
DNA Damage Response Mechanisms in Head and Neck Cancer: Significant Implications for Therapy and Survival
 , , and
, , and
Abstract
:1. Introduction
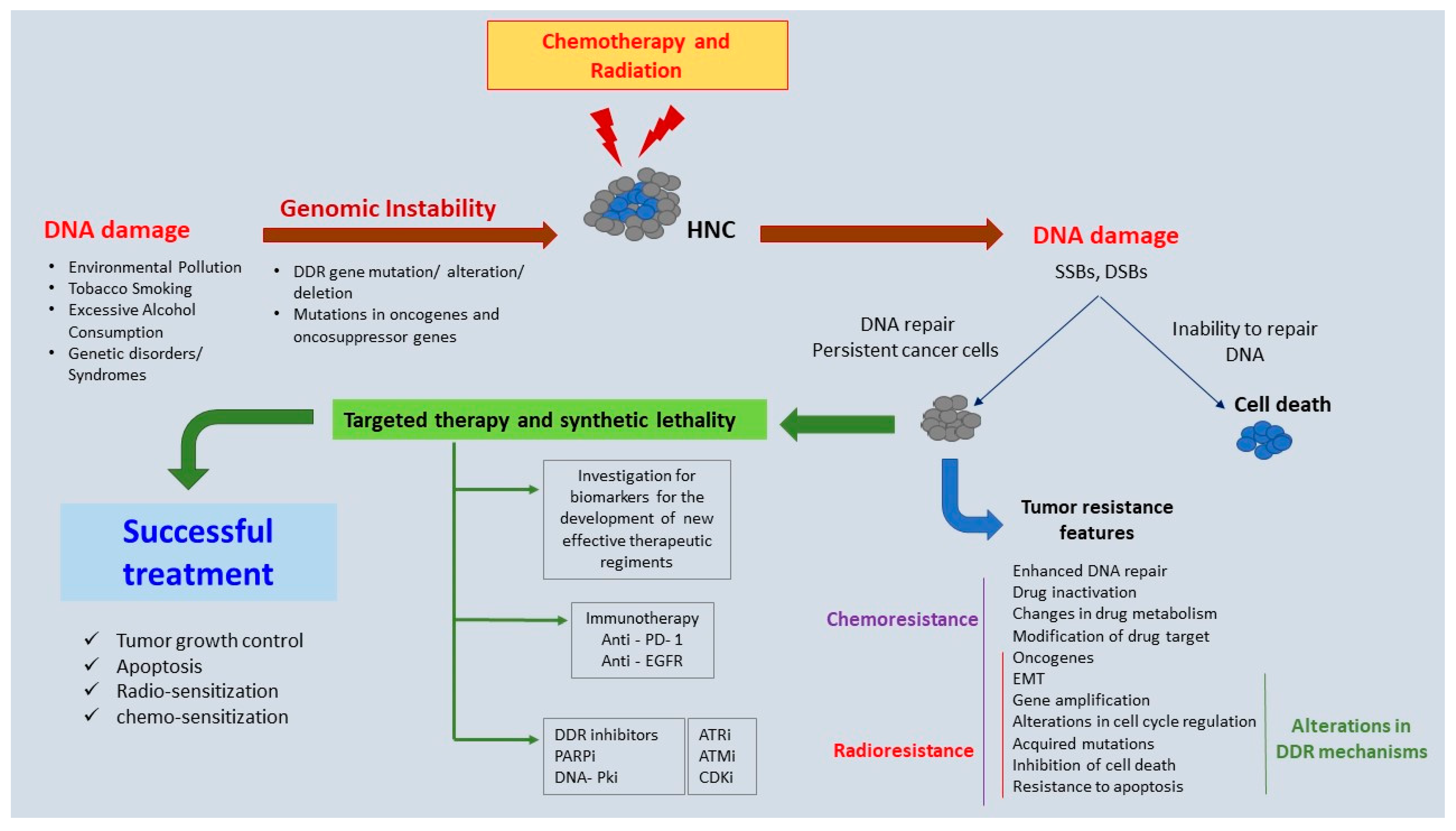
2. Genomic Instability in HNC
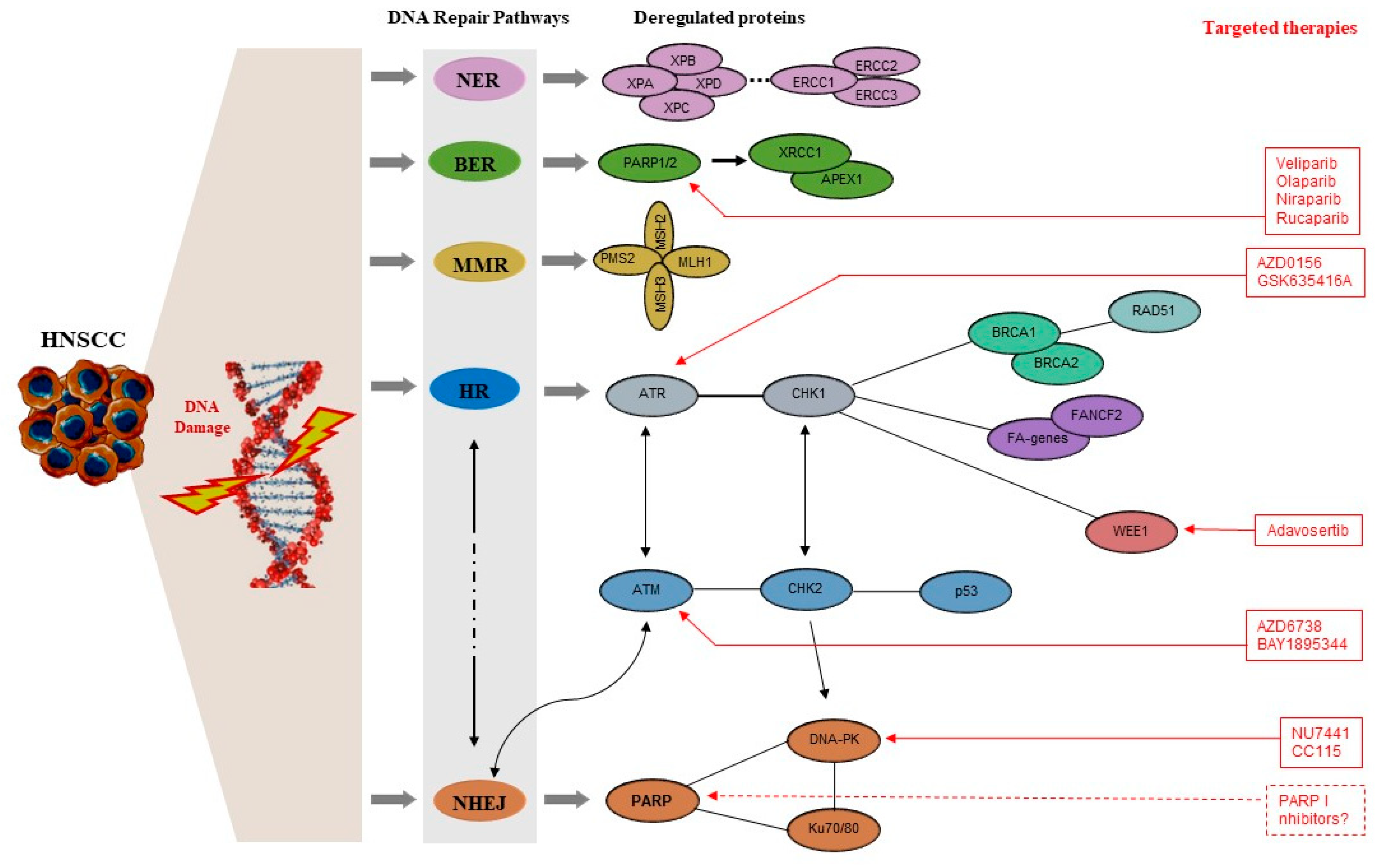
3. DDR Mechanisms—A Brief Overview
- (i)
- Alternative end joining (A-EJ) is a recently described mechanism that repairs DSBs through a subset of A-EJ pathways and relies on microhomology-mediated repair. The best characterized Alt-EJ pathway is the microhomology end joining (MMEJ) pathway; this requires 2 to 20 nucleotides of homologous sequence and shares the same first steps of DDR with HR, i.e., the repair process begins with end resection and involves many of the factors that contribute to the HR end resection machinery. These mechanisms are considered to be error-prone and highly mutagenic as they are associated with deletions flanking the original DSB, leading to chromosomal rearrangements and genomic instability [91,92,93,94].
- (ii)
- Nucleotide excision repair (NER) is a mechanism that repairs bulky DNA lesions that distort the DNA double helix and are usually caused by external mutagens, such as UV light and chemical carcinogenic substances [91,95]. NER is divided into two sub-pathways: global genome NER (GG-NER), which takes place throughout the genome, independently of transcriptional processes, and transcription-coupled NER, which only occurs in order to repair the damage on the transcribed strand [96,97,98].
- (iii)
- Mismatch repair (MMR) is a mechanism that repairs base–base mismatches, deletions, or insertions predominantly generated during DNA replication and recombination processes or mismatches that escape DNA polymerase proofreading activity. MMR is considered a highly conserved biological pathway and plays an important role in genome maintenance [91,99].
- (iv)
- Base excision repair (BER) is a mechanism that tends to repair DNA damage (small base lesions) that originate from endogenous sources, such as those attributed to ROS, alkylation, deamination, and methylation, and which do not create structural distortions of the DNA double helix [91,100]. BER is initiated by a DNA glycosylase that recognizes and removes the damaged base, leaving an empty space that is further processed by short-patch repair or long-patch repair to complete the repair mechanism. A study in a Pakistani population suggested that the deregulation of genes in the BER pathway may drive HNSCC progression [101].
4. DDR Molecules with Evidence of Implication in the Pathogenesis of HNC
4.1. DDR Signaling Kinases
4.1.1. Ataxia Telangiectasia Mutated (ATM)
4.1.2. ATM- and Rad3-Related (ATR)
4.1.3. Chk1 and Chk2
4.1.4. p53-Binding Protein 1 (53BP1)
4.2. Molecules of the Homologous Recombination (HR) Mechanism
4.2.1. FA Genes
4.2.2. BRCA1 and BRCA2
4.2.3. RAD51
4.3. Molecules of the Non-Homologous End Joining (NHEJ) Mechanism
4.3.1. DNA-Dependent Protein Kinase (DNA-PK)
4.3.2. Ku Protein
4.3.3. PARP Molecules
4.4. Molecules of the Nucleotide-Excision Repair (NER), Base-Excision Repair (BER), and Mismatch Repair (MMR) Mechanisms
4.4.1. NER-Associated Genes
4.4.2. BER-Associated Genes
4.4.3. MMR-Associated Genes
4.5. Genes That Have Been Functionally Related to HPV(+) HNSCCs
5. Novel Therapeutic Targets: Preclinical and Clinical Evidence
5.1. PARP Inhibitors
5.2. Immune Checkpoint Inhibitors
5.3. Tyrosine Kinase Inhibitors
5.4. CDK Inhibitors
5.5. DNA-PK Inhibitors
5.6. ATM and ATR Inhibitors
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Johnson, D.E.; Burtness, B.; Leemans, C.R.; Lui, V.W.Y.; Bauman, J.E.; Grandis, J.R. Head and neck squamous cell carcinoma. Nat. Rev. Dis. Primers 2020, 6, 92. [Google Scholar] [CrossRef] [PubMed]
- Auperin, A. Epidemiology of head and neck cancers: An update. Curr. Opin. Oncol. 2020, 32, 178–186. [Google Scholar] [CrossRef] [PubMed]
- Global Burden of Disease Cancer Collaboration; Fitzmaurice, C.; Abate, D.; Abbasi, N.; Abbastabar, H.; Abd-Allah, F.; Abdel-Rahman, O.; Abdelalim, A.; Abdoli, A.; Abdollahpour, I.; et al. Global, Regional, and National Cancer Incidence, Mortality, Years of Life Lost, Years Lived With Disability, and Disability-Adjusted Life-Years for 29 Cancer Groups, 1990 to 2017: A Systematic Analysis for the Global Burden of Disease Study. JAMA Oncol. 2019, 5, 1749–1768. [Google Scholar] [CrossRef] [PubMed]
- Torre, L.A.; Bray, F.; Siegel, R.L.; Ferlay, J.; Lortet-Tieulent, J.; Jemal, A. Global cancer statistics, 2012. CA Cancer J. Clin. 2015, 65, 87–108. [Google Scholar] [CrossRef]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.; Liu, H.; Lu, Y.; Li, H. Identification of a Novel Signature Predicting Overall Survival in Head and Neck Squamous Cell Carcinoma. Front. Surg. 2021, 8, 717084. [Google Scholar] [CrossRef]
- Baez, A. Genetic and environmental factors in head and neck cancer genesis. J. Environ. Sci. Health C Environ. Carcinog. Ecotoxicol. Rev. 2008, 26, 174–200. [Google Scholar] [CrossRef]
- Hashibe, M.; Brennan, P.; Benhamou, S.; Castellsague, X.; Chen, C.; Curado, M.P.; Dal Maso, L.; Daudt, A.W.; Fabianova, E.; Fernandez, L.; et al. Alcohol drinking in never users of tobacco, cigarette smoking in never drinkers, and the risk of head and neck cancer: Pooled analysis in the International Head and Neck Cancer Epidemiology Consortium. J. Natl. Cancer Inst. 2007, 99, 777–789. [Google Scholar] [CrossRef]
- Kawakita, D.; Matsuo, K. Alcohol and head and neck cancer. Cancer Metastasis Rev. 2017, 36, 425–434. [Google Scholar] [CrossRef]
- de Sanjose, S.; Serrano, B.; Tous, S.; Alejo, M.; Lloveras, B.; Quiros, B.; Clavero, O.; Vidal, A.; Ferrandiz-Pulido, C.; Pavon, M.A.; et al. Burden of Human Papillomavirus (HPV)-Related Cancers Attributable to HPVs 6/11/16/18/31/33/45/52 and 58. JNCI Cancer Spectr. 2018, 2, pky045. [Google Scholar] [CrossRef] [Green Version]
- Psyrri, A.; Gkotzamanidou, M.; Papaxoinis, G.; Krikoni, L.; Economopoulou, P.; Kotsantis, I.; Anastasiou, M.; Souliotis, V.L. The DNA damage response network in the treatment of head and neck squamous cell carcinoma. ESMO Open 2021, 6, 100075. [Google Scholar] [CrossRef]
- Cooper, J.S.; Pajak, T.F.; Forastiere, A.A.; Jacobs, J.; Campbell, B.H.; Saxman, S.B.; Kish, J.A.; Kim, H.E.; Cmelak, A.J.; Rotman, M.; et al. Postoperative concurrent radiotherapy and chemotherapy for high-risk squamous-cell carcinoma of the head and neck. N. Engl. J. Med. 2004, 350, 1937–1944. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, G.; O’Byrne, K.J.; Panizza, B.; Richard, D.J. Genome stability pathways in head and neck cancers. Int. J. Genom. 2013, 2013, 464720. [Google Scholar] [CrossRef] [PubMed]
- Osman, N.; Elamin, Y.Y.; Rafee, S.; O’Brien, C.; Stassen, L.F.; Timon, C.; Kinsella, J.; Brennan, S.; O’Byrne, K.J. Weekly cisplatin concurrently with radiotherapy in head and neck squamous cell cancer: A retrospective analysis of a tertiary institute experience. Eur. Arch. Otorhinolaryngol. 2014, 271, 2253–2259. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Galvan, K.J.; Bogard, R.D.; Peterson, C.E.; Shergill, A.; Crowe, D.L. DNA Double-strand Break Signaling Is a Therapeutic Target in Head and Neck Cancer. Anticancer Res. 2021, 41, 5393–5403. [Google Scholar] [CrossRef]
- Cramer, J.D.; Burtness, B.; Le, Q.T.; Ferris, R.L. The changing therapeutic landscape of head and neck cancer. Nat. Rev. Clin. Oncol. 2019, 16, 669–683. [Google Scholar] [CrossRef] [PubMed]
- Ferris, R.L. Immunology and Immunotherapy of Head and Neck Cancer. J. Clin. Oncol. 2015, 33, 3293–3304. [Google Scholar] [CrossRef]
- Chow, L.Q.M. Head and Neck Cancer. N. Engl. J. Med. 2020, 382, 60–72. [Google Scholar] [CrossRef]
- Mito, I.; Takahashi, H.; Kawabata-Iwakawa, R.; Ida, S.; Tada, H.; Chikamatsu, K. Comprehensive analysis of immune cell enrichment in the tumor microenvironment of head and neck squamous cell carcinoma. Sci. Rep. 2021, 11, 16134. [Google Scholar] [CrossRef]
- Wiseman, S.M.; Stoler, D.L.; Anderson, G.R. The role of genomic instability in the pathogenesis of squamous cell carcinoma of the head and neck. Surg. Oncol. Clin. N. Am. 2004, 13, 1–11. [Google Scholar] [CrossRef]
- Moon, J.J.; Lu, A.; Moon, C. Role of genomic instability in human carcinogenesis. Exp. Biol. Med. 2019, 244, 227–240. [Google Scholar] [CrossRef] [PubMed]
- Thompson, S.L.; Bakhoum, S.F.; Compton, D.A. Mechanisms of chromosomal instability. Curr. Biol. 2010, 20, R285–R295. [Google Scholar] [CrossRef] [PubMed]
- Gisselsson, D. Intratumor diversity and clonal evolution in cancer—A skeptical standpoint. Adv. Cancer Res. 2011, 112, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Loeb, L.A.; Bielas, J.H.; Beckman, R.A. Cancers exhibit a mutator phenotype: Clinical implications. Cancer Res. 2008, 68, 3551–3557. [Google Scholar] [CrossRef]
- Halazonetis, T.D.; Gorgoulis, V.G.; Bartek, J. An oncogene-induced DNA damage model for cancer development. Science 2008, 319, 1352–1355. [Google Scholar] [CrossRef] [PubMed]
- Gorgoulis, V.G.; Vassiliou, L.V.; Karakaidos, P.; Zacharatos, P.; Kotsinas, A.; Liloglou, T.; Venere, M.; Ditullio, R.A., Jr.; Kastrinakis, N.G.; Levy, B.; et al. Activation of the DNA damage checkpoint and genomic instability in human precancerous lesions. Nature 2005, 434, 907–913. [Google Scholar] [CrossRef]
- Alrawi, S.J.; Stoler, D.; Tan, D.; Dayton, M.; Loree, T.; Gibbs, J.F.; Rigual, N.; Sait, S.; Khoury, T.; Hicks, W., Jr.; et al. Genomic Instability, DNA Alterations and Tumor Eosinophilic Expression in Head and Neck Squamous Cell Carcinoma. Cancer Genom. Proteom. 2005, 2, 307–316. [Google Scholar]
- Walczak, A.; Rusin, P.; Dziki, L.; Zielinska-Blizniewska, H.; Olszewski, J.; Majsterek, I. Evaluation of DNA double strand breaks repair efficiency in head and neck cancer. DNA Cell Biol. 2012, 31, 298–305. [Google Scholar] [CrossRef]
- Quennet, V.; Beucher, A.; Barton, O.; Takeda, S.; Lobrich, M. CtIP and MRN promote non-homologous end-joining of etoposide-induced DNA double-strand breaks in G1. Nucleic Acids Res. 2011, 39, 2144–2152. [Google Scholar] [CrossRef]
- Zhao, X.; Wei, C.; Li, J.; Xing, P.; Li, J.; Zheng, S.; Chen, X. Cell cycle-dependent control of homologous recombination. Acta Biochim. Biophys. Sin. 2017, 49, 655–668. [Google Scholar] [CrossRef]
- Willers, H.; Azzoli, C.G.; Santivasi, W.L.; Xia, F. Basic mechanisms of therapeutic resistance to radiation and chemotherapy in lung cancer. Cancer J. 2013, 19, 200–207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Housman, G.; Byler, S.; Heerboth, S.; Lapinska, K.; Longacre, M.; Snyder, N.; Sarkar, S. Drug resistance in cancer: An overview. Cancers 2014, 6, 1769–1792. [Google Scholar] [CrossRef] [PubMed]
- Nie, X.; Guo, E.; Wu, C.; Liu, D.; Sun, W.; Zhang, L.; Long, G.; Mei, Q.; Wu, K.; Xiong, H.; et al. SALL4 induces radioresistance in nasopharyngeal carcinoma via the ATM/Chk2/p53 pathway. Cancer Med. 2019, 8, 1779–1792. [Google Scholar] [CrossRef] [PubMed]
- Di, M.; Wang, M.; Miao, J.; Chen, B.; Huang, H.; Lin, C.; Jian, Y.; Li, Y.; Ouyang, Y.; Chen, X.; et al. CHAF1B induces radioresistance by promoting DNA damage repair in nasopharyngeal carcinoma. Biomed. Pharmacother. 2020, 123, 109748. [Google Scholar] [CrossRef]
- Wang, Z.; Zuo, W.; Zeng, Q.; Li, Y.; Lu, T.; Bu, Y.; Hu, G. The Homologous Recombination Repair Pathway is Associated with Resistance to Radiotherapy in Nasopharyngeal Carcinoma. Int. J. Biol. Sci. 2020, 16, 408–419. [Google Scholar] [CrossRef]
- Lodovichi, S.; Cervelli, T.; Pellicioli, A.; Galli, A. Inhibition of DNA Repair in Cancer Therapy: Toward a Multi-Target Approach. Int. J. Mol. Sci. 2020, 21, 6684. [Google Scholar] [CrossRef]
- Curtin, N.J. Inhibiting the DNA damage response as a therapeutic manoeuvre in cancer. Br. J. Pharmacol. 2013, 169, 1745–1765. [Google Scholar] [CrossRef]
- Chow, J.P.; Man, W.Y.; Mao, M.; Chen, H.; Cheung, F.; Nicholls, J.; Tsao, S.W.; Li Lung, M.; Poon, R.Y. PARP1 is overexpressed in nasopharyngeal carcinoma and its inhibition enhances radiotherapy. Mol. Cancer Ther. 2013, 12, 2517–2528. [Google Scholar] [CrossRef]
- Chen, Y.; Zhao, Y.; Lu, R.; Zhao, H.; Guo, Y. Identification and Validation of a Novel Genomic Instability-Associated Long Non-Coding RNA Prognostic Signature in Head and Neck Squamous Cell Carcinoma. Front. Cell Dev. Biol. 2021, 9, 787766. [Google Scholar] [CrossRef]
- Pikor, L.; Thu, K.; Vucic, E.; Lam, W. The detection and implication of genome instability in cancer. Cancer Metastasis Rev. 2013, 32, 341–352. [Google Scholar] [CrossRef]
- Vaish, M.; Mittal, B. DNA mismatch repair, microsatellite instability and cancer. Indian J. Exp. Biol. 2002, 40, 989–994. [Google Scholar] [PubMed]
- Piccolo, S.R.; Frey, L.J. Somatic mutation signatures of cancer. AMIA Annu. Symp. Proc. 2008, 2008, 202–206. [Google Scholar] [PubMed]
- Hecht, S.S. Tobacco carcinogens, their biomarkers and tobacco-induced cancer. Nat. Rev. Cancer 2003, 3, 733–744. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; Jagirdar, B.R. Synthesis of mesoporous iridium nanosponge: A highly active, thermally stable and efficient olefin hydrogenation catalyst. Dalton Trans. 2017, 46, 11431–11439. [Google Scholar] [CrossRef] [PubMed]
- Jawa, D.S.; Sircar, K.; Somani, R.; Grover, N.; Jaidka, S.; Singh, S. Gorlin-Goltz syndrome. J. Oral Maxillofac. Pathol. 2009, 13, 89–92. [Google Scholar] [CrossRef]
- Joshi, P.S.; Deshmukh, V.; Golgire, S. Gorlin-Goltz syndrome. Dent. Res. J. 2012, 9, 100–106. [Google Scholar] [CrossRef]
- Moreno, O.M.; Paredes, A.C.; Suarez-Obando, F.; Rojas, A. An update on Fanconi anemia: Clinical, cytogenetic and molecular approaches (Review). Biomed. Rep. 2021, 15, 74. [Google Scholar] [CrossRef]
- Garcia-de-Teresa, B.; Rodriguez, A.; Frias, S. Chromosome Instability in Fanconi Anemia: From Breaks to Phenotypic Consequences. Genes 2020, 11, 1528. [Google Scholar] [CrossRef]
- Powell, S.F.; Vu, L.; Spanos, W.C.; Pyeon, D. The Key Differences between Human Papillomavirus-Positive and -Negative Head and Neck Cancers: Biological and Clinical Implications. Cancers 2021, 13, 5206. [Google Scholar] [CrossRef]
- Michor, F.; Iwasa, Y.; Nowak, M.A. Dynamics of cancer progression. Nat. Rev. Cancer 2004, 4, 197–205. [Google Scholar] [CrossRef]
- Ubhi, T.; Brown, G.W. Exploiting DNA Replication Stress for Cancer Treatment. Cancer Res. 2019, 79, 1730–1739. [Google Scholar] [CrossRef] [Green Version]
- Helmrich, A.; Ballarino, M.; Nudler, E.; Tora, L. Transcription-replication encounters, consequences and genomic instability. Nat. Struct. Mol. Biol. 2013, 20, 412–418. [Google Scholar] [CrossRef] [PubMed]
- Billard, P.; Poncet, D.A. Replication Stress at Telomeric and Mitochondrial DNA: Common Origins and Consequences on Ageing. Int. J. Mol. Sci. 2019, 20, 4959. [Google Scholar] [CrossRef] [PubMed]
- Zeman, M.K.; Cimprich, K.A. Causes and consequences of replication stress. Nat. Cell Biol. 2014, 16, 2–9. [Google Scholar] [CrossRef] [PubMed]
- Mazouzi, A.; Velimezi, G.; Loizou, J.I. DNA replication stress: Causes, resolution and disease. Exp. Cell Res. 2014, 329, 85–93. [Google Scholar] [CrossRef] [PubMed]
- Debatisse, M.; Le Tallec, B.; Letessier, A.; Dutrillaux, B.; Brison, O. Common fragile sites: Mechanisms of instability revisited. Trends Genet. 2012, 28, 22–32. [Google Scholar] [CrossRef]
- De Bont, R.; van Larebeke, N. Endogenous DNA damage in humans: A review of quantitative data. Mutagenesis 2004, 19, 169–185. [Google Scholar] [CrossRef]
- Wong, I.C.; Ng, Y.K.; Lui, V.W. Cancers of the lung, head and neck on the rise: Perspectives on the genotoxicity of air pollution. Chin. J. Cancer 2014, 33, 476–480. [Google Scholar] [CrossRef]
- Beynon, R.A.; Lang, S.; Schimansky, S.; Penfold, C.M.; Waylen, A.; Thomas, S.J.; Pawlita, M.; Waterboer, T.; Martin, R.M.; May, M.; et al. Tobacco smoking and alcohol drinking at diagnosis of head and neck cancer and all-cause mortality: Results from head and neck 5000, a prospective observational cohort of people with head and neck cancer. Int. J. Cancer 2018, 143, 1114–1127. [Google Scholar] [CrossRef]
- Di Credico, G.; Polesel, J.; Dal Maso, L.; Pauli, F.; Torelli, N.; Luce, D.; Radoi, L.; Matsuo, K.; Serraino, D.; Brennan, P.; et al. Alcohol drinking and head and neck cancer risk: The joint effect of intensity and duration. Br. J. Cancer 2020, 123, 1456–1463. [Google Scholar] [CrossRef]
- Rezapour, M.; Rezapour, H.A.; Chegeni, M.; Khanjani, N. Exposure to cadmium and head and neck cancers: A meta-analysis of observational studies. Rev. Environ. Health 2021, 36, 577–584. [Google Scholar] [CrossRef] [PubMed]
- Khariwala, S.S.; Ma, B.; Ruszczak, C.; Carmella, S.G.; Lindgren, B.; Hatsukami, D.K.; Hecht, S.S.; Stepanov, I. High Level of Tobacco Carcinogen-Derived DNA Damage in Oral Cells Is an Independent Predictor of Oral/Head and Neck Cancer Risk in Smokers. Cancer Prev. Res. 2017, 10, 507–513. [Google Scholar] [CrossRef] [PubMed]
- Vageli, D.P.; Doukas, P.G.; Doukas, S.G.; Tsatsakis, A.; Judson, B.L. Noxious Combination of Tobacco Smoke Nitrosamines with Bile, Deoxycholic Acid, Promotes Hypopharyngeal Squamous Cell Carcinoma, via NFkappaB, In Vivo. Cancer Prev. Res. 2022, 15, 297–308. [Google Scholar] [CrossRef] [PubMed]
- Schuch, L.F.; de Arruda, J.A.A.; Viana, K.S.S.; Caldeira, P.C.; Abreu, M.; Bernardes, V.F.; Aguiar, M.C.F. DNA damage-related proteins in smokers and non-smokers with oral cancer. Braz. Oral Res. 2022, 36, e027. [Google Scholar] [CrossRef] [PubMed]
- Baan, R.; Straif, K.; Grosse, Y.; Secretan, B.; El Ghissassi, F.; Bouvard, V.; Altieri, A.; Cogliano, V.; WHO International Agency for Research on Cancer Monograph Working Group. Carcinogenicity of alcoholic beverages. Lancet Oncol. 2007, 8, 292–293. [Google Scholar] [CrossRef]
- Denissoff, A.; Huusko, T.; Ventela, S.; Niemela, S.; Routila, J. Exposure to alcohol and overall survival in head and neck cancer: A regional cohort study. Head Neck 2022, 44, 2109–2117. [Google Scholar] [CrossRef]
- Seitz, H.K.; Mueller, S. Alcohol and cancer: An overview with special emphasis on the role of acetaldehyde and cytochrome P450 2E1. Adv. Exp. Med. Biol. 2015, 815, 59–70. [Google Scholar] [CrossRef]
- Wang, G.; Pan, C.; Cao, K.; Zhang, J.; Geng, H.; Wu, K.; Wen, J.; Liu, C. Impacts of Cigarette Smoking on the Tumor Immune Microenvironment in Esophageal Squamous Cell Carcinoma. J. Cancer 2022, 13, 413–425. [Google Scholar] [CrossRef]
- Li, Y.C.; Chang, J.T.; Chiu, C.; Lu, Y.C.; Li, Y.L.; Chiang, C.H.; You, G.R.; Lee, L.Y.; Cheng, A.J. Areca nut contributes to oral malignancy through facilitating the conversion of cancer stem cells. Mol. Carcinog. 2016, 55, 1012–1023. [Google Scholar] [CrossRef]
- Li, Y.C.; Cheng, A.J.; Lee, L.Y.; Huang, Y.C.; Chang, J.T. Multifaceted Mechanisms of Areca Nuts in Oral Carcinogenesis: The Molecular Pathology from Precancerous Condition to Malignant Transformation. J. Cancer 2019, 10, 4054–4062. [Google Scholar] [CrossRef]
- Wang, Y.C.; Lee, K.W.; Tsai, Y.S.; Lu, H.H.; Chen, S.Y.; Hsieh, H.Y.; Lin, C.S. Downregulation of ATM and BRCA1 Predicts Poor Outcome in Head and Neck Cancer: Implications for ATM-Targeted Therapy. J. Pers. Med. 2021, 11, 389. [Google Scholar] [CrossRef] [PubMed]
- Klussmann, J.P. Head and Neck Cancer—New Insights into a Heterogeneous Disease. Oncol. Res. Treat. 2017, 40, 318–319. [Google Scholar] [CrossRef] [PubMed]
- Mroz, E.A.; Rocco, J.W. Intra-tumor heterogeneity in head and neck cancer and its clinical implications. World J. Otorhinolaryngol. Head Neck Surg. 2016, 2, 60–67. [Google Scholar] [CrossRef]
- Sewell, A.; Brown, B.; Biktasova, A.; Mills, G.B.; Lu, Y.; Tyson, D.R.; Issaeva, N.; Yarbrough, W.G. Reverse-phase protein array profiling of oropharyngeal cancer and significance of PIK3CA mutations in HPV(−)associated head and neck cancer. Clin. Cancer Res. 2014, 20, 2300–2311. [Google Scholar] [CrossRef] [PubMed]
- Slebos, R.J.; Yi, Y.; Ely, K.; Carter, J.; Evjen, A.; Zhang, X.; Shyr, Y.; Murphy, B.M.; Cmelak, A.J.; Burkey, B.B.; et al. Gene expression differences associated with human papillomavirus status in head and neck squamous cell carcinoma. Clin. Cancer Res. 2006, 12, 701–709. [Google Scholar] [CrossRef]
- Schlecht, N.F.; Burk, R.D.; Adrien, L.; Dunne, A.; Kawachi, N.; Sarta, C.; Chen, Q.; Brandwein-Gensler, M.; Prystowsky, M.B.; Childs, G.; et al. Gene expression profiles in HPV(−)infected head and neck cancer. J. Pathol. 2007, 213, 283–293. [Google Scholar] [CrossRef]
- Pan, C.; Izreig, S.; Yarbrough, W.G.; Issaeva, N. NSD1 mutations by HPV status in head and neck cancer: Differences in survival and response to DNA-damaging agents. Cancers Head Neck 2019, 4, 3. [Google Scholar] [CrossRef]
- Gillison, M.L.; Akagi, K.; Xiao, W.; Jiang, B.; Pickard, R.K.L.; Li, J.; Swanson, B.J.; Agrawal, A.D.; Zucker, M.; Stache-Crain, B.; et al. Human papillomavirus and the landscape of secondary genetic alterations in oral cancers. Genom. Res. 2019, 29, 1–17. [Google Scholar] [CrossRef]
- Parfenov, M.; Pedamallu, C.S.; Gehlenborg, N.; Freeman, S.S.; Danilova, L.; Bristow, C.A.; Lee, S.; Hadjipanayis, A.G.; Ivanova, E.V.; Wilkerson, M.D.; et al. Characterization of HPV and host genome interactions in primary head and neck cancers. Proc. Natl. Acad. Sci. USA 2014, 111, 15544–15549. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Network. Comprehensive genomic characterization of head and neck squamous cell carcinomas. Nature 2015, 517, 576–582. [Google Scholar] [CrossRef]
- Bamps, M.; Dok, R.; Nuyts, S. The DNA Damage Response Is Differentially Involved in HPV(−)Positive and HPV(−)Negative Radioresistant Head and Neck Squamous Cell Carcinoma. Cancers 2021, 13, 3717. [Google Scholar] [CrossRef] [PubMed]
- Zhou, G.; Liu, Z.; Myers, J.N. TP53 Mutations in Head and Neck Squamous Cell Carcinoma and Their Impact on Disease Progression and Treatment Response. J. Cell Biochem. 2016, 117, 2682–2692. [Google Scholar] [CrossRef] [PubMed]
- Stransky, N.; Egloff, A.M.; Tward, A.D.; Kostic, A.D.; Cibulskis, K.; Sivachenko, A.; Kryukov, G.V.; Lawrence, M.S.; Sougnez, C.; McKenna, A.; et al. The mutational landscape of head and neck squamous cell carcinoma. Science 2011, 333, 1157–1160. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, N.; Frederick, M.J.; Pickering, C.R.; Bettegowda, C.; Chang, K.; Li, R.J.; Fakhry, C.; Xie, T.X.; Zhang, J.; Wang, J.; et al. Exome sequencing of head and neck squamous cell carcinoma reveals inactivating mutations in NOTCH1. Science 2011, 333, 1154–1157. [Google Scholar] [CrossRef] [PubMed]
- Pyeon, D.; Newton, M.A.; Lambert, P.F.; den Boon, J.A.; Sengupta, S.; Marsit, C.J.; Woodworth, C.D.; Connor, J.P.; Haugen, T.H.; Smith, E.M.; et al. Fundamental differences in cell cycle deregulation in human papillomavirus-positive and human papillomavirus-negative head/neck and cervical cancers. Cancer Res. 2007, 67, 4605–4619. [Google Scholar] [CrossRef] [PubMed]
- den Boon, J.A.; Pyeon, D.; Wang, S.S.; Horswill, M.; Schiffman, M.; Sherman, M.; Zuna, R.E.; Wang, Z.; Hewitt, S.M.; Pearson, R.; et al. Molecular transitions from papillomavirus infection to cervical precancer and cancer: Role of stromal estrogen receptor signaling. Proc. Natl. Acad. Sci. USA 2015, 112, E3255–E3264. [Google Scholar] [CrossRef]
- Hinic, S.; Rich, A.; Anayannis, N.V.; Cabarcas-Petroski, S.; Schramm, L.; Meneses, P.I. Gene Expression and DNA Methylation in Human Papillomavirus Positive and Negative Head and Neck Squamous Cell Carcinomas. Int. J. Mol. Sci. 2022, 23, 10967. [Google Scholar] [CrossRef]
- Ekanayake Weeramange, C.; Tang, K.D.; Vasani, S.; Langton-Lockton, J.; Kenny, L.; Punyadeera, C. DNA Methylation Changes in Human Papillomavirus-Driven Head and Neck Cancers. Cells 2020, 9, 1359. [Google Scholar] [CrossRef]
- Hajek, M.; Biktasova, A.; Sewell, A.; Gary, C.; Cantalupo, P.; Anderson, K.S.; Yarbrough, W.G.; Issaeva, N. Global Genome Demethylation Causes Transcription-Associated DNA Double Strand Breaks in HPV(−)Associated Head and Neck Cancer Cells. Cancers 2020, 13, 21. [Google Scholar] [CrossRef]
- Ceccaldi, R.; Sarangi, P.; D’Andrea, A.D. The Fanconi anaemia pathway: New players and new functions. Nat. Rev. Mol. Cell Biol. 2016, 17, 337–349. [Google Scholar] [CrossRef]
- Ye, Z.; Shi, Y.; Lees-Miller, S.P.; Tainer, J.A. Function and Molecular Mechanism of the DNA Damage Response in Immunity and Cancer Immunotherapy. Front. Immunol. 2021, 12, 797880. [Google Scholar] [CrossRef] [PubMed]
- Sfeir, A.; Symington, L.S. Microhomology-Mediated End Joining: A Back-up Survival Mechanism or Dedicated Pathway? Trends Biochem. Sci. 2015, 40, 701–714. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Xu, X. Microhomology-mediated end joining: New players join the team. Cell Biosci. 2017, 7, 6. [Google Scholar] [CrossRef] [PubMed]
- Caracciolo, D.; Montesano, M.; Tagliaferri, P.; Tassone, P. Alternative non-homologous end joining repair: A master regulator of genomic instability in cancer. Precis. Cancer Med. 2019, 2, 1–10. [Google Scholar] [CrossRef]
- Scharer, O.D. Nucleotide excision repair in eukaryotes. Cold Spring Harb. Perspect. Biol. 2013, 5, a012609. [Google Scholar] [CrossRef]
- Dworaczek, H.; Xiao, W. Xeroderma pigmentosum: A glimpse into nucleotide excision repair, genetic instability, and cancer. Crit. Rev. Oncog. 2007, 13, 159–177. [Google Scholar] [CrossRef]
- Vaughn, C.M.; Sancar, A. Chapter 16 Mechanisms and Maps of Nucleotide Excision Repair. In DNA Damage, DNA Repair and Disease: Volume 2; The Royal Society of Chemistry: Cambridge, UK, 2021; Volume 2, pp. 1–23. [Google Scholar]
- Fayyad, N.; Kobaisi, F.; Beal, D.; Mahfouf, W.; Ged, C.; Morice-Picard, F.; Fayyad-Kazan, M.; Fayyad-Kazan, H.; Badran, B.; Rezvani, H.R.; et al. Xeroderma Pigmentosum C (XPC) Mutations in Primary Fibroblasts Impair Base Excision Repair Pathway and Increase Oxidative DNA Damage. Front. Genet. 2020, 11, 561687. [Google Scholar] [CrossRef]
- Li, G.M. Mechanisms and functions of DNA mismatch repair. Cell Res. 2008, 18, 85–98. [Google Scholar] [CrossRef]
- Kocher, S.; Zech, H.B.; Krug, L.; Gatzemeier, F.; Christiansen, S.; Meyer, F.; Rietow, R.; Struve, N.; Mansour, W.Y.; Kriegs, M.; et al. A Lack of Effectiveness in the ATM-Orchestrated DNA Damage Response Contributes to the DNA Repair Defect of HPV(−)Positive Head and Neck Cancer Cells. Front. Oncol. 2022, 12, 765968. [Google Scholar] [CrossRef]
- Mahjabeen, I.; Ali, K.; Zhou, X.; Kayani, M.A. Deregulation of base excision repair gene expression and enhanced proliferation in head and neck squamous cell carcinoma. Tumour Biol. 2014, 35, 5971–5983. [Google Scholar] [CrossRef]
- Marechal, A.; Zou, L. DNA damage sensing by the ATM and ATR kinases. Cold Spring Harb. Perspect. Biol. 2013, 5, a012716. [Google Scholar] [CrossRef] [PubMed]
- Blackford, A.N.; Jackson, S.P. ATM, ATR, and DNA-PK: The Trinity at the Heart of the DNA Damage Response. Mol. Cell 2017, 66, 801–817. [Google Scholar] [CrossRef] [PubMed]
- Pedroza-Garcia, J.A.; Xiang, Y.; De Veylder, L. Cell cycle checkpoint control in response to DNA damage by environmental stresses. Plant J. 2022, 109, 490–507. [Google Scholar] [CrossRef] [PubMed]
- So, S.; Davis, A.J.; Chen, D.J. Autophosphorylation at serine 1981 stabilizes ATM at DNA damage sites. J. Cell. Biol. 2009, 187, 977–990. [Google Scholar] [CrossRef]
- Berkovich, E.; Monnat, R.J., Jr.; Kastan, M.B. Roles of ATM and NBS1 in chromatin structure modulation and DNA double-strand break repair. Nat. Cell Biol. 2007, 9, 683–690. [Google Scholar] [CrossRef] [PubMed]
- Shiloh, Y. ATM and related protein kinases: Safeguarding genome integrity. Nat. Rev. Cancer 2003, 3, 155–168. [Google Scholar] [CrossRef] [PubMed]
- Meier, A.; Fiegler, H.; Munoz, P.; Ellis, P.; Rigler, D.; Langford, C.; Blasco, M.A.; Carter, N.; Jackson, S.P. Spreading of mammalian DNA-damage response factors studied by ChIP-chip at damaged telomeres. EMBO J. 2007, 26, 2707–2718. [Google Scholar] [CrossRef]
- Savic, V.; Yin, B.; Maas, N.L.; Bredemeyer, A.L.; Carpenter, A.C.; Helmink, B.A.; Yang-Iott, K.S.; Sleckman, B.P.; Bassing, C.H. Formation of dynamic gamma-H2AX domains along broken DNA strands is distinctly regulated by ATM and MDC1 and dependent upon H2AX densities in chromatin. Mol. Cell 2009, 34, 298–310. [Google Scholar] [CrossRef]
- Chung, Y.M.; Park, S.H.; Tsai, W.B.; Wang, S.Y.; Ikeda, M.A.; Berek, J.S.; Chen, D.J.; Hu, M.C. FOXO3 signalling links ATM to the p53 apoptotic pathway following DNA damage. Nat. Commun. 2012, 3, 1000. [Google Scholar] [CrossRef]
- Abbotts, R.; Golato, T.; Wilson, D.M. Role of DNA Repair in Carcinogenesis and Cancer Therapeutics. In Encyclopedia of Cancer, 3rd ed.; Paolo Boffetta, P.H., Ed.; Academic Press: Cambridge, MA, USA, 2019; pp. 363–385. [Google Scholar] [CrossRef]
- Nalepa, G.; Clapp, D.W. Fanconi anaemia and cancer: An intricate relationship. Nat. Rev. Cancer 2018, 18, 168–185. [Google Scholar] [CrossRef]
- Kutler, D.I.; Auerbach, A.D.; Satagopan, J.; Giampietro, P.F.; Batish, S.D.; Huvos, A.G.; Goberdhan, A.; Shah, J.P.; Singh, B. High incidence of head and neck squamous cell carcinoma in patients with Fanconi anemia. Arch. Otolaryngol. Head Neck Surg. 2003, 129, 106–112. [Google Scholar] [CrossRef] [PubMed]
- Choi, M.; Kipps, T.; Kurzrock, R. ATM Mutations in Cancer: Therapeutic Implications. Mol. Cancer Ther. 2016, 15, 1781–1791. [Google Scholar] [CrossRef] [PubMed]
- Ai, L.; Vo, Q.N.; Zuo, C.; Li, L.; Ling, W.; Suen, J.Y.; Hanna, E.; Brown, K.D.; Fan, C.Y. Ataxia-telangiectasia-mutated (ATM) gene in head and neck squamous cell carcinoma: Promoter hypermethylation with clinical correlation in 100 cases. Cancer Epidemiol. Biomark. Prev. 2004, 13, 150–156. [Google Scholar] [CrossRef]
- Hier, J.; Vachon, O.; Bernstein, A.; Ibrahim, I.; Mlynarek, A.; Hier, M.; Alaoui-Jamali, M.A.; Maschietto, M.; da Silva, S.D. Portrait of DNA methylated genes predictive of poor prognosis in head and neck cancer and the implication for targeted therapy. Sci. Rep. 2021, 11, 10012. [Google Scholar] [CrossRef]
- Moody, C.A.; Laimins, L.A. Human papillomaviruses activate the ATM DNA damage pathway for viral genome amplification upon differentiation. PLoS Pathog. 2009, 5, e1000605. [Google Scholar] [CrossRef] [PubMed]
- Zou, L.; Elledge, S.J. Sensing DNA damage through ATRIP recognition of RPA-ssDNA complexes. Science 2003, 300, 1542–1548. [Google Scholar] [CrossRef] [PubMed]
- Saldivar, J.C.; Hamperl, S.; Bocek, M.J.; Chung, M.; Bass, T.E.; Cisneros-Soberanis, F.; Samejima, K.; Xie, L.; Paulson, J.R.; Earnshaw, W.C.; et al. An intrinsic S/G2 checkpoint enforced by ATR. Science 2018, 361, 806–810. [Google Scholar] [CrossRef]
- Consortium, A.P.G. AACR Project GENIE: Powering Precision Medicine through an International Consortium. Cancer Discov. 2017, 7, 818–831. [Google Scholar] [CrossRef]
- Bartek, J.; Lukas, J. Chk1 and Chk2 kinases in checkpoint control and cancer. Cancer Cell 2003, 3, 421–429. [Google Scholar] [CrossRef]
- Lukas, C.; Bartkova, J.; Latella, L.; Falck, J.; Mailand, N.; Schroeder, T.; Sehested, M.; Lukas, J.; Bartek, J. DNA damage-activated kinase Chk2 is independent of proliferation or differentiation yet correlates with tissue biology. Cancer Res. 2001, 61, 4990–4993. [Google Scholar]
- Smith, J.; Tho, L.M.; Xu, N.; Gillespie, D.A. The ATM-Chk2 and ATR-Chk1 pathways in DNA damage signaling and cancer. Adv. Cancer Res. 2010, 108, 73–112. [Google Scholar] [CrossRef]
- Choi, S.H.; Cho, K.; Kim, E.S.; Yoo, H.Y. Proline-serine-threonine-repeat region of MDC1 mediates Chk1 phosphorylation and the DNA double-strand break repair. Int. J. Biochem. Cell Biol. 2022, 143, 106152. [Google Scholar] [CrossRef] [PubMed]
- Her, J.; Ray, C.; Altshuler, J.; Zheng, H.; Bunting, S.F. 53BP1 Mediates ATR-Chk1 Signaling and Protects Replication Forks under Conditions of Replication Stress. Mol. Cell Biol. 2018, 38, e00472-17. [Google Scholar] [CrossRef] [PubMed]
- Chapman, J.R.; Barral, P.; Vannier, J.B.; Borel, V.; Steger, M.; Tomas-Loba, A.; Sartori, A.A.; Adams, I.R.; Batista, F.D.; Boulton, S.J. RIF1 Is Essential for 53BP1-Dependent Nonhomologous End Joining and Suppression of DNA Double-Strand Break Resection. Mol. Cell 2021, 81, 2868. [Google Scholar] [CrossRef] [PubMed]
- Gao, S.; Feng, S.; Ning, S.; Liu, J.; Zhao, H.; Xu, Y.; Shang, J.; Li, K.; Li, Q.; Guo, R.; et al. An OB-fold complex controls the repair pathways for DNA double-strand breaks. Nat. Commun. 2018, 9, 3925. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Xu, D. Repair pathway choice for double-strand breaks. Essays Biochem. 2020, 64, 765–777. [Google Scholar] [CrossRef]
- Panier, S.; Boulton, S.J. Double-strand break repair: 53BP1 comes into focus. Nat. Rev. Mol. Cell Biol. 2014, 15, 7–18. [Google Scholar] [CrossRef]
- Bartova, E.; Legartova, S.; Dundr, M.; Suchankova, J. A role of the 53BP1 protein in genome protection: Structural and functional characteristics of 53BP1-dependent DNA repair. Aging 2019, 11, 2488–2511. [Google Scholar] [CrossRef]
- Imaizumi, T.; Matsuda, K.; Tanaka, K.; Kondo, H.; Ueki, N.; Kurohama, H.; Otsubo, C.; Matsuoka, Y.; Akazawa, Y.; Miura, S.; et al. Detection of Endogenous DNA Double-strand Breaks in Oral Squamous Epithelial Lesions by P53-binding Protein 1. Anticancer Res. 2021, 41, 4771–4779. [Google Scholar] [CrossRef]
- Otsubo, R.; Matsuda, K.; Mussazhanova, Z.; Sato, A.; Matsumoto, M.; Yano, H.; Oikawa, M.; Kondo, H.; Ito, M.; Miyauchi, A.; et al. A Novel Diagnostic Method for Thyroid Follicular Tumors Based on Immunofluorescence Analysis of p53-Binding Protein 1 Expression: Detection of Genomic Instability. Thyroid 2019, 29, 657–665. [Google Scholar] [CrossRef]
- Yang, J.; Jing, L.; Liu, C.J.; Bai, W.W.; Zhu, S.C. 53BP1 regulates cell cycle arrest in esophageal cancer model. Eur. Rev. Med. Pharmacol. Sci. 2019, 23, 604–612. [Google Scholar] [CrossRef]
- Grigorova, M.; Staines, J.M.; Ozdag, H.; Caldas, C.; Edwards, P.A. Possible causes of chromosome instability: Comparison of chromosomal abnormalities in cancer cell lines with mutations in BRCA1, BRCA2, CHK2 and BUB1. Cytogenet. Genom. Res. 2004, 104, 333–340. [Google Scholar] [CrossRef] [PubMed]
- Sishc, B.J.; Davis, A.J. The Role of the Core Non-Homologous End Joining Factors in Carcinogenesis and Cancer. Cancers 2017, 9, 81. [Google Scholar] [CrossRef] [PubMed]
- Bishop, A.J.; Schiestl, R.H. Homologous Recombination and Its Role in Carcinogenesis. J. Biomed. Biotechnol. 2002, 2, 75–85. [Google Scholar] [CrossRef] [PubMed]
- Scheckenbach, K.; Wagenmann, M.; Freund, M.; Schipper, J.; Hanenberg, H. Squamous cell carcinomas of the head and neck in Fanconi anemia: Risk, prevention, therapy, and the need for guidelines. Klin. Padiatr. 2012, 224, 132–138. [Google Scholar] [CrossRef] [PubMed]
- Verhagen, C.V.M.; Vossen, D.M.; Borgmann, K.; Hageman, F.; Grenman, R.; Verwijs-Janssen, M.; Mout, L.; Kluin, R.J.C.; Nieuwland, M.; Severson, T.M.; et al. Fanconi anemia and homologous recombination gene variants are associated with functional DNA repair defects in vitro and poor outcome in patients with advanced head and neck squamous cell carcinoma. Oncotarget 2018, 9, 18198–18213. [Google Scholar] [CrossRef]
- Peake, J.D.; Noguchi, C.; Lin, B.; Theriault, A.; O’Connor, M.; Sheth, S.; Tanaka, K.; Nakagawa, H.; Noguchi, E. FANCD2 limits acetaldehyde-induced genomic instability during DNA replication in esophageal keratinocytes. Mol. Oncol. 2021, 15, 3109–3124. [Google Scholar] [CrossRef]
- Lee, J.M.; Ledermann, J.A.; Kohn, E.C. PARP Inhibitors for BRCA1/2 mutation-associated and BRCA-like malignancies. Ann. Oncol. 2014, 25, 32–40. [Google Scholar] [CrossRef]
- Feldman, R.; Gatalica, Z.; Knezetic, J.; Reddy, S.; Nathan, C.A.; Javadi, N.; Teknos, T. Molecular profiling of head and neck squamous cell carcinoma. Head Neck 2016, 38 (Suppl. S1), E1625–E1638. [Google Scholar] [CrossRef]
- Bugreev, D.V.; Mazin, A.V. Ca2+ activates human homologous recombination protein Rad51 by modulating its ATPase activity. Proc. Natl. Acad. Sci. USA 2004, 101, 9988–9993. [Google Scholar] [CrossRef]
- Brouwer, I.; Moschetti, T.; Candelli, A.; Garcin, E.B.; Modesti, M.; Pellegrini, L.; Wuite, G.J.; Peterman, E.J. Two distinct conformational states define the interaction of human RAD51-ATP with single-stranded DNA. EMBO J. 2018, 37, e98162. [Google Scholar] [CrossRef]
- Choi, J.; Kong, M.; Gallagher, D.N.; Li, K.; Bronk, G.; Cao, Y.; Greene, E.C.; Haber, J.E. Repair of mismatched templates during Rad51-dependent Break-Induced Replication. PLoS Genet. 2022, 18, e1010056. [Google Scholar] [CrossRef]
- Forget, A.L.; Bennett, B.T.; Knight, K.L. Xrcc3 is recruited to DNA double strand breaks early and independent of Rad51. J. Cell Biochem. 2004, 93, 429–436. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Zhang, X.; Zhang, Z.T. Genetic Polymorphisms in the RAD51 Gene with a Risk of Head and Neck Cancer and Esophageal Cancer: A Meta-Analysis. Int. J. Genom. 2019, 2019, 2789035. [Google Scholar] [CrossRef] [PubMed]
- Sliwinski, T.; Walczak, A.; Przybylowska, K.; Rusin, P.; Pietruszewska, W.; Zielinska-Blizniewska, H.; Olszewski, J.; Morawiec-Sztandera, A.; Jendrzejczyk, S.; Mlynarski, W.; et al. Polymorphisms of the XRCC3 C722T and the RAD51 G135C genes and the risk of head and neck cancer in a Polish population. Exp. Mol. Pathol. 2010, 89, 358–366. [Google Scholar] [CrossRef] [PubMed]
- Gresner, P.; Gromadzinska, J.; Polanska, K.; Twardowska, E.; Jurewicz, J.; Wasowicz, W. Genetic variability of Xrcc3 and Rad51 modulates the risk of head and neck cancer. Gene 2012, 504, 166–174. [Google Scholar] [CrossRef]
- Kong, F.; Wu, J.; Hu, L.; Du, Y.; Pan, Y. Association between RAD51 polymorphisms and susceptibility of head and neck cancer: A meta-analysis. Int. J. Clin. Exp. Med. 2015, 8, 6412–6419. [Google Scholar]
- Li, Y.; Li, J.; Sun, J.; Liu, Y.; Liu, D.; Du, L.; Wang, B.; Liu, W. Expression of RAD51 and Its Clinical Impact in Oral Squamous Cell Carcinoma. Anal. Cell Pathol. 2020, 2020, 1827676. [Google Scholar] [CrossRef]
- Lindemann, A.; Patel, A.A.; Tang, L.; Tanaka, N.; Gleber-Netto, F.O.; Bartels, M.D.; Wang, L.; McGrail, D.J.; Lin, S.Y.; Frank, S.J.; et al. Combined Inhibition of Rad51 and Wee1 Enhances Cell Killing in HNSCC Through Induction of Apoptosis Associated With Excessive DNA Damage and Replication Stress. Mol. Cancer Ther. 2021, 20, 1257–1269. [Google Scholar] [CrossRef]
- Mohiuddin, I.S.; Kang, M.H. DNA-PK as an Emerging Therapeutic Target in Cancer. Front. Oncol. 2019, 9, 635. [Google Scholar] [CrossRef]
- Yue, X.; Bai, C.; Xie, D.; Ma, T.; Zhou, P.K. DNA-PKcs: A Multi-Faceted Player in DNA Damage Response. Front. Genet. 2020, 11, 607428. [Google Scholar] [CrossRef] [PubMed]
- Allen, C.; Halbrook, J.; Nickoloff, J.A. Interactive competition between homologous recombination and non-homologous end joining. Mol. Cancer Res. 2003, 1, 913–920. [Google Scholar] [PubMed]
- Goodwin, J.F.; Kothari, V.; Drake, J.M.; Zhao, S.; Dylgjeri, E.; Dean, J.L.; Schiewer, M.J.; McNair, C.; Jones, J.K.; Aytes, A.; et al. DNA-PKcs-Mediated Transcriptional Regulation Drives Prostate Cancer Progression and Metastasis. Cancer Cell 2015, 28, 97–113. [Google Scholar] [CrossRef]
- Yan, S.S.; Liu, L.; Liu, Z.G.; Zeng, M.S.; Song, L.B.; Xia, Y.F. Expression and clinical significance of DNA-PKcs in nasopharyngeal carcinoma. Ai Zheng 2008, 27, 979–983. [Google Scholar] [PubMed]
- Yang, J.; Xu, X.; Hao, Y.; Chen, J.; Lu, H.; Qin, J.; Peng, L.; Chen, B. Expression of DNA-PKcs and BRCA1 as prognostic indicators in nasopharyngeal carcinoma following intensity-modulated radiation therapy. Oncol. Lett. 2013, 5, 1199–1204. [Google Scholar] [CrossRef]
- Shintani, S.; Mihara, M.; Li, C.; Nakahara, Y.; Hino, S.; Nakashiro, K.; Hamakawa, H. Up-regulation of DNA-dependent protein kinase correlates with radiation resistance in oral squamous cell carcinoma. Cancer Sci. 2003, 94, 894–900. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, Y. Development and Evolution of DNA-Dependent Protein Kinase Inhibitors toward Cancer Therapy. Int. J. Mol. Sci. 2022, 23, 4264. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.W.; Kim, S.Y.; Yi, S.L.; Son, S.H.; Song, D.Y.; Moon, S.Y.; Kim, J.H.; Choi, E.K.; Ahn, S.D.; Shin, S.S.; et al. Expression of Ku80 correlates with sensitivities to radiation in cancer cell lines of the head and neck. Oral Oncol. 2006, 42, 979–986. [Google Scholar] [CrossRef] [PubMed]
- Zahid, S.; Seif El Dahan, M.; Iehl, F.; Fernandez-Varela, P.; Le Du, M.H.; Ropars, V.; Charbonnier, J.B. The Multifaceted Roles of Ku70/80. Int. J. Mol. Sci. 2021, 22, 4134. [Google Scholar] [CrossRef]
- Wang, S.; Wang, Z.; Yang, Y.U.; Shi, M.O.; Sun, Z. Overexpression of Ku80 correlates with aggressive clinicopathological features and adverse prognosis in esophageal squamous cell carcinoma. Oncol. Lett. 2015, 10, 2705–2712. [Google Scholar] [CrossRef]
- van de Ven, S.; Bugter, O.; Hardillo, J.A.; Bruno, M.J.; Baatenburg de Jong, R.J.; Koch, A.D. Screening for head and neck second primary tumors in patients with esophageal squamous cell cancer: A systematic review and meta-analysis. United Eur. Gastroenterol. J. 2019, 7, 1304–1311. [Google Scholar] [CrossRef]
- Chang, H.W.; Nam, H.Y.; Kim, H.J.; Moon, S.Y.; Kim, M.R.; Lee, M.; Kim, G.C.; Kim, S.W.; Kim, S.Y. Effect of beta-catenin silencing in overcoming radioresistance of head and neck cancer cells by antagonizing the effects of AMPK on Ku70/Ku80. Head Neck 2016, 38 (Suppl. S1), E1909–E1917. [Google Scholar] [CrossRef] [PubMed]
- Kumazawa, T.; Mori, Y.; Sato, H.; Permata, T.B.M.; Uchihara, Y.; Noda, S.E.; Okada, K.; Kakoti, S.; Suzuki, K.; Ikota, H.; et al. Expression of non-homologous end joining factor, Ku80, is negatively correlated with PD-L1 expression in cancer cells after X-ray irradiation. Oncol. Lett. 2022, 23, 29. [Google Scholar] [CrossRef] [PubMed]
- Yang, G.; Liu, C.; Chen, S.H.; Kassab, M.A.; Hoff, J.D.; Walter, N.G.; Yu, X. Super-resolution imaging identifies PARP1 and the Ku complex acting as DNA double-strand break sensors. Nucleic Acids Res. 2018, 46, 3446–3457. [Google Scholar] [CrossRef]
- Lajud, S.A.; Nagda, D.A.; Yamashita, T.; Zheng, J.; Tanaka, N.; Abuzeid, W.M.; Civantos, A.; Bezpalko, O.; O’Malley, B.W., Jr.; Li, D. Dual disruption of DNA repair and telomere maintenance for the treatment of head and neck cancer. Clin. Cancer Res. 2014, 20, 6465–6478. [Google Scholar] [CrossRef]
- The Cancer Genome Atlas Research Network; Weinstein, J.N.; Collisson, E.A.; Mills, G.B.; Mills Shaw, K.R.; Ozenberger, B.A.; Ellrott, K.; Shmulevich, I.; Sander, C.; Stuart, J.M. The Cancer Genome Atlas Pan-Cancer analysis project. Nat. Genet. 2013, 45, 1113–1120. [Google Scholar] [CrossRef]
- Wu, J.; Crowe, D.L. PARP5B is required for nonhomologous end joining during tumorigenesis in vivo. Mol. Carcinog. 2022, 61, 85–98. [Google Scholar] [CrossRef]
- Bian, L.; Meng, Y.; Zhang, M.; Li, D. MRE11-RAD50-NBS1 complex alterations and DNA damage response: Implications for cancer treatment. Mol. Cancer 2019, 18, 169. [Google Scholar] [CrossRef]
- Li, S.; Wang, L.; Wang, Y.; Zhang, C.; Hong, Z.; Han, Z. The synthetic lethality of targeting cell cycle checkpoints and PARPs in cancer treatment. J. Hematol. Oncol. 2022, 15, 147. [Google Scholar] [CrossRef]
- Liu, Q.; Lopez, K.; Murnane, J.; Humphrey, T.; Barcellos-Hoff, M.H. Misrepair in Context: TGFbeta Regulation of DNA Repair. Front. Oncol. 2019, 9, 799. [Google Scholar] [CrossRef]
- Lord, C.J.; Ashworth, A. BRCAness revisited. Nat. Rev. Cancer 2016, 16, 110–120. [Google Scholar] [CrossRef] [PubMed]
- Hintelmann, K.; Berenz, T.; Kriegs, M.; Christiansen, S.; Gatzemeier, F.; Struve, N.; Petersen, C.; Betz, C.; Rothkamm, K.; Oetting, A.; et al. Dual Inhibition of PARP and the Intra-S/G2 Cell Cycle Checkpoints Results in Highly Effective Radiosensitization of HPV(−)Positive HNSCC Cells. Front. Oncol. 2021, 11, 683688. [Google Scholar] [CrossRef] [PubMed]
- Glorieux, M.; Dok, R.; Nuyts, S. Novel DNA targeted therapies for head and neck cancers: Clinical potential and biomarkers. Oncotarget 2017, 8, 81662–81678. [Google Scholar] [CrossRef] [PubMed]
- Ren, P.; Niu, X.; Liu, C.; Liu, J.; Li, H.; Zhao, Q.; Xing, J.; Bai, Y.; Liang, Y.; Han, P. Associations between expression levels of nine core nucleotide excision repair genes in lymphocytes and risk of head and neck squamous cell carcinomas in a Chinese population. Int. J. Clin. Oncol. 2020, 25, 660–669. [Google Scholar] [CrossRef]
- Han, P.; Liu, H.; Shi, Q.; Liu, Z.; Troy, J.D.; Lee, W.T.; Zevallos, J.P.; Li, G.; Sturgis, E.M.; Wei, Q. Associations between expression levels of nucleotide excision repair proteins in lymphoblastoid cells and risk of squamous cell carcinoma of the head and neck. Mol. Carcinog. 2018, 57, 784–793. [Google Scholar] [CrossRef]
- Prochnow, S.; Wilczak, W.; Bosch, V.; Clauditz, T.S.; Muenscher, A. ERCC1, XPF and XPA-locoregional differences and prognostic value of DNA repair protein expression in patients with head and neck squamous cell carcinoma. Clin. Oral Investig. 2019, 23, 3319–3329. [Google Scholar] [CrossRef] [PubMed]
- Lei, H.; He, A.; Jiang, Y.; Ruan, M.; Han, N. Targeting DNA damage response as a potential therapeutic strategy for head and neck squamous cell carcinoma. Front. Oncol. 2022, 12, 1031944. [Google Scholar] [CrossRef]
- Hasegawa, Y.; Goto, M.; Hanai, N.; Ozawa, T.; Hirakawa, H. Predictive biomarkers for combined chemotherapy with 5-fluorouracil and cisplatin in oro- and hypopharyngeal cancers. Mol. Clin. Oncol. 2018, 8, 378–386. [Google Scholar] [CrossRef]
- Santana, T.; Sa, M.C.; de Moura Santos, E.; Galvao, H.C.; Coletta, R.D.; Freitas, R.A. DNA base excision repair proteins APE-1 and XRCC-1 are overexpressed in oral tongue squamous cell carcinoma. J. Oral Pathol. Med. 2017, 46, 496–503. [Google Scholar] [CrossRef]
- Ming, R.; Wang, E.; Wei, J.; Shen, J.; Zong, S.; Xiao, H. The Prognostic Value of the DNA Repair Gene Signature in Head and Neck Squamous Cell Carcinoma. Front. Oncol. 2021, 11, 710694. [Google Scholar] [CrossRef]
- Tuteja, N.; Singh, M.B.; Misra, M.K.; Bhalla, P.L.; Tuteja, R. Molecular mechanisms of DNA damage and repair: Progress in plants. Crit. Rev. Biochem. Mol. Biol. 2001, 36, 337–397. [Google Scholar] [CrossRef]
- Park, N.H.; Min, B.M.; Li, S.L.; Huang, M.Z.; Cherick, H.M.; Doniger, J. Immortalization of normal human oral keratinocytes with type 16 human papillomavirus. Carcinogenesis 1991, 12, 1627–1631. [Google Scholar] [CrossRef] [PubMed]
- zur Hausen, H. Papillomaviruses and cancer: From basic studies to clinical application. Nat. Rev. Cancer 2002, 2, 342–350. [Google Scholar] [CrossRef] [PubMed]
- Smeets, S.J.; van der Plas, M.; Schaaij-Visser, T.B.; van Veen, E.A.; van Meerloo, J.; Braakhuis, B.J.; Steenbergen, R.D.; Brakenhoff, R.H. Immortalization of oral keratinocytes by functional inactivation of the p53 and pRb pathways. Int. J. Cancer 2011, 128, 1596–1605. [Google Scholar] [CrossRef] [PubMed]
- Park, J.W.; Nickel, K.P.; Torres, A.D.; Lee, D.; Lambert, P.F.; Kimple, R.J. Human papillomavirus type 16 E7 oncoprotein causes a delay in repair of DNA damage. Radiother. Oncol. 2014, 113, 337–344. [Google Scholar] [CrossRef]
- Liu, Q.; Ma, L.; Jones, T.; Palomero, L.; Pujana, M.A.; Martinez-Ruiz, H.; Ha, P.K.; Murnane, J.; Cuartas, I.; Seoane, J.; et al. Subjugation of TGFbeta Signaling by Human Papilloma Virus in Head and Neck Squamous Cell Carcinoma Shifts DNA Repair from Homologous Recombination to Alternative End Joining. Clin. Cancer Res. 2018, 24, 6001–6014. [Google Scholar] [CrossRef] [PubMed]
- Riess, C.; Irmscher, N.; Salewski, I.; Strüder, D.; Classen, C.-F.; Große-Thie, C.; Junghanss, C.; Maletzki, C. Cyclin-dependent kinase inhibitors in head and neck cancer and glioblastoma-backbone or add-on in immune-oncology? Cancer Metastasis Rev. 2021, 40, 153–171. [Google Scholar] [CrossRef]
- Golusinski, P.; Pazdrowski, J.; Szewczyk, M.; Misiolek, M.; Pietruszewska, W.; Klatka, J.; Okla, S.; Kazmierczak, H.; Marszalek, A.; Filas, V.; et al. Is immunohistochemical evaluation of p16 in oropharyngeal cancer enough to predict the HPV positivity? Rep. Pract. Oncol. Radiother. 2017, 22, 237–242. [Google Scholar] [CrossRef]
- Amin, M.B.; Greene, F.L.; Edge, S.B.; Compton, C.C.; Gershenwald, J.E.; Brookland, R.K.; Meyer, L.; Gress, D.M.; Byrd, D.R.; Winchester, D.P. The Eighth Edition AJCC Cancer Staging Manual: Continuing to build a bridge from a population-based to a more “personalized” approach to cancer staging. CA Cancer J. Clin. 2017, 67, 93–99. [Google Scholar] [CrossRef]
- Hammarstedt, L.; Holzhauser, S.; Zupancic, M.; Kapoulitsa, F.; Ursu, R.G.; Ramqvist, T.; Haeggblom, L.; Nasman, A.; Dalianis, T.; Marklund, L. The value of p16 and HPV DNA in non-tonsillar, non-base of tongue oropharyngeal cancer. Acta Otolaryngol. 2021, 141, 89–94. [Google Scholar] [CrossRef]
- Ramshankar, V.; Soundara, V.T.; Shyamsundar, V.; Ramani, P.; Krishnamurthy, A. Risk stratification of early stage oral tongue cancers based on HPV status and p16 immunoexpression. Asian Pac. J. Cancer Prev. 2014, 15, 8351–8359. [Google Scholar] [CrossRef]
- Alsharif, U.; Hofmann, M.; Gebhard, M.; Tharun, L.; Rades, D.; Sieg, P.; Hakim, S.G. Double Positivity for HPV DNA/P16(INK4a) Does Not Influence Survival of Patients With Oral Squamous Cell Carcinoma. Anticancer Res. 2021, 41, 5557–5568. [Google Scholar] [CrossRef]
- Ashworth, A. A synthetic lethal therapeutic approach: Poly(ADP) ribose polymerase inhibitors for the treatment of cancers deficient in DNA double-strand break repair. J. Clin. Oncol. 2008, 26, 3785–3790. [Google Scholar] [CrossRef]
- Forster, M.; Mendes, R.; Fedele, S. Synthetic lethality and PARP-inhibitors in oral and head & neck cancer. Curr. Pharm. Des. 2012, 18, 5431–5441. [Google Scholar] [CrossRef]
- Bhide, S.A.; Nutting, C.M. Advances in chemotherapy for head and neck cancer. Oral Oncol. 2010, 46, 436–438. [Google Scholar] [CrossRef]
- Specenier, P.M.; Vermorken, J.B. Current concepts for the management of head and neck cancer: Chemotherapy. Oral Oncol. 2009, 45, 409–415. [Google Scholar] [CrossRef]
- Ang, K.K.; Harris, J.; Wheeler, R.; Weber, R.; Rosenthal, D.I.; Nguyen-Tan, P.F.; Westra, W.H.; Chung, C.H.; Jordan, R.C.; Lu, C.; et al. Human papillomavirus and survival of patients with oropharyngeal cancer. N. Engl. J. Med. 2010, 363, 24–35. [Google Scholar] [CrossRef]
- Pascal, J.M. The comings and goings of PARP-1 in response to DNA damage. DNA Repair 2018, 71, 177–182. [Google Scholar] [CrossRef]
- Brody, L.C. Treating cancer by targeting a weakness. N. Engl. J. Med. 2005, 353, 949–950. [Google Scholar] [CrossRef]
- Couto, C.A.; Wang, H.Y.; Green, J.C.; Kiely, R.; Siddaway, R.; Borer, C.; Pears, C.J.; Lakin, N.D. PARP regulates nonhomologous end joining through retention of Ku at double-strand breaks. J. Cell Biol. 2011, 194, 367–375. [Google Scholar] [CrossRef]
- Caron, M.C.; Sharma, A.K.; O’Sullivan, J.; Myler, L.R.; Ferreira, M.T.; Rodrigue, A.; Coulombe, Y.; Ethier, C.; Gagne, J.P.; Langelier, M.F.; et al. Poly(ADP-ribose) polymerase-1 antagonizes DNA resection at double-strand breaks. Nat. Commun. 2019, 10, 2954. [Google Scholar] [CrossRef] [PubMed]
- Patel, A.G.; Sarkaria, J.N.; Kaufmann, S.H. Nonhomologous end joining drives poly(ADP-ribose) polymerase (PARP) inhibitor lethality in homologous recombination-deficient cells. Proc. Natl. Acad. Sci. USA 2011, 108, 3406–3411. [Google Scholar] [CrossRef] [Green Version]
- Gottipati, P.; Vischioni, B.; Schultz, N.; Solomons, J.; Bryant, H.E.; Djureinovic, T.; Issaeva, N.; Sleeth, K.; Sharma, R.A.; Helleday, T. Poly(ADP-ribose) polymerase is hyperactivated in homologous recombination-defective cells. Cancer Res. 2010, 70, 5389–5398. [Google Scholar] [CrossRef] [PubMed]
- Weaver, A.N.; Cooper, T.S.; Rodriguez, M.; Trummell, H.Q.; Bonner, J.A.; Rosenthal, E.L.; Yang, E.S. DNA double strand break repair defect and sensitivity to poly ADP-ribose polymerase (PARP) inhibition in human papillomavirus 16-positive head and neck squamous cell carcinoma. Oncotarget 2015, 6, 26995–27007. [Google Scholar] [CrossRef]
- Jelinek, M.J.; Foster, N.R.; Zoroufy, A.J.; Schwartz, G.K.; Munster, P.N.; Seiwert, T.Y.; de Souza, J.A.; Vokes, E.E. A phase I trial adding poly(ADP-ribose) polymerase inhibitor veliparib to induction carboplatin-paclitaxel in patients with head and neck squamous cell carcinoma: Alliance A091101. Oral Oncol. 2021, 114, 105171. [Google Scholar] [CrossRef]
- Hernandez, A.L.; Young, C.D.; Bian, L.; Weigel, K.; Nolan, K.; Frederick, B.; Han, G.; He, G.; Devon Trahan, G.; Rudolph, M.C.; et al. PARP Inhibition Enhances Radiotherapy of SMAD4-Deficient Human Head and Neck Squamous Cell Carcinomas in Experimental Models. Clin. Cancer Res. 2020, 26, 3058–3070. [Google Scholar] [CrossRef]
- Carter, R.J.; Nickson, C.M.; Thompson, J.M.; Kacperek, A.; Hill, M.A.; Parsons, J.L. Characterisation of Deubiquitylating Enzymes in the Cellular Response to High-LET Ionizing Radiation and Complex DNA Damage. Int. J. Radiat. Oncol. Biol. Phys. 2019, 104, 656–665. [Google Scholar] [CrossRef]
- Wang, L.; Cao, J.; Wang, X.; Lin, E.; Wang, Z.; Li, Y.; Li, Y.; Chen, M.; Wang, X.; Jiang, B.; et al. Proton and photon radiosensitization effects of niraparib, a PARP-1/-2 inhibitor, on human head and neck cancer cells. Head Neck 2020, 42, 2244–2256. [Google Scholar] [CrossRef]
- Molkentine, J.M.; Molkentine, D.P.; Bridges, K.A.; Xie, T.; Yang, L.; Sheth, A.; Heffernan, T.P.; Clump, D.A.; Faust, A.Z.; Ferris, R.L.; et al. Targeting DNA damage response in head and neck cancers through abrogation of cell cycle checkpoints. Int. J. Radiat. Biol. 2021, 97, 1121–1128. [Google Scholar] [CrossRef]
- Vidhya Karivedu, M.K.R.; Mathews, M.; Kurtzweil, N.; Monroe, I.; Romano, A.; Wise-Draper, T.M. A phase II study evaluating the efficacy of niraparib and dostarlimab (TSR-042) in recurrent/metastatic head and neck squamous cell carcinoma. J. Clin. Oncol. 2022, 40, TPS6105. [Google Scholar] [CrossRef]
- Liu, J.; Chen, Z.; Li, Y.; Zhao, W.; Wu, J.; Zhang, Z. PD-1/PD-L1 Checkpoint Inhibitors in Tumor Immunotherapy. Front. Pharmacol. 2021, 12, 731798. [Google Scholar] [CrossRef] [PubMed]
- Botticelli, A.; Cirillo, A.; Strigari, L.; Valentini, F.; Cerbelli, B.; Scagnoli, S.; Cerbelli, E.; Zizzari, I.G.; Rocca, C.D.; D’Amati, G.; et al. Anti-PD-1 and Anti-PD-L1 in Head and Neck Cancer: A Network Meta-Analysis. Front. Immunol. 2021, 12, 705096. [Google Scholar] [CrossRef] [PubMed]
- Sigismund, S.; Avanzato, D.; Lanzetti, L. Emerging functions of the EGFR in cancer. Mol. Oncol. 2018, 12, 3–20. [Google Scholar] [CrossRef] [PubMed]
- Byeon, H.K.; Ku, M.; Yang, J. Beyond EGFR inhibition: Multilateral combat strategies to stop the progression of head and neck cancer. Exp. Mol. Med. 2019, 51, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Volman, Y.; Hefetz, R.; Galun, E.; Rachmilewitz, J. DNA damage alters EGFR signaling and reprograms cellular response via Mre-11. Sci. Rep. 2022, 12, 5760. [Google Scholar] [CrossRef]
- Siano, M.; Molinari, F.; Martin, V.; Mach, N.; Fruh, M.; Freguia, S.; Corradino, I.; Ghielmini, M.; Frattini, M.; Espeli, V. Multicenter Phase II Study of Panitumumab in Platinum Pretreated, Advanced Head and Neck Squamous Cell Cancer. Oncologist 2017, 22, 782-e70. [Google Scholar] [CrossRef]
- Rehmani, H.S.; Issaeva, N. EGFR in head and neck squamous cell carcinoma: Exploring possibilities of novel drug combinations. Ann. Transl. Med. 2020, 8, 813. [Google Scholar] [CrossRef]
- Frederick, B.A.; Gupta, R.; Atilano-Roque, A.; Su, T.T.; Raben, D. Combined EGFR1 and PARP1 Inhibition Enhances the Effect of Radiation in Head and Neck Squamous Cell Carcinoma Models. Radiat. Res. 2020, 194, 519–531. [Google Scholar] [CrossRef]
- Karam, S.D.; Reddy, K.; Blatchford, P.J.; Waxweiler, T.; DeLouize, A.M.; Oweida, A.; Somerset, H.; Marshall, C.; Young, C.; Davies, K.D.; et al. Final Report of a Phase I Trial of Olaparib with Cetuximab and Radiation for Heavy Smoker Patients with Locally Advanced Head and Neck Cancer. Clin. Cancer Res. 2018, 24, 4949–4959. [Google Scholar] [CrossRef]
- Abourehab, M.A.S.; Alqahtani, A.M.; Youssif, B.G.M.; Gouda, A.M. Globally Approved EGFR Inhibitors: Insights into Their Syntheses, Target Kinases, Biological Activities, Receptor Interactions, and Metabolism. Molecules 2021, 26, 6677. [Google Scholar] [CrossRef]
- Mehta, V.K. Radiotherapy and erlotinib combined: Review of the preclinical and clinical evidence. Front. Oncol. 2012, 2, 31. [Google Scholar] [CrossRef] [PubMed]
- Kao, H.F.; Liao, B.C.; Huang, Y.L.; Huang, H.C.; Chen, C.N.; Chen, T.C.; Hong, Y.J.; Chan, C.Y.; Chia, J.S.; Hong, R.L. Afatinib and Pembrolizumab for Recurrent or Metastatic Head and Neck Squamous Cell Carcinoma (ALPHA Study): A Phase II Study with Biomarker Analysis. Clin. Cancer Res. 2022, 28, 1560–1571. [Google Scholar] [CrossRef]
- Wilson, G.D.; Wilson, T.G.; Hanna, A.; Dabjan, M.; Buelow, K.; Torma, J.; Marples, B.; Galoforo, S. Dacomitinib and gedatolisib in combination with fractionated radiation in head and neck cancer. Clin. Transl. Radiat. Oncol. 2021, 26, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Diab, A.; Kao, M.; Kehrli, K.; Kim, H.Y.; Sidorova, J.; Mendez, E. Multiple Defects Sensitize p53-Deficient Head and Neck Cancer Cells to the WEE1 Kinase Inhibition. Mol. Cancer Res. 2019, 17, 1115–1128. [Google Scholar] [CrossRef] [PubMed]
- Mendez, E.; Rodriguez, C.P.; Kao, M.C.; Raju, S.; Diab, A.; Harbison, R.A.; Konnick, E.Q.; Mugundu, G.M.; Santana-Davila, R.; Martins, R.; et al. A Phase I Clinical Trial of AZD1775 in Combination with Neoadjuvant Weekly Docetaxel and Cisplatin before Definitive Therapy in Head and Neck Squamous Cell Carcinoma. Clin. Cancer Res. 2018, 24, 2740–2748. [Google Scholar] [CrossRef] [PubMed]
- Kellogg, D.R. Wee1-dependent mechanisms required for coordination of cell growth and cell division. J. Cell Sci. 2003, 116, 4883–4890. [Google Scholar] [CrossRef]
- Esposito, F.; Giuffrida, R.; Raciti, G.; Puglisi, C.; Forte, S. Wee1 Kinase: A Potential Target to Overcome Tumor Resistance to Therapy. Int. J. Mol. Sci. 2021, 22, 10689. [Google Scholar] [CrossRef]
- Duda, H.; Arter, M.; Gloggnitzer, J.; Teloni, F.; Wild, P.; Blanco, M.G.; Altmeyer, M.; Matos, J. A Mechanism for Controlled Breakage of Under-replicated Chromosomes during Mitosis. Dev. Cell 2016, 39, 740–755. [Google Scholar] [CrossRef]
- Zhang, M.; Zhang, L.; Hei, R.; Li, X.; Cai, H.; Wu, X.; Zheng, Q.; Cai, C. CDK inhibitors in cancer therapy, an overview of recent development. Am. J. Cancer Res. 2021, 11, 1913–1935. [Google Scholar]
- van Caloen, G.; Schmitz, S.; El Baroudi, M.; Caignet, X.; Pyr Dit Ruys, S.; Roger, P.P.; Vertommen, D.; Machiels, J.P. Preclinical Activity of Ribociclib in Squamous Cell Carcinoma of the Head and Neck. Mol. Cancer Ther. 2020, 19, 777–789. [Google Scholar] [CrossRef]
- Seront, E.; Schmitz, S.; Papier, M.; van Maanen, A.; Henry, S.; Lonchay, C.; Rottey, S.; van Caloen, G.; Machiels, J.P. Phase 1 Study Evaluating the Association of the Cyclin-Dependent Kinase 4/6 Inhibitor Ribociclib and Cetuximab in Recurrent/Metastatic p16-Negative Squamous Cell Carcinoma of the Head and Neck. Front. Oncol. 2019, 9, 155. [Google Scholar] [CrossRef] [PubMed]
- Adkins, D.; Ley, J.; Neupane, P.; Worden, F.; Sacco, A.G.; Palka, K.; Grilley-Olson, J.E.; Maggiore, R.; Salama, N.N.; Trinkaus, K.; et al. Palbociclib and cetuximab in platinum-resistant and in cetuximab-resistant human papillomavirus-unrelated head and neck cancer: A multicentre, multigroup, phase 2 trial. Lancet Oncol. 2019, 20, 1295–1305. [Google Scholar] [CrossRef] [PubMed]
- Zeng, L.; Boggs, D.H.; Xing, C.; Zhang, Z.; Anderson, J.C.; Wajapeyee, N.; Veale, C.; Bredel, M.; Shi, L.Z.; Bonner, J.A.; et al. Combining PARP and DNA-PK Inhibitors With Irradiation Inhibits HPV(−)Negative Head and Neck Cancer Squamous Carcinoma Growth. Front. Genet. 2020, 11, 1036. [Google Scholar] [CrossRef] [PubMed]
- Faulhaber, E.M.; Jost, T.; Symank, J.; Scheper, J.; Burkel, F.; Fietkau, R.; Hecht, M.; Distel, L.V. Kinase Inhibitors of DNA-PK, ATM and ATR in Combination with Ionizing Radiation Can Increase Tumor Cell Death in HNSCC Cells While Sparing Normal Tissue Cells. Genes 2021, 12, 925. [Google Scholar] [CrossRef] [PubMed]
- Karukonda, P.; Odhiambo, D.; Mowery, Y.M. Pharmacologic inhibition of ataxia telangiectasia and Rad3-related (ATR) in the treatment of head and neck squamous cell carcinoma. Mol. Carcinog. 2022, 61, 225–238. [Google Scholar] [CrossRef]
- Leonard, B.C.; Lee, E.D.; Bhola, N.E.; Li, H.; Sogaard, K.K.; Bakkenist, C.J.; Grandis, J.R.; Johnson, D.E. ATR inhibition sensitizes HPV(−) and HPV(+) head and neck squamous cell carcinoma to cisplatin. Oral Oncol. 2019, 95, 35–42. [Google Scholar] [CrossRef]
- Dok, R.; Glorieux, M.; Bamps, M.; Nuyts, S. Effect of ATR Inhibition in RT Response of HPV(−)Negative and HPV(−)Positive Head and Neck Cancers. Int. J. Mol. Sci. 2021, 22, 1504. [Google Scholar] [CrossRef]
- Odhiambo, D.; Bassil, A.M.; Castillo, R.; Kirsch, D.G.; Mowery, Y.M. Evaluating the Therapeutic Potential of a Small Molecule ATR Inhibitor Combined with Radiotherapy for Head and Neck Squamous Cell Carcinoma. Radiat. Oncol. Biol. Phys. 2022, 112, E45–E46. [Google Scholar] [CrossRef]
- Patin, E.C.; Dillon, M.T.; Nenclares, P.; Grove, L.; Soliman, H.; Leslie, I.; Northcote, D.; Bozhanova, G.; Crespo-Rodriguez, E.; Baldock, H.; et al. Harnessing radiotherapy-induced NK-cell activity by combining DNA damage-response inhibition and immune checkpoint blockade. J. Immunother. Cancer 2022, 10, e004306. [Google Scholar] [CrossRef]
- Mowery, Y.M.; Niedzwiecki, D.; Choe, J.H.; Kirsch, D.G.; Brizel, D.M. Phase I trial of the ATR inhibitor BAY 1895344 combined with stereotactic body radiation therapy and pembrolizumab for recurrent head and neck squamous cell carcinoma. J. Clin. Oncol. 2022, 40, TPS6108. [Google Scholar] [CrossRef]
- Broustas, C.G.; Lieberman, H.B. DNA damage response genes and the development of cancer metastasis. Radiat. Res. 2014, 181, 111–130. [Google Scholar] [CrossRef] [PubMed]
- Dohmen, A.J.C.; Qiao, X.; Duursma, A.; Wijdeven, R.H.; Lieftink, C.; Hageman, F.; Morris, B.; Halonen, P.; Vens, C.; van den Brekel, M.W.M.; et al. Identification of a novel ATM inhibitor with cancer cell specific radiosensitization activity. Oncotarget 2017, 8, 73925–73937. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
| Gene | Pathway | Abnormal Expression/Alteration | Clinical Effect | Reference |
|---|---|---|---|---|
| Ku70/80 | NHEJ | Overexpression | Radioresistance | [160] |
| PARP5B | NHEJ | Downregulation/null | Impairment of NHEJ | [169] |
| DNA-PKcs | NHEJ | Overexpression | Decreased survival in NPC patients/poor outcomes in NPC patients undergoing intensity-modulated radiotherapy—potential biomarker for prediction of the response to therapy | [156,157] |
| RAD51 | HR | Overexpression | Lymph node metastasis, poorly differentiated tissues, worse prognosis in OSCC | [150] |
| BRCA2 | HR | Deficiency | Sensitivity to the PARP inhibitors | [195] |
| BRCA1 | HR | Downregulation | Poor outcome | [71] |
| FANCF and FA-related genes | Fanconi anemia/HR | Silencing or downregulation | MMC-hypersensitivity, G2-blockade, and olaparib (PARP-inhibitor) hypersensitivity | [138] |
| ATM | DNA repair (HR, NHEJ), cell cycle arrest, apoptosis | Downregulation | Poor prognosis | [71] |
| ATR | DNA repair (HR, NHEJ), cell cycle progression, apoptosis | Mutation | Unknown | [120] |
| TP53 | p53 pathway HR NHEJ | Deficiency | Short survival time and tumor resistance to radiotherapy and chemotherapy in HNSCC patients | [82] |
| XPB | NER | Downregulation | Increased risk for HNSCCs | [176] |
| ERCC1 | NER | Upregulation | Enhanced response to chemotherapy | [179] |
| XPA and ERCC1 | NER | Upregulation | Poor OS in patients with OSCC/exhibited better OS in patients with oropharyngeal SCC | [178] |
| XRCC1 | BER | Upregulation | Better clinical staging of OTSCC and negative lymph node metastasis | [181] |
| MLH1, MSH2, MLH3 and PMS2 | MMR | Downregulation | Reduced DNA repair capacity in OSCCs | [179] |
| Inhibitors | Target | Combination with Chemo or Radiotherapy | Cancer Type | Clinical Trial | Clinical Trial Stage |
|---|---|---|---|---|---|
| Veliparib (ABT-888) | PARP | Carboplatin + Paclitaxel | Locoregionally advanced HNSCCs | NCT01366144 | Phase I |
| Olaparib | PARP | Radiation | SMAD4-negative HNSCC | NCT02229656 | Phase I |
| Niraparib + Dostarlimab | PARP + PD-1 | Recurrent/metastatic HNSCC | NCT04681469 | Phase I (recruiting) | |
| Niraparib + Dostarlimab | PARP + PD-1 | Recurrent/metastatic HNSCC | NCT04313504 | Phase II | |
| Olaparib + Cetuximab | PARP + EGFR | Radiation | Heavy smokers with locally advanced HNC | NCT01758731 | Phase I |
| Panitumumab | EGFR | Platinum | Advanced HNSCC | NCT02643056 | Phase II |
| Afatinib + Pembrolizumab | EGFR + PD-1 | Recurrent or metastatic HNSCC | NCT03695510 | Phase II | |
| AZD1775 | Wee1 | Cisplatin + Docetaxel | Locally advanced HNSCC | NCT02508246 | Phase I |
| MSC2490484A | DNA-PK | Radiation + Cisplatin | Locally advanced solid tumors | NCT02516813 | Phase I |
| CC-115 | Dual mTOR/DNA-PK inhibitor | Advanced solid tumors | NCT01353625 | Phase Ia/Ib | |
| BAY 1895344 + Pembrolizumab | ATR + PD-1 | Radiation | Recurrent HNC | NCT04576091 | Phase I |
| Inhibitor | Target | Concomittant Administration of Radio and/or Chemotherapy | Cancer Type |
|---|---|---|---|
| NU7441 + Olaparib | DNA-PK + PARP | Radiotherapy | HPV(−) HNSCC cell lines/mouse models |
| AZD1775 (Adavosertib) | Wee1 | - | TP53-mutated or HPV(+) tumors |
| CC-115 | Dual mTOR/ DNA-PK inhibitor | Radiotherapy | HPV(+) and HPV(−) HNSCC cell lines |
| AZD6738 | ATR | Radiotherapy + Cisplatin | HPV(+) and HPV(−) HNSCC cell lines |
| AZD0156 | ATM | Radiotherapy | HPV(+) and HPV(−) HNSCC cell lines |
| GSK635416A + Olaparib | ATM + Olaparib | Radiotherapy | HNSCC cell lines |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Papalouka, C.; Adamaki, M.; Batsaki, P.; Zoumpourlis, P.; Tsintarakis, A.; Goulielmaki, M.; Fortis, S.P.; Baxevanis, C.N.; Zoumpourlis, V. DNA Damage Response Mechanisms in Head and Neck Cancer: Significant Implications for Therapy and Survival. Int. J. Mol. Sci. 2023, 24, 2760. https://doi.org/10.3390/ijms24032760
Papalouka C, Adamaki M, Batsaki P, Zoumpourlis P, Tsintarakis A, Goulielmaki M, Fortis SP, Baxevanis CN, Zoumpourlis V. DNA Damage Response Mechanisms in Head and Neck Cancer: Significant Implications for Therapy and Survival. International Journal of Molecular Sciences. 2023; 24(3):2760. https://doi.org/10.3390/ijms24032760
Chicago/Turabian StylePapalouka, Chara, Maria Adamaki, Panagiota Batsaki, Panagiotis Zoumpourlis, Antonis Tsintarakis, Maria Goulielmaki, Sotirios P. Fortis, Constantin N. Baxevanis, and Vassilis Zoumpourlis. 2023. "DNA Damage Response Mechanisms in Head and Neck Cancer: Significant Implications for Therapy and Survival" International Journal of Molecular Sciences 24, no. 3: 2760. https://doi.org/10.3390/ijms24032760