
Partial Reduction in BRCA1 Gene Dose Modulates DNA Replication Stress Level and Thereby Contributes to Sensitivity or Resistance
 , , ,
, , ,  , ,
, ,  , and
, and
Abstract
:1. Introduction
2. Results
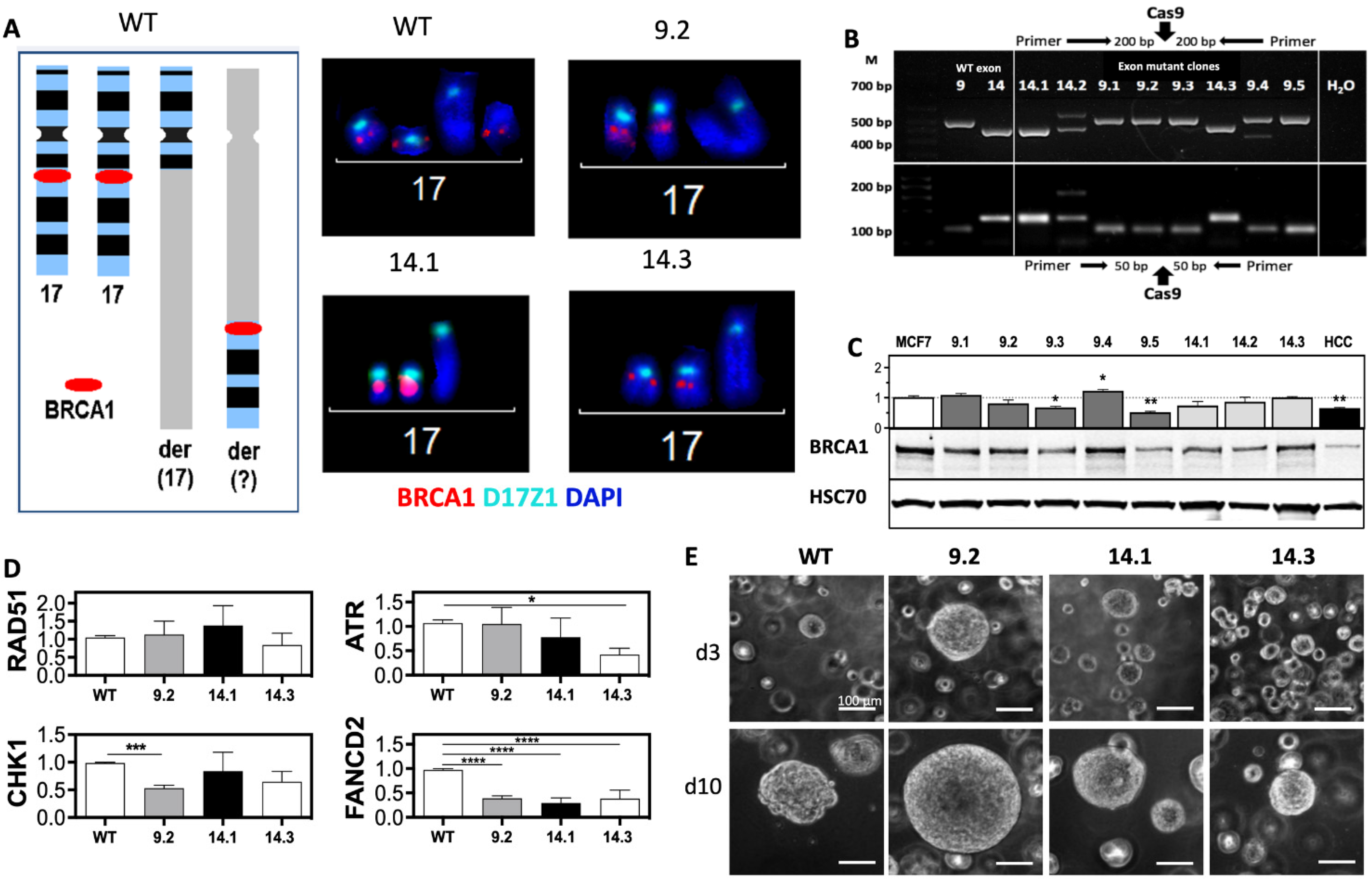
2.1. BRCA1 Mutations in Exon 9 and 14 Influence DDR Protein Expression and 3D Growth
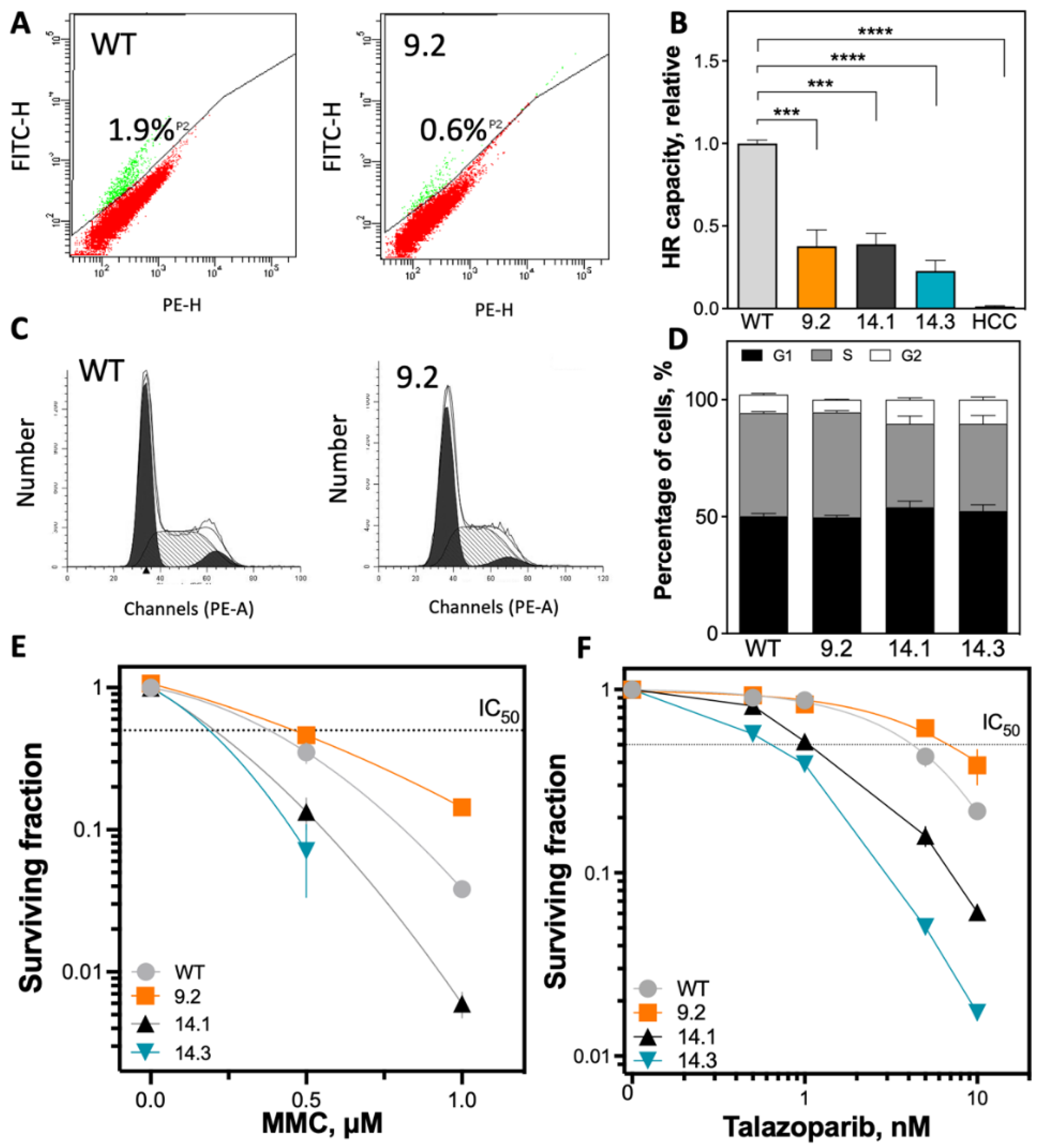
2.2. Significant Reduction of HR Capacity Does Not Result in Increased MMC and PARP1i Sensitivity
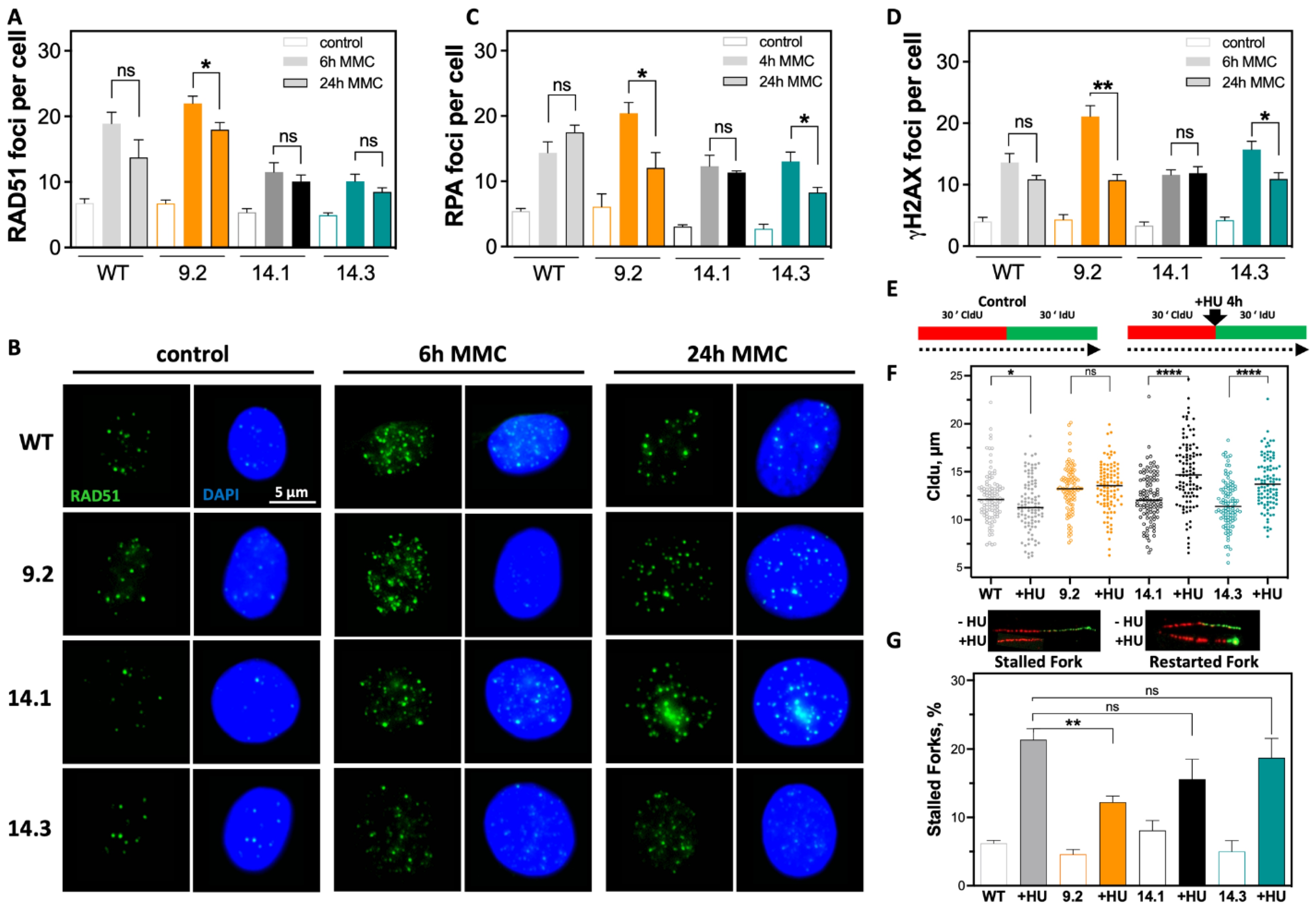
2.3. Efficient DSB Repair and Rapid DNA Replication Fork Restart Contributes to Therapy Resistance
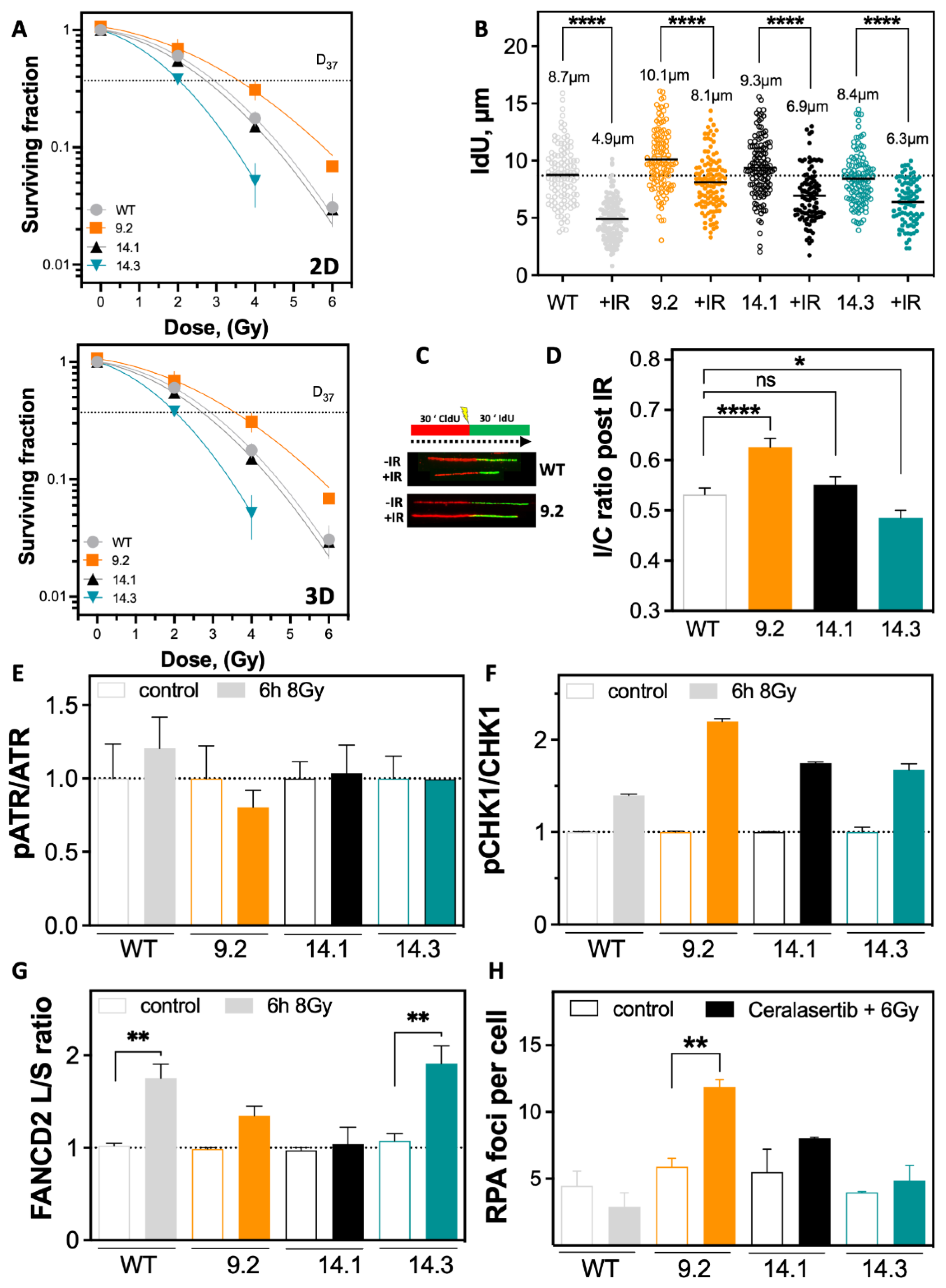
2.4. Resistance to Irradiation Emerges from Low Level of DNA Replication Stress
3. Discussion
4. Materials and Methods
4.1. Cell Lines, Culture and Treatment
4.2. CRISPR/Cas9-Mediated Modifications of BRCA1
4.3. PCR Screening
4.4. Amplicon Sequencing and Clone Characterization
4.5. FISH Analysis
4.6. Western Blot and Immunostaining
4.7. Homologous Recombination Assay
4.8. Cell Cycle
4.9. 2D and 3D Clonogenic Survival
4.10. DNA Fiber Assay
4.11. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wu, Z.; Li, S.; Tang, X.; Wang, Y.; Guo, W.; Cao, G.; Chen, K.; Zhang, M.; Guan, M.; Yang, D. Copy Number Amplification of DNA Damage Repair Pathways Potentiates Therapeutic Resistance in Cancer. Theranostics 2020, 10, 3939–3951. [Google Scholar] [CrossRef] [PubMed]
- Tung, N.; Lin, N.U.; Kidd, J.; Allen, B.A.; Singh, N.; Wenstrup, R.J.; Hartman, A.R.; Winer, E.P.; Garber, J.E. Frequency of Germline Mutations in 25 Cancer Susceptibility Genes in a Sequential Series of Patients With Breast Cancer. J. Clin. Oncol. 2016, 34, 1460–1468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, T.Y.; Park, K.S.; Nam, S.E.; Yoo, Y.B.; Park, W.S.; Yun, I.J. BRCA1/2 Serves as a Biomarker for Poor Prognosis in Breast Carcinoma. Int. J. Mol. Sci. 2022, 23, 3754. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Feng, H.; Cheng, X.; Liu, K.; Cai, D.; Zhao, R. Potential Therapeutic Targets of B7 Family in Colorectal Cancer. Front. Immunol. 2020, 11, 681. [Google Scholar] [CrossRef]
- Zhong, Q.; Peng, H.L.; Zhao, X.; Zhang, L.; Hwang, W.T. Effects of BRCA1- and BRCA2-related mutations on ovarian and breast cancer survival: A meta-analysis. Clin. Cancer Res. 2015, 21, 211–220. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Doty, T.; Gibson, B.; Heyer, W.D. Human BRCA2 protein promotes RAD51 filament formation on RPA-covered single-stranded DNA. Nat. Struct Mol. Biol. 2010, 17, 1260–1262. [Google Scholar] [CrossRef]
- Lord, C.J.; Ashworth, A. The DNA damage response and cancer therapy. Nature 2012, 481, 287–294. [Google Scholar] [CrossRef]
- Davies, H.; Glodzik, D.; Morganella, S.; Yates, L.R.; Staaf, J.; Zou, X.; Ramakrishna, M.; Martin, S.; Boyault, S.; Sieuwerts, A.M.; et al. HRDetect is a predictor of BRCA1 and BRCA2 deficiency based on mutational signatures. Nat. Med. 2017, 23, 517–525. [Google Scholar] [CrossRef]
- Wang, N.; Yang, Y.; Jin, D.; Zhang, Z.; Shen, K.; Yang, J.; Chen, H.; Zhao, X.; Yang, L.; Lu, H. PARP inhibitor resistance in breast and gynecological cancer: Resistance mechanisms and combination therapy strategies. Front. Pharmacol. 2022, 13, 967633. [Google Scholar] [CrossRef]
- Tutt, A.; Tovey, H.; Cheang, M.C.U.; Kernaghan, S.; Kilburn, L.; Gazinska, P.; Owen, J.; Abraham, J.; Barrett, S.; Barrett-Lee, P.; et al. Carboplatin in BRCA1/2-mutated and triple-negative breast cancer BRCAness subgroups: The TNT Trial. Nat. Med. 2018, 24, 628–637. [Google Scholar] [CrossRef]
- Telli, M.L.; Timms, K.M.; Reid, J.; Hennessy, B.; Mills, G.B.; Jensen, K.C.; Szallasi, Z.; Barry, W.T.; Winer, E.P.; Tung, N.M.; et al. Homologous Recombination Deficiency (HRD) Score Predicts Response to Platinum-Containing Neoadjuvant Chemotherapy in Patients with Triple-Negative Breast Cancer. Clin. Cancer Res. 2016, 22, 3764–3773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoppe, M.M.; Sundar, R.; Tan, D.S.P.; Jeyasekharan, A.D. Biomarkers for Homologous Recombination Deficiency in Cancer. J. Natl Cancer Inst. 2018, 110, 704–713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, R.E.; Leary, A.; Scott, C.L.; Serra, V.; Lord, C.J.; Bowtell, D.; Chang, D.K.; Garsed, D.W.; Jonkers, J.; Ledermann, J.A.; et al. ESMO recommendations on predictive biomarker testing for homologous recombination deficiency and PARP inhibitor benefit in ovarian cancer. Ann. Oncol 2020, 31, 1606–1622. [Google Scholar] [CrossRef] [PubMed]
- Schlacher, K.; Christ, N.; Siaud, N.; Egashira, A.; Wu, H.; Jasin, M. Double-strand break repair-independent role for BRCA2 in blocking stalled replication fork degradation by MRE11. Cell 2011, 145, 529–542. [Google Scholar] [CrossRef] [Green Version]
- Taglialatela, A.; Alvarez, S.; Leuzzi, G.; Sannino, V.; Ranjha, L.; Huang, J.W.; Madubata, C.; Anand, R.; Levy, B.; Rabadan, R.; et al. Restoration of Replication Fork Stability in BRCA1- and BRCA2-Deficient Cells by Inactivation of SNF2-Family Fork Remodelers. Mol. Cell 2017, 68, 414–430.e8. [Google Scholar] [CrossRef] [Green Version]
- Mutreja, K.; Krietsch, J.; Hess, J.; Ursich, S.; Berti, M.; Roessler, F.K.; Zellweger, R.; Patra, M.; Gasser, G.; Lopes, M. ATR-Mediated Global Fork Slowing and Reversal Assist Fork Traverse and Prevent Chromosomal Breakage at DNA Interstrand Cross-Links. Cell Rep. 2018, 24, 2629–2642.e5. [Google Scholar] [CrossRef] [Green Version]
- Rickman, K.; Smogorzewska, A. Advances in understanding DNA processing and protection at stalled replication forks. J. Cell Biol. 2019, 218, 1096–1107. [Google Scholar] [CrossRef] [Green Version]
- Pathania, S.; Bade, S.; Le Guillou, M.; Burke, K.; Reed, R.; Bowman-Colin, C.; Su, Y.; Ting, D.T.; Polyak, K.; Richardson, A.L.; et al. BRCA1 haploinsufficiency for replication stress suppression in primary cells. Nat. Commun. 2014, 5, 5496. [Google Scholar] [CrossRef] [Green Version]
- Couch, F.B.; Bansbach, C.E.; Driscoll, R.; Luzwick, J.W.; Glick, G.G.; Betous, R.; Carroll, C.M.; Jung, S.Y.; Qin, J.; Cimprich, K.A.; et al. ATR phosphorylates SMARCAL1 to prevent replication fork collapse. Genes Dev. 2013, 27, 1610–1623. [Google Scholar] [CrossRef] [Green Version]
- Eykelenboom, J.K.; Harte, E.C.; Canavan, L.; Pastor-Peidro, A.; Calvo-Asensio, I.; Llorens-Agost, M.; Lowndes, N.F. ATR activates the S-M checkpoint during unperturbed growth to ensure sufficient replication prior to mitotic onset. Cell Rep. 2013, 5, 1095–1107. [Google Scholar] [CrossRef]
- Maya-Mendoza, A.; Petermann, E.; Gillespie, D.A.; Caldecott, K.W.; Jackson, D.A. Chk1 regulates the density of active replication origins during the vertebrate S phase. EMBO J. 2007, 26, 2719–2731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petermann, E.; Woodcock, M.; Helleday, T. Chk1 promotes replication fork progression by controlling replication initiation. Proc. Natl. Acad. Sci. USA 2010, 107, 16090–16095. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Federico, M.B.; Campodonico, P.; Paviolo, N.S.; Gottifredi, V. Beyond interstrand crosslinks repair: Contribution of FANCD2 and other Fanconi Anemia proteins to the replication of DNA. Mutat Res. 2018, 808, 83–92. [Google Scholar] [CrossRef]
- Schlacher, K.; Wu, H.; Jasin, M. A distinct replication fork protection pathway connects Fanconi anemia tumor suppressors to RAD51-BRCA1/2. Cancer Cell 2012, 22, 106–116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaudhury, I.; Sareen, A.; Raghunandan, M.; Sobeck, A. FANCD2 regulates BLM complex functions independently of FANCI to promote replication fork recovery. Nucleic Acids Res. 2013, 41, 6444–6459. [Google Scholar] [CrossRef] [PubMed]
- Karnan, S.; Mohseni, M.; Konishi, Y.; Tamaki, A.; Hosokawa, Y.; Park, B.H.; Konishi, H. Controversial BRCA1 allelotypes in commonly used breast cancer cell lines. Breast Cancer Res. Treat 2010, 119, 249–251. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Silver, D.P.; Walpita, D.; Cantor, S.B.; Gazdar, A.F.; Tomlinson, G.; Couch, F.J.; Weber, B.L.; Ashley, T.; Livingston, D.M.; et al. Stable interaction between the products of the BRCA1 and BRCA2 tumor suppressor genes in mitotic and meiotic cells. Mol. Cell 1998, 2, 317–328. [Google Scholar] [CrossRef]
- Scully, R.; Ganesan, S.; Vlasakova, K.; Chen, J.; Socolovsky, M.; Livingston, D.M. Genetic analysis of BRCA1 function in a defined tumor cell line. Mol. Cell 1999, 4, 1093–1099. [Google Scholar] [CrossRef]
- Naipal, K.A.; Verkaik, N.S.; Ameziane, N.; van Deurzen, C.H.; Ter Brugge, P.; Meijers, M.; Sieuwerts, A.M.; Martens, J.W.; O’Connor, M.J.; Vrieling, H.; et al. Functional ex vivo assay to select homologous recombination-deficient breast tumors for PARP inhibitor treatment. Clin. Cancer Res. 2014, 20, 4816–4826. [Google Scholar] [CrossRef] [Green Version]
- Cruz, C.; Castroviejo-Bermejo, M.; Gutierrez-Enriquez, S.; Llop-Guevara, A.; Ibrahim, Y.H.; Gris-Oliver, A.; Bonache, S.; Morancho, B.; Bruna, A.; Rueda, O.M.; et al. RAD51 foci as a functional biomarker of homologous recombination repair and PARP inhibitor resistance in germline BRCA-mutated breast cancer. Ann. Oncol. 2018, 29, 1203–1210. [Google Scholar] [CrossRef]
- Dueva, R.; Iliakis, G. Replication protein A: A multifunctional protein with roles in DNA replication, repair and beyond. NAR Cancer 2020, 2, zcaa022. [Google Scholar] [CrossRef] [PubMed]
- Kais, Z.; Rondinelli, B.; Holmes, A.; O’Leary, C.; Kozono, D.; D’Andrea, A.D.; Ceccaldi, R. FANCD2 Maintains Fork Stability in BRCA1/2-Deficient Tumors and Promotes Alternative End-Joining DNA Repair. Cell Rep. 2016, 15, 2488–2499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Menolfi, D.; Jiang, W.; Lee, B.J.; Moiseeva, T.; Shao, Z.; Estes, V.; Frattini, M.G.; Bakkenist, C.J.; Zha, S. Kinase-dead ATR differs from ATR loss by limiting the dynamic exchange of ATR and RPA. Nat. Commun. 2018, 9, 5351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moynahan, M.E.; Chiu, J.W.; Koller, B.H.; Jasin, M. Brca1 controls homology-directed DNA repair. Mol. Cell 1999, 4, 511–518. [Google Scholar] [CrossRef]
- Foo, T.K.; Xia, B. BRCA1-Dependent and Independent Recruitment of PALB2-BRCA2-RAD51 in the DNA Damage Response and Cancer. Cancer Res. 2022, 82, 3191–3197. [Google Scholar] [CrossRef]
- Sy, S.M.; Huen, M.S.; Chen, J. PALB2 is an integral component of the BRCA complex required for homologous recombination repair. Proc. Natl. Acad. Sci. USA 2009, 106, 7155–7160. [Google Scholar] [CrossRef] [Green Version]
- Traven, A.; Heierhorst, J. SQ/TQ cluster domains: Concentrated ATM/ATR kinase phosphorylation site regions in DNA-damage-response proteins. Bioessays 2005, 27, 397–407. [Google Scholar] [CrossRef]
- Baer, R.; Ludwig, T. The BRCA1/BARD1 heterodimer, a tumor suppressor complex with ubiquitin E3 ligase activity. Curr. Opin. Genet. Dev. 2002, 12, 86–91. [Google Scholar] [CrossRef]
- Johnson, N.; Johnson, S.F.; Yao, W.; Li, Y.C.; Choi, Y.E.; Bernhardy, A.J.; Wang, Y.; Capelletti, M.; Sarosiek, K.A.; Moreau, L.A.; et al. Stabilization of mutant BRCA1 protein confers PARP inhibitor and platinum resistance. Proc. Natl. Acad. Sci. USA 2013, 110, 17041–17046. [Google Scholar] [CrossRef] [Green Version]
- Krais, J.J.; Wang, Y.; Patel, P.; Basu, J.; Bernhardy, A.J.; Johnson, N. RNF168-mediated localization of BARD1 recruits the BRCA1-PALB2 complex to DNA damage. Nat. Commun. 2021, 12, 5016. [Google Scholar] [CrossRef]
- Nacson, J.; Krais, J.J.; Bernhardy, A.J.; Clausen, E.; Feng, W.; Wang, Y.; Nicolas, E.; Cai, K.Q.; Tricarico, R.; Hua, X.; et al. BRCA1 Mutation-Specific Responses to 53BP1 Loss-Induced Homologous Recombination and PARP Inhibitor Resistance. Cell Rep. 2018, 25, 1384. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Bernhardy, A.J.; Cruz, C.; Krais, J.J.; Nacson, J.; Nicolas, E.; Peri, S.; van der Gulden, H.; van der Heijden, I.; O’Brien, S.W.; et al. The BRCA1-Delta11q Alternative Splice Isoform Bypasses Germline Mutations and Promotes Therapeutic Resistance to PARP Inhibition and Cisplatin. Cancer Res. 2016, 76, 2778–2790. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, B.; Matsuoka, S.; Ballif, B.A.; Zhang, D.; Smogorzewska, A.; Gygi, S.P.; Elledge, S.J. Abraxas and RAP80 form a BRCA1 protein complex required for the DNA damage response. Science 2007, 316, 1194–1198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roy, R.; Chun, J.; Powell, S.N. BRCA1 and BRCA2: Different roles in a common pathway of genome protection. Nat. Rev. Cancer 2011, 12, 68–78. [Google Scholar] [CrossRef] [Green Version]
- Parplys, A.C.; Seelbach, J.I.; Becker, S.; Behr, M.; Wrona, A.; Jend, C.; Mansour, W.Y.; Joosse, S.A.; Stuerzbecher, H.W.; Pospiech, H.; et al. High levels of RAD51 perturb DNA replication elongation and cause unscheduled origin firing due to impaired CHK1 activation. Cell Cycle 2015, 14, 3190–3202. [Google Scholar] [CrossRef]
- Saleh-Gohari, N.; Bryant, H.E.; Schultz, N.; Parker, K.M.; Cassel, T.N.; Helleday, T. Spontaneous homologous recombination is induced by collapsed replication forks that are caused by endogenous DNA single-strand breaks. Mol. Cell Biol. 2005, 25, 7158–7169. [Google Scholar] [CrossRef] [Green Version]
- Meyer, F.; Becker, S.; Classen, S.; Parplys, A.C.; Mansour, W.Y.; Riepen, B.; Timm, S.; Ruebe, C.; Jasin, M.; Wikman, H.; et al. Prevention of DNA Replication Stress by CHK1 Leads to Chemoresistance Despite a DNA Repair Defect in Homologous Recombination in Breast Cancer. Cells 2020, 9, 238. [Google Scholar] [CrossRef] [Green Version]
- Noordermeer, S.M.; van Attikum, H. PARP Inhibitor Resistance: A Tug-of-War in BRCA-Mutated Cells. Trends Cell Biol. 2019, 29, 820–834. [Google Scholar] [CrossRef] [Green Version]
- Ray Chaudhuri, A.; Callen, E.; Ding, X.; Gogola, E.; Duarte, A.A.; Lee, J.E.; Wong, N.; Lafarga, V.; Calvo, J.A.; Panzarino, N.J.; et al. Replication fork stability confers chemoresistance in BRCA-deficient cells. Nature 2016, 535, 382–387. [Google Scholar] [CrossRef] [Green Version]
- De Silva, I.U.; McHugh, P.J.; Clingen, P.H.; Hartley, J.A. Defining the roles of nucleotide excision repair and recombination in the repair of DNA interstrand cross-links in mammalian cells. Mol. Cell Biol. 2000, 20, 7980–7990. [Google Scholar] [CrossRef]
- Burrell, R.A.; McClelland, S.E.; Endesfelder, D.; Groth, P.; Weller, M.C.; Shaikh, N.; Domingo, E.; Kanu, N.; Dewhurst, S.M.; Gronroos, E.; et al. Replication stress links structural and numerical cancer chromosomal instability. Nature 2013, 494, 492–496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buisson, R.; Boisvert, J.L.; Benes, C.H.; Zou, L. Distinct but Concerted Roles of ATR, DNA-PK, and Chk1 in Countering Replication Stress during S Phase. Mol. Cell 2015, 59, 1011–1024. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ang, M.K.; Patel, M.R.; Yin, X.Y.; Sundaram, S.; Fritchie, K.; Zhao, N.; Liu, Y.; Freemerman, A.J.; Wilkerson, M.D.; Walter, V.; et al. High XRCC1 protein expression is associated with poorer survival in patients with head and neck squamous cell carcinoma. Clin. Cancer Res. 2011, 17, 6542–6552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, R.W.; Orelli, B.J.; Yamazoe, M.; Minn, A.J.; Takeda, S.; Bishop, D.K. RAD51 up-regulation bypasses BRCA1 function and is a common feature of BRCA1-deficient breast tumors. Cancer Res. 2007, 67, 9658–9665. [Google Scholar] [CrossRef] [Green Version]
- Smogorzewska, A.; Matsuoka, S.; Vinciguerra, P.; McDonald, E.R., 3rd; Hurov, K.E.; Luo, J.; Ballif, B.A.; Gygi, S.P.; Hofmann, K.; D’Andrea, A.D.; et al. Identification of the FANCI protein, a monoubiquitinated FANCD2 paralog required for DNA repair. Cell 2007, 129, 289–301. [Google Scholar] [CrossRef] [Green Version]
- Sims, A.E.; Spiteri, E.; Sims, R.J., 3rd; Arita, A.G.; Lach, F.P.; Landers, T.; Wurm, M.; Freund, M.; Neveling, K.; Hanenberg, H.; et al. FANCI is a second monoubiquitinated member of the Fanconi anemia pathway. Nat. Struct Mol. Biol. 2007, 14, 564–567. [Google Scholar] [CrossRef] [PubMed]
- Howlett, N.G.; Taniguchi, T.; Durkin, S.G.; D’Andrea, A.D.; Glover, T.W. The Fanconi anemia pathway is required for the DNA replication stress response and for the regulation of common fragile site stability. Hum. Mol. Genet. 2005, 14, 693–701. [Google Scholar] [CrossRef] [Green Version]
- Ran, F.A.; Hsu, P.D.; Wright, J.; Agarwala, V.; Scott, D.A.; Zhang, F. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 2013, 8, 2281–2308. [Google Scholar] [CrossRef] [Green Version]
- Bentley, D.R.; Balasubramanian, S.; Swerdlow, H.P.; Smith, G.P.; Milton, J.; Brown, C.G.; Hall, K.P.; Evers, D.J.; Barnes, C.L.; Bignell, H.R.; et al. Accurate whole human genome sequencing using reversible terminator chemistry. Nature 2008, 456, 53–59. [Google Scholar] [CrossRef] [Green Version]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EmbNet.J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Edgar, R.C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 2010, 26, 2460–2461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liehr, T. Molecular Cytogenetics in the Era of Chromosomics and Cytogenomic Approaches. Front. Genet. 2021, 12, 720507. [Google Scholar] [CrossRef] [PubMed]
- Horibata, S.; Vo, T.V.; Subramanian, V.; Thompson, P.R.; Coonrod, S.A. Utilization of the Soft Agar Colony Formation Assay to Identify Inhibitors of Tumorigenicity in Breast Cancer Cells. J. Vis. Exp. 2015, 99, e52727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meerz, A.; Deville, S.S.; Muller, J.; Cordes, N. Comparative Therapeutic Exploitability of Acute Adaptation Mechanisms to Photon and Proton Irradiation in 3D Head and Neck Squamous Cell Carcinoma Cell Cultures. Cancers (Basel) 2021, 13, 1190. [Google Scholar] [CrossRef] [PubMed]
- Parplys, A.C.; Petermann, E.; Petersen, C.; Dikomey, E.; Borgmann, K. DNA damage by X-rays and their impact on replication processes. Radiother. Oncol. 2012, 102, 466–471. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Line | Allele 1 | Allele 2 | Allele 3 |
|---|---|---|---|
| MCF7 WT | WT | WT | WT |
| MCF7 9.1 | WT | 7 bp deletion (c.620del7 → p.207fs23stop) | loss |
| MCF7 9.2 | WT | 7 bp deletion (c.620del7 → p.207fs23stop) | loss |
| MCF7 9.3 | WT | 7 bp deletion (c.620del7 → p.207fs23stop) | loss |
| MCF7 9.4 | In frame 4 aa exchange (p.QITP205LLQI) | 182 bp deletion (c.627del182 → p.209fs7stop) | loss |
| MCF7 9.5 | WT | 7 bp deletion (c.620del7 → p.207fs23stop) | loss |
| MCF7 14.1 | 6 bp exchange (p.RW1506GI) | 14 bp deletion (c.4516 del14 → p.1506fs9stop) | loss |
| MCF7 14.2 | WT | 183 bp insertion (c.4517ins183 → p.1506ins19stop) | loss |
| MCF7 14.3 | WT | 7 bp deletion (c.4511del7 → p.1505fs40stop) | loss |
| Exon | Direction | Sequence (5′ to 3′) |
|---|---|---|
| 9 | forward | CACCGTTGTTACAAATCACCCCTCA |
| reverse | AAACTGAGGGGTGATTTGTAACAAC | |
| 14 | forward | CACCGCCCATCATTAGATGATAGG |
| reverse | AAACCCTATCATCTAATGATGGGC |
| Exon | Length and Direction | Tm (°C) | Sequence (5′ to 3′) | Amplicon Size (bp) |
|---|---|---|---|---|
| 9 | short forward | 58.9 | TTCCCTATAGTGTGGGAGATCA | 101 |
| short reverse | 54.7 | CAAACTTTGCCATTACCCTTTT | ||
| long forward | 55.3 | CCACACCCAGCTACTGACCT | 494 | |
| long reverse | 55.3 | CTCTTCCAGCTGTTGCTCCT | ||
| 14 | short forward | 55.9 | CGATGGTTTTCTCCTTCCATT | 169 |
| short reverse | 55.3 | TTGCTCCTCCACATCAACAA | ||
| long forward | 61.4 | CCACACCCAGCTACTGACCT | 419 | |
| long reverse | 59.4 | CTCTTCCAGCTGTTGCTCCT |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Classen, S.; Rahlf, E.; Jungwirth, J.; Albers, N.; Hebestreit, L.P.; Zielinski, A.; Poole, L.; Groth, M.; Koch, P.; Liehr, T.; et al. Partial Reduction in BRCA1 Gene Dose Modulates DNA Replication Stress Level and Thereby Contributes to Sensitivity or Resistance. Int. J. Mol. Sci. 2022, 23, 13363. https://doi.org/10.3390/ijms232113363
Classen S, Rahlf E, Jungwirth J, Albers N, Hebestreit LP, Zielinski A, Poole L, Groth M, Koch P, Liehr T, et al. Partial Reduction in BRCA1 Gene Dose Modulates DNA Replication Stress Level and Thereby Contributes to Sensitivity or Resistance. International Journal of Molecular Sciences. 2022; 23(21):13363. https://doi.org/10.3390/ijms232113363
Chicago/Turabian StyleClassen, Sandra, Elena Rahlf, Johannes Jungwirth, Nina Albers, Luca Philipp Hebestreit, Alexandra Zielinski, Lena Poole, Marco Groth, Philipp Koch, Thomas Liehr, and et al. 2022. "Partial Reduction in BRCA1 Gene Dose Modulates DNA Replication Stress Level and Thereby Contributes to Sensitivity or Resistance" International Journal of Molecular Sciences 23, no. 21: 13363. https://doi.org/10.3390/ijms232113363