
Structural Insight into the Working Mechanism of the FAD Synthetase from the Human Pathogen Streptococcus pneumoniae: A Molecular Docking Simulation Study
Abstract
:1. Introduction
2. Results and Discussion
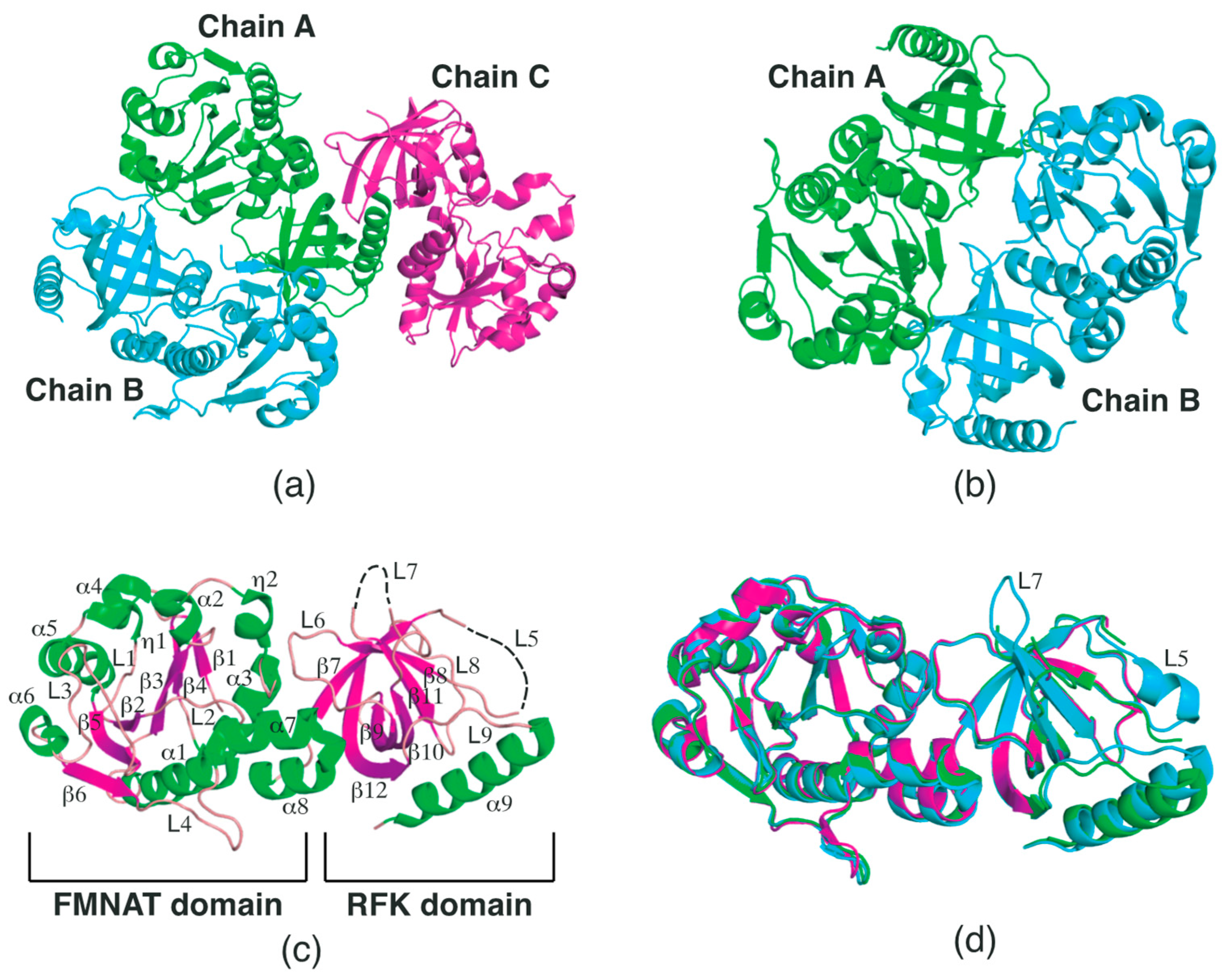
2.1. Structural Features of SpFADS
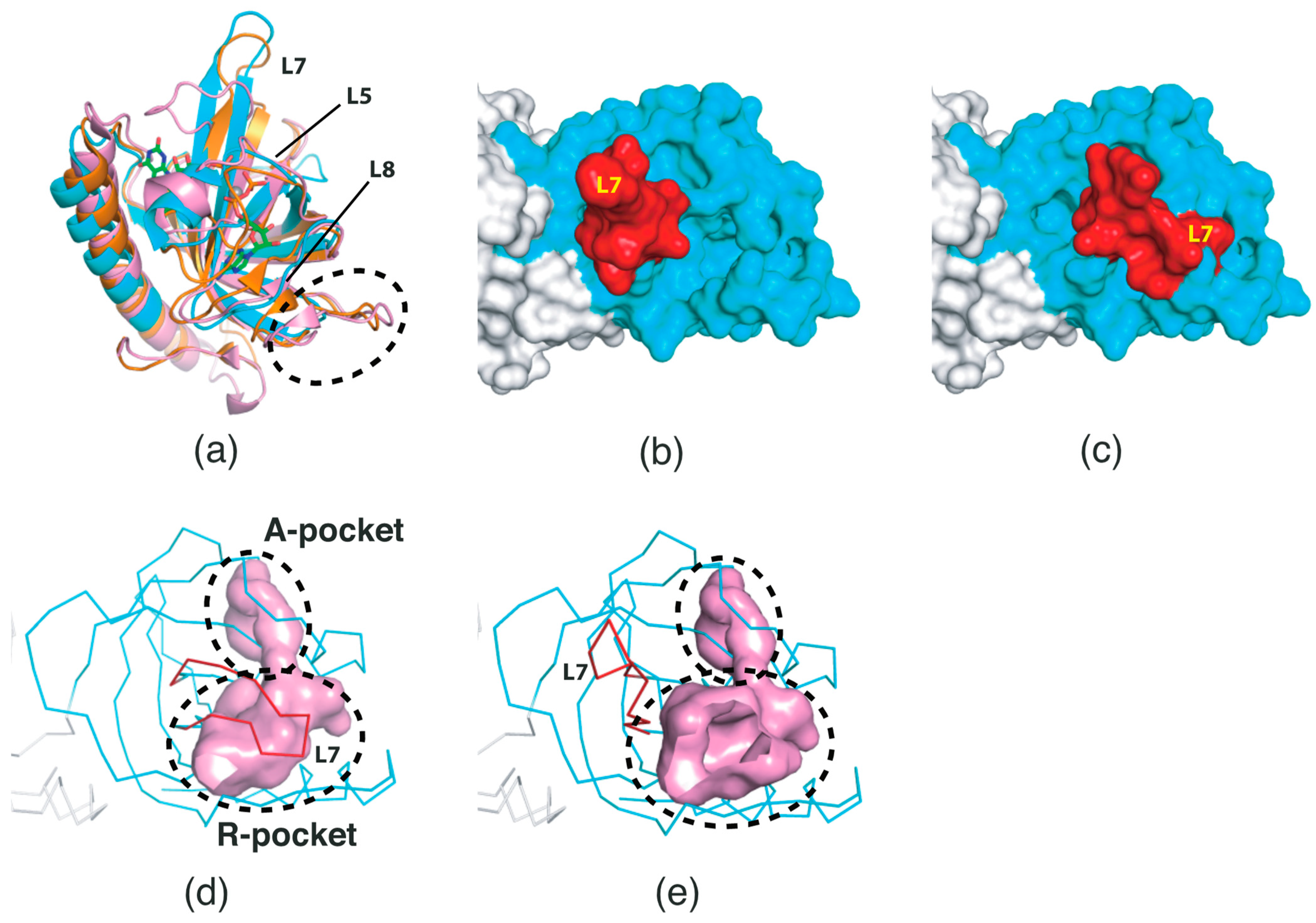
2.2. Conformational Changes in the RFK Domain
2.3. Surface Properties of SpFADS
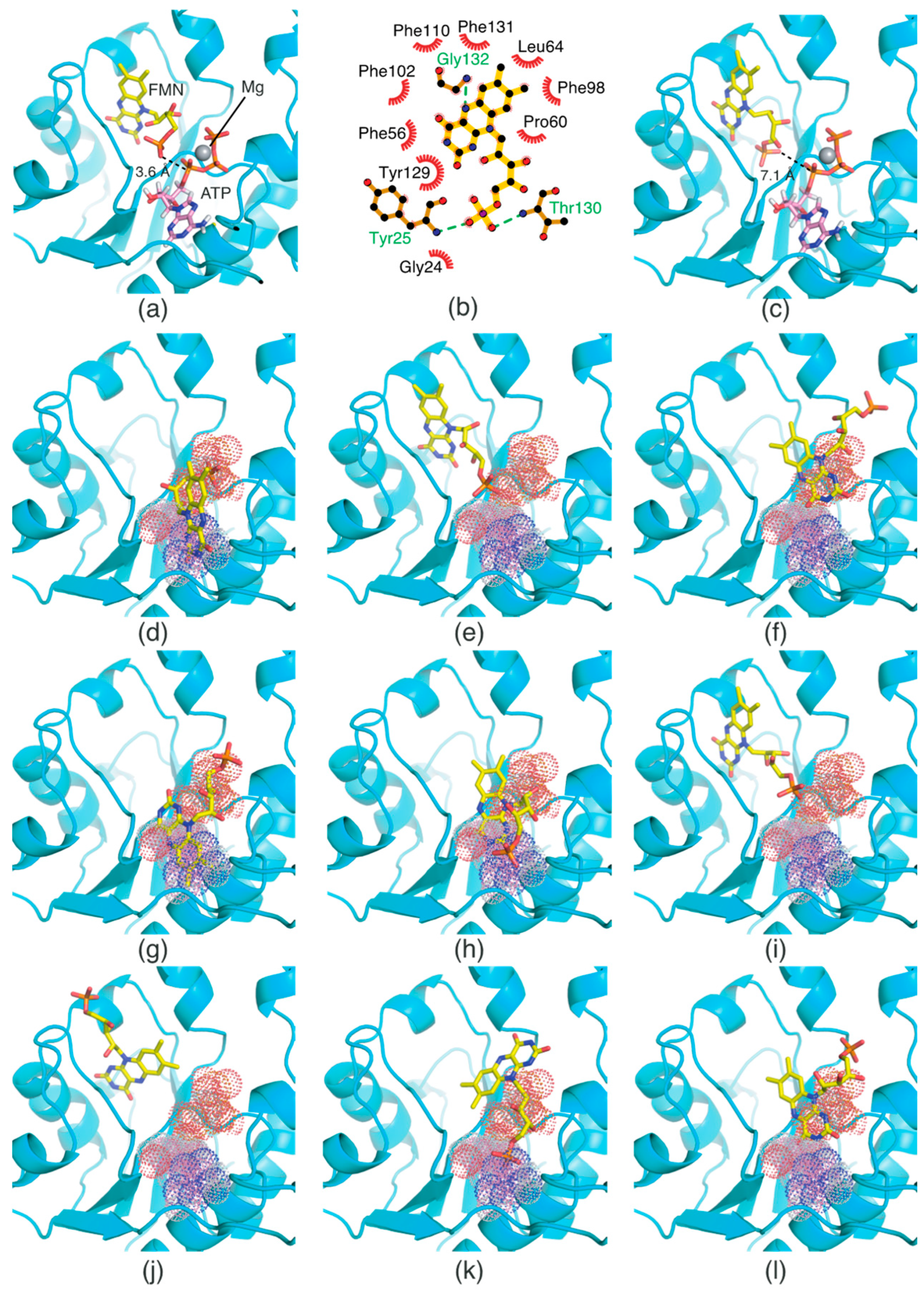
2.4. Oxidized and Reduced RF-Binding Modes in the RFK Domain
2.5. FMN-Binding Mode in the FMNAT Domain
2.6. Pharmacophore Model
3. Materials and Methods
3.1. Protein and Substrate Preparation
3.2. Molecular Docking Simulation
3.3. 3D-Pharmacophore Modeling
3.4. Structure Visualization
4. Conclusions and Future Perspectives
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mansoorabadi, S.O.; Thibodeaux, C.J.; Liu, H.W. The diverse roles of flavin coenzymes—nature’s most versatile thespians. J. Org. Chem. 2007, 72, 6329–6342. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Winge, D.R. Emerging concepts in the flavinylation of succinate dehydrogenase. Biochim. Biophys. Acta 2013, 1827, 627–636. [Google Scholar] [CrossRef] [PubMed]
- Garma, L.D.; Medina, M.; Juffer, A.H. Structure-based classification of FAD binding sites: A comparative study of structural alignment tools. Proteins 2016, 84, 1728–1747. [Google Scholar] [CrossRef] [PubMed]
- Manstein, D.J.; Pai, E.F. Purification and characterization of FAD synthetase from Brevibacterium ammoniagenes. J. Biol. Chem. 1986, 261, 16169–16173. [Google Scholar] [CrossRef]
- Efimov, I.; Kuusk, V.; Zhang, X.; McIntire, W.S. Proposed steady-state kinetic mechanism for Corynebacterium ammoniagenes FAD synthetase produced by Escherichia coli. J. Biol. Chem. 1998, 37, 9716–9723. [Google Scholar] [CrossRef] [PubMed]
- Barile, M.; Brizio, C.; Valenti, D.; De Virgilio, C.; Passarella, S. The riboflavin/FAD cycle in rat liver mitochondria. Eur. J. Biochem. 2000, 267, 4888–4900. [Google Scholar] [CrossRef] [PubMed]
- Sandoval, F.J.; Zhang, Y.; Roje, S. Flavin nucleotide metabolism in plants: Monofunctional enzymes synthesize fad in plastids. J. Biol. Chem. 2008, 283, 30890–30900. [Google Scholar] [CrossRef] [PubMed]
- Giancaspero, T.A.; Locato, V.; de Pinto, M.C.; De Gara, L.; Barile, M. The occurrence of riboflavin kinase and FAD synthetase ensures FAD synthesis in tobacco mitochondria and maintenance of cellular redox status. FEBS J. 2009, 276, 219–231. [Google Scholar] [CrossRef]
- Mashhadi, Z.; Xu, H.; Grochowski, L.L.; White, R.H. Archaeal RibL: A new FAD synthetase that is air sensitive. Biochemistry 2010, 49, 8748–8755. [Google Scholar] [CrossRef] [PubMed]
- Serrano, A.; Ferreira, P.; Martínez-Júlvez, M.; Medina, M. The prokaryotic FAD synthetase family: A potential drug target. Curr. Pharm. Des. 2013, 19, 2637–2648. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Kim, R.; Jancarik, J.; Yokota, H.; Kim, S.H. Crystal structure of a flavin-binding protein from Thermotoga maritima. Proteins 2003, 52, 633–635. [Google Scholar] [CrossRef]
- Wang, W.; Kim, R.; Yokota, H.; Kim, S.H. Crystal structure of flavin binding to FAD synthetase of Thermotoga maritima. Proteins 2005, 58, 246–248. [Google Scholar] [CrossRef]
- Herguedas, B.; Martínez-Júlvez, M.; Frago, S.; Medina, M.; Hermoso, J.A. Oligomeric state in the crystal structure of modular FAD synthetase provides insights into its sequential catalysis in prokaryotes. J. Mol. Biol. 2010, 400, 218–230. [Google Scholar] [CrossRef] [PubMed]
- Herguedas, B.; Lans, I.; Sebastián, M.; Hermoso, J.A.; Martínez-Júlvez, M.; Medina, M. Structural insights into the synthesis of FMN in prokaryotic organisms. Acta Crystallogr. D Biol. Crystallogr. 2015, 71, 2526–2542. [Google Scholar] [CrossRef] [PubMed]
- Marcuello, C.; Frempong, G.A.; Balsera, M.; Medina, M.; Lostao, A. Atomic Force Microscopy to Elicit Conformational Transitions of Ferredoxin-Dependent Flavin Thioredoxin Reductases. Antioxidants 2021, 10, 1437. [Google Scholar] [CrossRef]
- Villanueva, R.; Ferreira, P.; Marcuello, C.; Usón, A.; Miramar, M.D.; Peleato, M.L.; Lostao, A.; Susin, S.A.; Medina, M. Key Residues Regulating the Reductase Activity of the Human Mitochondrial Apoptosis Inducing Factor. Biochemistry. 2015, 54, 5175–5184. [Google Scholar] [CrossRef] [PubMed]
- Sebastián, M.; Lira-Navarrete, E.; Serrano, A.; Marcuello, C.; Velázquez-Campoy, A.; Lostao, A.; Hurtado-Guerrero, R.; Medina, M.; Martínez-Júlvez, M. The FAD synthetase from the human pathogen Streptococcus pneumoniae: A bifunctional enzyme exhibiting activity-dependent redox requirements. Sci. Rep. 2017, 7, 7609. [Google Scholar] [CrossRef]
- Krissinel, E.; Henrick, K. Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 2007, 372, 774–797. [Google Scholar] [CrossRef] [PubMed]
- Fogolari, F.; Brigo, A.; Molinari, H. The Poisson-Boltzmann equation for biomolecular electrostatics: A tool for structural biology. J. Mol. Recognit. 2002, 15, 377–392. [Google Scholar] [CrossRef] [PubMed]
- Teufel, R.; Miyanaga, A.; Michaudel, Q.; Stull, F.; Louie, G.; Noel, J.P.; Baran, P.S.; Palfey, B.; Moore, B.S. Flavin-mediated dual oxidation controls an enzymatic Favorskii-type rearrangement. Nature 2013, 503, 552–556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barile, M.; Giancaspero, T.A.; Brizio, C.; Panebianco, C.; Indiveri, C.; Galluccio, M.; Vergani, L.; Eberini, I.; Gianazza, E. Biosynthesis of flavin cofactors in man: Implications in health and disease. Curr. Pharm. Des. 2013, 19, 2649–2675. [Google Scholar] [CrossRef] [PubMed]
- Teufel, R.; Stull, F.; Meehan, M.J.; Michaudel, Q.; Dorrestein, P.C.; Palfey, B.; Moore, B.S. Biochemical Establishment and Characterization of EncM’s Flavin-N5-oxide Cofactor. J. Am. Chem. Soc. 2015, 137, 8078–8085. [Google Scholar] [CrossRef]
- Teufel, R.; Agarwal, V.; Moore, B.S. Unusual flavoenzyme catalysis in marine bacteria. Curr. Opin. Chem. Biol. 2016, 31, 31–39. [Google Scholar] [CrossRef]
- Sebastián, M.; Anoz-Carbonell, E.; Gracia, B.; Cossio, P.; Aínsa, J.A.; Lans, I.; Medina, M. Discovery of antimicrobial compounds targeting bacterial type FAD synthetases. J. Enzyme Inhib. Med. Chem. 2018, 33, 241–254. [Google Scholar] [CrossRef] [PubMed]
- Gasteiger, J.; Marsili, M. New model for calculating atomic charges in molecules. Tetrahedron Lett. 1978, 19, 3181–3184. [Google Scholar] [CrossRef]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [PubMed]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Eberhardt, J.; Santos-Martins, D.; Tillack, A.F.; Forli, S. AutoDock Vina 1.2.0: New Docking Methods, Expanded Force Field, and Python Bindings. J. Chem. Inf. Model. 2021, 61, 3891–3898. [Google Scholar] [CrossRef]
- Schneidman-Duhovny, D.; Dror, O.; Inbar, Y.; Nussinov, R.; Wolfson, H.J. PharmaGist: A webserver for ligand-based pharmacophore detection. Nucleic Acids Res. 2008, 36, W223–W228. [Google Scholar] [CrossRef] [PubMed]
- DeLano, W.L. The PyMOL Molecular Graphics System; DeLano Scientific: San Carlos, CA, USA, 2020; Available online: http://www.pymol.org (accessed on 26 November 2022).
- Laskowski, R.A.; Swindells, M.B. LigPlot+: Multiple ligand-protein interaction diagrams for drug discovery. J. Chem. Inf. Model. 2011, 51, 2778–2786. [Google Scholar] [CrossRef]
- Iamurri, S.M.; Daugherty, A.B.; Edmondson, D.E.; Lutz, S. Truncated FAD synthetase for direct biocatalytic conversion of riboflavin and analogs to their corresponding flavin mononucleotides. Protein Eng. Des. Sel. 2013, 26, 791–795. [Google Scholar] [CrossRef] [PubMed] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Rank | Binding Energy (kcal/mol) (Oxidized Form) | Binding Energy (kcal/mol) (Reduced Form) |
|---|---|---|
| 1 | −9.0 | −9.4 |
| 2 | −9.0 | −9.2 |
| 3 | −9.0 | −9.1 |
| 4 | −8.9 | −8.9 |
| 5 | −8.8 | −8.8 |
| 6 | −8.6 | −8.7 |
| 7 | −8.6 | −8.2 |
| 8 | −7.8 | −7.3 |
| 9 | −7.4 | −7.3 |
| 10 | −7.1 | −7.2 |
| Rank | Conformer | Binding Energy (kcal/mol) (with ATP) | Binding Energy (kcal/mol) (without ATP) |
|---|---|---|---|
| 1 | C1 | −9.1 | −8.9 |
| 2 | C2 | −9.1 | −8.8 |
| 3 | C3 | −9.0 | −8.8 |
| 4 | C4 | −8.8 | −8.7 |
| 5 | C5 | −8.8 | −8.6 |
| 6 | C6 | −8.7 | −8.5 |
| 7 | C7 | −8.6 | −8.1 |
| 8 | C8 | −8.4 | −8.1 |
| 9 | C9 | −8.3 | −8.1 |
| 10 | C10 | −8.1 | −8.1 |
| Compound | Chemical Structure |
|---|---|
| 1 |  |
| 2 |  |
| 3 |  |
| 4 |  |
| 5 |  |
| 6 |  |
| 7 |  |
| 8 |  |
| 9 |  |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kwon, S. Structural Insight into the Working Mechanism of the FAD Synthetase from the Human Pathogen Streptococcus pneumoniae: A Molecular Docking Simulation Study. Int. J. Mol. Sci. 2023, 24, 3121. https://doi.org/10.3390/ijms24043121
Kwon S. Structural Insight into the Working Mechanism of the FAD Synthetase from the Human Pathogen Streptococcus pneumoniae: A Molecular Docking Simulation Study. International Journal of Molecular Sciences. 2023; 24(4):3121. https://doi.org/10.3390/ijms24043121
Chicago/Turabian StyleKwon, Sunghark. 2023. "Structural Insight into the Working Mechanism of the FAD Synthetase from the Human Pathogen Streptococcus pneumoniae: A Molecular Docking Simulation Study" International Journal of Molecular Sciences 24, no. 4: 3121. https://doi.org/10.3390/ijms24043121