
Metabolic Effect of Blocking Sodium-Taurocholate Co-Transporting Polypeptide in Hypercholesterolemic Humans with a Twelve-Week Course of Bulevirtide—An Exploratory Phase I Clinical Trial
 , , , ,
, , , ,  , , ,
, , ,
Abstract
:1. Introduction
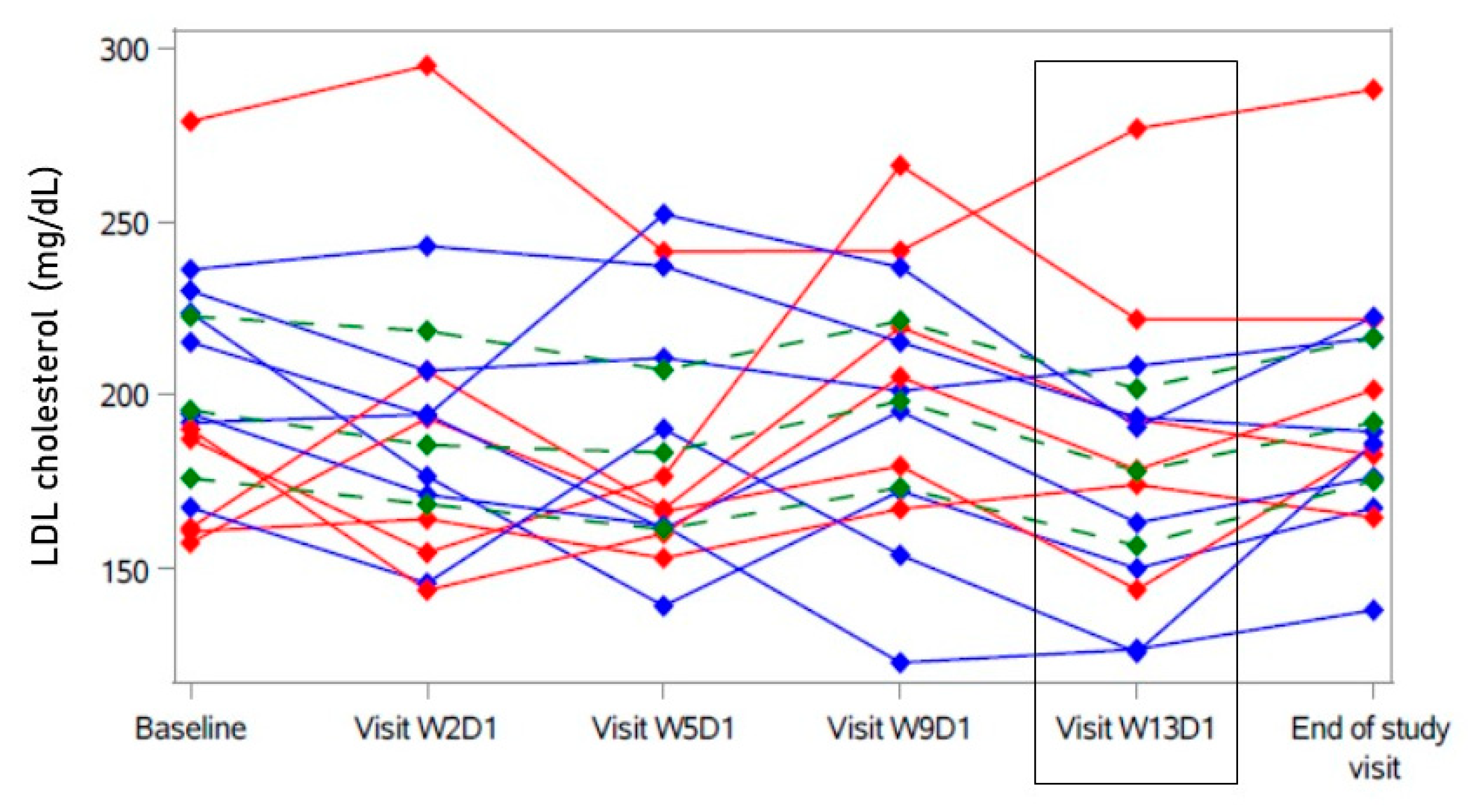
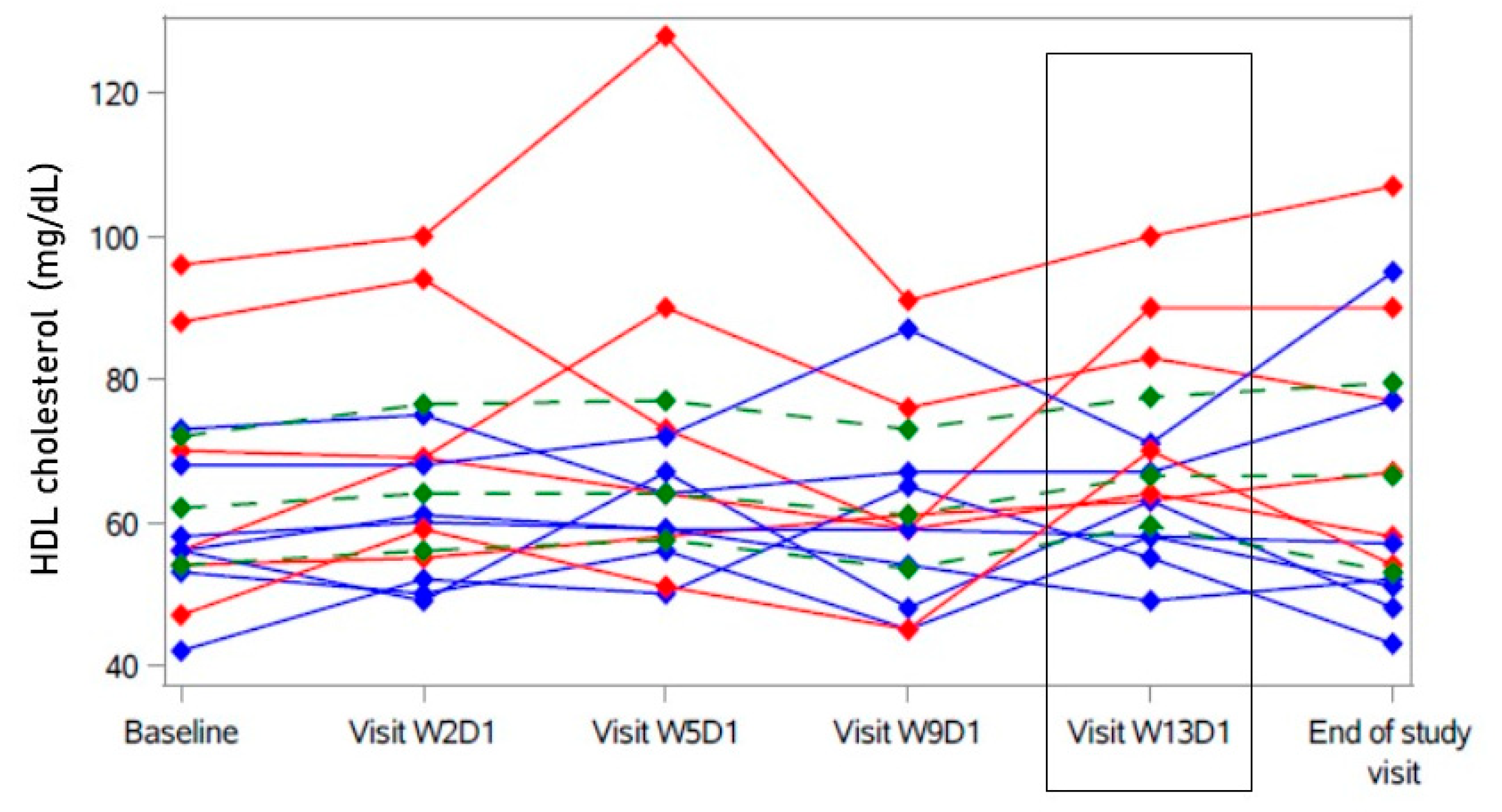
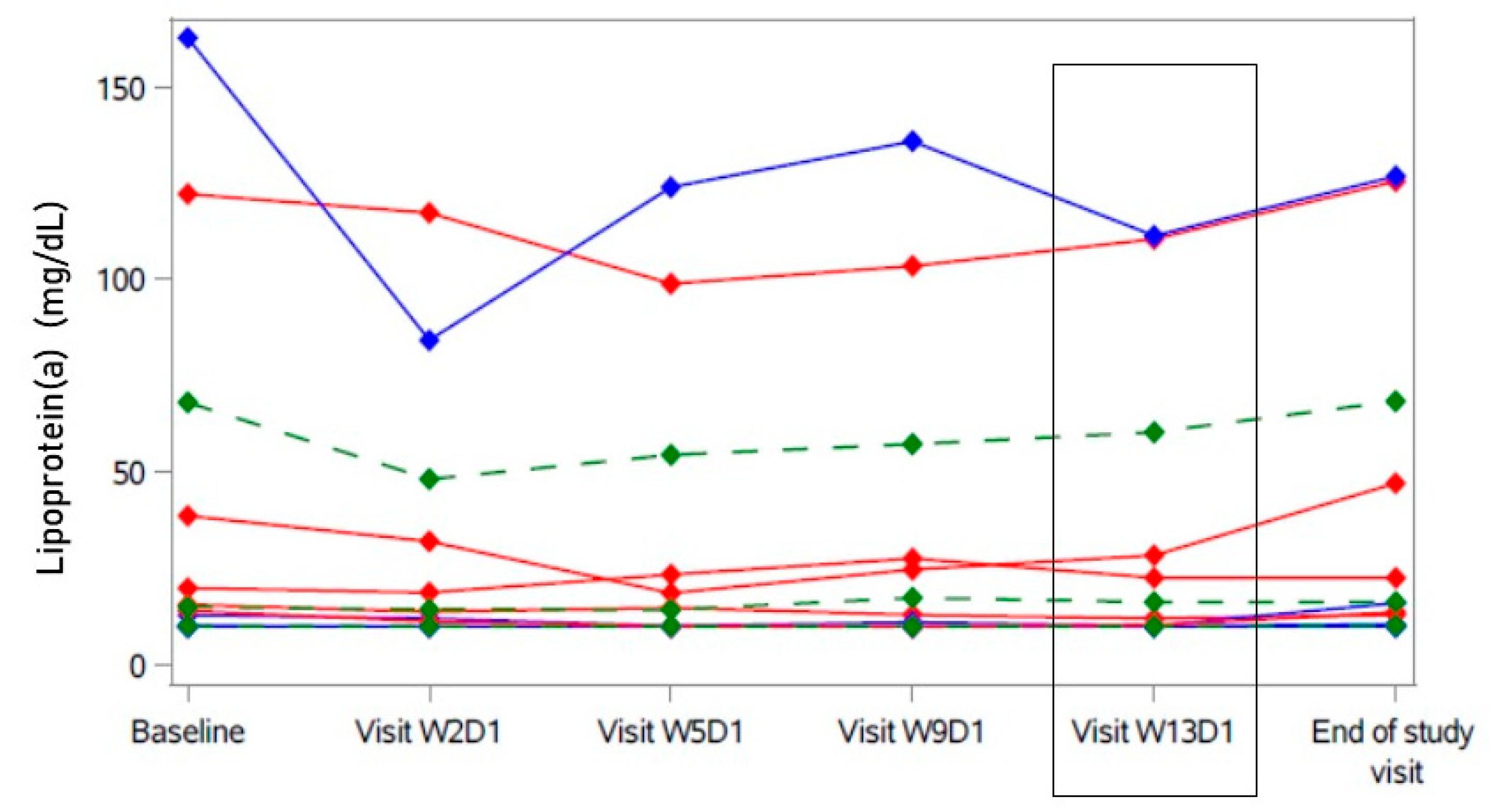
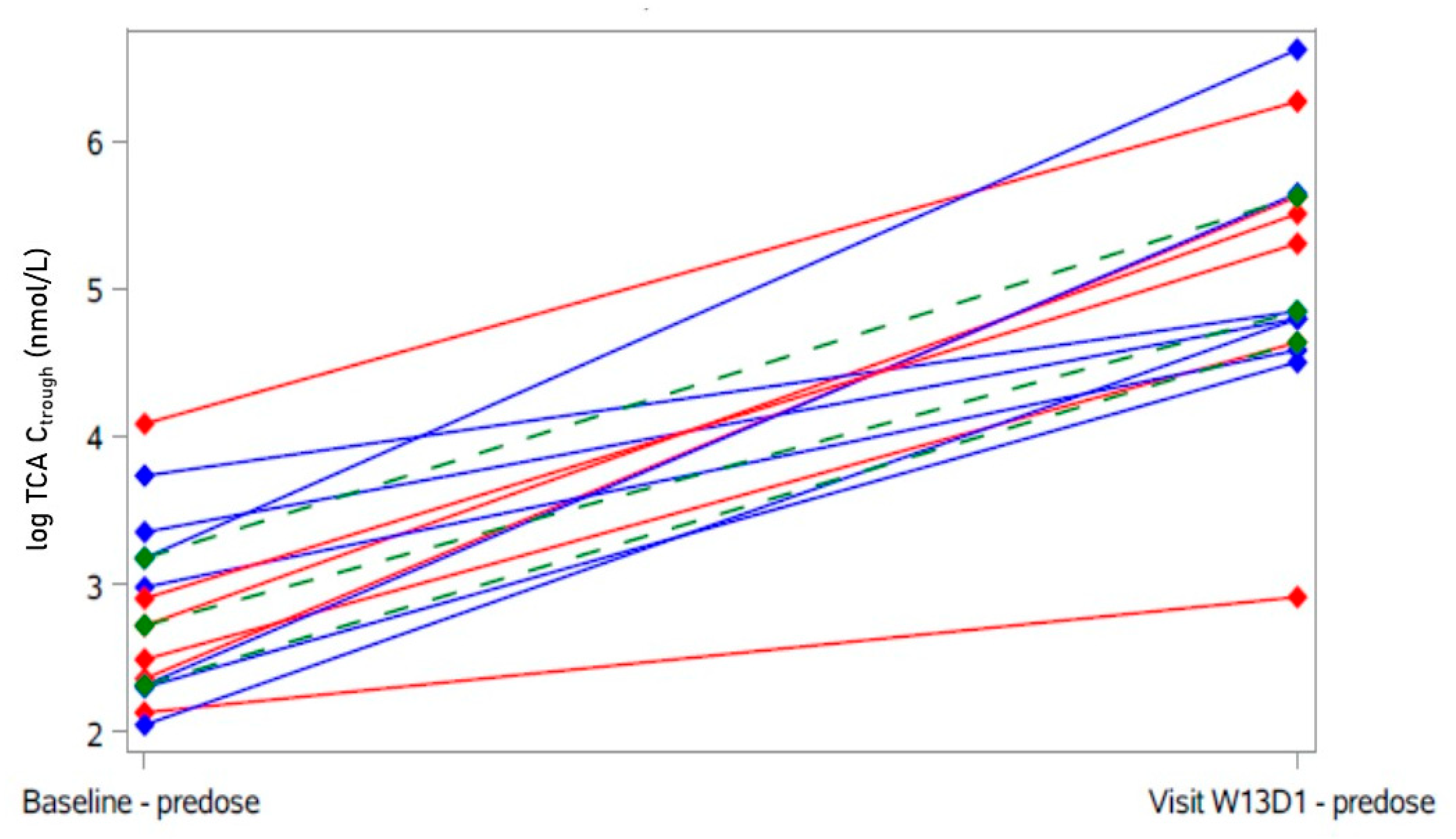
2. Results
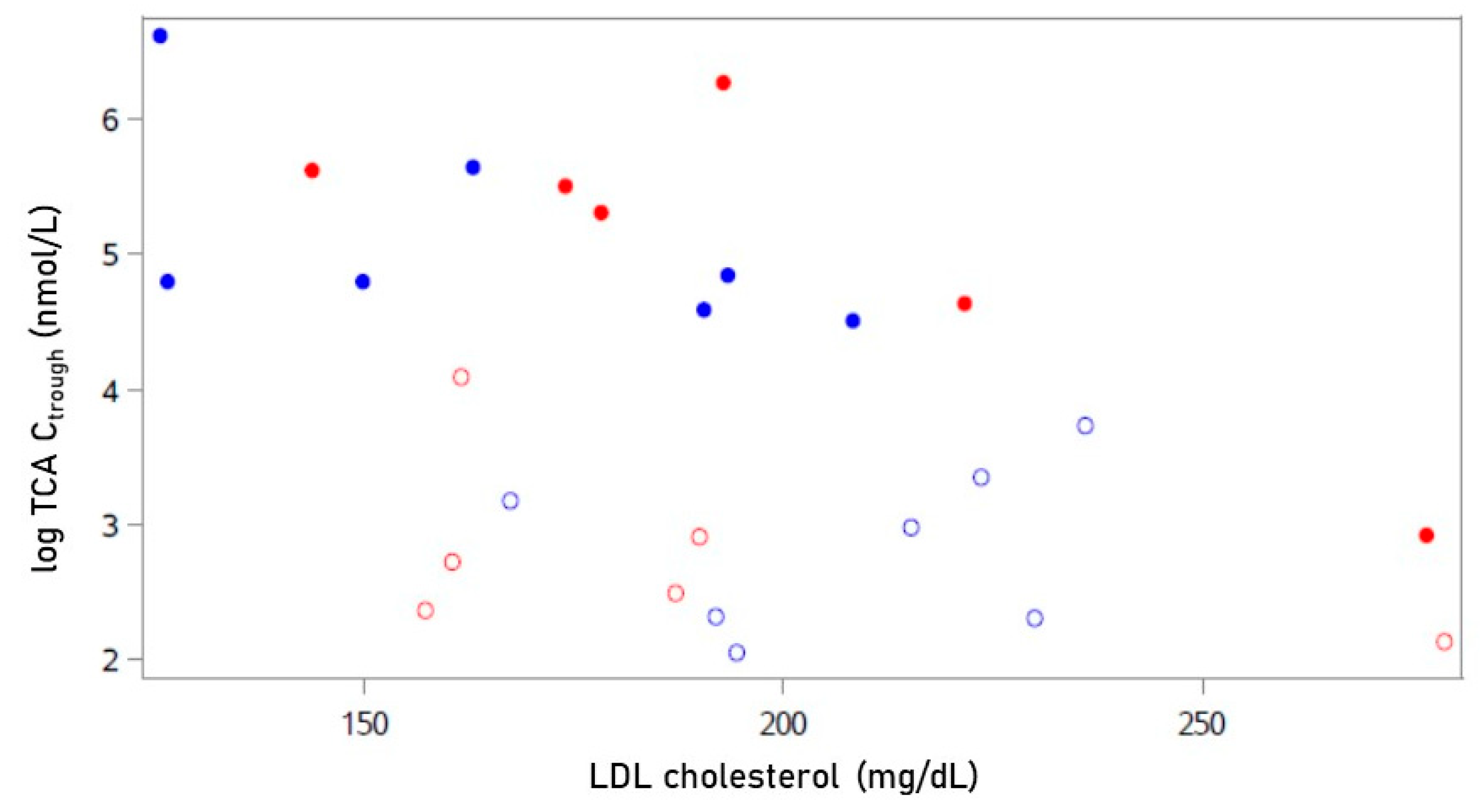
2.1. Baseline Characteristics
2.2. Primary Analysis
2.3. Secondary Analyses
2.4. Safety Assessment
3. Discussion
4. Methods
4.1. Study Design and Setting
4.2. Population
4.3. Intervention
4.4. Endpoints
4.5. Follow-Up Visits
4.6. Assessments
4.6.1. Lipid Profile
4.6.2. Biomarkers
4.6.3. Glucose Metabolism
4.6.4. Cardiac MRI
4.6.5. Pharmacokinetics
4.6.6. Bile Acids
4.6.7. Safety Evaluation
4.7. Statistics
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Timmis, A.; Townsend, N.; Gale, C.P.; Torbica, A.; Lettino, M.; Petersen, S.E.; Mossialos, E.A.; Maggioni, A.P.; Kazakiewicz, D.; May, H.T.; et al. European Society of Cardiology: Cardiovascular Disease Statistics 2019. Eur. Heart J. 2020, 41, 12–85. [Google Scholar] [CrossRef] [Green Version]
- Chiang, J.Y. Regulation of bile acid synthesis: Pathways, nuclear receptors, and mechanisms. J. Hepatol. 2004, 40, 539–551. [Google Scholar] [CrossRef] [PubMed]
- Zhou, E.; Hoeke, G.; Li, Z.; Eibergen, A.C.; Schonk, A.W.; Koehorst, M.; Boverhof, R.; Havinga, R.; Kuipers, F.; Coskun, T.; et al. Colesevelam enhances the beneficial effects of brown fat activation on hyperlipidaemia and atherosclerosis development. Cardiovasc. Res. 2020, 116, 1710–1720. [Google Scholar] [CrossRef] [Green Version]
- Chavez-Talavera, O.; Tailleux, A.; Lefebvre, P.; Staels, B. Bile Acid Control of Metabolism and Inflammation in Obesity, Type 2 Diabetes, Dyslipidemia, and Nonalcoholic Fatty Liver Disease. Gastroenterology 2017, 152, 1679–1694.e3. [Google Scholar] [CrossRef] [PubMed]
- Lefebvre, P.; Cariou, B.; Lien, F.; Kuipers, F.; Staels, B. Role of bile acids and bile acid receptors in metabolic regulation. Physiol. Rev. 2009, 89, 147–191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomas, C.; Gioiello, A.; Noriega, L.; Strehle, A.; Oury, J.; Rizzo, G.; Macchiarulo, A.; Yamamoto, H.; Mataki, C.; Pruzanski, M.; et al. TGR5-mediated bile acid sensing controls glucose homeostasis. Cell Metab. 2009, 10, 167–177. [Google Scholar] [CrossRef] [Green Version]
- Gerhard, G.S.; Styer, A.M.; Wood, G.C.; Roesch, S.L.; Petrick, A.T.; Gabrielsen, J.; Strodel, W.E.; Still, C.D.; Argyropoulos, G. A role for fibroblast growth factor 19 and bile acids in diabetes remission after Roux-en-Y gastric bypass. Diabetes Care 2013, 36, 1859–1864. [Google Scholar] [CrossRef] [Green Version]
- Pullinger, C.R.; Eng, C.; Salen, G.; Shefer, S.; Batta, A.K.; Erickson, S.K.; Verhagen, A.; Rivera, C.R.; Mulvihill, S.J.; Malloy, M.J.; et al. Human cholesterol 7alpha-hydroxylase (CYP7A1) deficiency has a hypercholesterolemic phenotype. J. Clin. Investig. 2002, 110, 109–117. [Google Scholar] [CrossRef]
- Neuschwander-Tetri, B.A.; Loomba, R.; Sanyal, A.J.; Lavine, J.E.; Van Natta, M.L.; Abdelmalek, M.F.; Chalasani, N.; Dasarathy, S.; Diehl, A.M.; Hameed, B.; et al. Farnesoid X nuclear receptor ligand obeticholic acid for non-cirrhotic, non-alcoholic steatohepatitis (FLINT): A multicentre, randomised, placebo-controlled trial. Lancet 2015, 385, 956–965. [Google Scholar] [CrossRef] [Green Version]
- Kullak-Ublick, G.A.; Stieger, B.; Meier, P.J. Enterohepatic bile salt transporters in normal physiology and liver disease. Gastroenterology 2004, 126, 322–342. [Google Scholar] [CrossRef]
- Blank, A.; Markert, C.; Hohmann, N.; Carls, A.; Mikus, G.; Lehr, T.; Alexandrov, A.; Haag, M.; Schwab, M.; Urban, S.; et al. First-in-human application of the novel hepatitis B and hepatitis D virus entry inhibitor myrcludex B. J. Hepatol. 2016, 65, 483–489. [Google Scholar] [CrossRef]
- Lutgehetmann, M.; Mancke, L.V.; Volz, T.; Helbig, M.; Allweiss, L.; Bornscheuer, T.; Pollok, J.M.; Lohse, A.W.; Petersen, J.; Urban, S.; et al. Humanized chimeric uPA mouse model for the study of hepatitis B and D virus interactions and preclinical drug evaluation. Hepatology 2012, 55, 685–694. [Google Scholar] [CrossRef]
- Ni, Y.; Lempp, F.A.; Mehrle, S.; Nkongolo, S.; Kaufman, C.; Falth, M.; Stindt, J.; Koniger, C.; Nassal, M.; Kubitz, R.; et al. Hepatitis B and D viruses exploit sodium taurocholate co-transporting polypeptide for species-specific entry into hepatocytes. Gastroenterology 2014, 146, 1070–1083. [Google Scholar] [CrossRef]
- Petersen, J.; Dandri, M.; Mier, W.; Lutgehetmann, M.; Volz, T.; von Weizsacker, F.; Haberkorn, U.; Fischer, L.; Pollok, J.M.; Erbes, B.; et al. Prevention of hepatitis B virus infection in vivo by entry inhibitors derived from the large envelope protein. Nat. Biotechnol. 2008, 26, 335–341. [Google Scholar] [CrossRef]
- Schulze, A.; Schieck, A.; Ni, Y.; Mier, W.; Urban, S. Fine mapping of pre-S sequence requirements for hepatitis B virus large envelope protein-mediated receptor interaction. J. Virol. 2010, 84, 1989–2000. [Google Scholar] [CrossRef] [Green Version]
- Yan, H.; Zhong, G.; Xu, G.; He, W.; Jing, Z.; Gao, Z.; Huang, Y.; Qi, Y.; Peng, B.; Wang, H.; et al. Sodium taurocholate cotransporting polypeptide is a functional receptor for human hepatitis B and D virus. Elife 2012, 1, e00049. [Google Scholar] [CrossRef]
- Oehler, N.; Volz, T.; Bhadra, O.D.; Kah, J.; Allweiss, L.; Giersch, K.; Bierwolf, J.; Riecken, K.; Pollok, J.M.; Lohse, A.W.; et al. Binding of hepatitis B virus to its cellular receptor alters the expression profile of genes of bile acid metabolism. Hepatology 2014, 60, 1483–1493. [Google Scholar] [CrossRef]
- Slijepcevic, D.; Roscam Abbing, R.L.P.; Katafuchi, T.; Blank, A.; Donkers, J.M.; van Hoppe, S.; de Waart, D.R.; Tolenaars, D.; van der Meer, J.H.M.; Wildenberg, M.; et al. Hepatic uptake of conjugated bile acids is mediated by both sodium taurocholate cotransporting polypeptide and organic anion transporting polypeptides and modulated by intestinal sensing of plasma bile acid levels in mice. Hepatology 2017, 66, 1631–1643. [Google Scholar] [CrossRef] [Green Version]
- Donkers, J.M.; Roscam Abbing, R.L.P.; van Weeghel, M.; Levels, J.H.M.; Boelen, A.; Schinkel, A.H.; Oude Elferink, R.P.J.; van de Graaf, S.F.J. Inhibition of Hepatic Bile Acid Uptake by Myrcludex B Promotes Glucagon-Like Peptide-1 Release and Reduces Obesity. Cell Mol. Gastroenterol. Hepatol. 2020, 10, 451–466. [Google Scholar] [CrossRef]
- Mach, F.; Baigent, C.; Catapano, A.L.; Koskinas, K.C.; Casula, M.; Badimon, L.; Chapman, M.J.; De Backer, G.G.; Delgado, V.; Ference, B.A.; et al. 2019 ESC/EAS Guidelines for the management of dyslipidaemias: Lipid modification to reduce cardiovascular risk. Eur. Heart J. 2020, 41, 111–188. [Google Scholar] [CrossRef]
- Blank, A.; Eidam, A.; Haag, M.; Hohmann, N.; Burhenne, J.; Schwab, M.; van de Graaf, S.; Meyer, M.R.; Maurer, H.H.; Meier, K.; et al. The NTCP-inhibitor Myrcludex B: Effects on Bile Acid Disposition and Tenofovir Pharmacokinetics. Clin. Pharm. 2018, 103, 341–348. [Google Scholar] [CrossRef] [PubMed]
- Sauter, M.; Blank, A.; Stoll, F.; Lutz, N.; Haefeli, W.E.; Burhenne, J. Intact plasma quantification of the large therapeutic lipopeptide bulevirtide. Anal. Bioanal. Chem. 2021, 413, 5645–5654. [Google Scholar] [CrossRef] [PubMed]
- Preiss, D.; Tobert, J.A.; Hovingh, G.K.; Reith, C. Lipid-Modifying Agents, From Statins to PCSK9 Inhibitors: JACC Focus Seminar. J. Am. Coll. Cardiol. 2020, 75, 1945–1955. [Google Scholar] [CrossRef] [PubMed]
- Gherondache, C.N.; Pincus, G. Metabolic Changes Induced in Elderly Patients with a Cholesterol Lowering Resin, Cholestyramine. Metabolism 1964, 13, 1462–1468. [Google Scholar] [CrossRef] [PubMed]
- Schneider, A.L.; Kohler, H.; Rothlisberger, B.; Grobholz, R.; McLin, V.A. Sodium taurocholate co-transporting polypeptide deficiency. Clin. Res. Hepatol. Gastroenterol. 2022, 46, 101824. [Google Scholar] [CrossRef]
- Vaz, F.M.; Paulusma, C.C.; Huidekoper, H.; de Ru, M.; Lim, C.; Koster, J.; Ho-Mok, K.; Bootsma, A.H.; Groen, A.K.; Schaap, F.G.; et al. Sodium taurocholate cotransporting polypeptide (SLC10A1) deficiency: Conjugated hypercholanemia without a clear clinical phenotype. Hepatology 2015, 61, 260–267. [Google Scholar] [CrossRef]
- Stein, E.; Kreisberg, R.; Miller, V.; Mantell, G.; Washington, L.; Shapiro, D.R. Effects of simvastatin and cholestyramine in familial and nonfamilial hypercholesterolemia. Multicenter Group I. Arch. Intern. Med. 1990, 150, 341–345. [Google Scholar] [CrossRef]
- Kullak-Ublick, G.A.; Beuers, U.; Fahney, C.; Hagenbuch, B.; Meier, P.J.; Paumgartner, G. Identification and functional characterization of the promoter region of the human organic anion transporting polypeptide gene. Hepatology 1997, 26, 991–997. [Google Scholar] [CrossRef]
- Trauner, M.; Meier, P.J.; Boyer, J.L. Molecular pathogenesis of cholestasis. N. Engl. J. Med. 1998, 339, 1217–1227. [Google Scholar] [CrossRef]
- Blank, A.; Meier, K.; Urban, S.; Haefeli, W.E.; Weiss, J. Drug-drug interaction potential of the HBV and HDV entry inhibitor myrcludex B assessed in vitro. Antivir. Ther. 2018, 23, 267–275. [Google Scholar] [CrossRef]
- Suga, T.; Yamaguchi, H.; Sato, T.; Maekawa, M.; Goto, J.; Mano, N. Preference of Conjugated Bile Acids over Unconjugated Bile Acids as Substrates for OATP1B1 and OATP1B3. PLoS ONE 2017, 12, e0169719. [Google Scholar] [CrossRef] [Green Version]
- Rohrl, C.; Eigner, K.; Fruhwurth, S.; Stangl, H. Bile acids reduce endocytosis of high-density lipoprotein (HDL) in HepG2 cells. PLoS ONE 2014, 9, e102026. [Google Scholar] [CrossRef] [Green Version]
- Gudbjartsson, D.F.; Thorgeirsson, G.; Sulem, P.; Helgadottir, A.; Gylfason, A.; Saemundsdottir, J.; Bjornsson, E.; Norddahl, G.L.; Jonasdottir, A.; Jonasdottir, A.; et al. Lipoprotein(a) Concentration and Risks of Cardiovascular Disease and Diabetes. J. Am. Coll. Cardiol. 2019, 74, 2982–2994. [Google Scholar] [CrossRef]
- Tsimikas, S.; Karwatowska-Prokopczuk, E.; Gouni-Berthold, I.; Tardif, J.C.; Baum, S.J.; Steinhagen-Thiessen, E.; Shapiro, M.D.; Stroes, E.S.; Moriarty, P.M.; Nordestgaard, B.G.; et al. Lipoprotein(a) Reduction in Persons with Cardiovascular Disease. N. Engl. J. Med. 2020, 382, 244–255. [Google Scholar] [CrossRef]
- Chennamsetty, I.; Claudel, T.; Kostner, K.M.; Baghdasaryan, A.; Kratky, D.; Levak-Frank, S.; Frank, S.; Gonzalez, F.J.; Trauner, M.; Kostner, G.M. Farnesoid X receptor represses hepatic human APOA gene expression. J. Clin. Investig. 2011, 121, 3724–3734. [Google Scholar] [CrossRef] [Green Version]
- Chennamsetty, I.; Claudel, T.; Kostner, K.M.; Trauner, M.; Kostner, G.M. FGF19 signaling cascade suppresses APOA gene expression. Arter. Thromb. Vasc. Biol. 2012, 32, 1220–1227. [Google Scholar] [CrossRef] [Green Version]
- Vasavan, T.; Ferraro, E.; Ibrahim, E.; Dixon, P.; Gorelik, J.; Williamson, C. Heart and bile acids—Clinical consequences of altered bile acid metabolism. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 1345–1355. [Google Scholar] [CrossRef]
- Zamorano, J.L.; Lancellotti, P.; Rodriguez Munoz, D.; Aboyans, V.; Asteggiano, R.; Galderisi, M.; Habib, G.; Lenihan, D.J.; Lip, G.Y.H.; Lyon, A.R.; et al. 2016 ESC Position Paper on cancer treatments and cardiovascular toxicity developed under the auspices of the ESC Committee for Practice Guidelines: The Task Force for cancer treatments and cardiovascular toxicity of the European Society of Cardiology (ESC). Eur. Heart J. 2016, 37, 2768–2801. [Google Scholar] [CrossRef]
- Kunst, R.F.; Verkade, H.J.; Oude Elferink, R.P.J.; van de Graaf, S.F.J. Targeting the Four Pillars of Enterohepatic Bile Salt Cycling; Lessons From Genetics and Pharmacology. Hepatology 2021, 73, 2577–2585. [Google Scholar] [CrossRef]
- Piepoli, M.F.; Hoes, A.W.; Agewall, S.; Albus, C.; Brotons, C.; Catapano, A.L.; Cooney, M.T.; Corra, U.; Cosyns, B.; Deaton, C.; et al. 2016 European Guidelines on cardiovascular disease prevention in clinical practice: The Sixth Joint Task Force of the European Society of Cardiology and Other Societies on Cardiovascular Disease Prevention in Clinical Practice (constituted by representatives of 10 societies and by invited experts)Developed with the special contribution of the European Association for Cardiovascular Prevention & Rehabilitation (EACPR). Eur. Heart J. 2016, 37, 2315–2381. [Google Scholar] [CrossRef]
- Haag, M.; Hofmann, U.; Murdter, T.E.; Heinkele, G.; Leuthold, P.; Blank, A.; Haefeli, W.E.; Alexandrov, A.; Urban, S.; Schwab, M. Quantitative bile acid profiling by liquid chromatography quadrupole time-of-flight mass spectrometry: Monitoring hepatitis B therapy by a novel Na-taurocholate cotransporting polypeptide inhibitor. Anal. Bioanal. Chem. 2015, 407, 6815–6825. [Google Scholar] [CrossRef] [PubMed]
- Han, L. Calculating the Point Estimate and Confidence Interval of Hodges-Lehmann’s Median Using SAS® Software. SESUG Proceedings, 2008. Paper ST-154. Available online: https://analytics.ncsu.edu/sesug/2008/ST-154.pdf (accessed on 20 October 2022).






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristic | ||
|---|---|---|
| Age (years; mean [min; max]) | 57.3 | [40; 64] |
| Gender | ||
| Male | 7 | 50% |
| Female | 7 | 50% |
| Body mass index (BMI) (kg/m2; mean [min; max]) | 27.1 | [21.3; 40.1] |
| Antihypertensives | ||
| ACE inhibitors | 2 | 14.3% |
| AT 1 antagonist | 1 | 7.14% |
| Beta blocker | 2 | 14.3% |
| HbA1c (mmol/mol) | ||
| <39 | 8 | 57.1% |
| 39–≤46 2 | 6 | 42.9% |
| Smoking status | ||
| Active smoker | 4 | 28.6% |
| Past smoker | 4 | 28.6% |
| Never smoker | 6 | 42.9% |
| Pseudo Median (HL Estimate) | 95% CI | Change (HL Estimate) | 95% CI | |
|---|---|---|---|---|
| Primary outcome: | ||||
| LDL cholesterol (mg/dL) | 196 | [176; 223] | −19.6 | [−41.8; 2.85] |
| Secondary outcomes: | ||||
| Total cholesterol (mg/dL) | 270 | [251; 295] | −7 | [−30.5; 15.0] |
| HDL cholesterol (mg/dL) | 62 | [54.0; 72.0] | 5.5 | [1.00; 10.5] |
| VLDL cholesterol (mg/dL) | 10.7 | [5.60; 15.8] | 2.4 | [−6.30; 19.1] |
| Non-HDL (mg/dL) | 206 | [181; 237] | −8 | [−30.0; 17.0] |
| Triglycerides (mg/dL) | 118 | [93.5; 141] | −17 | [−35.0; 5.0] |
| Apolipoprotein B (g/L) | 1.25 | [1.14; 1.43] | −0.045 | [−0.19; 0.11] |
| Lipoprotein(a) (mg/dL) | 15.0 | [10.1; 68.1] | −1.87 | [−7.65; 0.00] |
| Pseudo Median (HL Estimate) | 95% CI | Change (HL Estimate) | 95% CI | |
|---|---|---|---|---|
| hs-CRP (mg/L) | 1.06 | [0.66; 1.78] | −0.055 | [−0.22; 0.80] |
| IL-1b (pg/mL) | 0.12 | [0.10; 0.15] | 0.003 | [−0.02; 0.05] |
| IL-6 (pg/mL) | 1.01 | [0.68; 1.55] | −0.054 | [−0.33; 0.13] |
| TNF-α (pg/mL) | 7.64 | [6.09; 9.19] | 0.167 | [−0.24; 0.61] |
| E-selectin (pg/mL) | 32,654 | [28,403; 37,045] | −184 | [−2369; 2476] |
| ICAM-1 (pg/mL) | 295,745 | [249,257; 359,690] | −4158 | [−17,457; 13,677] |
| TGF-β1 (pg/mL) | 11,810 | [9420; 14,071] | −493 | [−3830; 4265] |
| Neopterin (µmol/L) | 8.08 | [7.17; 9.00] | 0.095 | [−0.61; 0.72] |
| LVEF (%) | 62.8 | [60.8; 64.5] | −0.25 | [−1.75; 1.00] |
| global circumferential strain (%) | −17.7 | [−18.8; −16.5] | −0.2 | [−1.48; 1.57] |
| global longitudinal strain (%) | −13.7 | [−14.9; −12.4] | 0.05 | [−1.74; 1.15] |
| global T1 time (ms) | 1236 | [1221; 1254] | 13.8 | [−0.26; 35.8] |
| global T2 time (ms) | 46.3 | [45.5; 47.9] | 1.37 * | [−0.43; 2.97] * |
| HbA1c (mmol/mol) | 38 | [34; 41] | −0.5 | [−3.50; 1.50] |
| HOMA | 2.15 | [1.19; 2.96] | −0.01 | [−0.35; 0.25] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stoll, F.; Seidel-Glätzer, A.; Burghaus, I.; Göring, O.; Sauter, M.; Rose, P.; Daniel, V.; Haag, M.; Schwab, M.; Riffel, J.; et al. Metabolic Effect of Blocking Sodium-Taurocholate Co-Transporting Polypeptide in Hypercholesterolemic Humans with a Twelve-Week Course of Bulevirtide—An Exploratory Phase I Clinical Trial. Int. J. Mol. Sci. 2022, 23, 15924. https://doi.org/10.3390/ijms232415924
Stoll F, Seidel-Glätzer A, Burghaus I, Göring O, Sauter M, Rose P, Daniel V, Haag M, Schwab M, Riffel J, et al. Metabolic Effect of Blocking Sodium-Taurocholate Co-Transporting Polypeptide in Hypercholesterolemic Humans with a Twelve-Week Course of Bulevirtide—An Exploratory Phase I Clinical Trial. International Journal of Molecular Sciences. 2022; 23(24):15924. https://doi.org/10.3390/ijms232415924
Chicago/Turabian StyleStoll, Felicitas, Andrea Seidel-Glätzer, Ina Burghaus, Oliver Göring, Max Sauter, Peter Rose, Volker Daniel, Mathias Haag, Matthias Schwab, Johannes Riffel, and et al. 2022. "Metabolic Effect of Blocking Sodium-Taurocholate Co-Transporting Polypeptide in Hypercholesterolemic Humans with a Twelve-Week Course of Bulevirtide—An Exploratory Phase I Clinical Trial" International Journal of Molecular Sciences 23, no. 24: 15924. https://doi.org/10.3390/ijms232415924