
Molecular Mechanisms in Genetic Aortopathy–Signaling Pathways and Potential Interventions
 , , and
, , and
Abstract
:1. Background
2. Cell Function in Normal and Aneurysmal Aorta
3. TGF-β Signaling
3.1. Regulation of TGF-β Signaling
3.2. TGF-β and VSMC
3.3. TGF-β and EC
4. WNT Signaling
4.1. Regulation of WNT Signaling
4.2. WNT and VSMC
4.3. WNT and EC
5. PI3K/AKT Signaling
5.1. Regulation of PI3K/AKT Signaling
5.2. PI3K/AKT and VSMC
5.3. PI3K/AKT and EC
6. NOTCH Signaling
6.1. Regulation of NOTCH Signaling
6.2. NOTCH and VSMC
6.3. NOTCH and EC
7. Angiotensin Signaling
7.1. Regulation of ANG II Signaling
7.2. Angiotensin II and VSMC
7.3. Angiotensin II and EC
8. Interactions between Pathways
Summary of Pathway Interactions
9. Role of Signaling Pathways in Pathogenesis of TAA
9.1. TGF-β and TAA
9.2. WNT and TAA
9.3. PI3K/AKT and TAA
9.4. NOTCH and TAA
9.5. Angiotensin II and TAA
10. Potential Therapeutic Interventions
10.1. TGF-β/BMP/SMAD Pathway
10.2. WNT Pathway
10.3. PI3K/AKT Pathway
10.4. NOTCH Pathway
10.5. Angiotensin II Pathway
10.6. Application of Pathway Therapeutics to TAA
11. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ACVRL1 | activin A receptor like type 1 |
| ADAM17 | A disintegrin and metalloproteinase 17 |
| ADNP | activity dependent neuroprotector homeobox transcription factor |
| ALK | ALK receptor tyrosine kinase |
| AKT | protein kinase B |
| ANG | Angiotensin |
| AP-1 | activator protein 1 |
| AT1(2)R | angiotensin type 1 (2) receptor |
| BAD | BCL2 associated agonist of cell death |
| BAEC | bovine aortic endothelial cell |
| BAMBI | BMP and activin membrane bound inhibitor |
| BAV | bicuspid aortic valve |
| BMP | bone morphogenic protein |
| CADASIL | cerebral autosomal dominant arteriopathy with sub-cortical infarcts and leukoencephalopathy |
| CAMKII | calcium/calmodulin dependent protein kinase 2 |
| CCND1 | cyclinD1 |
| COX2 | cyclooxygenase 2 |
| CREB | cAMP response element binding protein transcription factor |
| CSL/RBPJ | recombination binding protein for immunoglobulin Kappa J |
| CTGF | connective tissue growth factor |
| DKK | Dickkopf WNT signaling pathway inhibitor |
| DLL | delta like canonical Notch ligand |
| DVL1 | disheveled segment polarity protein 1 |
| EC | endothelial cell |
| ECM | extracellular matrix |
| EGFL7 | epidermal growth factor like domain multiple 7 |
| EGFR | epidermal growth factor receptor |
| ERK | extracellular signal-regulated kinase |
| FN1 | fibronectin 1 |
| FOXO | forkhead box gene group O |
| FZD | frizzled receptor |
| GSK-3β | glycogen synthase kinase 3 beta |
| H2O2 | hydrogen peroxide |
| HERP | homocysteine inducible ER protein |
| HES | hairy and enhancer of split-1 transcription factor |
| HEY | hairy ears Y linked transcription factor |
| HAEC | human aortic endothelial cell |
| HUVEC | human umbilical vein endothelial cell |
| hTAA | heritable thoracic aortic aneurysm |
| IGFBP | insulin-like growth factor binding protein |
| IGF-1R | insulin-like growth factor 1 receptor |
| IL-8 | interleukin 8 |
| JAK2 | janus kinase 2 |
| JNK | c-Jun N-terminal kinase |
| LDL | low density lipoprotein |
| LDS | Loeys-Dietz syndrome |
| LEF | lymphoid enhancer binding factor transcriptional activator |
| LRP | low-density lipoprotein receptor-related protein |
| LTBP | latent TGFbeta binding protein |
| MAGP-2 | microfibril associated glycoprotein 2 |
| MAML | mastermind-like protein 1 |
| MAPK | mitogen activated protein kinase |
| MAS | Mas-related G protein couple receptor |
| MDM2 | MDM2 proto-oncogene E3 ubiquitin protein ligase |
| MEF2 | myocyte enhancer factor 2C |
| MEK | MAPK/ERK kinase |
| MFS | Marfan syndrome |
| MK2 | mitogen-activated protein kinase-activated protein kinase 2 |
| MMP | matrix metalloproteinase |
| mTOR | mammalian target of rapamycin |
| NADPH | nicotinamide adenine dinucleotide phosphate |
| NEDD4 | neural precursor cell expressed developmentally downregulated protein 4 |
| NF-κB | nuclear factor kappa B |
| NICD | Notch intracellular domain |
| NO | nitric oxide |
| NOS | nitric oxide synthase |
| NOTCH | Notch receptor protein |
| NOTUM | Notum palmitoleoyl-protein-carboxylesterase |
| NOX | NADPH oxidase |
| PAI-1 | plasminogen activator inhibitor type-1 |
| PDGF | platelet-derived growth factor |
| PI3K | phosphoinositide 3-kinase |
| PIP3 | phosphatidyinositol (3,4,5)-triphosphate |
| PKC-δ | protein kinase C gamma |
| PLA(C) | phospholipase A (C) |
| PTEN | phosphatase and tensin homolog |
| p38 | p38 mitogen-activated protein kinase |
| ROCK | Rho associated coiled-coil containing protein kinase |
| ROS | reactive oxygen species |
| RTK | receptor tyrosine kinase |
| RUNX | RUNX family transcription factor |
| SARA | SMAD anchor for receptor activation |
| SKI | SKI proto-oncogene |
| SM22α | smooth muscle protein 22-alpha |
| SMA | smooth muscle actin |
| SMAD | small mothers against decapentaplegic |
| SMURF | SMAD ubiquitination regulatory factor |
| SNON | SKI-like proto-oncogene |
| SOD | superoxide dismutase |
| SOST | Sclerostin |
| SP1 | specificity protein 1 transcription factor |
| SPRY1 | sprouty RTK signaling antagonist 1 |
| TAA | thoracic aortic aneurysm |
| TAV | tricuspid aortic valve |
| TCF | T-cell factor enhancer binding factor transcriptional activator |
| TGF-β | transforming growth factor β |
| TGFBR | transforming growth factor beta receptor |
| TIMP1 | tissue inhibitor of metalloproteinases 1 |
| TNF-α | tumor necrosis factor alpha |
| TRABD2B | TraB domain containing 2B |
| VCAM-1 | vascular cell adhesion molecule 1 |
| VCAN | Versican |
| VEGF | vascular endothelial growth factor |
| VSMC | vascular smooth muscle cells |
| WIF-1 | WNT inhibitory factor 1 |
| WISP | WNT1 inducible signaling pathway protein |
| WNT | wingless-related integration site |
| ZNRF | zinc and ring finger ubiquitin protein ligands |
References
- Robertson, E.N.; Hambly, B.D.; Jeremy, R.W. Thoracic aortic dissection and heritability: Forensic implications. Forensic Sci. Med. Pathol. 2016, 12, 366–368. [Google Scholar] [CrossRef] [PubMed]
- Robertson, E.; Dilworth, C.; Lu, Y.; Hambly, B.D.; Jeremy, R.W. Molecular mechanisms of inherited thoracic aortic disease—From gene variant to surgical aneurysm. Biophys. Rev. 2015, 7, 105–115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davis, F.M.; Daugherty, A.; Lu, H.S. Updates of recent aortic aneurysm research. Arter. Thromb. Vasc. Biol. 2019, 39, e83–e90. [Google Scholar] [CrossRef] [PubMed]
- Michelena, H.I.; Khanna, A.D.; Mahoney, D.; Margaryan, D.; Topilsky, Y.; Suri, R.M.; Eidem, B.; Edwards, W.D.; Sundt, T.M.; Enriquez-Sarano, M. Incidence of aortic complications in patients with bicuspid aortic valves. JAMA 2011, 306, 1104–1112. [Google Scholar] [CrossRef] [Green Version]
- Portelli, S.S.; Robertson, E.N.; Malecki, C.; Liddy, K.A.; Hambly, B.D.; Jeremy, R.W. Epigenetic influences on genetically triggered thoracic aortic aneurysm. Biophys. Rev. 2018, 10, 1241–1256. [Google Scholar] [CrossRef]
- Quintana, R.A.; Taylor, W.R. Cellular mechanisms of aortic aneurysm formation. Circ. Res. 2019, 124, 607–618. [Google Scholar] [CrossRef]
- Malecki, C.; Hambly, B.D.; Jeremy, R.W.; Robertson, E.N. The role of inflammation and myeloperoxidase-related oxidative stress in the pathogenesis of genetically triggered thoracic aortic aneurysms. Int. J. Mol. Sci. 2020, 21, 7678. [Google Scholar] [CrossRef]
- Owens, G.K.; Kumar, M.S.; Wamhoff, B.R. Molecular regulation of vascular smooth muscle cell differentiation in development and disease. Physiol. Rev. 2004, 84, 767–801. [Google Scholar] [CrossRef]
- Caliceti, C.; Nigro, P.; Rizzo, P.; Ferrari, R. ROS, Notch, and Wnt signaling pathways: Crosstalk between three major regulators of cardiovascular biology. BioMed Res. Int. 2014, 2014, 318714. [Google Scholar] [CrossRef] [Green Version]
- Méndez-Barbero, N.; Gutiérrez-Muñoz, C.; Blanco-Colio, L.M. Cellular crosstalk between endothelial and smooth muscle cells in vascular wall remodeling. Int. J. Mol. Sci. 2021, 22, 7284. [Google Scholar] [CrossRef]
- Jeremy, R.W.; Robertson, E.; Lu, Y.; Hambly, B.D. Perturbations of mechanotransduction and aneurysm formation in heritable aortopathies. Int. J. Cardiol. 2013, 169, 7–16. [Google Scholar] [CrossRef]
- Daugherty, A.; Chen, Z.; Sawada, H.; Rateri, D.L.; Sheppard, M.B. Transforming growth factor-β in thoracic aortic aneurysms: Good, bad, or irrelevant? J. Am. Heart Assoc. 2017, 6, e005221. [Google Scholar] [CrossRef]
- El-Hamamsy, I.; Yacoub, M.H. Cellular and molecular mechanisms of thoracic aortic aneurysms. Nat. Rev. Cardiol. 2009, 6, 771–786. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Shen, Y.H.; Russell, L.; LeMaire, S.A. Molecular mechanisms of thoracic aortic dissection. J. Surg. Res. 2013, 184, 907–924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, K. Signaling cross talk between TGF-beta/Smad and other signaling pathways. Cold Spring Harb. Perspect. Biol. 2017, 9, a022137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bäck, M.; Gasser, T.C.; Michel, J.B.; Caligiuri, G. Biomechanical factors in the biology of aortic wall and aortic valve diseases. Cardiovasc. Res. 2013, 99, 232–241. [Google Scholar] [CrossRef] [Green Version]
- Majesky, M.W.; Dong, X.R.; Hoglund, V.; Mahoney, W.M.; Daum, G. The adventitia: A dynamic interface containing resident progenitor cells. Arter. Thromb. Vasc. Biol. 2011, 31, 1530–1539. [Google Scholar] [CrossRef] [Green Version]
- Kessler, K.; Borges, L.F.; Ho-Tin-Noe, B.; Jondeau, G.; Michel, J.B.; Vranckx, R. Angiogenesis and remodelling in human thoracic aortic aneurysms. Cardiovasc. Res. 2014, 104, 147–159. [Google Scholar] [CrossRef] [Green Version]
- Blunder, S.; Messner, B.; Aschacher, T.; Zeller, I.; Türkcan, A.; Wiedemann, D.; Andreas, M.; Blüschke, G.; Laufer, G.; Schachner, T.; et al. Characteristics of TAV- and BAV-associated thoracic aortic aneurysms—Smooth muscle cell biology, expression profiling, and histological analyses. Atherosclerosis 2012, 220, 355–361. [Google Scholar] [CrossRef]
- Fernandez-Moure, J.S.; Vykoukal, D.; Davies, M.G. Biology of aortic aneurysms and dissections. Methodist DeBakey Cardiovasc. J. 2011, 7, 2–7. [Google Scholar] [CrossRef]
- Rensen, S.S.; Doevendans, P.A.; van Eys, G.J. Regulation and characteristics of vascular smooth muscle cell phenotypic diversity. Neth. Heart J. 2007, 15, 100–108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rzucidlo, E.M.; Martin, K.A.; Powell, R.J. Regulation of vascular smooth muscle cell differentiation. J. Vasc. Surg. 2007, 45 (Suppl. A), A25–A32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, H.; Du, W.; Ren, L.; Hamblin, M.H.; Becker, R.C.; Chen, Y.E.; Fan, Y. Vascular smooth muscle cells in aortic aneurysm: From genetics to mechanisms. J. Am. Heart Assoc. 2021, 10, e023601. [Google Scholar] [CrossRef] [PubMed]
- Petsophonsakul, P.; Furmanik, M.; Forsythe, R.; Dweck, M.; Schurink, G.W.; Natour, E.; Reutelingsperger, C.; Jacobs, M.; Mees, B.; Schurgers, L. Role of vascular smooth muscle cell phenotypic switching and calcification in aortic aneurysm formation. Arter. Thromb. Vasc. Biol. 2019, 39, 1351–1368. [Google Scholar] [CrossRef]
- Esper, R.J.; Nordaby, R.A.; Vilariño, J.O.; Paragano, A.; Cacharrón, J.L.; Machado, R.A. Endothelial dysfunction: A comprehensive appraisal. Cardiovasc. Diabetol. 2006, 5, 4. [Google Scholar] [CrossRef] [Green Version]
- Malashicheva, A.; Kostina, D.; Kostina, A.; Irtyuga, O.; Voronkina, I.; Smagina, L.; Ignatieva, E.; Gavriliuk, N.; Uspensky, V.; Moiseeva, O.; et al. Phenotypic and functional changes of endothelial and smooth muscle cells in thoracic aortic aneurysms. Int. J. Vasc. Med. 2016, 2016, 3107879. [Google Scholar] [CrossRef] [Green Version]
- Sandoo, A.; van Zanten, J.J.; Metsios, G.S.; Carroll, D.; Kitas, G.D. The endothelium and its role in regulating vascular tone. Open Cardiovasc. Med. J. 2010, 4, 302–312. [Google Scholar] [CrossRef] [Green Version]
- Irace, F.G.; Cammisotto, V.; Valenti, V.; Forte, M.; Schirone, L.; Bartimoccia, S.; Iaccarino, A.; Peruzzi, M.; Schiavon, S.; Morelli, A.; et al. Role of oxidative stress and autophagy in thoracic aortic aneurysms. JACC Basic Transl. Sci. 2021, 6, 719–730. [Google Scholar] [CrossRef]
- Ali, O.A.; Chapman, M.; Nguyen, T.H.; Chirkov, Y.Y.; Heresztyn, T.; Mundisugih, J.; Horowitz, J.D. Interactions between inflammatory activation and endothelial dysfunction selectively modulate valve disease progression in patients with bicuspid aortic valve. Heart 2014, 100, 800–805. [Google Scholar] [CrossRef]
- Van de Pol, V.; Kurakula, K.; DeRuiter, M.C.; Goumans, M.J. Thoracic aortic aneurysm development in patients with bicuspid aortic valve: What is the role of endothelial cells? Front. Physiol. 2017, 8, 938. [Google Scholar] [CrossRef]
- Ten Dijke, P.; Goumans, M.J.; Itoh, F.; Itoh, S. Regulation of cell proliferation by Smad proteins. J. Cell. Physiol. 2002, 191, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Javelaud, D.; Mauviel, A. Mammalian transforming growth factor-βs: Smad signaling and physio-pathological roles. Int. J. Biochem. Cell Biol. 2004, 36, 1161–1165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruiz-Ortega, M.; Rodríguez-Vita, J.; Sanchez-Lopez, E.; Carvajal, G.; Egido, J. TGF-β signaling in vascular fibrosis. Cardiovasc. Res. 2007, 74, 196–206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Derynck, R.; Zhang, Y.E. Smad-dependent and Smad-independent pathways in TGF-β family signalling. Nature 2003, 425, 577–584. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Massagué, J. Mechanisms of TGF-β signaling from cell membrane to the nucleus. Cell 2003, 113, 685–700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akhurst, R.J.; Hata, A. Targeting the TGFβ signalling pathway in disease. Nat. Rev. Drug Discov. 2012, 11, 790–811. [Google Scholar] [CrossRef] [Green Version]
- Gordon, K.J.; Blobe, G.C. Role of transforming growth factor-β superfamily signaling pathways in human disease. Biochim. Biophys. Acta-Mol. Basis Dis. 2008, 1782, 197–228. [Google Scholar] [CrossRef] [Green Version]
- Zhao, B.; Chen, Y.G. Regulation of TGF-beta signal transduction. Scientifica 2014, 2014, 874065. [Google Scholar] [CrossRef] [Green Version]
- Abreu, J.G.; Ketpura, N.I.; Reversade, B.; De Robertis, E.M. Connective-tissue growth factor (CTGF) modulates cell signalling by BMP and TGF-β. Nat. Cell Biol. 2002, 4, 599–604. [Google Scholar] [CrossRef] [Green Version]
- Zuo, W.; Chen, Y.G. Specific activation of mitogen-activated protein kinase by transforming growth factor-beta receptors in lipid rafts is required for epithelial cell plasticity. Mol. Biol. Cell 2009, 20, 1020–1029. [Google Scholar] [CrossRef]
- Yan, X.; Lin, Z.; Chen, F.; Zhao, X.; Chen, H.; Ning, Y.; Chen, Y.G. Human BAMBI cooperates with Smad7 to inhibit transforming growth factor-beta signaling. J. Biol. Chem. 2009, 284, 30097–30104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tecalco-Cruz, A.C.; Sosa-Garrocho, M.; Vázquez-Victorio, G.; Ortiz-García, L.; Domínguez-Hüttinger, E.; Macías-Silva, M. Transforming growth factor-beta/SMAD Target gene SKIL is negatively regulated by the transcriptional cofactor complex SNON-SMAD4. J. Biol. Chem. 2012, 287, 26764–26776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Owens, G.K. Regulation of differentiation of vascular smooth muscle cells. Physiol. Rev. 1995, 75, 487–517. [Google Scholar] [CrossRef] [PubMed]
- Goumans, M.J.; Liu, Z.; ten Dijke, P. TGF-beta signaling in vascular biology and dysfunction. Cell Res. 2009, 19, 116–127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suwanabol, P.A.; Seedial, S.M.; Shi, X.; Zhang, F.; Yamanouchi, D.; Roenneburg, D.; Liu, B.; Kent, K.C. Transforming growth factor-beta increases vascular smooth muscle cell proliferation through the Smad3 and extracellular signal-regulated kinase mitogen-activated protein kinases pathways. J. Vasc. Surg. 2012, 56, 446–454. [Google Scholar] [CrossRef] [Green Version]
- Guo, X.; Chen, S.Y. Transforming growth factor-beta and smooth muscle differentiation. World J. Biol. Chem. 2012, 3, 41–52. [Google Scholar] [CrossRef]
- Pardali, E.; Goumans, M.J.; ten Dijke, P. Signaling by members of the TGF-β family in vascular morphogenesis and disease. Trends Cell Biol. 2010, 20, 556–567. [Google Scholar] [CrossRef]
- Shi, N.; Chen, S.Y. Mechanisms simultaneously regulate smooth muscle proliferation and differentiation. J. Biomed. Res. 2014, 28, 40–46. [Google Scholar] [CrossRef] [Green Version]
- Sinha, S.; Hoofnagle, M.H.; Kingston, P.A.; McCanna, M.E.; Owens, G.K. Transforming growth factor-β1 signaling contributes to development of smooth muscle cells from embryonic stem cells. Am. J. Physiol. Cell Physiol. 2004, 287, C1560–C1568. [Google Scholar] [CrossRef]
- Zhu, J.; Angelov, S.; Alp Yildirim, I.; Wei, H.; Hu, J.H.; Majesky, M.W.; Brozovich, F.V.; Kim, F.; Dichek, D.A. Loss of transforming growth factor beta signaling in aortic smooth muscle cells causes endothelial dysfunction and aortic hypercontractility. Arter. Thromb. Vasc. Biol. 2021, 41, 1956–1971. [Google Scholar] [CrossRef]
- Seay, U.; Sedding, D.; Krick, S.; Hecker, M.; Seeger, W.; Eickelberg, O. Transforming growth factor-beta-dependent growth inhibition in primary vascular smooth muscle cells is p38-dependent. J. Pharmacol. Exp. Ther. 2005, 315, 1005–1012. [Google Scholar] [CrossRef] [PubMed]
- Deaton, R.A.; Su, C.; Valencia, T.G.; Grant, S.R. Transforming growth factor-beta1-induced expression of smooth muscle marker genes involves activation of PKN and p38 MAPK. J. Biol. Chem. 2005, 280, 31172–31181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsai, S.; Hollenbeck, S.T.; Ryer, E.J.; Edlin, R.; Yamanouchi, D.; Kundi, R.; Wang, C.; Liu, B.; Kent, K.C. TGF-β through Smad3 signaling stimulates vascular smooth muscle cell proliferation and neointimal formation. Am. J. Physiol. Heart Circ. Physiol. 2009, 297, H540–H549. [Google Scholar] [CrossRef] [Green Version]
- Kundi, R.; Hollenbeck, S.T.; Yamanouchi, D.; Herman, B.C.; Edlin, R.; Ryer, E.J.; Wang, C.; Tsai, S.; Liu, B.; Kent, K.C. Arterial gene transfer of the TGF-beta signalling protein Smad3 induces adaptive remodelling following angioplasty: A role for CTGF. Cardiovasc. Res. 2009, 84, 326–335. [Google Scholar] [CrossRef] [PubMed]
- Samarakoon, R.; Higgins, S.P.; Higgins, C.E.; Higgins, P.J. TGF-beta1-induced plasminogen activator inhibitor-1 expression in vascular smooth muscle cells requires pp60(c-src)/EGFR(Y845) and Rho/ROCK signaling. J. Mol. Cell Cardiol. 2008, 44, 527–538. [Google Scholar] [CrossRef] [Green Version]
- Czekay, R.P.; Wilkins-Port, C.E.; Higgins, S.P.; Freytag, J.; Overstreet, J.M.; Klein, R.M.; Higgins, C.E.; Samarakoon, R.; Higgins, P.J. PAI-1: An integrator of cell signaling and migration. Int. J. Cell Biol. 2011, 2011, 562481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DiRenzo, D.M.; Chaudhary, M.A.; Shi, X.; Franco, S.R.; Zent, J.; Wang, K.; Guo, L.W.; Kent, K.C. A crosstalk between TGF-beta/Smad3 and Wnt/beta-catenin pathways promotes vascular smooth muscle cell proliferation. Cell Signal. 2016, 28, 498–505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goumans, M.-J.; Valdimarsdottir, G.; Itoh, S.; Rosendahl, A.; Sideras, P.; ten Dijke, P. Balancing the activation state of the endothelium via two distinct TGF-β type I receptors. EMBO J. 2002, 21, 1743–1753. [Google Scholar] [CrossRef]
- Goumans, M.J.; Valdimarsdottir, G.; Itoh, S.; Lebrin, F.; Larsson, J.; Mummery, C.; Karlsson, S.; ten Dijke, P. Activin receptor-like kinase (ALK)1 is an antagonistic mediator of lateral TGFβ/ALK5 signaling. Mol. Cell 2003, 12, 817–828. [Google Scholar] [CrossRef]
- Lebrin, F.; Deckers, M.; Bertolino, P.; ten Dijke, P. TGF-beta receptor function in the endothelium. Cardiovasc. Res. 2005, 65, 599–608. [Google Scholar] [CrossRef]
- Balsara, R.D.; Castellino, F.J.; Ploplis, V.A. A novel function of plasminogen activator inhibitor-1 in modulation of the AKT pathway in wild-type and plasminogen activator inhibitor-1-deficient endothelial cells. J. Biol. Chem. 2006, 281, 22527–22536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lebrin, F.; Goumans, M.J.; Jonker, L.; Carvalho, R.L.; Valdimarsdottir, G.; Thorikay, M.; Mummery, C.; Arthur, H.M.; ten Dijke, P. Endoglin promotes endothelial cell proliferation and TGF-β/ALK1 signal transduction. EMBO J. 2004, 23, 4018–4028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foulquier, S.; Daskalopoulos, E.P.; Lluri, G.; Hermans, K.C.M.; Deb, A.; Blankesteijn, W.M. WNT Signaling in Cardiac and Vascular Disease. Pharm. Rev. 2018, 70, 68–141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mill, C.; George, S.J. Wnt signalling in smooth muscle cells and its role in cardiovascular disorders. Cardiovasc. Res. 2012, 95, 233–240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- MacDonald, B.T.; Tamai, K.; He, X. Wnt/beta-catenin signaling: Components, mechanisms, and diseases. Dev. Cell 2009, 17, 9–26. [Google Scholar] [CrossRef] [Green Version]
- Komiya, Y.; Habas, R. Wnt signal transduction pathways. Organogenesis 2008, 4, 68–75. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Xiao, Q.; Xiao, J.; Niu, C.; Li, Y.; Zhang, X.; Zhou, Z.; Shu, G.; Yin, G. Wnt/β-catenin signalling: Function, biological mechanisms, and therapeutic opportunities. Signal. Transduct. Target. Ther. 2022, 7, 3. [Google Scholar] [CrossRef] [PubMed]
- proteinatlas.org. The Human Protein Atlas. 2022. Available online: https://www.proteinatlas.org/ (accessed on 19 September 2022).
- Topol, L.; Jiang, X.; Choi, H.; Garrett-Beal, L.; Carolan, P.J.; Yang, Y. Wnt-5a inhibits the canonical Wnt pathway by promoting GSK-3–independent β-catenin degradation. J. Cell Biol. 2003, 162, 899–908. [Google Scholar] [CrossRef]
- Quasnichka, H.; Slater, S.C.; Beeching, C.A.; Boehm, M.; Sala-Newby, G.B.; George, S.J. Regulation of smooth muscle cell proliferation by β-Catenin/T-cell factor signaling involves modulation of Cyclin D1 and p21 expression. Circ. Res. 2006, 99, 1329–1337. [Google Scholar] [CrossRef] [Green Version]
- Akoumianakis, I.; Polkinghorne, M.; Antoniades, C. Non-canonical WNT signalling in cardiovascular disease: Mechanisms and therapeutic implications. Nat. Rev. Cardiol. 2022, 12, 783–797. [Google Scholar] [CrossRef]
- Mao, C.; Malek, O.T.; Pueyo, M.T.; Steg, P.G.; Soubrier, F. Differential expression of rat frizzled-related frzb-1 and frizzled receptor fz1 and fz2 genes in the rat aorta after balloon injury. Arter. Thromb. Vasc. Biol. 2000, 20, 43–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malinauskas, T.; Aricescu, A.R.; Lu, W.; Siebold, C.; Jones, E.Y. Modular mechanism of Wnt signaling inhibition by Wnt inhibitory factor 1. Nat. Struct. Mol. Biol. 2011, 18, 886–893. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Cheong, S.M.; Amado, N.G.; Reis, A.H.; MacDonald, B.T.; Zebisch, M.; Jones, E.Y.; Abreu, J.G.; He, X. Notum is required for neural and head induction via Wnt deacylation, oxidation, and inactivation. Dev. Cell. 2015, 32, 719–730. [Google Scholar] [CrossRef] [Green Version]
- Cruciat, C.M.; Niehrs, C. Secreted and transmembrane wnt inhibitors and activators. Cold Spring Harb. Perspect. Biol. 2013, 5, a015081. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, W.; Shiojima, I.; Ito, Y.; Li, Z.; Ikeda, H.; Yoshida, M.; Naito, A.T.; Nishi, J.; Ueno, H.; Umezawa, A.; et al. IGFBP-4 is an inhibitor of canonical Wnt signalling required for cardiogenesis. Nature 2008, 454, 345–349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, X.; Charlat, O.; Zamponi, R.; Yang, Y.; Cong, F. Dishevelled promotes Wnt receptor degradation through recruitment of ZNRF3/RNF43 E3 ubiquitin ligases. Mol. Cell 2015, 58, 522–533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, X.; Peng, X.; Cao, Y.; Zhou, Y.; Sun, Y. ADNP promotes neural differentiation by modulating Wnt/β-catenin signaling. Nat. Commun. 2020, 11, 2984. [Google Scholar] [CrossRef]
- Ren, Z.; Spaargaren, M.; Pals, S.T. Syndecan-1 promotes Wnt/β-catenin signaling in multiple myeloma by presenting Wnts and R-spondins. Blood 2018, 131, 982–994. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Neogi, A.; Mani, A. The role of Wnt signalling in development of coronary artery disease and its risk factors. Open Biol. 2020, 10, 200128. [Google Scholar] [CrossRef]
- Mill, C.; Jeremy, J.Y.; George, S.J. Wnt5a signalling promotes VSMC survival via WISP-1: Consequences for VSMC viability in atherosclerotic plaques. Heart 2011, 97, e7. [Google Scholar] [CrossRef]
- Mill, C.; Monk, B.A.; Williams, H.; Simmonds, S.J.; Jeremy, J.Y.; Johnson, J.L.; George, S.J. Wnt5a-induced Wnt1-inducible secreted protein-1 suppresses vascular smooth muscle cell apoptosis induced by oxidative stress. Arter. Thromb. Vasc. Biol. 2014, 34, 2449–2456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, B.A.; Connolly, G.M.; Mill, C.E.J.; Williams, H.; Angelini, G.D.; Johnson, J.L.; George, S.J. Aging differentially modulates the Wnt pro-survival signalling pathways in vascular smooth muscle cells. Aging Cell 2019, 18, e12844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, L.; Zhuang, J.; Singh, S.; Wang, K.; Xiong, M.; Xu, D.; Chen, W.; Pang, J.; Xu, Y.; Li, X. XAV939 Inhibits Intima Formation by Decreasing Vascular Smooth Muscle Cell Proliferation and Migration Through Blocking Wnt Signaling. J. Cardiovasc. Pharmacol. 2016, 68, 414–424. [Google Scholar] [CrossRef] [PubMed]
- Reddy, V.S.; Valente, A.J.; Delafontaine, P.; Chandrasekar, B. Interleukin-18/WNT1-inducible signaling pathway protein-1 signaling mediates human saphenous vein smooth muscle cell proliferation. J. Cell. Physiol. 2011, 226, 3303–3315. [Google Scholar] [CrossRef] [Green Version]
- Tsaousi, A.; Williams, H.; Lyon, C.A.; Taylor, V.; Swain, A.; Johnson, J.L.; George, S.J. Wnt4/β-Catenin signaling induces VSMC proliferation and is associated with intimal thickening. Circ. Res. 2011, 108, 427–436. [Google Scholar] [CrossRef] [Green Version]
- Marinou, K.; Christodoulides, C.; Antoniades, C.; Koutsilieris, M. Wnt signaling in cardiovascular physiology. Trends Endocrinol. Metab. 2012, 23, 628–636. [Google Scholar] [CrossRef]
- Williams, H.; Mill, C.A.; Monk, B.A.; Hulin-Curtis, S.; Johnson, J.L.; George, S.J. Wnt2 and WISP-1/CCN4 induce intimal thickening via promotion of smooth muscle cell migration. Arter. Thromb. Vasc. Biol. 2016, 36, 1417–1424. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Xiao, Y.; Mou, Y.; Zhao, Y.; Blankesteijn, W.M.; Hall, J.L. A role for the β-catenin/T-cell factor signaling cascade in vascular remodeling. Circ. Res. 2002, 90, 340–347. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Xiao, Y.; Mou, Y.; Zhao, Y.; Blankesteijn, W.M.; Hall, J.L. LDL receptor-related protein LRP6 regulates proliferation and survival through the Wnt cascade in vascular smooth muscle cells. Am. J. Physiol. Heart Circ. Physiol. 2004, 287, H2376–H2383. [Google Scholar] [CrossRef] [Green Version]
- Dejana, E. The role of Wnt signaling in physiological and pathological angiogenesis. Circ. Res. 2010, 107, 943–952. [Google Scholar] [CrossRef]
- Masckauchan, T.N.; Agalliu, D.; Vorontchikhina, M.; Ahn, A.; Parmalee, N.L.; Li, C.M.; Khoo, A.; Tycko, B.; Brown, A.M.; Kitajewski, J. Wnt5a signaling induces proliferation and survival of endothelial cells in vitro and expression of MMP-1 and Tie-2. Mol. Biol. Cell 2006, 17, 5163–5172. [Google Scholar] [CrossRef] [PubMed]
- Cheng, C.W.; Yeh, J.C.; Fan, T.P.; Smith, S.K.; Charnock-Jones, D.S. Wnt5a-mediated non-canonical Wnt signalling regulates human endothelial cell proliferation and migration. Biochem. Biophys. Res. Commun. 2008, 365, 285–290. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Kim, J.; Kim, D.W.; Ha, Y.; Ihm, M.H.; Kim, H.; Song, K.; Lee, I. Wnt5a induces endothelial inflammation via beta-catenin-independent signaling. J. Immunol. 2010, 185, 1274–1282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guertin, D.A.; Stevens, D.M.; Thoreen, C.C.; Burds, A.A.; Kalaany, N.Y.; Moffat, J.; Brown, M.; Fitzgerald, K.J.; Sabatini, D.M. Ablation in mice of the mTORC components raptor, rictor, or mLST8 reveals that mTORC2 is required for signaling to Akt-FOXO and PKCalpha, but not S6K1. Dev. Cell. 2006, 11, 859–871. [Google Scholar] [CrossRef] [Green Version]
- Irie, H.Y.; Pearline, R.V.; Grueneberg, D.; Hsia, M.; Ravichandran, P.; Kothari, N.; Natesan, S.; Brugge, J.S. Distinct roles of Akt1 and Akt2 in regulating cell migration and epithelial–mesenchymal transition. J. Cell Biol. 2005, 171, 1023–1034. [Google Scholar] [CrossRef]
- Jin, Y.; Xie, Y.; Ostriker, A.C.; Zhang, X.; Liu, R.; Lee, M.Y.; Leslie, K.L.; Tang, W.; Du, J.; Lee, S.H.; et al. Opposing actions of AKT (Protein Kinase B) isoforms in vascular smooth muscle injury and therapeutic response. Arter. Thromb. Vasc. Biol. 2017, 37, 2311–2321. [Google Scholar] [CrossRef] [Green Version]
- Morello, F.; Perino, A.; Hirsch, E. Phosphoinositide 3-kinase signalling in the vascular system. Cardiovasc. Res. 2009, 82, 261–271. [Google Scholar] [CrossRef] [Green Version]
- Karar, J.; Maity, A. PI3K/AKT/mTOR pathway in angiogenesis. Front. Mol. Neurosci. 2011, 4, 51. [Google Scholar] [CrossRef] [Green Version]
- Hemmings, B.A.; Restuccia, D.F. PI3K-PKB/Akt pathway. Cold Spring Harb. Perspect. Biol. 2012, 4, a011189. [Google Scholar] [CrossRef] [Green Version]
- Chow, J.Y.C.; Quach, K.T.; Cabrera, B.L.; Cabral, J.A.; Beck, S.E.; Carethers, J.M. RAS/ERK modulates TGFβ-regulated PTEN expression in human pancreatic adenocarcinoma cells. Carcinogenesis 2007, 28, 2321–2327. [Google Scholar] [CrossRef]
- Vidal, S.; Bouzaher, Y.H.; El Motiam, A.; Seoane, R.; Rivas, C. Overview of the regulation of the class IA PI3K/AKT pathway by SUMO. Semin. Cell Dev. Biol. 2022, 132, 51–61. [Google Scholar] [CrossRef] [PubMed]
- Stabile, E.; Zhou, Y.F.; Saji, M.; Castagna, M.; Shou, M.; Kinnaird, T.D.; Baffour, R.; Ringel, M.D.; Epstein, S.E.; Fuchs, S. Akt controls vascular smooth muscle cell proliferation in vitro and in vivo by delaying G1/S exit. Circ. Res. 2003, 93, 1059–1065. [Google Scholar] [CrossRef] [Green Version]
- Cross, M.; Dexter, T.M. Growth factors in development, transformation, and tumorigenesis. Cell Press 1991, 64, 271–280. [Google Scholar] [CrossRef]
- Brunet, A.; Bonni, A.; Zigmond, M.J.; Lin, M.Z.; Juo, P.; Hu, L.S.; Anderson, M.J.; Arden, K.C.; Blenis, J.; Greenberg, M.E. AKT promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell Press 1999, 96, 857–868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abid, M.R.; Yano, K.; Guo, S.; Patel, V.I.; Shrikhande, G.; Spokes, K.C.; Ferran, C.; Aird, W.C. Forkhead transcription factors inhibit vascular smooth muscle cell proliferation and neointimal hyperplasia. J. Biol. Chem. 2005, 280, 29864–29873. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, X.; Gong, Y.; Tang, Y.; Li, H.; He, Q.; Gower, L.; Liaw, L.; Friesel, R.E. Spry1 and Spry4 differentially regulate human aortic smooth muscle cell phenotype via Akt/FoxO/myocardin signaling. PLoS ONE 2013, 8, e58746. [Google Scholar] [CrossRef] [Green Version]
- Tucka, J.; Yu, H.; Gray, K.; Figg, N.; Maguire, J.; Lam, B.; Bennett, M.; Littlewood, T. Akt1 regulates vascular smooth muscle cell apoptosis through FoxO3a and Apaf1 and protects against arterial remodeling and atherosclerosis. Arter. Thromb. Vasc. Biol. 2014, 34, 2421–2428. [Google Scholar] [CrossRef] [Green Version]
- Allard, D.; Figg, N.; Bennett, M.R.; Littlewood, T.D. Akt regulates the survival of vascular smooth muscle cells via inhibition of FoxO3a and GSK3. J. Biol. Chem. 2008, 283, 19739–19747. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Qian, Y.; Sun, Z.; Shen, X.; Cai, Y.; Li, L.; Wang, Z. Role of PI3K in the progression and regression of atherosclerosis. Front. Pharmacol. 2021, 12, 632378. [Google Scholar] [CrossRef]
- Diebold, I.; Petry, A.; Burger, M.; Hess, J.; Görlach, A. NOX4 mediates activation of FoxO3a and matrix metalloproteinase-2 expression by urotensin-II. Mol. Biol. Cell 2011, 22, 4424–4434. [Google Scholar] [CrossRef]
- Yu, H.; Fellows, A.; Foote, K.; Yang, Z.; Figg, N.; Littlewood, T.; Bennett, M. FOXO3a (Forkhead Transcription Factor O Subfamily Member 3a) links vascular smooth muscle cell apoptosis, matrix breakdown, atherosclerosis, and vascular remodeling through a novel pathway involving MMP13 (Matrix Metalloproteinase 13). Arterioscler. Thromb. Vasc. Biol. 2018, 38, 555–565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abid, M.R.; Guo, S.; Minami, T.; Spokes, K.C.; Ueki, K.; Skurk, C.; Walsh, K.; Aird, W.C. Vascular endothelial growth factor activates PI3K/Akt/forkhead signaling in endothelial cells. Arter. Thromb. Vasc. Biol. 2004, 24, 294–300. [Google Scholar] [CrossRef] [Green Version]
- Skurk, C.; Maatz, H.; Kim, H.S.; Yang, J.; Abid, M.R.; Aird, W.C.; Walsh, K. The Akt-regulated forkhead transcription factor FOXO3a controls endothelial cell viability through modulation of the caspase-8 inhibitor FLIP. J. Biol. Chem. 2004, 279, 1513–1525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taddei, A.; Giampietro, C.; Conti, A.; Orsenigo, F.; Breviario, F.; Pirazzoli, V.; Potente, M.; Daly, C.; Dimmeler, S.; Dejana, E. Endothelial adherens junctions control tight junctions by VE-cadherin-mediated upregulation of claudin-5. Nat. Cell Biol. 2008, 10, 923–934. [Google Scholar] [CrossRef]
- LaFoya, B.; Munroe, J.A.; Mia, M.M.; Detweiler, M.A.; Crow, J.J.; Wood, T.; Roth, S.; Sharma, B.; Albig, A.R. Notch: A multi-functional integrating system of microenvironmental signals. Dev. Biol. 2016, 418, 227–241. [Google Scholar] [CrossRef] [PubMed]
- Fiúza, U.M.; Arias, A.M. Cell and molecular biology of Notch. J. Endocrinol. 2007, 194, 459–474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Falo-Sanjuan, J.; Bray, S.J. Decoding the Notch signal. Dev. Growth Differ. 2020, 62, 4–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Souza, B.; Meloty-Kapella, L.; Weinmaster, G. Canonical and Non-Canonical Notch Ligands. In Current Topics in Developmental Biology; Kopan, R., Ed.; Academic Press: Cambridge, MA, USA, 2010; pp. 73–129. [Google Scholar]
- Baeten, J.T.; Lilly, B. Differential regulation of NOTCH2 and NOTCH3 contribute to their unique functions in vascular smooth muscle cells. J. Biol. Chem. 2015, 290, 16226–16237. [Google Scholar] [CrossRef] [Green Version]
- Andersen, P.; Uosaki, H.; Shenje, L.T.; Kwon, C. Non-canonical Notch signaling: Emerging role and mechanism. Trends Cell Biol. 2012, 22, 257–265. [Google Scholar] [CrossRef] [Green Version]
- Zhou, B.; Lin, W.; Long, Y.; Yang, Y.; Zhang, H.; Wu, K.; Chu, Q. Notch signaling pathway: Architecture, disease, and therapeutics. Signal. Transduct. Target. Ther. 2022, 7, 95. [Google Scholar] [CrossRef]
- Zavadil, J.; Cermak, L.; Soto-Nieves, N.; Böttinger, E.P. Integration of TGF-β/Smad and Jagged1/Notch signalling in epithelial-to-mesenchymal transition. EMBO J. 2004, 23, 1155–1165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moretti, J.; Brou, C. Ubiquitinations in the notch signaling pathway. Int. J. Mol. Sci. 2013, 14, 6359–6381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Contreras-Cornejo, H.; Saucedo-Correa, G.; Oviedo-Boyso, J.; Valdez-Alarcón, J.J.; Baizabal-Aguirre, V.M.; Cajero-Juárez, M.; Bravo-Patiño, A. The CSL proteins, versatile transcription factors and context dependent corepressors of the notch signaling pathway. Cell Div. 2016, 11, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyamoto, A.; Lau, R.; Hein, P.W.; Shipley, J.M.; Weinmaster, G. Microfibrillar proteins MAGP-1 and MAGP-2 induce Notch1 extracellular domain dissociation and receptor activation. J. Biol. Chem. 2006, 281, 10089–10097. [Google Scholar] [CrossRef] [Green Version]
- Deford, P.; Brown, K.; Richards, R.L.; King, A.; Newburn, K.; Westover, K.; Albig, A.R. MAGP2 controls Notch via interactions with RGD binding integrins: Identification of a novel ECM-integrin-Notch signaling axis. Exp. Cell Res. 2016, 341, 84–91. [Google Scholar] [CrossRef] [Green Version]
- Meng, H.; Zhang, X.; Hankenson, K.D.; Wang, M.M. Thrombospondin 2 potentiates notch3/jagged1 signaling. J. Biol. Chem. 2009, 284, 7866–7874. [Google Scholar] [CrossRef] [Green Version]
- Zhao, N.; Liu, H.H.; Lilly, B. Reciprocal regulation of syndecan-2 and Notch signaling in vascular smooth muscle cells. J. Biol. Chem. 2012, 287, 16111–16120. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Meng, H.; Wang, M.M. Collagen represses canonical Notch signaling and binds to Notch ectodomain. Int. J. Biochem. Cell Biol. 2013, 45, 1274–1280. [Google Scholar] [CrossRef] [Green Version]
- Nandhu, M.S.; Hu, B.; Cole, S.E.; Erdreich-Epstein, A.; Rodriguez-Gil, D.J.; Viapiano, M.S. Novel paracrine modulation of Notch-DLL4 signaling by fibulin-3 promotes angiogenesis in high-grade gliomas. Cancer Res. 2014, 74, 5435–5448. [Google Scholar] [CrossRef] [Green Version]
- High, F.A.; Lu, M.M.; Pear, W.S.; Loomes, K.M.; Kaestner, K.H.; Epstein, J.A. Endothelial expression of the Notch ligand Jagged1 is required for vascular smooth muscle development. Proc. Natl. Acad. Sci. USA 2008, 105, 1955–1959. [Google Scholar] [CrossRef]
- Morrow, D.; Guha, S.; Sweeney, C.; Birney, Y.; Walshe, T.; O’Brien, C.; Walls, D.; Redmond, E.M.; Cahill, P.A. Notch and vascular smooth muscle cell phenotype. Circ. Res. 2008, 103, 1370–1382. [Google Scholar] [CrossRef] [Green Version]
- Boucher, J.; Gridley, T.; Liaw, L. Molecular pathways of notch signaling in vascular smooth muscle cells. Front. Physiol. 2012, 3, 81. [Google Scholar] [CrossRef] [Green Version]
- Pedrosa, A.R.; Trindade, A.; Fernandes, A.C.; Carvalho, C.; Gigante, J.; Tavares, A.T.; Diéguez-Hurtado, R.; Yagita, H.; Adams, R.H.; Duarte, A. Endothelial Jagged1 antagonizes Dll4 regulation of endothelial branching and promotes vascular maturation downstream of Dll4/Notch1. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 1134–1146. [Google Scholar] [CrossRef] [Green Version]
- Miele, L.; Osborne, B. Arbiter of differentiation and death: Notch signaling meets apoptosis. J. Cell. Physiol. 1999, 181, 393–409. [Google Scholar] [CrossRef]
- Sweeney, C.; Morrow, D.; Birney, Y.A.; Coyle, S.; Hennessy, C.; Scheller, A.; Cummins, P.M.; Walls, D.; Redmond, E.M.; Cahill, P.A. Notch 1 and 3 receptor signaling modulates vascular smooth muscle cell growth, apoptosis, and migration via a CBF-1/RBP-Jk dependent pathway. FASEB J. 2004, 18, 1421–1423. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.; Li, Y.; Chen, H.; Wei, W.; An, Y.; Zhu, G. RNA interference-mediated NOTCH3 knockdown induces phenotype switching of vascular smooth muscle cells in vitro. Int. J. Clin. Exp. Med. 2015, 8, 12674–12684. [Google Scholar] [PubMed]
- Boucher, J.M.; Harrington, A.; Rostama, B.; Lindner, V.; Liaw, L. A receptor-specific function for Notch2 in mediating vascular smooth muscle cell growth arrest through cyclin-dependent kinase inhibitor 1B. Circ Res. 2013, 113, 975–985. [Google Scholar] [CrossRef] [Green Version]
- Tang, Y.; Urs, S.; Boucher, J.; Bernaiche, T.; Venkatesh, D.; Spicer, D.B.; Vary, C.P.; Liaw, L. Notch and transforming growth factor-beta (TGFbeta) signaling pathways cooperatively regulate vascular smooth muscle cell differentiation. J. Biol. Chem. 2010, 285, 17556–17563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, C.H.; Lilly, B. Notch signaling governs phenotypic modulation of smooth muscle cells. Vasc. Pharmacol. 2014, 63, 88–96. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Kennard, S.; Lilly, B. NOTCH3 expression is induced in mural cells through an autoregulatory loop that requires endothelial-expressed JAGGED1. Circ. Res. 2009, 104, 466–475. [Google Scholar] [CrossRef]
- Proweller, A.; Pear, W.S.; Parmacek, M.S. Notch signaling represses myocardin-induced smooth muscle cell differentiation. J. Biol. Chem. 2005, 280, 8994–9004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Takeshita, K.; Liu, P.Y.; Satoh, M.; Oyama, N.; Mukai, Y.; Chin, M.T.; Krebs, L.; Kotlikoff, M.I.; Radtke, F.; et al. Smooth muscle Notch1 mediates neointimal formation after vascular injury. Circulation 2009, 119, 2686–2692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baeten, J.T.; Lilly, B. Notch signaling in vascular smooth muscle cells. Adv. Pharmacol. 2017, 78, 351–382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leslie, J.D.; Ariza-McNaughton, L.; Bermange, A.L.; McAdow, R.; Johnson, S.L.; Lewis, J. Endothelial signalling by the Notch ligand Delta-like 4 restricts angiogenesis. Development 2007, 134, 839–844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mack, J.J.; Mosqueiro, T.S.; Archer, B.J.; Jones, W.M.; Sunshine, H.; Faas, G.C.; Briot, A.; Aragón, R.L.; Su, T.; Romay, M.C.; et al. NOTCH1 is a mechanosensor in adult arteries. Nat. Commun. 2017, 8, 1620. [Google Scholar] [CrossRef] [Green Version]
- Mack, J.J.; Iruela-Arispe, M.L. NOTCH regulation of the endothelial cell phenotype. Curr. Opin. Hematol. 2018, 25, 212–218. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.J.; Xiao, M.; Balint, K.; Soma, A.; Pinnix, C.C.; Capobianco, A.J.; Velazquez, O.C.; Herlyn, M. Inhibition of endothelial cell proliferation by Notch1 signaling is mediated by repressing MAPK and PI3K/Akt pathways and requires MAML1. FASEB J. 2006, 20, 1009–1011. [Google Scholar] [CrossRef] [Green Version]
- Benedito, R.; Trindade, A.; Hirashima, M.; Henrique, D.; da Costa, L.L.; Rossant, J.; Gill, P.S.; Duarte, A. Loss of Notch signalling induced by Dll4 causes arterial calibre reduction by increasing endothelial cell response to angiogenic stimuli. BMC Dev. Biol. 2008, 8, 117. [Google Scholar] [CrossRef] [Green Version]
- Yang, K.; Proweller, A. Vascular smooth muscle Notch signals regulate endothelial cell sensitivity to angiogenic stimulation. J. Biol. Chem. 2011, 286, 13741–13753. [Google Scholar] [CrossRef] [Green Version]
- Polacheck, W.J.; Kutys, M.L.; Yang, J.; Eyckmans, J.; Wu, Y.; Vasavada, H.; Hirschi, K.K.; Chen, C.S. A non-canonical Notch complex regulates adherens junctions and vascular barrier function. Nature 2017, 552, 258–262. [Google Scholar] [CrossRef]
- Griendling, K.K.; Lassègue, B.; Alexander, R.W. Angiotensin receptors and their therapeutic implications. Annu. Rev. Pharm. Toxicol. 1996, 36, 281–306. [Google Scholar] [CrossRef] [PubMed]
- Mehta, P.K.; Griendling, K.K. Angiotensin II cell signaling: Physiological and pathological effects in the cardiovascular system. Am. J. Physiol.-Cell Physiol. 2007, 292, C82–C97. [Google Scholar] [CrossRef] [PubMed]
- Forrester, S.J.; Kawai, T.; O’Brien, S.; Thomas, W.; Harris, R.C.; Eguchi, S. Epidermal growth factor receptor transactivation: Mechanisms, pathophysiology, and potential therapies in the cardiovascular system. Annu. Rev. Pharm. Toxicol. 2016, 56, 627–653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forrester, S.J.; Booz, G.W.; Sigmund, C.D.; Coffman, T.M.; Kawai, T.; Rizzo, V.; Scalia, R.; Eguchi, S. Angiotensin II signal transduction: An update on mechanisms of physiology and pathophysiology. Physiol. Rev. 2018, 98, 1627–1738. [Google Scholar] [CrossRef]
- Lassègue, B.; Alexander, R.W.; Nickenig, G.; Clark, M.; Murphy, T.J.; Griendling, K.K. Angiotensin II down-regulates the vascular smooth muscle AT1 receptor by transcriptional and post-transcriptional mechanisms: Evidence for homologous and heterologous regulation. Mol. Pharmacol. 1995, 48, 601–609. [Google Scholar]
- Gunther, S.; Gimbrone, M.A.; Alexander, R.W. Regulation by angiotensin II of its receptors in resistance blood vessels. Nature 1980, 287, 230–232. [Google Scholar] [CrossRef]
- Nishida, M.; Ogushi, M.; Suda, R.; Toyotaka, M.; Saiki, S.; Kitajima, N.; Nakaya, M.; Kim, K.M.; Ide, T.; Sato, Y.; et al. Heterologous down-regulation of angiotensin type 1 receptors by purinergic P2Y2 receptor stimulation through S-nitrosylation of NF-kappaB. Proc. Natl. Acad. Sci. USA 2011, 108, 6662–6667. [Google Scholar] [CrossRef] [Green Version]
- Dzau, V.J. Theodore Cooper Lecture: Tissue angiotensin and pathobiology of vascular disease: A unifying hypothesis. Hypertension 2001, 37, 1047–1052. [Google Scholar] [CrossRef] [Green Version]
- Gao, B.B.; Stuart, L.; Feener, E.P. Label-free quantitative analysis of one-dimensional PAGE LC/MS/MS proteome: Application on angiotensin II-stimulated smooth muscle cells secretome. Mol. Cell Proteom. 2008, 7, 2399–2409. [Google Scholar] [CrossRef] [Green Version]
- Kranzhöfer, R.; Schmidt, J.; Pfeiffer, C.A.; Hagl, S.; Libby, P.; Kübler, W. Angiotensin induces inflammatory activation of human vascular smooth muscle cells. Arter. Thromb. Vasc. Biol. 1999, 19, 1623–1629. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.; Ohta, K.; Hamaguchi, A.; Yukimura, T.; Miura, K.; Iwao, H. Angiotensin II induces cardiac phenotypic modulation and remodeling in vivo in rats. Hypertension 1995, 25, 1252–1259. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, V.; Golledge, J.; Heywood, E.B.; Bruemmer, D.; Daugherty, A. Regulation of peroxisome proliferator-activated receptor-γ by angiotensin II via transforming growth factor-β1-activated p38 mitogen-activated protein kinase in aortic smooth muscle cells. Arter. Thromb. Vasc. Biol. 2012, 32, 397–405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kirabo, A.; Oh, S.P.; Kasahara, H.; Wagner, K.U.; Sayeski, P.P. Vascular smooth muscle Jak2 deletion prevents angiotensin II-mediated neointima formation following injury in mice. J. Mol. Cell Cardiol. 2011, 50, 1026–1034. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Li, W.; Gupta, A.K.; Mohler, P.J.; Anderson, M.E.; Grumbach, I.M. Calmodulin kinase II is required for angiotensin II-mediated vascular smooth muscle hypertrophy. Am. J. Physiol.-Heart Circ. Physiol. 2010, 298, H688–H698. [Google Scholar] [CrossRef] [Green Version]
- Prasad, A.M.; Ketsawatsomkron, P.; Nuno, D.W.; Koval, O.M.; Dibbern, M.E.; Venema, A.N.; Sigmund, C.D.; Lamping, K.G.; Grumbach, I.M. Role of CaMKII in Ang-II-dependent small artery remodeling. Vasc. Pharmacol. 2016, 87, 172–179. [Google Scholar] [CrossRef] [Green Version]
- Dikalova, A.; Clempus, R.; Lassègue, B.; Cheng, G.; McCoy, J.; Dikalov, S.; San Martin, A.; Lyle, A.; Weber, D.S.; Weiss, D.; et al. Nox1 overexpression potentiates angiotensin II-induced hypertension and vascular smooth muscle hypertrophy in transgenic mice. Circulation 2005, 112, 2668–2676. [Google Scholar] [CrossRef] [Green Version]
- Carlström, M.; Lai, E.Y.; Ma, Z.; Steege, A.; Patzak, A.; Eriksson, U.J.; Lundberg, J.O.; Wilcox, C.S.; Persson, A.E. Superoxide dismutase 1 limits renal microvascular remodeling and attenuates arteriole and blood pressure responses to angiotensin II via modulation of nitric oxide bioavailability. Hypertension 2010, 56, 907–913. [Google Scholar] [CrossRef] [Green Version]
- Schröder, K.; Zhang, M.; Benkhoff, S.; Mieth, A.; Pliquett, R.; Kosowski, J.; Kruse, C.; Luedike, P.; Michaelis, U.R.; Weissmann, N.; et al. Nox4 is a protective reactive oxygen species generating vascular NADPH oxidase. Circ. Res. 2012, 110, 1217–1225. [Google Scholar] [CrossRef] [Green Version]
- Yaghini, F.A.; Song, C.Y.; Lavrentyev, E.N.; Ghafoor, H.U.; Fang, X.R.; Estes, A.M.; Campbell, W.B.; Malik, K.U. Angiotensin II-induced vascular smooth muscle cell migration and growth are mediated by cytochrome P450 1B1-dependent superoxide generation. Hypertension 2010, 55, 1461–1467. [Google Scholar] [CrossRef] [Green Version]
- Dzau, V.J.; Gibbons, G.H. Endothelium and growth factors in vascular remodeling of hypertension. Hypertension 1991, 18, III115. [Google Scholar] [CrossRef]
- Mahmud, A.; Feely, J. Arterial stiffness and the renin-angiotensin-aldosterone system. J. Renin-Angiotensin-Aldosterone Syst. 2004, 5, 102–108. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Kim, S.H.; Monticone, R.E.; Lakatta, E.G. Matrix metalloproteinases promote arterial remodeling in aging, hypertension, and atherosclerosis. Hypertension 2015, 65, 698–703. [Google Scholar] [CrossRef] [PubMed]
- Nguyen Dinh Cat, A.; Montezano, A.C.; Burger, D.; Touyz, R.M. Angiotensin II, NADPH oxidase, and redox signaling in the vasculature. Antioxid. Redox Signal. 2013, 19, 1110–1120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pueyo, M.E.; Gonzalez, W.; Nicoletti, A.; Savoie, F.; Arnal, J.F.; Michel, J.B. Angiotensin II stimulates endothelial vascular cell adhesion molecule-1 via nuclear factor-kappaB activation induced by intracellular oxidative stress. Arter. Thromb. Vasc. Biol. 2000, 20, 645–651. [Google Scholar] [CrossRef] [Green Version]
- Arenas, I.A.; Xu, Y.; Lopez-Jaramillo, P.; Davidge, S.T. Angiotensin II-induced MMP-2 release from endothelial cells is mediated by TNF-alpha. Am. J. Physiol.-Cell Physiol. 2004, 286, C779–C784. [Google Scholar] [CrossRef] [PubMed]
- Dimmeler, S.; Rippmann, V.; Weiland, U.; Haendeler, J.; Zeiher, A.M. Angiotensin II induces apoptosis of human endothelial cells. Protective effect of nitric oxide. Circ. Res. 1997, 81, 970–976. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, T.; Barker, T.A.; Berk, B.C. Angiotensin II and the endothelium: Diverse signals and effects. Hypertension 2005, 45, 163–169. [Google Scholar] [CrossRef]
- Han, C.; Liu, J.; Liu, X.; Li, M. Angiotensin II induces C-reactive protein expression through ERK1/2 and JNK signaling in human aortic endothelial cells. Atherosclerosis 2010, 212, 206–212. [Google Scholar] [CrossRef]
- Kopaliani, I.; Martin, M.; Zatschler, B.; Bortlik, K.; Müller, B.; Deussen, A. Cell-specific and endothelium-dependent regulations of matrix metalloproteinase-2 in rat aorta. Basic Res. Cardiol. 2014, 109, 419. [Google Scholar] [CrossRef]
- Wei, H.; Hu, J.H.; Angelov, S.N.; Fox, K.; Yan, J.; Enstrom, R.; Smith, A.; Dichek, D.A. Aortopathy in a Mouse Model of Marfan Syndrome Is Not Mediated by Altered Transforming Growth Factor β Signaling. J. Am. Heart Assoc. 2017, 6, e004968. [Google Scholar] [CrossRef]
- Guo, X.; Wang, X.F. Signaling cross-talk between TGF-beta/BMP and other pathways. Cell Res. 2009, 19, 71–88. [Google Scholar] [CrossRef]
- Holm, T.M.; Habashi, J.P.; Doyle, J.J.; Bedja, D.; Chen, Y.; van Erp, C.; Lindsay, M.E.; Kim, D.; Schoenhoff, F.; Cohn, R.D.; et al. Noncanonical TGFβ signaling contributes to aortic aneurysm progression in Marfan syndrome mice. Science 2011, 332, 358–361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsuura, I.; Wang, G.; He, D.; Liu, F. Identification and characterization of ERK MAP kinase phosphorylation sites in Smad3. Biochemistry 2005, 44, 12546–12553. [Google Scholar] [CrossRef] [PubMed]
- Millet, C.; Yamashita, M.; Heller, M.; Yu, L.R.; Veenstra, T.D.; Zhang, Y.E. A negative feedback control of transforming growth factor-beta signaling by glycogen synthase kinase 3-mediated Smad3 linker phosphorylation at Ser-204. J. Biol. Chem. 2009, 284, 19808–19816. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kretzschmar, M.; Doody, J.; Massagué, J. Opposing BMP and EGF signalling pathways converge on the TGF-beta family mediator Smad1. Nature 1997, 389, 618–622. [Google Scholar] [CrossRef] [PubMed]
- Kosinski, C.; Li, V.S.; Chan, A.S.; Zhang, J.; Ho, C.; Tsui, W.Y.; Chan, T.L.; Mifflin, R.C.; Powell, D.W.; Yuen, S.T.; et al. Gene expression patterns of human colon tops and basal crypts and BMP antagonists as intestinal stem cell niche factors. Proc. Natl. Acad. Sci. USA 2007, 104, 15418–15423. [Google Scholar] [CrossRef] [Green Version]
- Guo, X.; Ramirez, A.; Waddell, D.S.; Li, Z.; Liu, X.; Wang, X.F. Axin and GSK3- control Smad3 protein stability and modulate TGF- signaling. Genes Dev. 2008, 22, 106–120. [Google Scholar] [CrossRef] [Green Version]
- Furuhashi, M.; Yagi, K.; Yamamoto, H.; Furukawa, Y.; Shimada, S.; Nakamura, Y.; Kikuchi, A.; Miyazono, K.; Kato, M. Axin facilitates Smad3 activation in the transforming growth factor beta signaling pathway. Mol. Cell Biol. 2001, 21, 5132–5141. [Google Scholar] [CrossRef] [Green Version]
- Han, G.; Li, A.G.; Liang, Y.Y.; Owens, P.; He, W.; Lu, S.; Yoshimatsu, Y.; Wang, D.; Ten Dijke, P.; Lin, X.; et al. Smad7-induced beta-catenin degradation alters epidermal appendage development. Dev. Cell 2006, 11, 301–312. [Google Scholar] [CrossRef] [Green Version]
- Liu, W.; Rui, H.; Wang, J.; Lin, S.; He, Y.; Chen, M.; Li, Q.; Ye, Z.; Zhang, S.; Chan, S.C.; et al. Axin is a scaffold protein in TGF-beta signaling that promotes degradation of Smad7 by Arkadia. EMBO J. 2006, 25, 1646–1658. [Google Scholar] [CrossRef] [Green Version]
- Dao, D.Y.; Yang, X.; Chen, D.; Zuscik, M.; O’Keefe, R.J. Axin1 and Axin2 are regulated by TGF- and mediate cross-talk between TGF- and Wnt signaling pathways. Ann. N. Y. Acad Sci. 2007, 1116, 82–99. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Jho, E.H. The protein stability of Axin, a negative regulator of Wnt signaling, is regulated by Smad ubiquitination regulatory factor 2 (Smurf2). J. Biol. Chem. 2010, 285, 36420–36426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Labbé, E.; Letamendia, A.; Attisano, L. Association of Smads with lymphoid enhancer binding factor 1/T cell-specific factor mediates cooperative signaling by the transforming growth factor-beta and wnt pathways. Proc. Natl. Acad. Sci. USA 2000, 97, 8358–8363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akhmetshina, A.; Palumbo, K.; Dees, C.; Bergmann, C.; Venalis, P.; Zerr, P.; Horn, A.; Kireva, T.; Beyer, C.; Zwerina, J.; et al. Activation of canonical Wnt signalling is required for TGF-β-mediated fibrosis. Nat. Commun. 2012, 3, 735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamiya, N.; Ye, L.; Kobayashi, T.; Mochida, Y.; Yamauchi, M.; Kronenberg, H.M.; Feng, J.Q.; Mishina, Y. BMP signaling negatively regulates bone mass through sclerostin by inhibiting the canonical Wnt pathway. Development 2008, 135, 3801–3811. [Google Scholar] [CrossRef] [Green Version]
- Perez, V.A.; Ali, Z.; Alastalo, T.P.; Ikeno, F.; Sawada, H.; Lai, Y.J.; Kleisli, T.; Spiekerkoetter, E.; Qu, X.; Rubinos, L.H.; et al. BMP promotes motility and represses growth of smooth muscle cells by activation of tandem Wnt pathways. J. Cell Biol. 2011, 192, 171–188. [Google Scholar] [CrossRef] [Green Version]
- Ruiz-Ortega, M.; Ruperez, M.; Esteban, V.; Egido, J. Molecular mechanisms of angiotensin II-induced vascular injury. Curr. Hypertens. Rep. 2003, 5, 73–79. [Google Scholar] [CrossRef]
- Border, W.A.; Noble, N.A. Interactions of transforming growth factor-beta and angiotensin II in renal fibrosis. Hypertension 1998, 31, 181–188. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Huang, X.R.; Canlas, E.; Oka, K.; Truong, L.D.; Deng, C.; Bhowmick, N.A.; Ju, W.; Bottinger, E.P.; Lan, H.Y. Essential role of Smad3 in angiotensin II-induced vascular fibrosis. Circ. Res. 2006, 98, 1032–1039. [Google Scholar] [CrossRef] [Green Version]
- Sriramula, S.; Francis, J. Tumor Necrosis Factor—Alpha Is Essential for Angiotensin II-Induced Ventricular Remodeling: Role for Oxidative Stress. PLoS ONE 2015, 10, e0138372. [Google Scholar] [CrossRef]
- Zhang, X.H.; Zheng, B.; Gu, C.; Fu, J.R.; Wen, J.K. TGF-β1 downregulates AT1 receptor expression via PKC-δ-mediated Sp1 dissociation from KLF4 and Smad-mediated PPAR-γ association with KLF4. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 1015–1023. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Vita, J.; Sánchez-López, E.; Esteban, V.; Rupérez, M.; Egido, J.; Ruiz-Ortega, M. Angiotensin II activates the Smad pathway in vascular smooth muscle cells by a transforming growth factor-beta-independent mechanism. Circulation 2005, 111, 2509–2517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baurand, A.; Zelarayan, L.; Betney, R.; Gehrke, C.; Dunger, S.; Noack, C.; Busjahn, A.; Huelsken, J.; Taketo, M.M.; Birchmeier, W.; et al. Beta-catenin downregulation is required for adaptive cardiac remodeling. Circ. Res. 2007, 100, 1353–1362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shanmugam, P.; Valente, A.J.; Prabhu, S.D.; Venkatesan, B.; Yoshida, T.; Delafontaine, P.; Chandrasekar, B. Angiotensin-II type 1 receptor and NOX2 mediate TCF/LEF and CREB dependent WISP1 induction and cardiomyocyte hypertrophy. J. Mol. Cell Cardiol. 2011, 50, 928–938. [Google Scholar] [CrossRef] [Green Version]
- Ozasa, Y.; Akazawa, H.; Qin, Y.; Tateno, K.; Ito, K.; Kudo-Sakamoto, Y.; Yano, M.; Yabumoto, C.; Naito, A.T.; Oka, T.; et al. Notch activation mediates angiotensin II-induced vascular remodeling by promoting the proliferation and migration of vascular smooth muscle cells. Hypertens. Res. 2013, 36, 859–865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Basu, S.; Srinivasan, D.K.; Yang, K.; Raina, H.; Banerjee, S.; Zhang, R.; Fisher, S.A.; Proweller, A. Notch transcriptional control of vascular smooth muscle regulatory gene expression and function. J. Biol. Chem. 2013, 288, 11191–11202. [Google Scholar] [CrossRef] [Green Version]
- Klüppel, M.; Wrana, J.L. Turning it up a Notch: Cross-talk between TGF beta and Notch signaling. Bioessays 2005, 27, 115–118. [Google Scholar] [CrossRef]
- Blokzijl, A.; Dahlqvist, C.; Reissmann, E.; Falk, A.; Moliner, A.; Lendahl, U.; Ibáñez, C.F. Cross-talk between the Notch and TGF-beta signaling pathways mediated by interaction of the Notch intracellular domain with Smad3. J. Cell Biol. 2003, 163, 723–728. [Google Scholar] [CrossRef]
- Fu, Y.; Chang, A.; Chang, L.; Niessen, K.; Eapen, S.; Setiadi, A.; Karsan, A. Differential regulation of transforming growth factor beta signaling pathways by Notch in human endothelial cells. J. Biol. Chem. 2009, 284, 19452–19462. [Google Scholar] [CrossRef] [Green Version]
- Itoh, F.; Itoh, S.; Goumans, M.J.; Valdimarsdottir, G.; Iso, T.; Dotto, G.P.; Hamamori, Y.; Kedes, L.; Kato, M.; ten Dijke, P. Synergy and antagonism between Notch and BMP receptor signaling pathways in endothelial cells. EMBO J. 2004, 23, 541–551. [Google Scholar] [CrossRef] [Green Version]
- Masuda, S.; Kumano, K.; Shimizu, K.; Imai, Y.; Kurokawa, M.; Ogawa, S.; Miyagishi, M.; Taira, K.; Hirai, H.; Chiba, S. Notch1 oncoprotein antagonizes TGF-beta/Smad-mediated cell growth suppression via sequestration of coactivator p300. Cancer Sci. 2005, 96, 274–282. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Lowther, W.; Kato, K.; Bianco, C.; Kenney, N.; Strizzi, L.; Raafat, D.; Hirota, M.; Khan, N.I.; Bargo, S.; et al. Notch4 intracellular domain binding to Smad3 and inhibition of the TGF-beta signaling. Oncogene 2005, 24, 5365–5374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niimi, H.; Pardali, K.; Vanlandewijck, M.; Heldin, C.H.; Moustakas, A. Notch signaling is necessary for epithelial growth arrest by TGF-beta. J. Cell Biol. 2007, 176, 695–707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nyhan, K.C.; Faherty, N.; Murray, G.; Cooey, L.B.; Godson, C.; Crean, J.K.; Brazil, D.P. Jagged/Notch signalling is required for a subset of TGFβ1 responses in human kidney epithelial cells. Biochim. Biophys. Acta. 2010, 1803, 1386–1395. [Google Scholar] [CrossRef] [Green Version]
- Xiao, Z.; Zhang, J.; Peng, X.; Dong, Y.; Jia, L.; Li, H.; Du, J. The Notch γ-secretase inhibitor ameliorates kidney fibrosis via inhibition of TGF-β/Smad2/3 signaling pathway activation. Int. J. Biochem. Cell Biol. 2014, 55, 65–71. [Google Scholar] [CrossRef]
- Borggrefe, T.; Lauth, M.; Zwijsen, A.; Huylebroeck, D.; Oswald, F.; Giaimo, B.D. The Notch intracellular domain integrates signals from Wnt, Hedgehog, TGFβ/BMP and hypoxia pathways. Biochim. Biophys. Acta 2016, 1863, 303–313. [Google Scholar] [CrossRef]
- Phng, L.K.; Potente, M.; Leslie, J.D.; Babbage, J.; Nyqvist, D.; Lobov, I.; Ondr, J.K.; Rao, S.; Lang, R.A.; Thurston, G.; et al. Nrarp coordinates endothelial Notch and Wnt signaling to control vessel density in angiogenesis. Dev. Cell 2009, 16, 70–82. [Google Scholar] [CrossRef]
- Ann, E.J.; Kim, H.Y.; Seo, M.S.; Mo, J.S.; Kim, M.Y.; Yoon, J.H.; Ahn, J.S.; Park, H.S. Wnt5a controls Notch1 signaling through CaMKII-mediated degradation of the SMRT corepressor protein. J. Biol. Chem. 2012, 287, 36814–36829. [Google Scholar] [CrossRef] [Green Version]
- Corada, M.; Nyqvist, D.; Orsenigo, F.; Caprini, A.; Giampietro, C.; Taketo, M.M.; Iruela-Arispe, M.L.; Adams, R.H.; Dejana, E. The Wnt/beta-catenin pathway modulates vascular remodeling and specification by upregulating Dll4/Notch signaling. Dev. Cell 2010, 18, 938–949. [Google Scholar] [CrossRef] [Green Version]
- Acar, A.; Hidalgo-Sastre, A.; Leverentz, M.K.; Mills, C.G.; Woodcock, S.; Baron, M.; Collu, G.M.; Brennan, K. Inhibition of Wnt signalling by Notch via two distinct mechanisms. Sci. Rep. 2021, 11, 9096. [Google Scholar] [CrossRef]
- Ross, D.A.; Kadesch, T. The notch intracellular domain can function as a coactivator for LEF-1. Mol. Cell Biol. 2001, 21, 7537–7544. [Google Scholar] [CrossRef]
- Yamamizu, K.; Matsunaga, T.; Uosaki, H.; Fukushima, H.; Katayama, S.; Hiraoka-Kanie, M.; Mitani, K.; Yamashita, J.K. Convergence of Notch and beta-catenin signaling induces arterial fate in vascular progenitors. J. Cell Biol. 2010, 189, 325–338, Erratum in J. Cell Biol. 2013, 202, 179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, K.; Cornelius, S.C.; Reiss, M.; Danielpour, D. Insulin-like growth factor-I inhibits transcriptional responses of transforming growth factor-beta by phosphatidylinositol 3-kinase/Akt-dependent suppression of the activation of Smad3 but not Smad2. J. Biol. Chem. 2003, 278, 38342–38351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, K.; Wang, H.; Krebs, T.L.; Danielpour, D. Novel roles of Akt and mTOR in suppressing TGF-beta/ALK5-mediated Smad3 activation. EMBO J. 2006, 25, 58–69. [Google Scholar] [CrossRef] [Green Version]
- Atfi, A.; Abécassis, L.; Bourgeade, M.F. Bcr-Abl activates the AKT/Fox O3 signalling pathway to restrict transforming growth factor-beta-mediated cytostatic signals. EMBO Rep. 2005, 6, 985–991. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Zhou, F.; Drabsch, Y.; Gao, R.; Snaar-Jagalska, B.E.; Mickanin, C.; Huang, H.; Sheppard, K.A.; Porter, J.A.; Lu, C.X.; et al. USP4 is regulated by AKT phosphorylation and directly deubiquitylates TGF-β type I receptor. Nat. Cell Biol. 2012, 14, 717–726. [Google Scholar] [CrossRef]
- Runyan, C.E.; Schnaper, H.W.; Poncelet, A.C. The phosphatidylinositol 3-kinase/Akt pathway enhances Smad3-stimulated mesangial cell collagen I expression in response to transforming growth factor-beta1. J. Biol. Chem. 2004, 279, 2632–2639. [Google Scholar] [CrossRef] [Green Version]
- Karagiannis, G.S.; Weile, J.; Bader, G.D.; Minta, J. Integrative pathway dissection of molecular mechanisms of moxLDL-induced vascular smooth muscle phenotype transformation. BMC Cardiovasc. Disord. 2013, 13, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, S.B.; Zhu, J.; Zhou, Z.Z.; Xi, E.P.; Wang, R.P.; Zhang, Y. TGF-β1 induces human aortic vascular smooth muscle cell phenotype switch through PI3K/AKT/ID2 signaling. Am. J. Transl. Res. 2015, 7, 2764–2774. [Google Scholar]
- Fukumoto, S.; Hsieh, C.M.; Maemura, K.; Layne, M.D.; Yet, S.F.; Lee, K.H.; Matsui, T.; Rosenzweig, A.; Taylor, W.G.; Rubin, J.S.; et al. Akt participation in the Wnt signaling pathway through Dishevelled. J. Biol. Chem. 2001, 276, 17479–17483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saward, L.; Zahradka, P. Angiotensin II activates phosphatidylinositol 3-kinase in vascular smooth muscle cells. Circ. Res. 1997, 81, 249–257. [Google Scholar] [CrossRef] [PubMed]
- Gervais, M.; Dugourd, C.; Muller, L.; Ardidie, C.; Canton, B.; Loviconi, L.; Corvol, P.; Chneiweiss, H.; Monnot, C. Akt down-regulates ERK1/2 nuclear localization and angiotensin II-induced cell proliferation through PEA-15. Mol. Biol. Cell 2006, 17, 3940–3951. [Google Scholar] [CrossRef]
- Gillis, E.; Van Laer, L.; Loeys, B.L. Genetics of thoracic aortic aneurysm: At the crossroad of transforming growth factor-β signaling and vascular smooth muscle cell contractility. Circ. Res. 2013, 113, 327–340. [Google Scholar] [CrossRef] [Green Version]
- Loeys, B.L.; Schwarze, U.; Holm, T.; Callewaert, B.L.; Thomas, G.H.; Pannu, H.; De Backer, J.F.; Oswald, G.L.; Symoens, S.; Manouvrier, S.; et al. Aneurysm syndromes caused by mutations in the TGF-beta receptor. N. Engl. J. Med. 2006, 355, 788–798. [Google Scholar] [CrossRef] [PubMed]
- Pyeritz, R.E. Heritable thoracic aortic disorders. Curr. Opin. Cardiol. 2014, 29, 97–102. [Google Scholar] [CrossRef] [PubMed]
- Lindsay, M.E.; Dietz, H.C. The genetic basis of aortic aneurysm. Cold Spring Harb. Perspect. Med. 2014, 4, a015909. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Habashi, J.P.; Judge, D.P.; Holm, T.M.; Cohn, R.D.; Loeys, B.L.; Cooper, T.K.; Myers, L.; Klein, E.C.; Liu, G.; Calvi, C.; et al. Losartan, an AT1 antagonist, prevents aortic aneurysm in a mouse model of Marfan syndrome. Science 2006, 312, 117–121. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Rateri, D.L.; Howatt, D.A.; Balakrishnan, A.; Moorleghen, J.J.; Cassis, L.A.; Daugherty, A. TGF-β Neutralization Enhances AngII-Induced Aortic Rupture and Aneurysm in Both Thoracic and Abdominal Regions. PLoS ONE 2016, 11, e0153811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, W.; Li, Q.; Jiao, Y.; Qin, L.; Ali, R.; Zhou, J.; Ferruzzi, J.; Kim, R.W.; Geirsson, A.; Dietz, H.C.; et al. Tgfbr2 disruption in postnatal smooth muscle impairs aortic wall homeostasis. J. Clin. Investig. 2014, 124, 755–767. [Google Scholar] [CrossRef] [Green Version]
- Yang, P.; Schmit, B.M.; Fu, C.; DeSart, K.; Oh, S.P.; Berceli, S.A.; Jiang, Z. Smooth muscle cell-specific Tgfbr1 deficiency promotes aortic aneurysm formation by stimulating multiple signaling events. Sci. Rep. 2016, 6, 35444. [Google Scholar] [CrossRef] [Green Version]
- Cook, J.R.; Clayton, N.P.; Carta, L.; Galatioto, J.; Chiu, E.; Smaldone, S.; Nelson, C.A.; Cheng, S.H.; Wentworth, B.M.; Ramirez, F. Dimorphic effects of transforming growth factor-β signaling during aortic aneurysm progression in mice suggest a combinatorial therapy for Marfan syndrome. Arter. Thromb. Vasc. Biol. 2015, 35, 911–917. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paloschi, V.; Gådin, J.R.; Khan, S.; Björck, H.M.; Du, L.; Maleki, S.; Roy, J.; Lindeman, J.H.; Mohamed, S.A.; Tsuda, T.; et al. Aneurysm development in patients with a bicuspid aortic valve is not associated with transforming growth factor-β activation. Arter. Thromb. Vasc. Biol. 2015, 35, 973–980. [Google Scholar] [CrossRef] [PubMed]
- Rocchiccioli, S.; Cecchettini, A.; Panesi, P.; Farneti, P.A.; Mariani, M.; Ucciferri, N.; Citti, L.; Andreassi, M.G.; Foffa, I. Hypothesis-free secretome analysis of thoracic aortic aneurysm reinforces the central role of TGF-β cascade in patients with bicuspid aortic valve. J. Cardiol. 2017, 69, 570–576. [Google Scholar] [CrossRef] [Green Version]
- Hillebrand, M.; Millot, N.; Sheikhzadeh, S.; Rybczynski, M.; Gerth, S.; Kölbel, T.; Keyser, B.; Kutsche, K.; Robinson, P.N.; Berger, J.; et al. Total serum transforming growth factor-β1 is elevated in the entire spectrum of genetic aortic syndromes. Clin. Cardiol. 2014, 37, 672–679. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mallat, Z.; Daugherty, A. AT1 receptor antagonism to reduce aortic expansion in Marfan syndrome: Lost in translation or in need of different interpretation? Arterioscler. Thromb. Vasc. Biol. 2015, 35, e10–e12. [Google Scholar] [CrossRef] [Green Version]
- Alfieri, C.M.; Cheek, J.; Chakraborty, S.; Yutzey, K.E. Wnt signaling in heart valve development and osteogenic gene induction. Dev. Biol. 2010, 338, 127–135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bosada, F.M.; Devasthali, V.; Jones, K.A.; Stankunas, K. Wnt/β-catenin signaling enables developmental transitions during valvulogenesis. Development 2016, 143, 1041–1054. [Google Scholar] [CrossRef] [Green Version]
- Hulin, A.; Moore, V.; James, J.M.; Yutzey, K.E. Loss of Axin2 results in impaired heart valve maturation and subsequent myxomatous valve disease. Cardiovasc. Res. 2017, 113, 40–51. [Google Scholar] [CrossRef]
- Caira, F.C.; Stock, S.R.; Gleason, T.G.; McGee, E.C.; Huang, J.; Bonow, R.O.; Spelsberg, T.C.; McCarthy, P.M.; Rahimtoola, S.H.; Rajamannan, N.M. Human degenerative valve disease is associated with up-regulation of low-density lipoprotein receptor-related protein 5 receptor-mediated bone formation. J. Am. Coll Cardiol. 2006, 47, 1707–1712. [Google Scholar] [CrossRef] [Green Version]
- Albanese, I.; Khan, K.; Barratt, B.; Al-Kindi, H.; Schwertani, A. Atherosclerotic Calcification: Wnt Is the Hint. J. Am. Heart Assoc. 2018, 7, e007356. [Google Scholar] [CrossRef] [Green Version]
- Bayle, J.; Fitch, J.; Jacobsen, K.; Kumar, R.; Lafyatis, R.; Lemaire, R. Increased expression of Wnt2 and SFRP4 in Tsk mouse skin: Role of Wnt signaling in altered dermal fibrillin deposition and systemic sclerosis. J. Investig. Dermatol. 2008, 128, 871–881. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lemaire, R.; Farina, G.; Bayle, J.; Dimarzio, M.; Pendergrass, S.A.; Milano, A.; Perbal, B.; Whitfield, M.L.; Lafyatis, R. Antagonistic effect of the matricellular signaling protein CCN3 on TGF-beta- and Wnt-mediated fibrillinogenesis in systemic sclerosis and Marfan syndrome. J. Investig. Dermatol. 2010, 130, 1514–1523. [Google Scholar] [CrossRef] [PubMed]
- Działo, E.; Czepiel, M.; Tkacz, K.; Siedlar, M.; Kania, G.; Błyszczuk, P. WNT/β-Catenin signaling promotes TGF-β-mediated activation of human cardiac fibroblasts by enhancing IL-11 production. Int. J. Mol. Sci. 2021, 22, 10072. [Google Scholar] [CrossRef] [PubMed]
- Kostina, A.; Bjork, H.; Ignatieva, E.; Irtyuga, O.; Uspensky, V.; Semenova, D.; Maleki, S.; Tomilin, A.; Moiseeva, O.; Franco-Cereceda, A.; et al. Notch, BMP and WNT/β-catenin network is impaired in endothelial cells of the patients with thoracic aortic aneurysm. Atheroscler. Suppl. 2018, 35, e6–e13. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, R.; Rolyan, H.; Xie, Y.; Li, N.; Bhat, N.; Hong, L.; Esteghamat, F.; Adeniran, A.; Geirsson, A.; Zhang, J.; et al. TCF7L2 (Transcription Factor 7-Like 2) regulation of GATA6 (GATA-Binding Protein 6)-dependent and -independent vascular smooth muscle cell plasticity and intimal hyperplasia. Arter. Thromb. Vasc. Biol. 2019, 39, 250–262. [Google Scholar] [CrossRef]
- Roychowdhury, T.; Lu, H.; Hornsby, W.E.; Crone, B.; Wang, G.T.; Guo, D.C.; Sendamarai, A.K.; Devineni, P.; Lin, M.; Zhou, W.; et al. Regulatory variants in TCF7L2 are associated with thoracic aortic aneurysm. Am. J. Hum. Genet. 2021, 108, 1578–1589. [Google Scholar] [CrossRef]
- Durdu, S.; Deniz, G.C.; Balci, D.; Zaim, C.; Dogan, A.; Can, A.; Akcali, K.C.; Akar, A.R. Apoptotic vascular smooth muscle cell depletion via BCL2 family of proteins in human ascending aortic aneurysm and dissection. Cardiovasc. Ther. 2012, 30, 308–316. [Google Scholar] [CrossRef] [PubMed]
- Trollope, A.F.; Golledge, J. Angiopoietins, abdominal aortic aneurysm and atherosclerosis. Atherosclerosis 2011, 214, 237–243. [Google Scholar] [CrossRef] [Green Version]
- Ghosh, A.; Lu, G.; Su, G.; McEvoy, B.; Sadiq, O.; DiMusto, P.D.; Laser, A.; Futchko, J.S.; Henke, P.K.; Eliason, J.L.; et al. Phosphorylation of AKT and abdominal aortic aneurysm formation. Am. J. Pathol. 2014, 184, 148–158. [Google Scholar] [CrossRef] [Green Version]
- Liu, Q.; Shan, P.; Li, H. Gambogic acid prevents angiotensin II-induced abdominal aortic aneurysm through inflammatory and oxidative stress dependent targeting the PI3K/Akt/mTOR and NF-κB signaling pathways. Mol. Med. Rep. 2019, 19, 1396–1402. [Google Scholar] [CrossRef] [Green Version]
- Estrada, A.C.; Irons, L.; Rego, B.V.; Li, G.; Tellides, G.; Humphrey, J.D. Roles of mTOR in thoracic aortopathy understood by complex intracellular signaling interactions. PLoS Comput. Biol. 2021, 17, e1009683. [Google Scholar] [CrossRef] [PubMed]
- Arcucci, A.; Ruocco, M.R.; Albano, F.; Granato, G.; Romano, V.; Corso, G.; Bancone, C.; De Vendittis, E.; Della Corte, A.; Montagnani, S. Analysis of extracellular superoxide dismutase and Akt in ascending aortic aneurysm with tricuspid or bicuspid aortic valve. Eur. J. Histochem. 2014, 58, 2383, Erratum in Eur. J. Histochem. 2015, 59, 2517. [Google Scholar] [CrossRef] [PubMed]
- Hirata, Y.; Aoki, H.; Shojima, T.; Takagi, K.; Takaseya, T.; Akasu, K.; Tobinaga, S.; Fukumoto, Y.; Tanaka, H. Activation of the AKT pathway in the ascending aorta with bicuspid aortic valve. Circ. J. 2018, 82, 2485–2492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chung, A.W.; Au Yeung, K.; Cortes, S.F.; Sandor, G.G.; Judge, D.P.; Dietz, H.C.; van Breemen, C. Endothelial dysfunction and compromised eNOS/Akt signaling in the thoracic aorta during the progression of Marfan syndrome. Br. J. Pharmacol. 2007, 150, 1075–1083. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.H.; Kim, J.M.; Chum, E.; van Breemen, C.; Chung, A.W. Long-term effects of losartan on structure and function of the thoracic aorta in a mouse model of Marfan syndrome. Br. J. Pharmacol. 2009, 158, 1503–1512. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.; Chen, H.; Yu, C.; Lu, R.; Song, T.; Wang, X.; Tang, W.; Gao, Y. Long noncoding RNA myocardial infarction associated transcript promotes the development of thoracic aortic by targeting microRNA-145 via the PI3K/Akt signaling pathway. J. Cell Biochem. 2019, 120, 14405–14413. [Google Scholar] [CrossRef]
- Garg, V.; Muth, A.N.; Ransom, J.F.; Schluterman, M.K.; Barnes, R.; King, I.N.; Grossfeld, P.D.; Srivastava, D. Mutations in NOTCH1 cause aortic valve disease. Nature 2005, 437, 270–274. [Google Scholar] [CrossRef]
- Koenig, S.N.; Bosse, K.; Majumdar, U.; Bonachea, E.M.; Radtke, F.; Garg, V. Endothelial Notch1 is required for proper development of the semilunar valves and cardiac outflow tract. J. Am. Heart Assoc. 2016, 5, e003075. [Google Scholar] [CrossRef] [Green Version]
- Malashicheva, A.; Kostina, A.; Kostareva, A.; Irtyuga, O.; Gordeev, M.; Uspensky, V. Notch signaling in the pathogenesis of thoracic aortic aneurysms: A bridge between embryonic and adult states. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165631, Erratum in Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165732. [Google Scholar] [CrossRef]
- Mohamed, S.A.; Aherrahrou, Z.; Liptau, H.; Erasmi, A.W.; Hagemann, C.; Wrobel, S.; Borzym, K.; Schunkert, H.; Sievers, H.H.; Erdmann, J. Novel missense mutations (p.T596M and p.P1797H) in NOTCH1 in patients with bicuspid aortic valve. Biochem. Biophys. Res. Commun. 2006, 345, 1460–1465. [Google Scholar] [CrossRef]
- McKellar, S.H.; Tester, D.J.; Yagubyan, M.; Majumdar, R.; Ackerman, M.J.; Sundt, T.M. Novel NOTCH1 mutations in patients with bicuspid aortic valve disease and thoracic aortic aneurysms. J. Thorac Cardiovasc. Surg. 2007, 134, 290–296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koenig, S.N.; Lincoln, J.; Garg, V. Genetic basis of aortic valvular disease. Curr. Opin. Cardiol. 2017, 32, 239–245. [Google Scholar] [CrossRef] [PubMed]
- Bonderman, D.; Gharehbaghi-Schnell, E.; Wollenek, G.; Maurer, G.; Baumgartner, H.; Lang, I.M. Mechanisms underlying aortic dilatation in congenital aortic valve malformation. Circulation 1999, 99, 2138–2143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zou, S.; Ren, P.; Nguyen, M.; Coselli, J.S.; Shen, Y.H.; LeMaire, S.A. Notch signaling in descending thoracic aortic aneurysm and dissection. PLoS ONE 2012, 7, e52833. [Google Scholar] [CrossRef] [Green Version]
- Sciacca, S.; Pilato, M.; Mazzoccoli, G.; Pazienza, V.; Vinciguerra, M. Anti-correlation between longevity gene SirT1 and Notch signaling in ascending aorta biopsies from patients with bicuspid aortic valve disease. Heart Vessel. 2013, 28, 268–275. [Google Scholar] [CrossRef]
- Maleki, S.; Kjellqvist, S.; Paloschi, V.; Magné, J.; Branca, R.M.; Du, L.; Hultenby, K.; Petrini, J.; Fuxe, J.; MIBAVA Leducq Consortium; et al. Mesenchymal state of intimal cells may explain higher propensity to ascending aortic aneurysm in bicuspid aortic valves. Sci. Rep. 2016, 25, 35712. [Google Scholar] [CrossRef] [Green Version]
- Kostina, A.S.; Uspensky, V.E.; Irtyuga, O.B.; Ignatieva, E.V.; Freylikhman, O.; Gavriliuk, N.D.; Moiseeva, O.M.; Zhuk, S.; Tomilin, A.; Kostareva, A.A.; et al. Notch-dependent EMT is attenuated in patients with aortic aneurysm and bicuspid aortic valve. Biochim. Biophys. Acta 2016, 1862, 733–740. [Google Scholar] [CrossRef]
- Balistreri, C.R.; Crapanzano, F.; Schirone, L.; Allegra, A.; Pisano, C.; Ruvolo, G.; Forte, M.; Greco, E.; Cavarretta, E.; Marullo, A.G.M.; et al. Deregulation of Notch1 pathway and circulating endothelial progenitor cell (EPC) number in patients with bicuspid aortic valve with and without ascending aorta aneurysm. Sci. Rep. 2018, 8, 13834. [Google Scholar] [CrossRef]
- Jespersen, K.; Li, C.; Batra, R.; Stephenson, C.A.; Harding, P.; Sestak, K.; Foley, R.T.; Greene, H.; Meisinger, T.; Cook, J.R.; et al. Impact of Notch3 Activation on Aortic Aneurysm Development in Marfan Syndrome. J. Immunol. Res. 2022, 2022, 7538649. [Google Scholar] [CrossRef]
- Shimizu-Hirota, R.; Sasamura, H.; Mifune, M.; Nakaya, H.; Kuroda, M.; Hayashi, M.; Saruta, T. Regulation of vascular proteoglycan synthesis by angiotensin II type 1 and type 2 receptors. J. Am. Soc. Nephrol. 2001, 12, 2609–2615. [Google Scholar] [CrossRef]
- Lu, H.; Rateri, D.L.; Bruemmer, D.; Cassis, L.A.; Daugherty, A. Involvement of the renin-angiotensin system in abdominal and thoracic aortic aneurysms. Clin. Sci. 2012, 123, 531–543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rateri, D.L.; Moorleghen, J.J.; Balakrishnan, A.; Owens, A.P.; Howatt, D.A.; Subramanian, V.; Poduri, A.; Charnigo, R.; Cassis, L.A.; Daugherty, A. Endothelial cell-specific deficiency of Ang II type 1a receptors attenuates Ang II-induced ascending aortic aneurysms in LDL receptor-/- mice. Circ. Res. 2011, 108, 574–581. [Google Scholar] [CrossRef] [PubMed]
- Davis, F.M.; Rateri, D.L.; Balakrishnan, A.; Howatt, D.A.; Strickland, D.K.; Muratoglu, S.C.; Haggerty, C.M.; Fornwalt, B.K.; Cassis, L.A.; Daugherty, A. Smooth muscle cell deletion of low-density lipoprotein receptor-related protein 1 augments angiotensin II-induced superior mesenteric arterial and ascending aortic aneurysms. Arter. Thromb. Vasc. Biol. 2015, 35, 155–162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galatioto, J.; Caescu, C.I.; Hansen, J.; Cook, J.R.; Miramontes, I.; Iyengar, R.; Ramirez, F. Cell type-specific contributions of the angiotensin II type 1a receptor to aorta homeostasis and aneurysmal disease-Brief report. Arter. Thromb. Vasc. Biol. 2018, 38, 588–591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Habashi, J.P.; Doyle, J.J.; Holm, T.M.; Aziz, H.; Schoenhoff, F.; Bedja, D.; Chen, Y.; Modiri, A.N.; Judge, D.P.; Dietz, H.C. Angiotensin II type 2 receptor signaling attenuates aortic aneurysm in mice through ERK antagonism. Science 2011, 332, 361–365. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Lu, H.; Rateri, D.L.; Cassis, L.A.; Daugherty, A. Conundrum of angiotensin II and TGF-β interactions in aortic aneurysms. Curr. Opin. Pharmacol. 2013, 13, 180–185. [Google Scholar] [CrossRef] [Green Version]
- Nagasawa, A.; Yoshimura, K.; Suzuki, R.; Mikamo, A.; Yamashita, O.; Ikeda, Y.; Tsuchida, M.; Hamano, K. Important role of the angiotensin II pathway in producing matrix metalloproteinase-9 in human thoracic aortic aneurysms. J. Surg. Res. 2013, 183, 472–477. [Google Scholar] [CrossRef]
- Gallo, E.M.; Loch, D.C.; Habashi, J.P.; Calderon, J.F.; Chen, Y.; Bedja, D.; van Erp, C.; Gerber, E.E.; Parker, S.J.; Sauls, K.; et al. Angiotensin II-dependent TGF-β signaling contributes to Loeys-Dietz syndrome vascular pathogenesis. J. Clin. Investig. 2014, 124, 448–460. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Peters, A.; Papke, C.L.; Villamizar, C.; Ringuette, L.J.; Cao, J.; Wang, S.; Ma, S.; Gong, L.; Byanova, K.L.; et al. Loss of smooth muscle α-Actin leads to NF-κB-dependent increased sensitivity to angiotensin II in smooth muscle cells and aortic enlargement. Circ. Res. 2017, 120, 1903–1915. [Google Scholar] [CrossRef]
- Shoja, M.M.; Agutter, P.S.; Tubbs, R.S.; Payner, T.D.; Ghabili, K.; Cohen-Gadol, A.A. The role of the renin-angiotensin system in the pathogenesis of intracranial aneurysms. J. Renin-Angiotensin-Aldosterone Syst. 2011, 12, 262–273. [Google Scholar] [CrossRef] [Green Version]
- Huang, J.; Yamashiro, Y.; Papke, C.L.; Ikeda, Y.; Lin, Y.; Patel, M.; Inagami, T.; Le, V.P.; Wagenseil, J.E.; Yanagisawa, H. Angiotensin-converting enzyme-induced activation of local angiotensin signaling is required for ascending aortic aneurysms in fibulin-4-deficient mice. Sci. Transl. Med. 2013, 5, 183ra58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, Y.H.; Zhang, L.; Ren, P.; Nguyen, M.T.; Zou, S.; Wu, D.; Wang, X.L.; Coselli, J.S.; LeMaire, S.A. AKT2 confers protection against aortic aneurysms and dissections. Circ. Res. 2013, 112, 618–632. [Google Scholar] [CrossRef] [PubMed]
- Shen, M.; Lee, J.; Basu, R.; Sakamuri, S.S.; Wang, X.; Fan, D.; Kassiri, Z. Divergent roles of matrix metalloproteinase 2 in pathogenesis of thoracic aortic aneurysm. Arter. Thromb. Vasc. Biol. 2015, 35, 888–898. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benetos, A.; Topouchian, J.; Ricard, S.; Gautier, S.; Bonnardeaux, A.; Asmar, R.; Poirier, O.; Soubrier, F.; Safar, M.; Cambien, F. Influence of angiotensin II type 1 receptor polymorphism on aortic stiffness in never-treated hypertensive patients. Hypertension 1995, 26, 44–47. [Google Scholar] [CrossRef]
- Boon, R.A.; Seeger, T.; Heydt, S.; Fischer, A.; Hergenreider, E.; Horrevoets, A.J.; Vinciguerra, M.; Rosenthal, N.; Sciacca, S.; Pilato, M.; et al. MicroRNA-29 in aortic dilation: Implications for aneurysm formation. Circ. Res. 2011, 109, 1115–1119. [Google Scholar] [CrossRef] [Green Version]
- Brooke, B.S.; Habashi, J.P.; Judge, D.P.; Patel, N.; Loeys, B.; Dietz, H.C. Angiotensin II blockade and aortic-root dilation in Marfan’s syndrome. N. Engl. J. Med. 2008, 358, 2787–2795. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forteza, A.; Evangelista, A.; Sánchez, V.; Teixidó-Turà, G.; Sanz, P.; Gutiérrez, L.; Gracia, T.; Centeno, J.; Rodríguez-Palomares, J.; Rufilanchas, J.J.; et al. Efficacy of losartan vs. atenolol for the prevention of aortic dilation in Marfan syndrome: A randomized clinical trial. Eur. Heart J. 2016, 37, 978–985. [Google Scholar] [CrossRef] [Green Version]
- Lacro, R.V.; Dietz, H.C.; Sleeper, L.A.; Yetman, A.T.; Bradley, T.J.; Colan, S.D.; Pearson, G.D.; Selamet Tierney, E.S.; Levine, J.C.; Atz, A.M.; et al. Atenolol versus losartan in children and young adults with Marfan’s syndrome. N. Engl. J. Med. 2014, 371, 2061–2071. [Google Scholar] [CrossRef] [Green Version]
- Groenink, M.; den Hartog, A.W.; Franken, R.; Radonic, T.; de Waard, V.; Timmermans, J.; Scholte, A.J.; van den Berg, M.P.; Spijkerboer, A.M.; Marquering, H.A.; et al. Losartan reduces aortic dilatation rate in adults with Marfan syndrome: A randomized controlled trial. Eur. Heart J. 2013, 34, 3491–3500. [Google Scholar] [CrossRef] [Green Version]
- Mullen, M.; Jin, X.Y.; Child, A.; Stuart, A.G.; Dodd, M.; Aragon-Martin, J.A.; Gaze, D.; Kiotsekoglou, A.; Yuan, L.; Hu, J.; et al. Irbesartan in Marfan syndrome (AIMS): A double-blind, placebo-controlled randomised trial. Lancet 2019, 394, 2263–2270. [Google Scholar] [CrossRef] [Green Version]
- Al-Abcha, A.; Saleh, Y.; Mujer, M.; Boumegouas, M.; Herzallah, K.; Charles, L.; Elkhatib, L.; Abdelkarim, O.; Kehdi, M.; Abela, G.S. Meta-analysis examining the usefulness of angiotensin receptor blockers for the prevention of aortic root dilation in patients with the Marfan Syndrome. Am. J. Cardiol. 2020, 128, 101–106. [Google Scholar] [CrossRef] [PubMed]
- King, T.E.; Bradford, W.Z.; Castro-Bernardini, S.; Fagan, E.A.; Glaspole, I.; Glassberg, M.K.; Gorina, E.; Hopkins, P.M.; Kardatzke, D.; Lancaster, L.; et al. A phase 3 trial of pirfenidone in patients with idiopathic pulmonary fibrosis. N. Engl. J. Med. 2014, 370, 2083–2092, Erratum in N. Engl. J. Med. 2014, 371, 1172. [Google Scholar] [CrossRef] [PubMed]
- Noble, P.W.; Albera, C.; Bradford, W.Z.; Costabel, U.; Glassberg, M.K.; Kardatzke, D.; King, T.E.; Lancaster, L.; Sahn, S.A.; Szwarcberg, J.; et al. Pirfenidone in patients with idiopathic pulmonary fibrosis (CAPACITY): Two randomised trials. Lancet 2011, 377, 1760–1769. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.Y.; Chung, C.L.; Hu, T.H.; Chen, J.J.; Liu, P.F.; Chen, C.L. Recent progress in TGF-β inhibitors for cancer therapy. Biomed. Pharmacother. 2021, 134, 111046. [Google Scholar] [CrossRef]
- Xiao, H.; Zhang, J.; Xu, Z.; Feng, Y.; Zhang, M.; Liu, J.; Chen, R.; Shen, J.; Wu, J.; Lu, Z.; et al. Metformin is a novel suppressor for transforming growth factor (TGF)-β1. Sci. Rep. 2016, 6, 28597. [Google Scholar] [CrossRef]
- Teixeira, A.F.; Ten Dijke, P.; Zhu, H.J. On-Target anti-TGF-β therapies are not succeeding in clinical cancer treatments: What are remaining challenges? Front. Cell Dev. Biol. 2020, 8, 605. [Google Scholar] [CrossRef]
- Zehender, A.; Li, Y.N.; Lin, N.Y.; Stefanica, A.; Nüchel, J.; Chen, C.W.; Hsu, H.H.; Zhu, H.; Ding, X.; Huang, J.; et al. TGFβ promotes fibrosis by MYST1-dependent epigenetic regulation of autophagy. Nat. Commun. 2021, 12, 4404. [Google Scholar] [CrossRef]
- Jenke, A.; Kistner, J.; Saradar, S.; Chekhoeva, A.; Yazdanyar, M.; Bergmann, A.K.; Rötepohl, M.V.; Lichtenberg, A.; Akhyari, P. Transforming growth factor-β1 promotes fibrosis but attenuates calcification of valvular tissue applied as a three-dimensional calcific aortic valve disease model. Am. J. Physiol.-Heart Circ. Physiol. 2020, 319, H1123–H1141. [Google Scholar] [CrossRef]
- Herbertz, S.; Sawyer, J.S.; Stauber, A.J.; Gueorguieva, I.; Driscoll, K.E.; Estrem, S.T.; Cleverly, A.L.; Desaiah, D.; Guba, S.C.; Benhadji, K.A.; et al. Clinical development of galunisertib (LY2157299 monohydrate), a small molecule inhibitor of transforming growth factor-beta signaling pathway. Drug Des. Dev. Ther. 2015, 9, 4479–4499. [Google Scholar] [CrossRef] [Green Version]
- Yamazaki, T.; Gunderson, A.J.; Gilchrist, M.; Whiteford, M.; Kiely, M.X.; Hayman, A.; O’Brien, D.; Ahmad, R.; Manchio, J.V.; Fox, N.; et al. Galunisertib plus neoadjuvant chemoradiotherapy in patients with locally advanced rectal cancer: A single-arm, phase 2 trial. Lancet Oncol. 2022, 23, 1189–1200. [Google Scholar] [CrossRef]
- Melisi, D.; Oh, D.Y.; Hollebecque, A.; Calvo, E.; Varghese, A.; Borazanci, E.; Macarulla, T.; Merz, V.; Zecchetto, C.; Zhao, Y.; et al. Safety and activity of the TGFβ receptor I kinase inhibitor galunisertib plus the anti-PD-L1 antibody durvalumab in metastatic pancreatic cancer. J. Immunother. Cancer 2021, 9, e002068. [Google Scholar] [CrossRef]
- Santini, V.; Valcárcel, D.; Platzbecker, U.; Komrokji, R.S.; Cleverly, A.L.; Lahn, M.M.; Janssen, J.; Zhao, Y.; Chiang, A.; Giagounidis, A.; et al. Phase II Study of the ALK5 Inhibitor Galunisertib in Very Low-, Low-, and Intermediate-Risk Myelodysplastic Syndromes. Clin. Cancer Res. 2019, 25, 6976–6985. [Google Scholar] [CrossRef]
- Voss, M.H.; Bhatt, R.S.; Vogelzang, N.J.; Fishman, M.; Alter, R.S.; Rini, B.I.; Beck, J.T.; Joshi, M.; Hauke, R.; Atkins, M.B.; et al. A phase 2, randomized trial evaluating the combination of dalantercept plus axitinib in patients with advanced clear cell renal cell carcinoma. Cancer 2019, 125, 2400–2408. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Wang, X. Circular RNA ITCH mediates H2O2-induced myocardial cell apoptosis by targeting miR-17-5p via wnt/β-catenin signalling pathway. Int. J. Exp. Pathol. 2020, 102, 22–31. [Google Scholar] [CrossRef] [PubMed]
- Proffitt, K.D.; Madan, B.; Ke, Z.; Pendharkar, V.; Ding, L.; Lee, M.A.; Hannoush, R.N.; Virshup, D.M. Pharmacological inhibition of the Wnt acyltransferase PORCN prevents growth of WNT-driven mammary cancer. Cancer Res. 2013, 73, 502–507. [Google Scholar] [CrossRef] [Green Version]
- Torres, V.I.; Godoy, J.A.; Inestrosa, N.C. Modulating Wnt signaling at the root: Porcupine and Wnt acylation. Pharm. Ther. 2019, 198, 34–45. [Google Scholar] [CrossRef]
- Rodon, J.; Argilés, G.; Connolly, R.M.; Vaishampayan, U.; de Jonge, M.; Garralda, E.; Giannakis, M.; Smith, D.C.; Dobson, J.R.; McLaughlin, M.E.; et al. Phase 1 study of single-agent WNT974, a first-in-class Porcupine inhibitor, in patients with advanced solid tumours. Br. J. Cancer 2021, 125, 28–37. [Google Scholar] [CrossRef] [PubMed]
- Bastakoty, D.; Saraswati, S.; Joshi, P.; Atkinson, J.; Feoktistov, I.; Liu, J.; Harris, J.L.; Young, P.P. Temporary, systemic inhibition of the WNT/β-Catenin pathway promotes regenerative cardiac repair following myocardial infarct. Cell Stem Cells Regen. Med. 2016, 2, 1–24. [Google Scholar] [CrossRef]
- Neiheisel, A.; Kaur, M.; Ma, N.; Havard, P.; Shenoy, A.K. Wnt pathway modulators in cancer therapeutics: An update on completed and ongoing clinical trials. Int. J. Cancer 2022, 150, 727–740. [Google Scholar] [CrossRef]
- Dunbar, K.; Valanciute, A.; Lima, A.C.S.; Vinuela, P.F.; Jamieson, T.; Rajasekaran, V.; Blackmur, J.; Ochocka-Fox, A.M.; Guazzelli, A.; Cammareri, P.; et al. Aspirin rescues Wnt-driven stem-like phenotype in human intestinal organoids and increases the Wnt antagonist Dickkopf-1. Cell Mol. Gastroenterol. Hepatol. 2021, 11, 465–489. [Google Scholar] [CrossRef]
- Tai, W.P.; Hu, P.J.; Wu, J.; Lin, X.C. The inhibition of Wnt/β-catenin signaling pathway in human colon cancer cells by sulindac. Tumori J. 2014, 100, 97–101. [Google Scholar] [CrossRef]
- Dihlmann, S.; Siermann, A.; von Knebel Doeberitz, M. The nonsteroidal anti-inflammatory drugs aspirin and indomethacin attenuate beta-catenin/TCF-4 signaling. Oncogene 2001, 20, 645–653. [Google Scholar] [CrossRef]
- Jung, Y.S.; Park, J.I. Wnt signaling in cancer: Therapeutic targeting of Wnt signaling beyond β-catenin and the destruction complex. Exp. Mol. Med. 2020, 52, 183–191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.; Nie, J.; Ma, X.; Wei, Y.; Peng, Y.; Wei, X. Targeting PI3K in cancer: Mechanisms and advances in clinical trials. Mol. Cancer 2019, 18, 26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, J.; Liu, R.; Huang, J.; Wang, L.; Wang, W. Inhibition of Phosphatidylinositol 3-kinease suppresses formation and progression of experimental abdominal aortic aneurysms. Sci. Rep. 2017, 7, 15208. [Google Scholar] [CrossRef] [Green Version]
- Gharbi, S.I.; Zvelebil, M.J.; Shuttleworth, S.J.; Hancox, T.; Saghir, N.; Timms, J.F.; Waterfield, M.D. Exploring the specificity of the PI3K family inhibitor LY294002. Biochem. J. 2007, 404, 15–21. [Google Scholar] [CrossRef] [Green Version]
- Eisenreich, A.; Rauch, U. PI3K inhibitors in cardiovascular disease. Cardiovasc. Ther. 2011, 29, 29–36. [Google Scholar] [CrossRef]
- Majumder, S.; Crabtree, J.S.; Golde, T.E.; Minter, L.M.; Osborne, B.A.; Miele, L. Targeting Notch in oncology: The path forward. Nat. Rev. Drug Discov. 2021, 20, 125–144. [Google Scholar] [CrossRef]
- Katoh, M.; Katoh, M. Precision medicine for human cancers with Notch signaling dysregulation (Review). Int. J. Mol. Med. 2020, 45, 279–297. [Google Scholar] [CrossRef] [Green Version]
- Takebe, N.; Nguyen, D.; Yang, S.X. Targeting notch signaling pathway in cancer: Clinical development advances and challenges. Pharm. Ther. 2014, 141, 140–149. [Google Scholar] [CrossRef] [Green Version]
- Sharma, N.; Dev, R.; Ruiz-Rosado, J.D.; Partida-Sanchez, S.; Guerau-de-Arellano, M.; Dhakal, P.; Kuivaniemi, H.; Hans, C.P. Pharmacological inhibition of Notch signaling regresses pre-established abdominal aortic aneurysm. Sci. Rep. 2019, 9, 13458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hans, C.P.; Sharma, N.; Dev, R.; Blain, J.M.; Tonniges, J.; Agarwal, G. DAPT, a potent Notch inhibitor regresses actively growing abdominal aortic aneurysm via divergent pathways. Clin. Sci. 2020, 134, 1555–1572. [Google Scholar] [CrossRef] [PubMed]
- Van Dorst, D.C.H.; de Wagenaar, N.P.; van der Pluijm, I.; Roos-Hesselink, J.W.; Essers, J.; Danser, A.H.J. Transforming growth factor-β and the renin-angiotensin system in syndromic thoracic aortic aneurysms: Implications for treatment. Cardiovasc. Drugs Ther. 2021, 35, 1233–1252. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, T.; Suzuki, J.; Yamawaki, H.; Sharma, V.K.; Sheu, S.S.; Berk, B.C. Losartan metabolite EXP3179 activates Akt and endothelial nitric oxide synthase via vascular endothelial growth factor receptor-2 in endothelial cells: Angiotensin II type 1 receptor-independent effects of EXP3179. Circulation 2005, 112, 1798–1805. [Google Scholar] [CrossRef] [PubMed]













{kind=link}
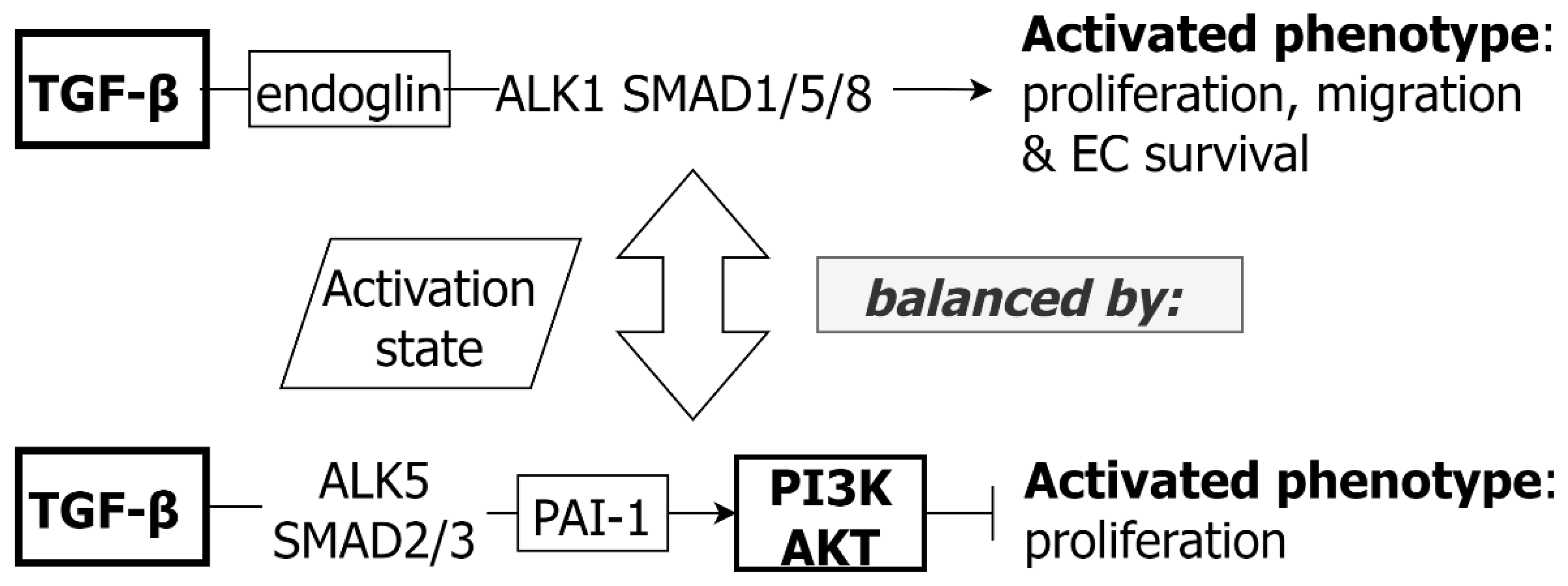{kind=link}
{kind=link}
{kind=link}
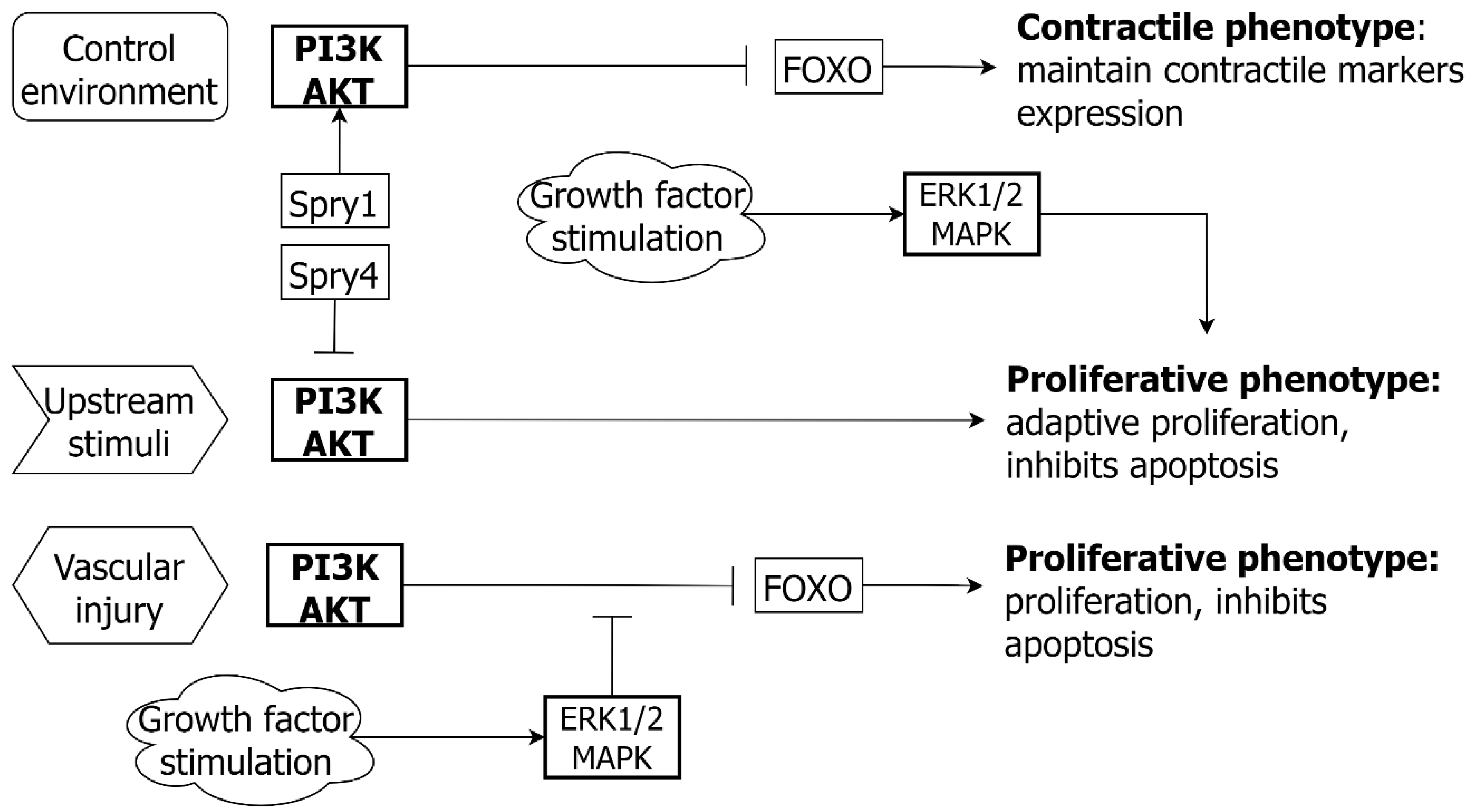{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Vascular Smooth Muscle Cell | ||||||
| Pathway | Promote contractile phenotype and survival | Promote proliferative response | Promote synthetic phenotype, apoptosis, proliferation, and migration | |||
| Direct | Indirect | Direct | Indirect | Direct | Indirect | |
| TGF-β | + | + | + | + | ||
| WNT | + | + | + | |||
| PI3K/AK | + | + | + | |||
| NOTCH | + | + | + | + | ||
| ANGII | + | + | ||||
| Endothelial Cell | ||||||
| Pathway | Promote survival and EC quiescence | Promote proliferation and migration | Promote EC dysregulation, apoptosis, and inflammation | |||
| Direct | Indirect | Direct | Indirect | Direct | Indirect | |
| TGF-β | + | + | + | |||
| WNT | + | + | + | + | + | |
| PI3K/AK | + | + | + | + | ||
| NOTCH | + | |||||
| ANGII | + | + | + | |||
| Pathway | Experimental | Human |
|---|---|---|
| TGF-ß/SMAD | Murine Murine MFS/LDS | Isolated aortic VSMC Proteomics Clinical Genetics |
| Angiotensin II | Murine Murine MFS | Clinical |
| PI3K/AKT | Murine Murine MFS | Isolated aortic VSMC TAA tissue |
| WNT | Murine Murine MFS Aortic interstitial cells | Cardiac fibroblasts Isolated EC Isolated aortic VSMC GWAS TAA tissue |
| NOTCH | Murine Murine MFS | Isolated EC Isolated aortic VSMC Proteomics TAA tissue Clinical Genetics |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dong, C.X.; Malecki, C.; Robertson, E.; Hambly, B.; Jeremy, R. Molecular Mechanisms in Genetic Aortopathy–Signaling Pathways and Potential Interventions. Int. J. Mol. Sci. 2023, 24, 1795. https://doi.org/10.3390/ijms24021795
Dong CX, Malecki C, Robertson E, Hambly B, Jeremy R. Molecular Mechanisms in Genetic Aortopathy–Signaling Pathways and Potential Interventions. International Journal of Molecular Sciences. 2023; 24(2):1795. https://doi.org/10.3390/ijms24021795
Chicago/Turabian StyleDong, Charlotte Xue, Cassandra Malecki, Elizabeth Robertson, Brett Hambly, and Richmond Jeremy. 2023. "Molecular Mechanisms in Genetic Aortopathy–Signaling Pathways and Potential Interventions" International Journal of Molecular Sciences 24, no. 2: 1795. https://doi.org/10.3390/ijms24021795