
Impact of Dysfunctional Adipose Tissue Depots on the Cardiovascular System
 ,
,  , , and
, , and 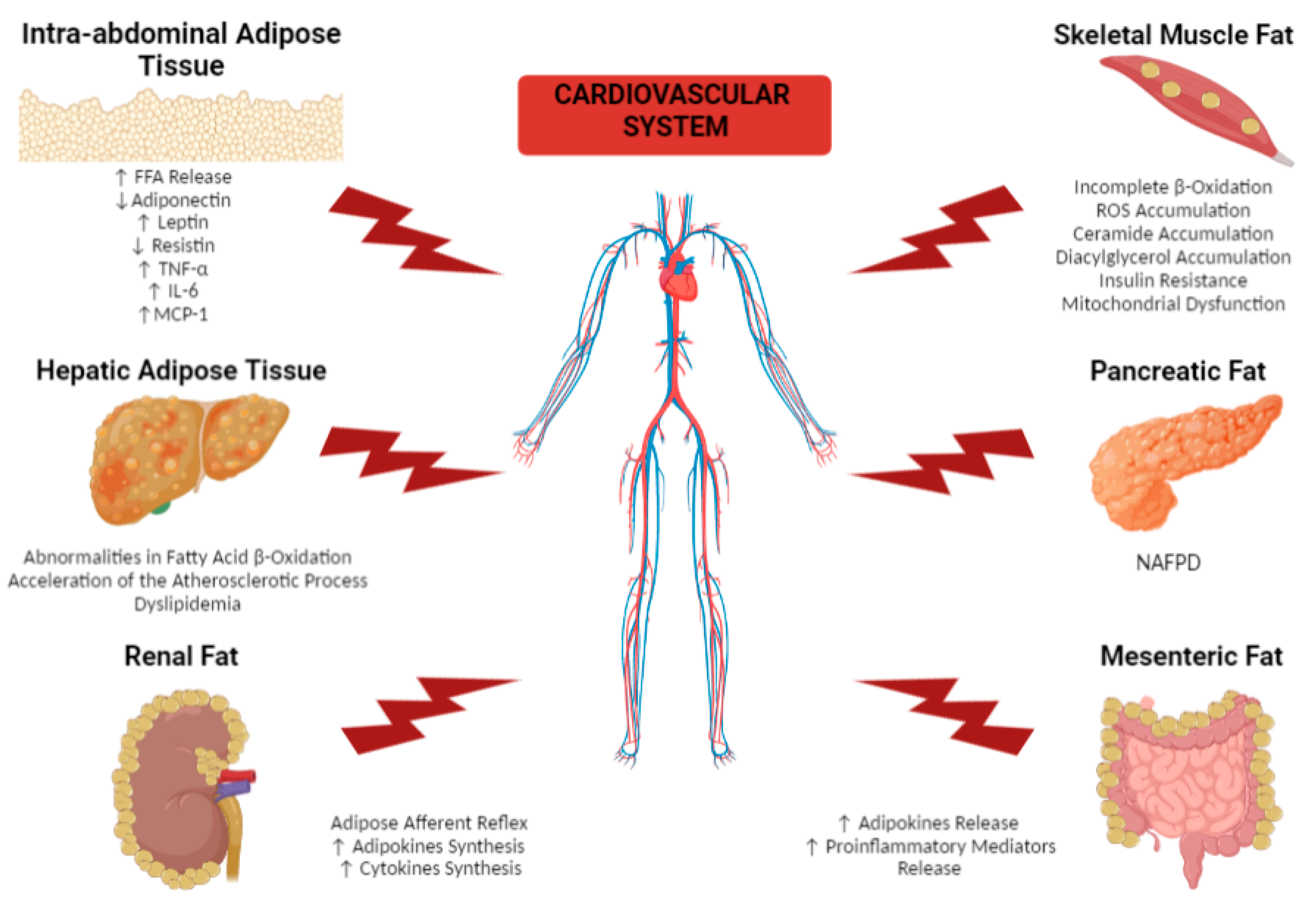{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Effects of Different Dysfunctional Adipose Tissue Depots on the Cardiovascular System
2.1. Remote Adipose Tissue Depots
2.1.1. Intra-Abdominal Adipose Tissue
2.1.2. Hepatic Adipose Tissue
2.1.3. Skeletal Muscle Fat
2.1.4. Pancreatic Fat
2.1.5. Renal Fat
2.1.6. Mesenteric Fat
2.2. Cardiac Fat: Focus on Epicardial Adipose Tissue
3. Strategies for Reducing Cardiovascular Risk That Impact Adipose Tissue
3.1. Lifestyle Modifications: Restricting Food Intake and Increasing Energy Expenditure
3.2. Pharmacotherapy: Focus on GLP-1 Receptor Agonists and SGLT2 Inhibitors
3.3. Bariatric Surgery
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Chooi, Y.C.; Ding, C.; Magkos, F. The Epidemiology of Obesity. Metabolism 2019, 92, 6–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, G.M.; Danaei, G.; Farzadfar, F.; Stevens, G.A.; Woodward, M.; Wormser, D.; Kaptoge, S.; Whitlock, G.; Qiao, Q.; Lewington, S.; et al. The Age-Specific Quantitative Effects of Metabolic Risk Factors on Cardiovascular Diseases and Diabetes: A Pooled Analysis. PLoS ONE 2013, 8, e65174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Czernichow, S.; Kengne, A.P.; Stamatakis, E.; Hamer, M.; Batty, G.D. Body Mass Index, Waist Circumference and Waist-Hip Ratio: Which Is the Better Discriminator of Cardiovascular Disease Mortality Risk? Evidence from an Individual-Participant Meta-Analysis of 82 864 Participants from Nine Cohort Studies. Obes. Rev. 2011, 12, 680–687. [Google Scholar] [CrossRef] [PubMed]
- Lauby-Secretan, B.; Scoccianti, C.; Loomis, D.; Grosse, Y.; Bianchini, F.; Straif, K. Body Fatness and Cancer—Viewpoint of the IARC Working Group. N. Engl. J. Med. 2016, 375, 794–798. [Google Scholar] [CrossRef] [Green Version]
- Anstey, K.J.; Cherbuin, N.; Budge, M.; Young, J. Body mass index in midlife and late-life as a risk factor for dementia: A meta-analysis of prospective studies. Obes. Rev. 2011, 12, e426–e437. [Google Scholar] [CrossRef]
- Anandacoomarasamy, A.; Caterson, I.; Sambrook, P.; Fransen, M.; March, L. The impact of obesity on the musculoskeletal system. Int. J. Obes. 2008, 32, 211–222. [Google Scholar] [CrossRef] [Green Version]
- Poirier, P.; Giles, T.D.; Bray, G.A.; Hong, Y.; Stern, J.S.; Pi-Sunyer, F.X.; Eckel, R.H. Obesity and Cardiovascular Disease: Pathophysiology, Evaluation, and Effect of Weight Loss: An Update of the 1997 American Heart Association Scientific Statement on Obesity and Heart Disease from the Obesity Committee of the Council on Nutrition, Physical Activity, and Metabolism. Circulation 2006, 113, 898–918. [Google Scholar] [CrossRef] [Green Version]
- Bastien, M.; Poirier, P.; Lemieux, I.; Després, J.-P. Overview of Epidemiology and Contribution of Obesity to Cardiovascular Disease. Prog. Cardiovasc. Dis. 2013, 56, 369–381. [Google Scholar] [CrossRef]
- Chait, A.; den Hartigh, L.J. Adipose Tissue Distribution, Inflammation and Its Metabolic Consequences, Including Diabetes and Cardiovascular Disease. Front. Cardiovasc. Med. 2020, 7, 22. [Google Scholar] [CrossRef] [Green Version]
- Wajchenberg, B.L. Subcutaneous and Visceral Adipose Tissue: Their Relation to the Metabolic Syndrome. Endocr. Rev. 2000, 21, 697–738. [Google Scholar] [CrossRef]
- Britton, K.A.; Massaro, J.M.; Murabito, J.M.; Kreger, B.E.; Hoffmann, U.; Fox, C.S. Body Fat Distribution, Incident Cardiovascular Disease, Cancer, and All-Cause Mortality. J. Am. Coll. Cardiol. 2013, 62, 921–925. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iacobellis, G. Local and Systemic Effects of the Multifaceted Epicardial Adipose Tissue Depot. Nat. Rev. Endocrinol. 2015, 11, 363–371. [Google Scholar] [CrossRef] [PubMed]
- Oikonomou, E.K.; Antoniades, C. The Role of Adipose Tissue in Cardiovascular Health and Disease. Nat. Rev. Cardiol. 2018, 16, 83–99. [Google Scholar] [CrossRef] [PubMed]
- Salgado-Somoza, A.; Teijeira-Fernández, E.; Fernández, Á.L.; González-Juanatey, J.R.; Eiras, S. Proteomic analysis of epicardial and subcutaneous adipose tissue reveals differences in proteins involved in oxidative stress. Am. J. Physiol. Circ. Physiol. 2010, 299, H202–H209. [Google Scholar] [CrossRef] [Green Version]
- Vohl, M.C.; Sladek, R.; Robitaille, J.; Gurd, S.; Marceau, P.; Richard, D.; Hudson, T.J.; Tchernof, A. A Survey of Genes Differentially Expressed in Subcutaneous and Visceral Adipose Tissue in Men. Obes. Res. 2004, 12, 1217–1222. [Google Scholar] [CrossRef]
- Silva, K.R.; Côrtes, I.; Liechocki, S.; Carneiro, J.R.I.; Souza, A.A.P.; Borojevic, R.; Maya-Monteiro, C.M.; Baptista, L.S. Characterization of stromal vascular fraction and adipose stem cells from subcutaneous, preperitoneal and visceral morbidly obese human adipose tissue depots. PLoS ONE 2017, 12, e0174115. [Google Scholar] [CrossRef] [Green Version]
- Koenen, M.; Hill, M.A.; Cohen, P.; Sowers, J.R. Obesity, Adipose Tissue and Vascular Dysfunction. Circ. Res. 2021, 128, 951–968. [Google Scholar] [CrossRef]
- McGavock, J.M.; Lingvay, I.; Zib, I.; Tillery, T.; Salas, N.; Unger, R.; Levine, B.D.; Raskin, P.; Victor, R.G.; Szczepaniak, L.S. Cardiac Steatosis in Diabetes Mellitus: A 1H-Magnetic Resonance Spectroscopy Study. Circulation 2007, 116, 1170–1175. [Google Scholar] [CrossRef] [Green Version]
- Hankiewicz, J.H.; Banke, N.H.; Farjah, M.; Lewandowski, E.D. Early Impairment of Transmural Principal Strains in the Left Ventricular Wall after Short-Term, High-Fat Feeding of Mice Predisposed to Cardiac Steatosis. Circ. Cardiovasc. Imaging 2010, 3, 710–717. [Google Scholar] [CrossRef] [Green Version]
- Lavie, C.J.; Milani, R.V.; Ventura, H.O. Obesity and Cardiovascular Disease: Risk Factor, Paradox, and Impact of Weight Loss. J. Am. Coll. Cardiol. 2009, 53, 1925–1932. [Google Scholar] [CrossRef]
- Shibata, R.; Murohara, T.; Ouchi, N. Protective Role of Adiponectin in Cardiovascular Disease. Curr. Med. Chem. 2012, 19, 5459–5546. [Google Scholar] [CrossRef] [PubMed]
- Morínigo, R.; Musri, M.; Vidal, J.; Casamitjana, R.; Delgado, S.; Lacy, A.M.; Ayuso, C.; Gomis, R.; Corominola, H. Intra-Abdominal Fat Adiponectin Receptors Expression and Cardiovascular Metabolic Risk Factors in Obesity and Diabetes. Obes. Surg. 2006, 16, 745–751. [Google Scholar] [CrossRef] [PubMed]
- Verdeş, G.A.B.R.I.E.L.; DuŢă, C.C.; Popescu, R.; MituleŢu, M.I.H.A.I.; Ursoniu, S.; Lazăr, O.F. Correlation between leptin and ghrelin expression in adipose visceral tissue and clinical-biological features in malignant obesity. Rom. J. Morphol. Embryol. 2017, 58, 923–929. [Google Scholar] [PubMed]
- Landecho, M.F.; Tuero, C.; Valentí, V.; Bilbao, I.; De La Higuera, M.; Frühbeck, G. Relevance of Leptin and Other Adipokines in Obesity-Associated Cardiovascular Risk. Nutrients 2019, 11, 2664. [Google Scholar] [CrossRef] [Green Version]
- Gómez-Ambrosi, J.; Salvador, J.; Páramo, J.A.; Orbe, J.; de Irala, J.; Diez-Caballero, A.; Gil, M.J.; Cienfuegos, J.A.; Frühbeck, G. Involvement of Leptin in the Association between Percentage of Body Fat and Cardiovascular Risk Factors. Clin. Biochem. 2002, 35, 315–320. [Google Scholar] [CrossRef] [Green Version]
- Rodríguez, A.; Gómez-Ambrosi, J.; Catalán, V.; Fortuño, A.; Frühbeck, G. Leptin Inhibits the Proliferation of Vascular Smooth Muscle Cells Induced by Angiotensin II through Nitric Oxide-Dependent Mechanisms. Mediat. Inflamm. 2010, 2010, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Dirksen, C.; Jørgensen, N.B.; Bojsen-Møller, K.N.; Jacobsen, S.H.; Hansen, D.L.; Worm, D.; Holst, J.J.; Madsbad, S. Mechanisms of Improved Glycaemic Control after Roux-En-Y Gastric Bypass. Diabetologia 2012, 55, 1890–1901. [Google Scholar] [CrossRef] [Green Version]
- Adams, T.D.; Davidson, L.E.; Litwin, S.E.; Kim, J.; Kolotkin, R.L.; Nanjee, M.N.; Gutierrez, J.M.; Frogley, S.J.; Ibele, A.R.; Brinton, E.A.; et al. Weight and Metabolic Outcomes 12 Years after Gastric Bypass. N. Engl. J. Med. 2017, 377, 1143–1155. [Google Scholar] [CrossRef]
- Smitka, K.; Marešová, D. Adipose Tissue as an Endocrine Organ: An Update on Pro-Inflammatory and Anti-Inflammatory Microenvironment. Prague Med. Rep. 2015, 116, 87–111. [Google Scholar] [CrossRef] [Green Version]
- van Berendoncks, A.M.; Garnier, A.; Beckers, P.; Hoymans, V.Y.; Possemiers, N.; Fortin, D.; Martinet, W.; van Hoof, V.; Vrints, C.J.; Ventura-Clapier, R.; et al. Functional Adiponectin Resistance at the Level of the Skeletal Muscle in Mild to Moderate Chronic Heart Failure. Circ. Heart Fail. 2010, 3, 185–194. [Google Scholar] [CrossRef]
- Verma, S.; Li, S.H.; Wang, C.H.; Fedak, P.W.M.; Li, R.K.; Weisel, R.D.; Mickle, D.A.G. Resistin Promotes Endothelial Cell Activation: Further Evidence of Adipokine-Endothelial Interaction. Circulation 2003, 108, 736–740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calabro, P.; Samudio, I.; Willerson, J.T.; Yeh, E.T.H. Resistin Promotes Smooth Muscle Cell Proliferation through Activation of Extracellular Signal-Regulated Kinase 1/2 and Phosphatidylinositol 3-Kinase Pathways. Circulation 2004, 110, 3335–3340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reilly, M.P.; Lehrke, M.; Wolfe, M.L.; Rohatgi, A.; Lazar, M.A.; Rader, D.J. Resistin Is an Inflammatory Marker of Atherosclerosis in Humans. Circulation 2005, 111, 932–939. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiao, N.; Yin, M.; Zhang, L.; Qu, X.; Du, H.; Sun, X.; Mao, L.; Ren, G.; Zhang, C.; Geng, Y.; et al. Tumor Necrosis Factor-Alpha Deficiency Retards Early Fatty-Streak Lesion by Influencing the Expression of Inflammatory Factors in ApoE-Null Mice. Mol. Genet. Metab. 2009, 96, 239–244. [Google Scholar] [CrossRef]
- Brånén, L.; Hovgaard, L.; Nitulescu, M.; Bengtsson, E.; Nilsson, J.; Jovinge, S. Inhibition of Tumor Necrosis Factor-α Reduces Atherosclerosis in Apolipoprotein E Knockout Mice. Arter. Thromb. Vasc. Biol. 2004, 24, 2137–2142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boesten, L.S.M.; Zadelaar, A.S.M.; van Nieuwkoop, A.; Gijbels, M.J.J.; de Winther, M.P.J.; Havekes, L.M.; van Vlijmen, B.J.M. Tumor Necrosis Factor-Alpha Promotes Atherosclerotic Lesion Progression in APOE*3-Leiden Transgenic Mice. Cardiovasc. Res. 2005, 66, 179–185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bergh, N.; Ulfhammer, E.; Glise, K.; Jern, S.; Karlsson, L. Influence of TNF-α and biomechanical stress on endothelial anti- and prothrombotic genes. Biochem. Biophys. Res. Commun. 2009, 385, 314–318. [Google Scholar] [CrossRef] [Green Version]
- Ohta, H.; Wada, H.; Niwa, T.; Kirii, H.; Iwamoto, N.; Fujii, H.; Saito, K.; Sekikawa, K.; Seishima, M. Disruption of Tumor Necrosis Factor-Alpha Gene Diminishes the Development of Atherosclerosis in ApoE-Deficient Mice. Atherosclerosis 2005, 180, 11–17. [Google Scholar] [CrossRef]
- Kleinbongard, P.; Heusch, G.; Schulz, R. TNFalpha in Atherosclerosis, Myocardial Ischemia/Reperfusion and Heart Failure. Pharmacol. Ther. 2010, 127, 295–314. [Google Scholar] [CrossRef]
- Goetze, S.; Xi, X.P.; Kawano, Y.; Kawano, H.; Fleck, E.; Hsueh, W.A.; Law, R.E. TNF-Alpha-Induced Migration of Vascular Smooth Muscle Cells Is MAPK Dependent. Hypertension 1999, 33, 183–189. [Google Scholar] [CrossRef]
- Goetze, S.; Kintscher, U.; Kaneshiro, K.; Meehan, W.P.; Collins, A.; Fleck, E.; Hsueh, W.A.; Law, R.E. TNFalpha Induces Expression of Transcription Factors C-Fos, Egr-1, and Ets-1 in Vascular Lesions through Extracellular Signal-Regulated Kinases 1/2. Atherosclerosis 2001, 159, 93–101. [Google Scholar] [CrossRef]
- Boyle, J.J.; Weissberg, P.L.; Bennett, M.R. Tumor Necrosis Factor-α Promotes Macrophage-Induced Vascular Smooth Muscle Cell Apoptosis by Direct and Autocrine Mechanisms. Arter. Thromb. Vasc. Biol. 2003, 23, 1553–1558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weisberg, S.P.; McCann, D.; Desai, M.; Rosenbaum, M.; Leibel, R.L.; Ferrante, A.W. Obesity Is Associated with Macrophage Accumulation in Adipose Tissue. J. Clin. Investig. 2003, 112, 1796–1808. [Google Scholar] [CrossRef] [PubMed]
- El-Mikkawy, D.M.E.; EL-Sadek, M.A.; EL-Badawy, M.A.; Samaha, D. Circulating Level of Interleukin-6 in Relation to Body Mass Indices and Lipid Profile in Egyptian Adults with Overweight and Obesity. Egypt. Rheumatol. Rehabil. 2020, 47, 1–7. [Google Scholar] [CrossRef]
- Wedell-Neergaard, A.S.; Lang Lehrskov, L.; Christensen, R.H.; Legaard, G.E.; Dorph, E.; Larsen, M.K.; Launbo, N.; Fagerlind, S.R.; Seide, S.K.; Nymand, S.; et al. Exercise-Induced Changes in Visceral Adipose Tissue Mass Are Regulated by IL-6 Signaling: A Randomized Controlled Trial. Cell Metab. 2019, 29, 844–855. [Google Scholar] [CrossRef] [PubMed]
- Malavazos, A.E.; Cereda, E.; Morricone, L.; Coman, C.; Corsi, M.M.; Ambrosi, B. Monocyte Chemoattractant Protein 1: A Possible Link between Visceral Adipose Tissue-Associated Inflammation and Subclinical Echocardiographic Abnormalities in Uncomplicated Obesity. Eur. J. Endocrinol. 2005, 153, 871–877. [Google Scholar] [CrossRef] [Green Version]
- Öhman, M.K.; Wright, A.P.; Wickenheiser, K.J.; Luo, W.; Russo, H.M.; Eitzman, D.T. Monocyte Chemoattractant Protein-1 Deficiency Protects Against Visceral Fat-Induced Atherosclerosis. Arter. Thromb. Vasc. Biol. 2010, 30, 1151–1158. [Google Scholar] [CrossRef] [Green Version]
- Anstee, Q.M.; Mantovani, A.; Tilg, H.; Targher, G. Risk of cardiomyopathy and cardiac arrhythmias in patients with nonalcoholic fatty liver disease. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 425–439. [Google Scholar] [CrossRef]
- Ismaiel, A.; Dumitraşcu, D.L. Cardiovascular Risk in Fatty Liver Disease: The Liver-Heart Axis—Literature Review. Front. Med. 2019, 6, 202. [Google Scholar] [CrossRef] [Green Version]
- Ferrara, D.; Montecucco, F.; Dallegri, F.; Carbone, F. Impact of Different Ectopic Fat Depots on Cardiovascular and Metabolic Diseases. J. Cell Physiol. 2019, 234, 21630–21641. [Google Scholar] [CrossRef]
- Anstee, Q.M.; Targher, G.; Day, C.P. Progression of NAFLD to Diabetes Mellitus, Cardiovascular Disease or Cirrhosis. Nat Rev Gastroenterol. Hepatol. 2013, 10, 330–344. [Google Scholar] [CrossRef] [PubMed]
- Dabravolski, S.; Bezsonov, E.; Baig, M.; Popkova, T.; Orekhov, A. Mitochondrial Lipid Homeostasis at the Crossroads of Liver and Heart Diseases. Int. J. Mol. Sci. 2021, 22, 6949. [Google Scholar] [CrossRef] [PubMed]
- Deprince, A.; Haas, J.T.; Staels, B. Dysregulated lipid metabolism links NAFLD to cardiovascular disease. Mol. Metab. 2020, 42, 101092. [Google Scholar] [CrossRef] [PubMed]
- Bhatia, L.S.; Curzen, N.P.; Byrne, C.D. Nonalcoholic Fatty Liver Disease and Vascular Risk. Curr. Opin. Cardiol. 2012, 27, 420–428. [Google Scholar] [CrossRef]
- Liu, H.; Lu, H.Y. Nonalcoholic Fatty Liver Disease and Cardiovascular Disease. World J. Gastroenterol. 2014, 20, 8407–8415. [Google Scholar] [CrossRef]
- Petersen, M.C.; Vatner, D.F.; Shulman, G.I. Regulation of Hepatic Glucose Metabolism in Health and Disease. Nat. Rev. Endocrinol. 2017, 13, 572–587. [Google Scholar] [CrossRef] [Green Version]
- Hashizume, H.; Sato, K.; Yamazaki, Y.; Horiguchi, N.; Kakizaki, S.; Mori, M. A prospective study of long-term outcomes in female patients with nonalcoholic steatohepatitis using age- and body mass index-matched cohorts. Acta Med. Okayama 2013, 67, 45–53. [Google Scholar] [CrossRef]
- Song, Z.; Xiaoli, A.M.; Yang, F. Regulation and Metabolic Significance of De Novo Lipogenesis in Adipose Tissues. Nutrients 2018, 10, 1383. [Google Scholar] [CrossRef] [Green Version]
- Hausman, G.J.; Basu, U.; Du, M.; Fernyhough-Culver, M.; Dodson, M.V. Intermuscular and Intramuscular Adipose Tissues: Bad vs. Good Adipose Tissues. Adipocyte 2014, 3, 242–255. [Google Scholar] [CrossRef] [Green Version]
- Coen, P.M.; Goodpaster, B.H. Role of Intramyocelluar Lipids in Human Health. Trends Endocrinol. Metab. 2012, 23, 391. [Google Scholar] [CrossRef]
- Adams, J.M.; Pratipanawatr, T.; Berria, R.; Wang, E.; DeFronzo, R.A.; Sullards, M.C.; Mandarino, L.J. Ceramide Content Is Increased in Skeletal Muscle from Obese Insulin-Resistant Humans. Diabetes 2004, 53, 25–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chavez, J.A.; Knotts, T.A.; Wang, L.P.; Li, G.; Dobrowsky, R.T.; Florant, G.L.; Summers, S.A. A Role for Ceramide, but Not Diacylglycerol, in the Antagonism of Insulin Signal Transduction by Saturated Fatty Acids. J. Biol. Chem. 2003, 278, 10297–10303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stratford, S.; Hoehn, K.L.; Liu, F.; Summers, S.A. Regulation of Insulin Action by Ceramide: Dual Mechanisms Linking Ceramide Accumulation to the Inhibition of Akt/Protein Kinase B. J. Biol. Chem. 2004, 279, 36608–36615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Paola, M.; Cocco, T.; Lorusso, M. Ceramide Interaction with the Respiratory Chain of Heart Mitochondria. Biochemistry 2000, 39, 6660–6668. [Google Scholar] [CrossRef]
- García-Ruiz, C.; Colell, A.; Marí, M.; Morales, A.; Fernández-Checa, J.C. Direct Effect of Ceramide on the Mitochondrial Electron Transport Chain Leads to Generation of Reactive Oxygen Species. Role of Mitochondrial Glutathione. J. Biol. Chem. 1997, 272, 11369–11377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holland, W.L.; Brozinick, J.T.; Wang, L.P.; Hawkins, E.D.; Sargent, K.M.; Liu, Y.; Narra, K.; Hoehn, K.L.; Knotts, T.A.; Siesky, A.; et al. Inhibition of Ceramide Synthesis Ameliorates Glucocorticoid-, Saturated-Fat-, and Obesity-Induced Insulin Resistance. Cell Metab. 2007, 5, 167–179. [Google Scholar] [CrossRef] [Green Version]
- Yu, C.; Chen, Y.; Cline, G.W.; Zhang, D.; Zong, H.; Wang, Y.; Bergeron, R.; Kim, J.K.; Cushman, S.W.; Cooney, G.J.; et al. Mechanism by Which Fatty Acids Inhibit Insulin Activation of Insulin Receptor Substrate-1 (IRS-1)-Associated Phosphatidylinositol 3-Kinase Activity in Muscle. J. Biol. Chem. 2002, 277, 50230–50236. [Google Scholar] [CrossRef] [Green Version]
- Itani, S.I.; Ruderman, N.B.; Schmieder, F.; Boden, G. Lipid-Induced Insulin Resistance in Human Muscle Is Associated with Changes in Diacylglycerol, Protein Kinase C, and IkappaB-Alpha. Diabetes 2002, 51, 2005–2011. [Google Scholar] [CrossRef] [Green Version]
- Bergman, B.C.; Hunerdosse, D.M.; Kerege, A.; Playdon, M.C.; Perreault, L. Localisation and Composition of Skeletal Muscle Diacylglycerol Predicts Insulin Resistance in Humans. Diabetologia 2012, 55, 1140–1150. [Google Scholar] [CrossRef] [Green Version]
- Timmers, S.; Schrauwen, P.; de Vogel, J. Muscular Diacylglycerol Metabolism and Insulin Resistance. Physiol. Behav. 2008, 94, 242–251. [Google Scholar] [CrossRef]
- Terry, J.G.; Hartley, K.G.; Steffen, L.M.; Nair, S.; Alman, A.C.; Wellons, M.F.; Jacobs, D.R.; Tindle, H.A.; Carr, J.J. Association of smoking with abdominal adipose deposition and muscle composition in Coronary Artery Risk Development in Young Adults (CARDIA) participants at mid-life: A population-based cohort study. PLoS Med. 2020, 17, e1003223. [Google Scholar] [CrossRef] [PubMed]
- Haykowsky, M.J.; Nicklas, B.J.; Brubaker, P.H.; Hundley, W.G.; Brinkley, T.E.; Upadhya, B.; Becton, J.T.; Nelson, M.D.; Chen, H.; Kitzman, D.W. Regional Adipose Distribution and Its Relationship to Exercise Intolerance in Older Obese Patients Who Have Heart Failure with Preserved Ejection Fraction. JACC Heart Fail. 2018, 6, 640–649. [Google Scholar] [CrossRef] [PubMed]
- Schwenzer, N.F.; Machann, J.; Martirosian, P.; Stefan, N.; Schraml, C.; Fritsche, A.; Claussen, C.D.; Schick, F. Quantification of Pancreatic Lipomatosis and Liver Steatosis by MRI: Comparison of in/Opposed-Phase and Spectral-Spatial Excitation Techniques. Investig. Radiol. 2008, 43, 330–337. [Google Scholar] [CrossRef] [PubMed]
- Ji, J.; Petropavlovskaia, M.; Khatchadourian, A.; Patapas, J.; Makhlin, J.; Rosenberg, L.; Maysinger, D. Type 2 Diabetes Is Associated with Suppression of Autophagy and Lipid Accumulation in β-Cells. J. Cell Mol. Med. 2019, 23, 2890–2900. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tong, X.; Dai, C.; Walker, J.T.; Nair, G.G.; Kennedy, A.; Carr, R.M.; Hebrok, M.; Powers, A.C.; Stein, R. Lipid Droplet Accumulation in Human Pancreatic Islets Is Dependent on Both Donor Age and Health. Diabetes 2020, 69, 342–354. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Promes, J.A.; Harata, M.; Mishra, A.; Stephens, S.B.; Taylor, E.B.; Burand, A.J.; Sivitz, W.I.; Fink, B.D.; Ankrum, J.A.; et al. Adipose Triglyceride Lipase Is a Key Lipase for the Mobilization of Lipid Droplets in Human β-Cells and Critical for the Maintenance of Syntaxin 1A Levels in β-Cells. Diabetes 2020, 69, 1178–1192. [Google Scholar] [CrossRef]
- Saisho, Y.; Butler, A.; Meier, J.; Monchamp, T.; Allen-Auerbach, M.; Rizza, R.; Butler, P. Pancreas volumes in humans from birth to age one hundred taking into account sex, obesity, and presence of type-2 diabetes. Clin. Anat. 2007, 20, 933–942. [Google Scholar] [CrossRef] [Green Version]
- Tariq, H.; Nayudu, S.; Akella, S.; Glandt, M.; Chilimuri, S. Non-Alcoholic Fatty Pancreatic Disease: A Review of Literature. Gastroenterol. Res. 2016, 9, 87–91. [Google Scholar] [CrossRef]
- Dite, P.; Blaho, M.; Bojkova, M.; Jabandziev, P.; Kunovsky, L. Nonalcoholic Fatty Pancreas Disease: Clinical Consequences. Dig. Dis. 2020, 38, 143–149. [Google Scholar] [CrossRef]
- Petrov, M.S.; Taylor, R. Intra-pancreatic fat deposition: Bringing hidden fat to the fore. Nat. Rev. Gastroenterol. Hepatol. 2021, 19, 153–168. [Google Scholar] [CrossRef]
- Ozturk, K.; Dogan, T.; Celikkanat, S.; Ozen, A.; Demirci, H.; Kurt, O.; Turker, T.; Yilmaz, Y.; Uygun, A. The Association of Fatty Pancreas with Subclinical Atherosclerosis in Nonalcoholic Fatty Liver Disease. Eur. J. Gastroenterol. Hepatol. 2018, 30, 411–417. [Google Scholar] [CrossRef] [PubMed]
- Kul, S.; Karadeniz, A.; Dursun, I.; Şahin, S.; Çirakoglu, Ö.F.; Sayin, M.R.; Turan, T.; Ateş, A.H. Non-Alcoholic Fatty Pancreas Disease Is Associated with Increased Epicardial Adipose Tissue and Aortic Intima-Media Thickness. Acta Cardiol. Sin. 2019, 35, 118–125. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.H.; Kim, J.Y.; Yang, H.R. Ectopic Pancreatic Fat as a Risk Factor for Hypertension in Children and Adolescents with Nonalcoholic Fatty Liver Disease. J. Clin. Hypertens. 2021, 23, 1506–1515. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.X.; Sun, W.; Kong, X.Q. Perirenal Fat: A Unique Fat Pad and Potential Target for Cardiovascular Disease. Angiology 2019, 70, 584–593. [Google Scholar] [CrossRef]
- Ricci, M.A.; Scavizzi, M.; Ministrini, S.; De Vuono, S.; Pucci, G.; Lupattelli, G. Morbid obesity and hypertension: The role of perirenal fat. J. Clin. Hypertens. 2018, 20, 1430–1437. [Google Scholar] [CrossRef] [Green Version]
- De Pergola, G.; Campobasso, N.; Nardecchia, A.; Triggiani, V.; Caccavo, D.; Gesualdo, L.; Silvestris, F.; Manno, C. Para- and perirenal ultrasonographic fat thickness is associated with 24-hours mean diastolic blood pressure levels in overweight and obese subjects. BMC Cardiovasc. Disord. 2015, 15, 108. [Google Scholar] [CrossRef] [Green Version]
- Manno, C.; Campobasso, N.; Nardecchia, A.; Triggiani, V.; Zupo, R.; Gesualdo, L.; Silvestris, F.; de Pergola, G. Relationship of Para- and Perirenal Fat and Epicardial Fat with Metabolic Parameters in Overweight and Obese Subjects. Eat. Weight Disord. 2019, 24, 67–72. [Google Scholar] [CrossRef]
- Vlachos, I.S.; Hatziioannou, A.; Perelas, A.; Perrea, D.N. Sonographic Assessment of Regional Adiposity. AJR Am. J. Roentgenol. 2007, 189, 1545–1553. [Google Scholar] [CrossRef]
- Rebuffé-Scrive, M.; Anderson, B.; Olbe, L.; Björntorp, P. Metabolism of Adipose Tissue in Intraabdominal Depots in Severely Obese Men and Women. Metabolism 1990, 39, 1021–1025. [Google Scholar] [CrossRef]
- Tchernof, A.; Després, J.P. Pathophysiology of Human Visceral Obesity: An Update. Physiol. Rev. 2013, 93, 359–404. [Google Scholar] [CrossRef] [Green Version]
- Liu, K.H.; Chan, Y.L.; Chan, W.B.; Kong, W.L.; Kong, M.O.; Chan, J.C.N. Sonographic Measurement of Mesenteric Fat Thickness Is a Good Correlate with Cardiovascular Risk Factors: Comparison with Subcutaneous and Preperitoneal Fat Thickness, Magnetic Resonance Imaging and Anthropometric Indexes. Int. J. Obes. Relat. Metab. Disord. 2003, 27, 1267–1273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kin, H.L.; Yu, L.C.; Wing, B.C.; Chan, J.C.N.; Chu, C.W.W. Mesenteric Fat Thickness Is an Independent Determinant of Metabolic Syndrome and Identifies Subjects with Increased Carotid Intima-Media Thickness. Diabetes Care 2006, 29, 379–384. [Google Scholar] [CrossRef] [Green Version]
- Wagdy, W.; Mohamad, M.A.; El Hamed, W.R.A.; Motaweih, A.K. Correlation between Mesenteric Fat Thickness and Characteristics of Coronary Artery Disease in Patients with Metabolic Syndrome. J. Clin. Exp. Cardiol. 2018, 9, 1–6. [Google Scholar] [CrossRef]
- Mathieu, P.; Pibarot, P.; Larose, É.; Poirier, P.; Marette, A.; Després, J.P. Visceral Obesity and the Heart. Int. J. Biochem. Cell Biol. 2008, 40, 821–836. [Google Scholar] [CrossRef] [PubMed]
- Mazurek, T.; Zhang, L.F.; Zalewski, A.; Mannion, J.D.; Diehl, J.T.; Arafat, H.; Sarov-Blat, L.; O’Brien, S.; Keiper, E.A.; Johnson, A.G.; et al. Human Epicardial Adipose Tissue Is a Source of Inflammatory Mediators. Circulation 2003, 108, 2460–2466. [Google Scholar] [CrossRef] [Green Version]
- Després, J.P.; Lemieux, I. Abdominal Obesity and Metabolic Syndrome. Nature 2006, 444, 881–887. [Google Scholar] [CrossRef] [PubMed]
- Bornachea, O.; Vea, A.; Llorente-Cortes, V. Interplay between Epicardial Adipose Tissue, Metabolic and Cardiovascular Diseases. Clin. Investig. Arterioscler. 2018, 30, 230–239. [Google Scholar] [CrossRef]
- Sacks, H.S.; Fain, J.N.; Holman, B.; Cheema, P.; Chary, A.; Parks, F.; Karas, J.; Optican, R.; Bahouth, S.W.; Garrett, E.; et al. Uncoupling Protein-1 and Related Messenger Ribonucleic Acids in Human Epicardial and Other Adipose Tissues: Epicardial Fat Functioning as Brown Fat. J. Clin. Endocrinol. Metab. 2009, 94, 3611–3615. [Google Scholar] [CrossRef]
- Iacobellis, G.; Bianco, A.C. Epicardial Adipose Tissue: Emerging Physiological, Pathophysiological and Clinical Features. Trends Endocrinol. Metab. 2011, 22, 450–457. [Google Scholar] [CrossRef] [Green Version]
- Iozzo, P. Metabolic Toxicity of the Heart: Insights from Molecular Imaging. Nutr. Metab. Cardiovasc. Dis. 2010, 20, 147–156. [Google Scholar] [CrossRef]
- Deng, G.; Long, Y.; Yu, Y.R.; Li, M.R. Adiponectin Directly Improves Endothelial Dysfunction in Obese Rats through the AMPK-ENOS Pathway. Int. J. Obes. 2010, 34, 165–171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, R.; Wang, W.-Q.; Zhang, H.; Yang, X.; Fan, Q.; Christopher, T.A.; Lopez, B.L.; Tao, L.; Goldstein, B.J.; Gao, F.; et al. Adiponectin improves endothelial function in hyperlipidemic rats by reducing oxidative/nitrative stress and differential regulation of eNOS/iNOS activity. Am. J. Physiol. Metab. 2007, 293, E1703–E1708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheung, B.M.Y.; Li, C.Y.Y.; Wong, L.Y.F. Adrenomedullin: Its Role in the Cardiovascular System. Semin. Vasc. Med. 2004, 4, 129–134. [Google Scholar] [CrossRef] [PubMed]
- Dzielińska, Z.; Januszewicz, A.; Wiȩcek, A.; Demkow, M.; Makowiecka-Cieśla, M.; Prejbisz, A.; Kądziela, J.; Mielniczuk, R.; Florczak, E.; Janas, J.; et al. Decreased Plasma Concentration of a Novel Anti-Inflammatory Protein—Adiponectin—In Hypertensive Men with Coronary Artery Disease. Thromb. Res. 2003, 110, 365–369. [Google Scholar] [CrossRef]
- Chen, W.J.; Greulich, S.; van der Meer, R.W.; Rijzewijk, L.J.; Lamb, H.J.; de Roos, A.; Smit, J.W.; Romijn, J.A.; Ruige, J.B.; Lammertsma, A.A.; et al. Activin a is associated with impaired myocardial glucose metabolism and left ventricular remodeling in patients with uncomplicated type 2 diabetes. Cardiovasc. Diabetol. 2013, 12, 150. [Google Scholar] [CrossRef] [Green Version]
- Smith, C.; Yndestad, A.; Halvorsen, B.; Ueland, T.; Wæhre, T.; Otterdal, K.; Scholz, H.; Endresen, K.; Gullestad, L.; Frøland, S.S.; et al. Potential Anti-Inflammatory Role of Activin A in Acute Coronary Syndromes. J. Am. Coll. Cardiol. 2004, 44, 369–375. [Google Scholar] [CrossRef] [Green Version]
- Mastaitis, J.; Eckersdorff, M.; Min, S.; Xin, Y.; Cavino, K.; Aglione, J.; Okamoto, H.; Na, E.; Stitt, T.; Dominguez, M.G.; et al. Loss of SFRP4 Alters Body Size, Food Intake, and Energy Expenditure in Diet-Induced Obese Male Mice. Endocrinology 2015, 156, 4502–4510. [Google Scholar] [CrossRef] [Green Version]
- Ji, Q.; Zhang, J.; Du, Y.; Zhu, E.; Wang, Z.; Que, B.; Miao, H.; Shi, S.; Qin, X.; Zhao, Y.; et al. Human epicardial adipose tissue-derived and circulating secreted frizzled-related protein 4 (SFRP4) levels are increased in patients with coronary artery disease. Cardiovasc. Diabetol. 2017, 16, 133. [Google Scholar] [CrossRef] [Green Version]
- Patel, V.B.; Shah, S.; Verma, S.; Oudit, G.Y. Epicardial Adipose Tissue as a Metabolic Transducer: Role in Heart Failure and Coronary Artery Disease. Heart Fail. Rev. 2017, 22, 889–902. [Google Scholar] [CrossRef]
- Packer, M. Epicardial Adipose Tissue May Mediate Deleterious Effects of Obesity and Inflammation on the Myocardium. J. Am. Coll. Cardiol. 2018, 71, 2360–2372. [Google Scholar] [CrossRef]
- Konwerski, M.; Gąsecka, A.; Opolski, G.; Grabowski, M.; Mazurek, T. Role of Epicardial Adipose Tissue in Cardiovascular Diseases: A Review. Biology 2022, 11, 355. [Google Scholar] [CrossRef] [PubMed]
- Gaborit, B.; Abdesselam, I.; Dutour, A. Epicardial Fat: More than Just an “Epi” Phenomenon? Horm. Metab. Res. 2013, 45, 991–1001. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.F.; Zalewski, A.; Liu, Y.; Mazurek, T.; Cowan, S.; Martin, J.L.; Hofmann, S.M.; Vlassara, H.; Shi, Y. Diabetes-Induced Oxidative Stress and Low-Grade Inflammation in Porcine Coronary Arteries. Circulation 2003, 108, 472–478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eiras, S.; Teijeira-Fernández, E.; Shamagian, L.G.; Fernandez, A.L.; Vazquez-Boquete, A.; Gonzalez-Juanatey, J.R. Extension of Coronary Artery Disease Is Associated with Increased IL-6 and Decreased Adiponectin Gene Expression in Epicardial Adipose Tissue. Cytokine 2008, 43, 174–180. [Google Scholar] [CrossRef]
- Iacobellis, G.; Pistilli, D.; Gucciardo, M.; Leonetti, F.; Miraldi, F.; Brancaccio, G.; Gallo, P.; Di Gioia, C.R.T. Adiponectin Expression in Human Epicardial Adipose Tissue In Vivo Is Lower in Patients with Coronary Artery Disease. Cytokine 2005, 29, 251–255. [Google Scholar] [CrossRef]
- Iacobellis, G.; Malavazos, A.E.; Corsi, M.M. Epicardial Fat: From the Biomolecular Aspects to the Clinical Practice. Int. J. Biochem. Cell Biol. 2011, 43, 1651–1654. [Google Scholar] [CrossRef]
- Raman, P.; Khanal, S. Leptin in Atherosclerosis: Focus on Macrophages, Endothelial and Smooth Muscle Cells. Int. J. Mol. Sci. 2021, 22, 5446. [Google Scholar] [CrossRef]
- Zhao, S.; Kusminski, C.M.; Elmquist, J.K.; Scherer, P.E. Leptin: Less Is More. Diabetes 2020, 69, 823–829. [Google Scholar] [CrossRef]
- Katsiki, N.; Mikhailidis, D.P.; Banach, M. Leptin, Cardiovascular Diseases and Type 2 Diabetes Mellitus. Acta Pharmacol. Sin. 2018, 39, 1176–1188. [Google Scholar] [CrossRef] [Green Version]
- Kitzman, D.W.; Shah, S.J. The HFpEF Obesity Phenotype: The Elephant in the Room. J. Am. Coll. Cardiol. 2016, 68, 200–203. [Google Scholar] [CrossRef]
- van Woerden, G.; Gorter, T.M.; Westenbrink, B.D.; Willems, T.P.; van Veldhuisen, D.J.; Rienstra, M. Epicardial Fat in Heart Failure Patients with Mid-Range and Preserved Ejection Fraction. Eur. J. Heart Fail. 2018, 20, 1559–1566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fontes-Carvalho, R.; Fontes-Oliveira, M.; Sampaio, F.; Mancio, J.; Bettencourt, N.; Teixeira, M.; Rocha Gonçalves, F.; Gama, V.; Leite-Moreira, A. Influence of Epicardial and Visceral Fat on Left Ventricular Diastolic and Systolic Functions in Patients after Myocardial Infarction. Am. J. Cardiol. 2014, 114, 1663–1669. [Google Scholar] [CrossRef] [PubMed]
- Doesch, C.; Haghi, D.; Flüchter, S.; Suselbeck, T.; Schoenberg, S.O.; Michaely, H.; Borggrefe, M.; Papavassiliu, T. Epicardial adipose tissue in patients with heart failure. J. Cardiovasc. Magn. Reson. 2010, 12, 40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doesch, C.; Streitner, F.; Bellm, S.; Suselbeck, T.; Haghi, D.; Heggemann, F.; Schoenberg, S.O.; Michaely, H.; Borggrefe, M.; Papavassiliu, T. Epicardial Adipose Tissue Assessed by Cardiac Magnetic Resonance Imaging in Patients with Heart Failure Due to Dilated Cardiomyopathy. Obesity 2013, 21, E253–E261. [Google Scholar] [CrossRef] [Green Version]
- Iacobellis, G. Epicardial adipose tissue in contemporary cardiology. Nat. Rev. Cardiol. 2022, 19, 593–606. [Google Scholar] [CrossRef]
- Zhou, M.; Wang, H.; Chen, J.; Zhao, L. Epicardial Adipose Tissue and Atrial Fibrillation: Possible Mechanisms, Potential Therapies, and Future Directions. Pacing Clin. Electrophysiol. 2020, 43, 133–145. [Google Scholar] [CrossRef]
- Agra, R.M.; Teijeira-Fernández, E.; Pascual-Figal, D.; Sánchez-Más, J.; Fernández-Trasancos, Á.; González-Juanatey, J.R.; Eiras, S. Adiponectin and P53 MRNA in Epicardial and Subcutaneous Fat from Heart Failure Patients. Eur. J. Clin. Investig. 2014, 44, 29–37. [Google Scholar] [CrossRef]
- Wu, C.K.; Tsai, H.Y.; Su, M.Y.M.; Wu, Y.F.; Hwang, J.J.; Lin, J.L.; Lin, L.Y.; Chen, J.J. Evolutional Change in Epicardial Fat and Its Correlation with Myocardial Diffuse Fibrosis in Heart Failure Patients. J. Clin. Lipidol. 2017, 11, 1421–1431. [Google Scholar] [CrossRef]
- Parisi, V.; Rengo, G.; Perrone-Filardi, P.; Pagano, G.; Femminella, G.D.; Paolillo, S.; Petraglia, L.; Gambino, G.; Caruso, A.; Grimaldi, M.G.; et al. Increased Epicardial Adipose Tissue Volume Correlates with Cardiac Sympathetic Denervation in Patients with Heart Failure. Circ. Res. 2016, 118, 1244–1253. [Google Scholar] [CrossRef] [Green Version]
- Nerlekar, N.; Muthalaly, R.G.; Wong, N.; Thakur, U.; Wong, D.T.L.; Brown, A.J.; Marwick, T.H. Association of Volumetric Epicardial Adipose Tissue Quantification and Cardiac Structure and Function. J. Am. Heart Assoc. 2018, 7, e009975. [Google Scholar] [CrossRef]
- Obokata, M.; Reddy, Y.N.V.; Pislaru, S.V.; Melenovsky, V.; Borlaug, B.A. Evidence Supporting the Existence of a Distinct Obese Phenotype of Heart Failure with Preserved Ejection Fraction. Circulation 2017, 136, 6–19. [Google Scholar] [CrossRef] [PubMed]
- Koepp, K.E.; Obokata, M.; Reddy, Y.N.V.; Olson, T.P.; Borlaug, B.A. Hemodynamic and Functional Impact of Epicardial Adipose Tissue in Heart Failure with Preserved Ejection Fraction. JACC Heart Fail. 2020, 8, 657–666. [Google Scholar] [CrossRef] [PubMed]
- Kallistratos, M.S.; Poulimenos, L.E.; Manolis, A.J. Atrial Fibrillation and Arterial Hypertension. Pharmacol. Res. 2018, 128, 322–326. [Google Scholar] [CrossRef] [PubMed]
- Kurokawa, S.; Okumura, Y. Atrial Fibrillation with Valvular Heart Disease—New Insight into Clinical Outcomes. Circ. J. 2020, 84, 697–699. [Google Scholar] [CrossRef] [Green Version]
- Hu, Y.F.; Chen, Y.J.; Lin, Y.J.; Chen, S.A. Inflammation and the Pathogenesis of Atrial Fibrillation. Nat. Rev. Cardiol. 2015, 12, 230–243. [Google Scholar] [CrossRef]
- Karam, B.S.; Chavez-Moreno, A.; Koh, W.; Akar, J.G.; Akar, F.G. Oxidative stress and inflammation as central mediators of atrial fibrillation in obesity and diabetes. Cardiovasc. Diabetol. 2017, 16, 1–9. [Google Scholar] [CrossRef]
- Harada, M.; Nattel, S. Implications of Inflammation and Fibrosis in Atrial Fibrillation Pathophysiology. Card. Electrophysiol. Clin. 2021, 13, 25–35. [Google Scholar] [CrossRef]
- Liu, Q.; Zhang, F.; Yang, M.; Zhong, J. Increasing Level of Interleukin-1β in Epicardial Adipose Tissue Is Associated with Persistent Atrial Fibrillation. J. Interf. Cytokine Res. 2020, 40, 64–69. [Google Scholar] [CrossRef]
- Tse, G.; Yan, B.P.; Chan, Y.W.F.; Tian, X.Y.; Huang, Y. Reactive Oxygen Species, Endoplasmic Reticulum Stress and Mitochondrial Dysfunction: The Link with Cardiac Arrhythmogenesis. Front. Physiol. 2016, 7, 313. [Google Scholar] [CrossRef] [Green Version]
- Haemers, P.; Hamdi, H.; Guedj, K.; Suffee, N.; Farahmand, P.; Popovic, N.; Claus, P.; LePrince, P.; Nicoletti, A.; Jalife, J.; et al. Atrial Fibrillation Is Associated with the Fibrotic Remodelling of Adipose Tissue in the Subepicardium of Human and Sheep Atria. Eur. Heart J. 2017, 38, 53–61. [Google Scholar] [CrossRef]
- Iacobellis, G.; Singh, N.; Wharton, S.; Sharma, A.M. Substantial Changes in Epicardial Fat Thickness After Weight Loss in Severely Obese Subjects. Obesity 2008, 16, 1693–1697. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.K.; Tanaka, K.; Kim, M.J.; Matuso, T.; Endo, T.; Tomita, T.; Maeda, S.; Ajisaka, R. Comparison of Epicardial, Abdominal and Regional Fat Compartments in Response to Weight Loss. Nutr. Metab. Cardiovasc. Dis. 2009, 19, 760–766. [Google Scholar] [CrossRef] [PubMed]
- Borges, J.H.; Carter, S.J.; Bryan, D.R.; Hunter, G.R. Exercise Training and/or Diet on Reduction of Intra-Abdominal Adipose Tissue and Risk Factors for Cardiovascular Disease. Eur. J. Clin. Nutr. 2019, 73, 1063. [Google Scholar] [CrossRef]
- Colonetti, T.; Grande, A.J.; Amaral, M.C.; Colonetti, L.; Uggioni, M.L.; da Rosa, M.I.; Hernandez, A.V.; Tse, G.; Liu, T.; Nerlekar, N.; et al. Effect of Exercise on Epicardial Adipose Tissue in Adults: A Systematic Review and Meta-Analyses. Heart Fail. Rev. 2021, 26, 1399–1411. [Google Scholar] [CrossRef]
- Christensen, R.H.; Wedell-Neergaard, A.S.; Lehrskov, L.L.; Legaard, G.E.; Dorph, E.; Larsen, M.K.; Launbo, N.; Fagerlind, S.R.; Seide, S.K.; Nymand, S.; et al. Effect of Aerobic and Resistance Exercise on Cardiac Adipose Tissues: Secondary Analyses from a Randomized Clinical Trial. JAMA Cardiol. 2019, 4, 778–787. [Google Scholar] [CrossRef] [PubMed]
- Ard, J.; Fitch, A.; Fruh, S.; Herman, L. Weight Loss and Maintenance Related to the Mechanism of Action of Glucagon-Like Peptide 1 Receptor Agonists. Adv. Ther. 2021, 38, 2821. [Google Scholar] [CrossRef]
- Bertoccini, L.; Baroni, M.G. GLP-1 Receptor Agonists and SGLT2 Inhibitors for the Treatment of Type 2 Diabetes: New Insights and Opportunities for Cardiovascular Protection. Adv. Exp. Med. Biol. 2021, 1307, 193–212. [Google Scholar] [CrossRef]
- Ma, X.; Liu, Z.; Ilyas, I.; Little, P.J.; Kamato, D.; Sahebka, A.; Chen, Z.; Luo, S.; Zheng, X.; Weng, J.; et al. GLP-1 Receptor Agonists (GLP-1RAs): Cardiovascular Actions and Therapeutic Potential. Int. J. Biol. Sci. 2021, 17, 2050–2068. [Google Scholar] [CrossRef]
- Iacobellis, G.; Camarena, V.; Sant, D.W.; Wang, G. Human Epicardial Fat Expresses Glucagon-Like Peptide 1 and 2 Receptors Genes. Horm. Metab. Res. 2017, 49, 625–630. [Google Scholar] [CrossRef]
- Dozio, E.; Vianello, E.; Malavazos, A.E.; Tacchini, L.; Schmitz, G.; Iacobellis, G.; Corsi Romanelli, M.M. Epicardial Adipose Tissue GLP-1 Receptor Is Associated with Genes Involved in Fatty Acid Oxidation and White-to-Brown Fat Differentiation: A Target to Modulate Cardiovascular Risk? Int. J. Cardiol. 2019, 292, 218–224. [Google Scholar] [CrossRef]
- Iacobellis, G.; Mohseni, M.; Bianco, S.D.; Banga, P.K. Liraglutide Causes Large and Rapid Epicardial Fat Reduction. Obesity 2017, 25, 311–316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morano, S.; Romagnoli, E.; Filardi, T.; Nieddu, L.; Mandosi, E.; Fallarino, M.; Turinese, I.; Dagostino, M.P.; Lenzi, A.; Carnevale, V. Short-Term Effects of Glucagon-like Peptide 1 (GLP-1) Receptor Agonists on Fat Distribution in Patients with Type 2 Diabetes Mellitus: An Ultrasonography Study. Acta Diabetol. 2015, 52, 727–732. [Google Scholar] [CrossRef] [PubMed]
- Iacobellis, G.; Villasante Fricke, A.C. Effects of Semaglutide Versus Dulaglutide on Epicardial Fat Thickness in Subjects with Type 2 Diabetes and Obesity. J. Endocr. Soc. 2020, 4, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Ludvik, B.; Giorgino, F.; Jódar, E.; Frias, J.P.; Fernández Landó, L.; Brown, K.; Bray, R.; Rodríguez, Á. Once-Weekly Tirzepatide versus Once-Daily Insulin Degludec as Add-on to Metformin with or without SGLT2 Inhibitors in Patients with Type 2 Diabetes (SURPASS-3): A Randomised, Open-Label, Parallel-Group, Phase 3 Trial. Lancet 2021, 398, 583–598. [Google Scholar] [CrossRef]
- Giugliano, D.; Longo, M.; Scappaticcio, L.; Bellastella, G.; Maiorino, M.I.; Esposito, K. SGLT-2 inhibitors and cardiorenal outcomes in patients with or without type 2 diabetes: A meta-analysis of 11 CVOTs. Cardiovasc. Diabetol. 2021, 20, 1–11. [Google Scholar] [CrossRef] [PubMed]
- McMurray, J.J.V.; Solomon, S.D.; Inzucchi, S.E.; Køber, L.; Kosiborod, M.N.; Martinez, F.A.; Ponikowski, P.; Sabatine, M.S.; Anand, I.S.; Bělohlávek, J.; et al. Dapagliflozin in Patients with Heart Failure and Reduced Ejection Fraction. N. Engl. J. Med. 2019, 381, 1995–2008. [Google Scholar] [CrossRef] [Green Version]
- Yagi, S.; Hirata, Y.; Ise, T.; Kusunose, K.; Yamada, H.; Fukuda, D.; Salim, H.M.; Maimaituxun, G.; Nishio, S.; Takagawa, Y.; et al. Canagliflozin reduces epicardial fat in patients with type 2 diabetes mellitus. Diabetol. Metab. Syndr. 2017, 9, 78. [Google Scholar] [CrossRef] [Green Version]
- Sato, T.; Aizawa, Y.; Yuasa, S.; Kishi, S.; Fuse, K.; Fujita, S.; Ikeda, Y.; Kitazawa, H.; Takahashi, M.; Sato, M.; et al. The effect of dapagliflozin treatment on epicardial adipose tissue volume. Cardiovasc. Diabetol. 2018, 17, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Iacobellis, G.; Gra-Menendez, S. Effects of Dapagliflozin on Epicardial Fat Thickness in Patients with Type 2 Diabetes and Obesity. Obesity 2020, 28, 1068–1074. [Google Scholar] [CrossRef]
- Masson, W.; Lavalle-Cobo, A.; Nogueira, J.P. Effect of SGLT2-Inhibitors on Epicardial Adipose Tissue: A Meta-Analysis. Cells 2021, 10, 2150. [Google Scholar] [CrossRef]
- Kenngott, H.G.; Nickel, F.; Wise, P.A.; Wagner, F.; Billeter, A.T.; Nattenmüller, J.; Nabers, D.; Maier-Hein, K.; Kauczor, H.U.; Fischer, L.; et al. Weight Loss and Changes in Adipose Tissue and Skeletal Muscle Volume after Laparoscopic Sleeve Gastrectomy and Roux-En-Y Gastric Bypass: A Prospective Study with 12-Month Follow-Up. Obes. Surg. 2019, 29, 4018–4028. [Google Scholar] [CrossRef] [PubMed]
- Melchor-López, A.; Suárez-Cuenca, J.A.; Banderas-Lares, D.Z.; Peña-Sosa, G.D.L.; Salamanca-García, M.; Vera-Gómez, E.; Hernández-Patricio, A.; Gutiérrez-Buendía, J.A.; Zamora-Alemán, C.R.; Alcaráz-Estrada, S.L.; et al. Identification of Adipose Tissue-Related Predictors of the Reduction in Cardiovascular Risk Induced by Metabolic Surgery. J. Int. Med. Res. 2021, 49, 03000605211012569. [Google Scholar] [CrossRef] [PubMed]
- Willens, H.J.; Byers, P.; Chirinos, J.A.; Labrador, E.; Hare, J.M.; de Marchena, E. Effects of Weight Loss after Bariatric Surgery on Epicardial Fat Measured Using Echocardiography. Am. J. Cardiol. 2007, 99, 1242–1245. [Google Scholar] [CrossRef] [PubMed]
- Altin, C.; Erol, V.; Aydin, E.; Yilmaz, M.; Tekindal, M.A.; Sade, L.E.; Gulay, H.; Muderrisoglu, H. Impact of Weight Loss on Epicardial Fat and Carotid Intima Media Thickness after Laparoscopic Sleeve Gastrectomy: A Prospective Study. Nutr. Metab. Cardiovasc. Dis. 2018, 28, 501–509. [Google Scholar] [CrossRef] [PubMed]
- Voglino, C.; Tirone, A.; Ciuoli, C.; Benenati, N.; Paolini, B.; Croce, F.; Gaggelli, I.; Vuolo, M.L.; Cuomo, R.; Grimaldi, L.; et al. Cardiovascular Benefits and Lipid Profile Changes 5 Years After Bariatric Surgery: A Comparative Study Between Sleeve Gastrectomy and Roux-En-Y Gastric Bypass. J. Gastrointest. Surg. 2020, 24, 2722–2729. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
D’Oria, R.; Genchi, V.A.; Caccioppoli, C.; Calderoni, I.; Marrano, N.; Biondi, G.; Borrelli, A.; Di Gioia, L.; Giorgino, F.; Laviola, L. Impact of Dysfunctional Adipose Tissue Depots on the Cardiovascular System. Int. J. Mol. Sci. 2022, 23, 14296. https://doi.org/10.3390/ijms232214296
D’Oria R, Genchi VA, Caccioppoli C, Calderoni I, Marrano N, Biondi G, Borrelli A, Di Gioia L, Giorgino F, Laviola L. Impact of Dysfunctional Adipose Tissue Depots on the Cardiovascular System. International Journal of Molecular Sciences. 2022; 23(22):14296. https://doi.org/10.3390/ijms232214296
Chicago/Turabian StyleD’Oria, Rossella, Valentina Annamaria Genchi, Cristina Caccioppoli, Isabella Calderoni, Nicola Marrano, Giuseppina Biondi, Anna Borrelli, Ludovico Di Gioia, Francesco Giorgino, and Luigi Laviola. 2022. "Impact of Dysfunctional Adipose Tissue Depots on the Cardiovascular System" International Journal of Molecular Sciences 23, no. 22: 14296. https://doi.org/10.3390/ijms232214296