disease. Cells 2019, 8, 1294. [Google Scholar]
- McLoughlin, E.; O’Boyle, N. Colchicine-Binding Site Inhibitors from Chemistry to Clinic: A Review. Pharmaceuticals 2020, 13, 8. [Google Scholar] [CrossRef]
- Malik, H.S.; Bilal, A.; Ullah, R.; Iqbal, M.; Khan, S.; Ahmed, I.; Krohn, K.; Saleem, R.S.Z.; Hussain, H.; Faisal, A. Natural and Semisynthetic Chalcones as Dual FLT3 and Microtubule Polymerization Inhibitors. J. Nat. Prod. 2020, 83, 3111–3121. [Google Scholar] [CrossRef]
- Wang, G.; Liu, W.; Gong, Z.; Huang, Y.; Li, Y.; Peng, Z. Synthesis, biological evaluation, and molecular modelling of new naphthalene-chalcone derivatives as potential anticancer agents on MCF-7 breast cancer cells by targeting tubulin colchicine binding site. J. Enzyme Inhib. Med. Chem. 2019, 35, 139–144. [Google Scholar] [CrossRef]
- Wang, G.; Liu, W.; Gong, Z.; Huang, Y.; Li, Y.; Peng, Z. Design, synthesis, biological evaluation and molecular docking studies of new chalcone derivatives containing diaryl ether moiety as potential anticancer agents and tubulin polymerization inhibitors. Bioorg. Chem. 2020, 95, 103565. [Google Scholar] [CrossRef]
- Du, S.; Sarver, J.; Trabbic, C.; Erhardt, P.; Schroering, A.; Maltese, W. 6-MOMIPP, a novel brain-penetrant anti-mitotic indolyl-chalcone, inhibits glioblastoma growth and viability. Cancer Chemother. Pharmacol. 2018, 83, 237–254. [Google Scholar] [CrossRef]
- Yan, J.; Xu, Y.; Jin, X.; Zhang, Q.; Ouyang, F.; Han, L.; Zhan, M.; Li, X.; Liang, B.; Huang, X. Structure modification and biological evaluation of indole-chalcone derivatives as anti-tumor agents through dual targeting tubulin and TrxR. Eur. J. Med. Chem. 2022, 227, 113897. [Google Scholar] [CrossRef]
- Canela, M.; Noppen, S.; Bueno, O.; Prota, A.; Sáez-Calvo, G.; Jimeno, M.; Benkheil, M.; Ribatti, D.; Valázquez, S.; Camarasa, M.J.; et al. Antivascular and antitumor properties of the tubulin-binding chalcone TUB091. Oncotarget 2017, 8, 14325–14342. [Google Scholar] [CrossRef] [Green Version]
- Lindamulage, K.; Vu, H.Y.; Karthikeyan, C.; Knockleby, J.; Lee, Y.F.; Trivedi, P.; Lee, H. Novel quinolone chalcones targeting colchicine-binding pocket kill multidrug-resistant cancer cells by inhibiting tubulin activity and MRP1 function. Sci. Rep. 2017, 7, 10298. [Google Scholar] [CrossRef]
- Shankaraiah, N.; Nekkanti, S.; Rani, U.; Praveen, N.; Deshpande, N.; Prasanna, D.; Senwar, K.R.; Lankshmi, U.J. Synthesis of different heterocycles-linked chalcone conjugates as cytotoxic agents and tubulin polymerization inhibitors. Bioorg. Med. Chem. 2017, 25, 4805–4816. [Google Scholar] [CrossRef]
- Qiu, H.; Wang, F.; Wang, X.; Sun, W.; Qi, J.; Pang, Y.; Yang, R.; Lu, G.; Wang, X.; Yang, Y. Design, Synthesis, and Biological Evaluation of Chalcone-Containing Shikonin Derivatives as Inhibitors of Tubulin Polymerization. Chem. Med. Chem. 2017, 12, 399–406. [Google Scholar] [CrossRef]
- Yan, W.; Xiangyu, C.; Ya, L.; Yu, W.; Feng, X. An orally antitumor chalcone hybrid inhibited HepG2 cells growth and migration as the tubulin binding agent. Investig. New Drugs 2019, 37, 784–790. [Google Scholar] [CrossRef]
Figure 1.
Structure of Licochalcone A.

Figure 2.
Structure of Licochalcone B.

Figure 3.
General structure of chalcones 14–47.

Figure 4.
General structure of naphthochalcones (Compounds 58–61).

Figure 5.
General structure of quinazoline chalcones (Compounds 66–71).

Figure 6.
General structure of tariquidar-chalcones (Compounds 132–137).

Figure 7.
Structure of a semisynthetic chalcone (Compound 138).

Figure 8.
General structure of naphthalenchalcones (Compounds 142–161).

Figure 9.
General structure of chalcones 162–177.

Figure 10.
General structure of chalcones 179–187.

Figure 11.
General structure of quinoline chalcones (Compounds 190–214).

Figure 12.
General structure of shikonium-derived chalcones (Compounds 215–232).

Figure 13.
The two mechanisms and most active compounds.

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Constantinescu, T.; Mihis, A.G. Two Important Anticancer Mechanisms of Natural and Synthetic Chalcones. Int. J. Mol. Sci. 2022, 23, 11595. https://doi.org/10.3390/ijms231911595
AMA Style
Constantinescu T, Mihis AG. Two Important Anticancer Mechanisms of Natural and Synthetic Chalcones. International Journal of Molecular Sciences. 2022; 23(19):11595. https://doi.org/10.3390/ijms231911595
Chicago/Turabian StyleConstantinescu, Teodora, and Alin Grig Mihis. 2022. "Two Important Anticancer Mechanisms of Natural and Synthetic Chalcones" International Journal of Molecular Sciences 23, no. 19: 11595. https://doi.org/10.3390/ijms231911595
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.