
Emerging Targeted Therapeutic Strategies to Overcome Imatinib Resistance of Gastrointestinal Stromal Tumors
 , , ,
, , ,
Abstract
:1. Introduction
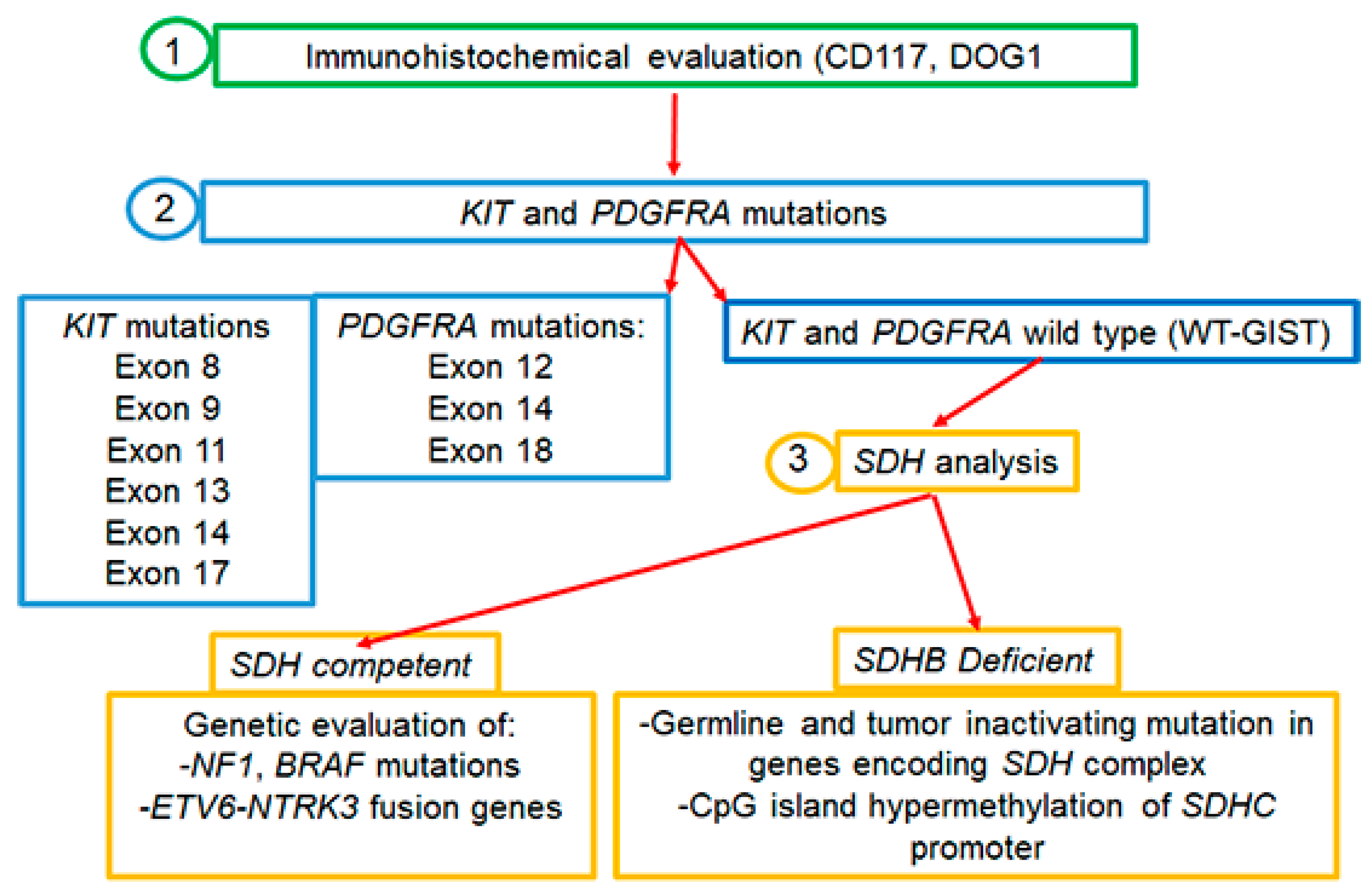
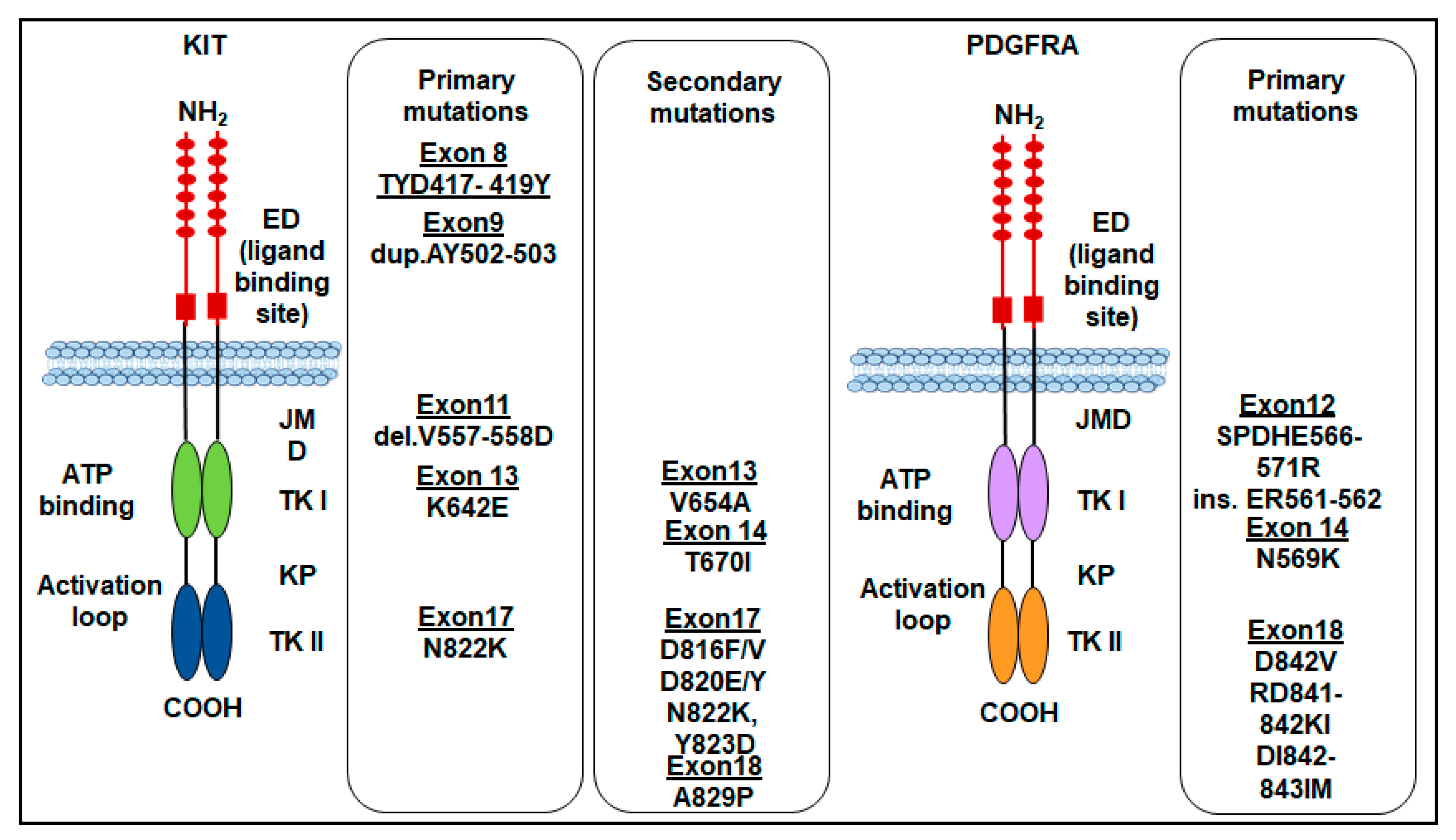
2. KIT Mutations in GISTs
3. PDGFRA Mutations in GISTs
4. Other Mutations in GISTs
4.1. SDH Mutations
4.2. BRAF Mutations
4.3. NF1 Mutations
4.4. NTRK Mutations
5. Imatinib for the First-Line Therapy of GISTs
6. Sunitinib for Second-Line Therapy of Resistant GISTs
7. Third-Line Therapy for Resistant GISTs
7.1. Regorafenib
7.2. Nilotinib
7.3. Pazopanib
7.4. Sorafenib
7.5. Dasatinib
8. Fourth-line Therapy for Resistant GISTs
Ripretinib
9. New TKIs Targeting GIST Mutations
9.1. Bezuclastinib
9.2. Cabozantinib
9.3. Avapritinib

{kind=link}
{kind=link}
| Line of Therapy | Drug | Study | ID | Phase | Endpoints | Findings | Refs |
|---|---|---|---|---|---|---|---|
| 2nd | Sunitinib | A study of SU011248 administered on a continuous daily dosing schedule in patients with a gastrointestinal stromal tumor | NCT00137449 | II | Safety, pharmacokinetic, and plasma biomarker levels, PR, SD (>24 weeks), PFS, and OS | Efficacy and safety of daily oral administration of 37.5 mg sunitinib for 28 days | [114] |
| A study to assess the safety and efficacy of SU11248 in patients with gastrointestinal stromal tumor (GIST) | NCT00075218 | III | TTP, PFS, OS, OR, CR, and PR | Good tolerability, longer PD (27.3 weeks) compared to placebo | [116] | ||
| 3rd | Regorafenib | A phase II trial of regorafenib in patients with imatinib-resistant gastrointestinal stromal tumor | UMIN000016115 | II | PFS rate (>24 weeks), ORR, RR, DCR, and AE | Efficacy in 2nd line therapy for imatinib-resistant GISTs. Secondary mutation in KIT can be predictive of regorafenib efficacy | [128] |
| A study of regorafenib as a 3rd-line or beyond treatment for GISTs (GRID) | NCT01271712 | III | PSF and OS | Regorafenib PSF > placebo | [129,130] | ||
| Nilotinib | A phase II Study aiming to evaluate the efficacy and safety of nilotinib patients with gastrointestinal stromal tumors (GIST) resistant or intolerant to imatinib and or to 2nd-line tyrosine kinas (TK) inhibitor | NCT00633295 | II | Efficacy in GISTs resistant to imatinib and/or 2nd-line TKI, safety, and tolerability | Ongoing | ||
| Nilotinib | Phase III Study of nilotinib versus best supportive Care with or without a TKI in patients with gastrointestinal stromal tumors resistant to or intolerant of imatinib and sunitinib | Open-Label Trial | III | Efficacy in patients with advanced GISTs following imatinib and sunitinib failure, and PFS | Longer median OS | [134] | |
| Pazopanib | Efficacy of pazopanib in gastrointestinal stromal tumors (GIST) (PAZOGIST) | NCT01323400 | II | OS, PFS, and tolerance profile | Effective on 50% of patients with metastatic or locally advanced GISTs | [136] | |
| Sorafenib | Sorafenib in treating patients with malignant gastrointestinal stromal tumor that progressed during or after previous treatment with imatinib mesylate and sunitinib malate | NCT00265798 | II | ORR and OS | Ongoing | ||
| 4th | Ripretinib | Phase 3 study of DCC-2618 vs. placebo in advanced GIST patients who have been treated with prior anticancer therapies (INVICTUS) | NCT03353753 | III | Safety profile, PFS, and ORR | Acceptable safety profile and Ripretinib PSF > placebo | [148] |
| A study of DCC-2618 vs. sunitinib in advanced GIST patients after treatment with imatinib (INTRIGUE) | NCT03673501 | III | PFS and ORR | ORR ripretinib > sunitinib | [149] | ||
| Preoperative treatment of potentially resectable locally advanced and recurrent metastatic GIST after failure of imatinib therapy | NCT05132738 | Single arm | NED Rate, surgery rate (proportion of patients who can undergo surgery), and ORR | Ongoing | |||
| Ripretinib combined with surgery in advanced GIST that have failed imatinib therapy: A multicenter, observational study (NAVIGATOR) | NCT05354388 | Observational | PFS (at 12 months), ORR (at 12 months), TTP, and the 2-year overall survival rate | Ongoing | |||
| NEW TKI Drugs | Cabozantinib (Selective for the T670I mutation) | Ph II CABOGIST in GIST | NCT02216578 | II | PFS, OS, and ORR | Potential drug for metastatic, imatinib, and sunitinib-resistant GISTs | [159] |
| Avapritinib (Selective for the D842V mutation) | Study of BLU-285 in patients with gastrointestinal stromal tumors (GIST) and other relapsed and refractory solid tumors | NCT02508532 | I | MTD and ORR | 86% response rate by RECIST 1.1 in patients with the PDGFRA D842V mutation | [161] | |
| Study of avapritinib vs. regorafenib in patients with locally advanced unresectable or metastatic GIST (VOYAGER) | NCT03465722 | III | PFS, ORR, CR, and OR | The primary endpoint (PFS by central radiology per RECIST) has not been yet reached | [164] |
10. BAF Inhibitors and GISTs
11. Serum Markers for Precision Medicine in GISTs
12. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
Abbreviations
| AE | adverse event |
| BAF | nuclear assembly factor 1 |
| BRAF | serine-threonine protein kinase |
| BRD9 | bromodomain containing 9 |
| CR | complete response |
| DCR | disease control rate |
| DOG1 | discovered on GIST-1 |
| dPCR | droplet digital PCR |
| ED | extracellular domain |
| ESMO | European Society for Medical Oncology |
| ETV6-NTRK3 | variant transcription factor 6 |
| FDA | Food and Drug Administration |
| GIST | gastrointestinal stromal tumor |
| IGF1R | insulin-like growth factor 1 receptor |
| JMD | juxtamembrane domain |
| KP | kinase pocket |
| MTD | maximum tolerated dose |
| NCCN | National Comprehensive Cancer Network |
| NED | no evidence of disease |
| NF1 | neurofibromatosis type 1 |
| NGS | next-generation sequencing |
| NTRK | neurotrophic tyrosine receptor kinase |
| OR | overall response |
| ORR | objective response rate |
| OS | overall survival |
| PDGFRA | platelet-derived growth factor receptor-alpha |
| PFS | progression-free survival |
| PI3K | phosphoinositide 3-kinase |
| PIK3CA | phosphatidylinositol 3-kinase, catalytic, alpha |
| PR | partial response |
| PUMA | p53 upregulated modulator of apoptosis |
| RECIST | response evaluation criteria in solid tumors |
| RFS | recurrence-free survival |
| RR | response rate |
| SCF | stem cell factor |
| SDH | succinate dehydrogenase |
| TKI | tyrosine kinase inhibitors |
| TKR | tyrosine kinase receptor |
| TTP | time to progression |
| TTR | time to tumor response |
| WT-GIST | wild-type GIST |
References
- Huizinga, J.D.; Thuneberg, L.; Klüppel, M.; Malysz, J.; Mikkelsen, H.B.; Bernstein, A. W/Kit Gene Required for Interstitial Cells of Cajal and for Intestinal Pacemaker Activity. Nature 1995, 373, 347–349. [Google Scholar] [CrossRef] [PubMed]
- Kindblom, L.G.; Remotti, H.E.; Aldenborg, F.; Meis-Kindblom, J.M. Gastrointestinal Pacemaker Cell Tumor (GIPACT): Gastrointestinal Stromal Tumors Show Phenotypic Characteristics of the Interstitial Cells of Cajal. Am. J. Pathol. 1998, 152, 1259–1269. [Google Scholar] [PubMed]
- Sawaki, A. Rare Gastrointestinal Stromal Tumors (GIST): Omentum and Retroperitoneum. Transl. Gastroenterol. Hepatol. 2017, 2, 116. [Google Scholar] [CrossRef] [PubMed]
- Miettinen, M.; Lasota, J. Gastrointestinal Stromal Tumors. Gastroenterol. Clin. N. Am. 2013, 42, 399–415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miettinen, M.; Lasota, J. Gastrointestinal Stromal Tumors--Definition, Clinical, Histological, Immunohistochemical, and Molecular Genetic Features and Differential Diagnosis. Virchows Arch. 2001, 438, 1–12. [Google Scholar] [CrossRef]
- DeMatteo, R.P.; Lewis, J.J.; Leung, D.; Mudan, S.S.; Woodruff, J.M.; Brennan, M.F. Two Hundred Gastrointestinal Stromal Tumors: Recurrence Patterns and Prognostic Factors for Survival. Ann. Surg. 2000, 231, 51–58. [Google Scholar] [CrossRef]
- Sbaraglia, M.; Businello, G.; Bellan, E.; Fassan, M.; Dei Tos, A.P. Mesenchymal Tumours of the Gastrointestinal Tract. Pathologica 2021, 113, 230–251. [Google Scholar] [CrossRef]
- Hirota, S. Differential Diagnosis of Gastrointestinal Stromal Tumor by Histopathology and Immunohistochemistry. Transl. Gastroenterol. Hepatol. 2018, 3, 27. [Google Scholar] [CrossRef]
- Hirota, S.; Isozaki, K.; Moriyama, Y.; Hashimoto, K.; Nishida, T.; Ishiguro, S.; Kawano, K.; Hanada, M.; Kurata, A.; Takeda, M.; et al. Gain-of-Function Mutations of c-Kit in Human Gastrointestinal Stromal Tumors. Science 1998, 279, 577–580. [Google Scholar] [CrossRef]
- Heinrich, M.C.; Corless, C.L.; Duensing, A.; McGreevey, L.; Chen, C.-J.; Joseph, N.; Singer, S.; Griffith, D.J.; Haley, A.; Town, A.; et al. PDGFRA Activating Mutations in Gastrointestinal Stromal Tumors. Science 2003, 299, 708–710. [Google Scholar] [CrossRef]
- Corless, C.; Fletcher, J.; Heinrich, M. Biology of Gastrointestinal Stromal Tumors. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2004, 22, 3813–3825. [Google Scholar] [CrossRef]
- Janeway, K.A.; Liegl, B.; Harlow, A.; Le, C.; Perez-Atayde, A.; Kozakewich, H.; Corless, C.L.; Heinrich, M.C.; Fletcher, J.A. Pediatric KIT Wild-Type and Platelet-Derived Growth Factor Receptor Alpha-Wild-Type Gastrointestinal Stromal Tumors Share KIT Activation but Not Mechanisms of Genetic Progression with Adult Gastrointestinal Stromal Tumors. Cancer Res. 2007, 67, 9084–9088. [Google Scholar] [CrossRef] [Green Version]
- Kays, J.K.; Sohn, J.D.; Kim, B.J.; Goze, K.; Koniaris, L.G. Approach to Wild-Type Gastrointestinal Stromal Tumors. Transl. Gastroenterol. Hepatol. 2018, 3, 92. [Google Scholar] [CrossRef] [Green Version]
- Moosavi, B.; Berry, E.A.; Zhu, X.-L.; Yang, W.-C.; Yang, G.-F. The Assembly of Succinate Dehydrogenase: A Key Enzyme in Bioenergetics. Cell. Mol. Life Sci. 2019, 76, 4023–4042. [Google Scholar] [CrossRef]
- Janeway, K.A.; Kim, S.Y.; Lodish, M.; Nosé, V.; Rustin, P.; Gaal, J.; Dahia, P.L.M.; Liegl, B.; Ball, E.R.; Raygada, M.; et al. Defects in Succinate Dehydrogenase in Gastrointestinal Stromal Tumors Lacking KIT and PDGFRA Mutations. Proc. Natl. Acad. Sci. USA 2011, 108, 314–318. [Google Scholar] [CrossRef] [Green Version]
- Killian, J.K.; Kim, S.Y.; Miettinen, M.; Smith, C.; Merino, M.; Tsokos, M.; Quezado, M.; Smith, W.I.; Jahromi, M.S.; Xekouki, P.; et al. Succinate Dehydrogenase Mutation Underlies Global Epigenomic Divergence in Gastrointestinal Stromal Tumor. Cancer Discov. 2013, 3, 648–657. [Google Scholar] [CrossRef] [Green Version]
- Boikos, S.A.; Pappo, A.S.; Killian, J.K.; LaQuaglia, M.P.; Weldon, C.B.; George, S.; Trent, J.C.; von Mehren, M.; Wright, J.A.; Schiffman, J.D.; et al. Molecular Subtypes of KIT/PDGFRA Wild-Type Gastrointestinal Stromal Tumors: A Report From the National Institutes of Health Gastrointestinal Stromal Tumor Clinic. JAMA Oncol. 2016, 2, 922–928. [Google Scholar] [CrossRef] [Green Version]
- TO Nielsen WHO Classification of Tumours 5th Ed, Soft Tissue and Bone Tumours; International Agency for Research on Cancer (IARC): Lyon, France, 2020; ISBN 978-92-832-4502-5.
- Sbaraglia, M.; Bellan, E.; Dei Tos, A.P. The 2020 WHO Classification of Soft Tissue Tumours: News and Perspectives. Pathologica 2021, 113, 70–84. [Google Scholar] [CrossRef]
- Casali, P.G.; Abecassis, N.; Aro, H.T.; Bauer, S.; Biagini, R.; Bielack, S.; Bonvalot, S.; Boukovinas, I.; Bovee, J.V.M.G.; Brodowicz, T.; et al. Gastrointestinal Stromal Tumours: ESMO-EURACAN Clinical Practice Guidelines for Diagnosis, Treatment and Follow-Up. Ann. Oncol. 2018, 29, iv68–iv78. [Google Scholar] [CrossRef]
- ESMO/European Sarcoma Network Working Group Gastrointestinal Stromal Tumours: ESMO Clinical Practice Guidelines for Diagnosis, Treatment and Follow-Up. Ann. Oncol. 2014, 25 (Suppl. S3), iii21–iii26. [CrossRef]
- Emile, J.F.; Brahimi, S.; Coindre, J.M.; Bringuier, P.P.; Monges, G.; Samb, P.; Doucet, L.; Hostein, I.; Landi, B.; Buisine, M.P.; et al. Frequencies of KIT and PDGFRA Mutations in the MolecGIST Prospective Population-Based Study Differ from Those of Advanced GISTs. Med. Oncol. 2012, 29, 1765–1772. [Google Scholar] [CrossRef] [PubMed]
- Mei, L.; Smith, S.C.; Faber, A.C.; Trent, J.; Grossman, S.R.; Stratakis, C.A.; Boikos, S.A. Gastrointestinal Stromal Tumors: The GIST of Precision Medicine. Trends Cancer 2018, 4, 74–91. [Google Scholar] [CrossRef] [PubMed]
- Rubin, B.P.; Singer, S.; Tsao, C.; Duensing, A.; Lux, M.L.; Ruiz, R.; Hibbard, M.K.; Chen, C.J.; Xiao, S.; Tuveson, D.A.; et al. KIT Activation Is a Ubiquitous Feature of Gastrointestinal Stromal Tumors. Cancer Res. 2001, 61, 8118–8121. [Google Scholar] [PubMed]
- Tuveson, D.A.; Willis, N.A.; Jacks, T.; Griffin, J.D.; Singer, S.; Fletcher, C.D.; Fletcher, J.A.; Demetri, G.D. STI571 Inactivation of the Gastrointestinal Stromal Tumor C-KIT Oncoprotein: Biological and Clinical Implications. Oncogene 2001, 20, 5054–5058. [Google Scholar] [CrossRef] [Green Version]
- Nishida, T.; Hirota, S.; Taniguchi, M.; Hashimoto, K.; Isozaki, K.; Nakamura, H.; Kanakura, Y.; Tanaka, T.; Takabayashi, A.; Matsuda, H.; et al. Familial Gastrointestinal Stromal Tumours with Germline Mutation of the KIT Gene. Nat. Genet. 1998, 19, 323–324. [Google Scholar] [CrossRef]
- Lev, S.; Blechman, J.; Nishikawa, S.; Givol, D.; Yarden, Y. Interspecies Molecular Chimeras of Kit Help Define the Binding Site of the Stem Cell Factor. Mol. Cell Biol. 1993, 13, 2224–2234. [Google Scholar] [CrossRef]
- Besmer, P.; Murphy, J.E.; George, P.C.; Qiu, F.H.; Bergold, P.J.; Lederman, L.; Snyder, H.W.; Brodeur, D.; Zuckerman, E.E.; Hardy, W.D. A New Acute Transforming Feline Retrovirus and Relationship of Its Oncogene V-Kit with the Protein Kinase Gene Family. Nature 1986, 320, 415–421. [Google Scholar] [CrossRef]
- Zsebo, K.M.; Williams, D.A.; Geissler, E.N.; Broudy, V.C.; Martin, F.H.; Atkins, H.L.; Hsu, R.Y.; Birkett, N.C.; Okino, K.H.; Murdock, D.C. Stem Cell Factor Is Encoded at the Sl Locus of the Mouse and Is the Ligand for the C-Kit Tyrosine Kinase Receptor. Cell 1990, 63, 213–224. [Google Scholar] [CrossRef]
- Hemesath, T.J.; Price, E.R.; Takemoto, C.; Badalian, T.; Fisher, D.E. MAP Kinase Links the Transcription Factor Microphthalmia to C-Kit Signalling in Melanocytes. Nature 1998, 391, 298–301. [Google Scholar] [CrossRef]
- Timokhina, I.; Kissel, H.; Stella, G.; Besmer, P. Kit Signaling through PI 3-Kinase and Src Kinase Pathways: An Essential Role for Rac1 and JNK Activation in Mast Cell Proliferation. EMBO J. 1998, 17, 6250–6262. [Google Scholar] [CrossRef] [Green Version]
- Szucs, Z.; Thway, K.; Fisher, C.; Bulusu, R.; Constantinidou, A.; Benson, C.; van der Graaf, W.T.; Jones, R.L. Molecular Subtypes of Gastrointestinal Stromal Tumors and Their Prognostic and Therapeutic Implications. Future Oncol. 2017, 13, 93–107. [Google Scholar] [CrossRef] [Green Version]
- Corless, C.L.; Heinrich, M.C. Molecular Pathobiology of Gastrointestinal Stromal Sarcomas. Annu. Rev. Pathol. 2008, 3, 557–586. [Google Scholar] [CrossRef]
- Lasota, J.; Miettinen, M. Clinical Significance of Oncogenic KIT and PDGFRA Mutations in Gastrointestinal Stromal Tumours. Histopathology 2008, 53, 245–266. [Google Scholar] [CrossRef]
- Heinrich, M.C.; Corless, C.L.; Demetri, G.D.; Blanke, C.D.; von Mehren, M.; Joensuu, H.; McGreevey, L.S.; Chen, C.-J.; Van den Abbeele, A.D.; Druker, B.J.; et al. Kinase Mutations and Imatinib Response in Patients with Metastatic Gastrointestinal Stromal Tumor. J. Clin. Oncol. 2003, 21, 4342–4349. [Google Scholar] [CrossRef]
- Gounder, M.M.; Maki, R.G. Molecular Basis for Primary and Secondary Tyrosine Kinase Inhibitor Resistance in Gastrointestinal Stromal Tumor. Cancer Chemother. Pharmacol. 2011, 67 (Suppl. S1), S25–S43. [Google Scholar] [CrossRef] [Green Version]
- Gramza, A.W.; Corless, C.L.; Heinrich, M.C. Resistance to Tyrosine Kinase Inhibitors in Gastrointestinal Stromal Tumors. Clin. Cancer Res. 2009, 15, 7510–7518. [Google Scholar] [CrossRef] [Green Version]
- Debiec-Rychter, M.; Sciot, R.; Le Cesne, A.; Schlemmer, M.; Hohenberger, P.; van Oosterom, A.T.; Blay, J.-Y.; Leyvraz, S.; Stul, M.; Casali, P.G.; et al. KIT Mutations and Dose Selection for Imatinib in Patients with Advanced Gastrointestinal Stromal Tumours. Eur. J. Cancer 2006, 42, 1093–1103. [Google Scholar] [CrossRef]
- von Mehren, M.; Kane, J.; Bui, M.; Choy, E.; Connelly, M.; Dry, S.; Ganjoo, K.; George, S.; Gonzalez, R.; Heslin, M.; et al. NCCN Guidelines Insights: Soft Tissue Sarcoma, Version 1.2021. J. Natl. Compr. Cancer Netw. JNCCN 2020, 18, 1604–1612. [Google Scholar] [CrossRef]
- Lasota, J.; Corless, C.L.; Heinrich, M.C.; Debiec-Rychter, M.; Sciot, R.; Wardelmann, E.; Merkelbach-Bruse, S.; Schildhaus, H.-U.; Steigen, S.E.; Stachura, J.; et al. Clinicopathologic Profile of Gastrointestinal Stromal Tumors (GISTs) with Primary KIT Exon 13 or Exon 17 Mutations: A Multicenter Study on 54 Cases. Mod. Pathol. 2008, 21, 476–484. [Google Scholar] [CrossRef] [Green Version]
- Oppelt, P.J.; Hirbe, A.C.; Van Tine, B.A. Gastrointestinal Stromal Tumors (GISTs): Point Mutations Matter in Management, a Review. J. Gastrointest. Oncol. 2017, 8, 466–473. [Google Scholar] [CrossRef] [Green Version]
- Ito, T.; Yamamura, M.; Hirai, T.; Ishikawa, T.; Kanda, T.; Nakai, T.; Ohkouchi, M.; Hashikura, Y.; Isozaki, K.; Hirota, S. Gastrointestinal Stromal Tumors with Exon 8 C-Kit Gene Mutation Might Occur at Extragastric Sites and Have Metastasis-Prone Nature. Int. J. Clin. Exp. Pathol. 2014, 7, 8024–8031. [Google Scholar] [PubMed]
- Hirota, S.; Ohashi, A.; Nishida, T.; Isozaki, K.; Kinoshita, K.; Shinomura, Y.; Kitamura, Y. Gain-of-Function Mutations of Platelet-Derived Growth Factor Receptor α Gene in Gastrointestinal Stromal Tumors. Gastroenterology 2003, 125, 660–667. [Google Scholar] [CrossRef] [PubMed]
- Wozniak, A.; Rutkowski, P.; Piskorz, A.; Ciwoniuk, M.; Osuch, C.; Bylina, E.; Sygut, J.; Chosia, M.; Rys, J.; Urbanczyk, K.; et al. Prognostic Value of KIT/PDGFRA Mutations in Gastrointestinal Stromal Tumours (GIST): Polish Clinical GIST Registry Experience. Ann. Oncol. 2012, 23, 353–360. [Google Scholar] [CrossRef] [PubMed]
- Cassier, P.A.; Fumagalli, E.; Rutkowski, P.; Schöffski, P.; Van Glabbeke, M.; Debiec-Rychter, M.; Emile, J.-F.; Duffaud, F.; Martin-Broto, J.; Landi, B.; et al. Outcome of Patients with Platelet-Derived Growth Factor Receptor Alpha-Mutated Gastrointestinal Stromal Tumors in the Tyrosine Kinase Inhibitor Era. Clin. Cancer Res. 2012, 18, 4458–4464. [Google Scholar] [CrossRef] [Green Version]
- Subramanian, S.; West, R.; Corless, C.; Ou, W.; Rubin, B.; Chu, K.; Leung, S.; Yuen, S.; Zhu, S.; Hernandez-Boussard, T.; et al. Gastrointestinal Stromal Tumors (GISTs) with KIT and PDGFRA Mutations Have Distinct Gene Expression Profiles. Oncogene 2004, 23, 7780–7790. [Google Scholar] [CrossRef] [Green Version]
- Wasag, B.; Debiec-Rychter, M.; Pauwels, P.; Stul, M.; Vranckx, H.; Oosterom, A.V.; Hagemeijer, A.; Sciot, R. Differential Expression of KIT/PDGFRA Mutant Isoforms in Epithelioid and Mixed Variants of Gastrointestinal Stromal Tumors Depends Predominantly on the Tumor Site. Mod. Pathol. 2004, 17, 889–894. [Google Scholar] [CrossRef] [Green Version]
- Corless, C.L.; Schroeder, A.; Griffith, D.; Town, A.; McGreevey, L.; Harrell, P.; Shiraga, S.; Bainbridge, T.; Morich, J.; Heinrich, M.C. PDGFRA Mutations in Gastrointestinal Stromal Tumors: Frequency, Spectrum and in Vitro Sensitivity to Imatinib. J. Clin. Oncol. 2005, 23, 5357–5364. [Google Scholar] [CrossRef]
- Indio, V.; Ravegnini, G.; Astolfi, A.; Urbini, M.; Saponara, M.; De Leo, A.; Gruppioni, E.; Tarantino, G.; Angelini, S.; Pession, A.; et al. Gene Expression Profiling of PDGFRA Mutant GIST Reveals Immune Signatures as a Specific Fingerprint of D842V Exon 18 Mutation. Front. Immunol. 2020, 11, 851. [Google Scholar] [CrossRef]
- Lasota, J.; Dansonka-Mieszkowska, A.; Sobin, L.H.; Miettinen, M. A Great Majority of GISTs with PDGFRA Mutations Represent Gastric Tumors of Low or No Malignant Potential. Lab. Investig. 2004, 84, 874–883. [Google Scholar] [CrossRef] [Green Version]
- Rutter, J.; Winge, D.R.; Schiffman, J.D. Succinate Dehydrogenase—Assembly, Regulation and Role in Human Disease. Mitochondrion 2010, 10, 393–401. [Google Scholar] [CrossRef] [Green Version]
- Miettinen, M.; Wang, Z.-F.; Sarlomo-Rikala, M.; Osuch, C.; Rutkowski, P.; Lasota, J. Succinate Dehydrogenase-Deficient GISTs: A Clinicopathologic, Immunohistochemical, and Molecular Genetic Study of 66 Gastric GISTs with Predilection to Young Age. Am. J. Surg. Pathol. 2011, 35, 1712–1721. [Google Scholar] [CrossRef] [Green Version]
- Pantaleo, M.A.; Astolfi, A.; Urbini, M.; Nannini, M.; Paterini, P.; Indio, V.; Saponara, M.; Formica, S.; Ceccarelli, C.; Casadio, R.; et al. Analysis of All Subunits, SDHA, SDHB, SDHC, SDHD, of the Succinate Dehydrogenase Complex in KIT/PDGFRA Wild-Type GIST. Eur. J. Hum. Genet. 2014, 22, 32–39. [Google Scholar] [CrossRef] [Green Version]
- Nannini, M.; Rizzo, A.; Indio, V.; Schipani, A.; Astolfi, A.; Pantaleo, M.A. Targeted Therapy in SDH-Deficient GIST. Ther. Adv. Med. Oncol. 2021, 13, 17588359211023278. [Google Scholar] [CrossRef]
- Belinsky, M.G.; Rink, L.; von Mehren, M. Succinate Dehydrogenase Deficiency in Pediatric and Adult Gastrointestinal Stromal Tumors. Front. Oncol. 2013, 3, 117. [Google Scholar] [CrossRef] [Green Version]
- Weldon, C.B.; Madenci, A.L.; Boikos, S.A.; Janeway, K.A.; George, S.; von Mehren, M.; Pappo, A.S.; Schiffman, J.D.; Wright, J.; Trent, J.C.; et al. Surgical Management of Wild-Type Gastrointestinal Stromal Tumors: A Report From the National Institutes of Health Pediatric and Wildtype GIST Clinic. J. Clin. Oncol. 2017, 35, 523–528. [Google Scholar] [CrossRef] [Green Version]
- Glod, J.; Arnaldez, F.I.; Wiener, L.; Spencer, M.; Killian, J.K.; Meltzer, P.; Dombi, E.; Derse-Anthony, C.; Derdak, J.; Srinivasan, R.; et al. A Phase II Trial of Vandetanib in Children and Adults with Succinate Dehydrogenase-Deficient Gastrointestinal Stromal Tumor. Clin. Cancer Res. 2019, 25, 6302–6308. [Google Scholar] [CrossRef] [Green Version]
- Davies, H.; Bignell, G.R.; Cox, C.; Stephens, P.; Edkins, S.; Clegg, S.; Teague, J.; Woffendin, H.; Garnett, M.J.; Bottomley, W.; et al. Mutations of the BRAF Gene in Human Cancer. Nature 2002, 417, 949–954. [Google Scholar] [CrossRef] [Green Version]
- Agaram, N.P.; Wong, G.C.; Guo, T.; Maki, R.G.; Singer, S.; Dematteo, R.P.; Besmer, P.; Antonescu, C.R. Novel V600E BRAF Mutations in Imatinib-Naive and Imatinib-Resistant Gastrointestinal Stromal Tumors. Genes Chromosomes Cancer 2008, 47, 853–859. [Google Scholar] [CrossRef] [Green Version]
- Hostein, I.; Faur, N.; Primois, C.; Boury, F.; Denard, J.; Emile, J.-F.; Bringuier, P.-P.; Scoazec, J.-Y.; Coindre, J.-M. BRAF Mutation Status in Gastrointestinal Stromal Tumors. Am. J. Clin. Pathol. 2010, 133, 141–148. [Google Scholar] [CrossRef] [Green Version]
- Agaimy, A.; Terracciano, L.M.; Dirnhofer, S.; Tornillo, L.; Foerster, A.; Hartmann, A.; Bihl, M.P. V600E BRAF Mutations Are Alternative Early Molecular Events in a Subset of KIT/PDGFRA Wild-Type Gastrointestinal Stromal Tumours. J. Clin. Pathol. 2009, 62, 613–616. [Google Scholar] [CrossRef] [Green Version]
- Huss, S.; Pasternack, H.; Ihle, M.A.; Merkelbach-Bruse, S.; Heitkötter, B.; Hartmann, W.; Trautmann, M.; Gevensleben, H.; Büttner, R.; Schildhaus, H.-U.; et al. Clinicopathological and Molecular Features of a Large Cohort of Gastrointestinal Stromal Tumors (GISTs) and Review of the Literature: BRAF Mutations in KIT/PDGFRA Wild-Type GISTs Are Rare Events. Hum. Pathol. 2017, 62, 206–214. [Google Scholar] [CrossRef] [PubMed]
- Klug, L.R.; Khosroyani, H.M.; Kent, J.D.; Heinrich, M.C. New Treatment Strategies for Advanced-Stage Gastrointestinal Stromal Tumours. Nat. Rev. Clin. Oncol. 2022, 19, 328–341. [Google Scholar] [CrossRef] [PubMed]
- Jašek, K.; Váňová, B.; Grendár, M.; Štanclová, A.; Szépe, P.; Hornáková, A.; Holubeková, V.; Plank, L.; Lasabová, Z. BRAF Mutations in KIT/PDGFRA Positive Gastrointestinal Stromal Tumours (GISTs): Is Their Frequency Underestimated? Pathol. Res. Pract. 2020, 216, 153171. [Google Scholar] [CrossRef] [PubMed]
- Torrence, D.; Xie, Z.; Zhang, L.; Chi, P.; Antonescu, C.R. Gastrointestinal Stromal Tumors with BRAF Gene Fusions. A Report of Two Cases Showing Low or Absent KIT Expression Resulting in Diagnostic Pitfalls. Genes Chromosomes Cancer 2021, 60, 789–795. [Google Scholar] [CrossRef]
- Zwarthoff, E.C. Neurofibromatosis and Associated Tumour Suppressor Genes. Pathol. Res. Pract. 1996, 192, 647–657. [Google Scholar] [CrossRef]
- Schubbert, S.; Shannon, K.; Bollag, G. Hyperactive Ras in Developmental Disorders and Cancer. Nat. Rev. Cancer 2007, 7, 295–308. [Google Scholar] [CrossRef]
- Guertin, D.A.; Sabatini, D.M. Defining the Role of MTOR in Cancer. Cancer Cell 2007, 12, 9–22. [Google Scholar] [CrossRef] [Green Version]
- Andersson, J.; Sihto, H.; Meis-Kindblom, J.M.; Joensuu, H.; Nupponen, N.; Kindblom, L.-G. NF1-Associated Gastrointestinal Stromal Tumors Have Unique Clinical, Phenotypic, and Genotypic Characteristics. Am. J. Surg. Pathol. 2005, 29, 1170–1176. [Google Scholar] [CrossRef]
- Ferner, R.E. Neurofibromatosis 1 and Neurofibromatosis 2: A Twenty First Century Perspective. Lancet Neurol. 2007, 6, 340–351. [Google Scholar] [CrossRef]
- Ferner, R.E.; Huson, S.M.; Thomas, N.; Moss, C.; Willshaw, H.; Evans, D.G.; Upadhyaya, M.; Towers, R.; Gleeson, M.; Steiger, C.; et al. Guidelines for the Diagnosis and Management of Individuals with Neurofibromatosis 1. J. Med. Genet. 2007, 44, 81–88. [Google Scholar] [CrossRef] [Green Version]
- Salvi, P.F.; Lorenzon, L.; Caterino, S.; Antolino, L.; Antonelli, M.S.; Balducci, G. Gastrointestinal Stromal Tumors Associated with Neurofibromatosis 1: A Single Centre Experience and Systematic Review of the Literature Including 252 Cases. Int. J. Surg. Oncol. 2013, 2013, 398570. [Google Scholar] [CrossRef]
- Ricci, R. Syndromic Gastrointestinal Stromal Tumors. Hered. Cancer Clin. Pract. 2016, 14, 15. [Google Scholar] [CrossRef]
- Agaimy, A.; Vassos, N.; Croner, R. Gastrointestinal Manifestations of Neurofibromatosis Type 1 (Recklinghausen’s Disease): Clinicopathological Spectrum with Pathogenetic Considerations. Int. J. Clin. Exp. Pathol. 2012, 5, 852–862. [Google Scholar]
- Nishida, T.; Hirota, S. Biological and Clinical Review of Stromal Tumors in the Gastrointestinal Tract. Histol. Histopathol. 2000, 15, 1293–1301. [Google Scholar] [CrossRef]
- Burgoyne, A.M.; De Siena, M.; Alkhuziem, M.; Tang, C.-M.; Medina, B.; Fanta, P.T.; Belinsky, M.G.; von Mehren, M.; Thorson, J.A.; Madlensky, L.; et al. Duodenal-Jejunal Flexure GI Stromal Tumor Frequently Heralds Somatic NF1 and Notch Pathway Mutations. JCO Precis. Oncol. 2017, 1, PO.17.00014. [Google Scholar] [CrossRef]
- Ho, M.Y.; Blanke, C.D. Gastrointestinal Stromal Tumors: Disease and Treatment Update. Gastroenterology 2011, 140, 1372–1376.e2. [Google Scholar] [CrossRef]
- Maertens, O.; Prenen, H.; Debiec-Rychter, M.; Wozniak, A.; Sciot, R.; Pauwels, P.; De Wever, I.; Vermeesch, J.R.; de Raedt, T.; De Paepe, A.; et al. Molecular Pathogenesis of Multiple Gastrointestinal Stromal Tumors in NF1 Patients. Hum. Mol. Genet. 2006, 15, 1015–1023. [Google Scholar] [CrossRef] [Green Version]
- Kinoshita, K.; Hirota, S.; Isozaki, K.; Ohashi, A.; Nishida, T.; Kitamura, Y.; Shinomura, Y.; Matsuzawa, Y. Absence of C-Kit Gene Mutations in Gastrointestinal Stromal Tumours from Neurofibromatosis Type 1 Patients. J. Pathol. 2004, 202, 80–85. [Google Scholar] [CrossRef]
- Mussi, C.; Schildhaus, H.-U.; Gronchi, A.; Wardelmann, E.; Hohenberger, P. Therapeutic Consequences from Molecular Biology for Gastrointestinal Stromal Tumor Patients Affected by Neurofibromatosis Type 1. Clin. Cancer Res. 2008, 14, 4550–4555. [Google Scholar] [CrossRef] [Green Version]
- Valencia, E.; Saif, M.W. Neurofibromatosis Type 1 and GIST: Is There a Correlation? Anticancer Res. 2014, 34, 5609–5612. [Google Scholar]
- Gasparotto, D.; Rossi, S.; Polano, M.; Tamborini, E.; Lorenzetto, E.; Sbaraglia, M.; Mondello, A.; Massani, M.; Lamon, S.; Bracci, R.; et al. Quadruple-Negative GIST Is a Sentinel for Unrecognized Neurofibromatosis Type 1 Syndrome. Clin. Cancer Res. 2017, 23, 273–282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Sullivan Coyne, G.H.; Gross, A.M.; Dombi, E.; Tibery, C.; Carbonell, A.; Takebe, N.; Derdak, J.; Pichard, D.; Srivastava, A.K.; Herrick, W.; et al. Phase II Trial of the MEK 1/2 Inhibitor Selumetinib (AZD6244, ARRY-142886 Hydrogen Sulfate) in Adults with Neurofibromatosis Type 1 (NF1) and Inoperable Plexiform Neurofibromas (PN). JCO 2020, 38, 3612. [Google Scholar] [CrossRef]
- Weier, H.U.; Rhein, A.P.; Shadravan, F.; Collins, C.; Polikoff, D. Rapid Physical Mapping of the Human Trk Protooncogene (NTRK1) to Human Chromosome 1q21-Q22 by P1 Clone Selection, Fluorescence in Situ Hybridization (FISH), and Computer-Assisted Microscopy. Genomics 1995, 26, 390–393. [Google Scholar] [CrossRef] [PubMed]
- D’Alpino Peixoto, R.; Medeiros, B.A.; Cronemberger, E.H. Resected High-Risk Rectal GIST Harboring NTRK1 Fusion: A Case Report and Review of the Literature. J. Gastrointest Cancer 2021, 52, 316–319. [Google Scholar] [CrossRef]
- Davis, J.L.; Lockwood, C.M.; Stohr, B.; Boecking, C.; Al-Ibraheemi, A.; DuBois, S.G.; Vargas, S.O.; Black, J.O.; Cox, M.C.; Luquette, M.; et al. Expanding the Spectrum of Pediatric NTRK-Rearranged Mesenchymal Tumors. Am. J. Surg. Pathol. 2019, 43, 435–445. [Google Scholar] [CrossRef]
- Cao, Z.; Li, J.; Sun, L.; Xu, Z.; Ke, Y.; Shao, B.; Guo, Y.; Sun, Y. GISTs with NTRK Gene Fusions: A Clinicopathological, Immunophenotypic, and Molecular Study. Cancers 2022, 15, 105. [Google Scholar] [CrossRef]
- Brenca, M.; Rossi, S.; Polano, M.; Gasparotto, D.; Zanatta, L.; Racanelli, D.; Valori, L.; Lamon, S.; Dei Tos, A.P.; Maestro, R. Transcriptome Sequencing Identifies ETV6–NTRK3 as a Gene Fusion Involved in GIST. J. Pathol. 2016, 238, 543–549. [Google Scholar] [CrossRef]
- Atiq, M.A.; Davis, J.L.; Hornick, J.L.; Dickson, B.C.; Fletcher, C.D.M.; Fletcher, J.A.; Folpe, A.L.; Mariño-Enríquez, A. Mesenchymal Tumors of the Gastrointestinal Tract with NTRK Rearrangements: A Clinicopathological, Immunophenotypic, and Molecular Study of Eight Cases, Emphasizing Their Distinction from Gastrointestinal Stromal Tumor (GIST). Mod. Pathol. 2021, 34, 95–103. [Google Scholar] [CrossRef]
- Marchiò, C.; Scaltriti, M.; Ladanyi, M.; Iafrate, A.J.; Bibeau, F.; Dietel, M.; Hechtman, J.F.; Troiani, T.; López-Rios, F.; Douillard, J.-Y.; et al. ESMO Recommendations on the Standard Methods to Detect NTRK Fusions in Daily Practice and Clinical Research. Ann. Oncol. 2019, 30, 1417–1427. [Google Scholar] [CrossRef] [Green Version]
- Drilon, A.; Laetsch, T.W.; Kummar, S.; DuBois, S.G.; Lassen, U.N.; Demetri, G.D.; Nathenson, M.; Doebele, R.C.; Farago, A.F.; Pappo, A.S.; et al. Efficacy of Larotrectinib in TRK Fusion-Positive Cancers in Adults and Children. N. Engl. J. Med. 2018, 378, 731–739. [Google Scholar] [CrossRef]
- Laetsch, T.W.; DuBois, S.G.; Mascarenhas, L.; Turpin, B.; Federman, N.; Albert, C.M.; Nagasubramanian, R.; Davis, J.L.; Rudzinski, E.; Feraco, A.M.; et al. Larotrectinib for Paediatric Solid Tumours Harbouring NTRK Gene Fusions: Phase 1 Results from a Multicentre, Open-Label, Phase 1/2 Study. Lancet Oncol. 2018, 19, 705–714. [Google Scholar] [CrossRef]
- Doebele, R.C.; Drilon, A.; Paz-Ares, L.; Siena, S.; Shaw, A.T.; Farago, A.F.; Blakely, C.M.; Seto, T.; Cho, B.C.; Tosi, D.; et al. Entrectinib in Patients with Advanced or Metastatic NTRK Fusion-Positive Solid Tumours: Integrated Analysis of Three Phase 1–2 Trials. Lancet Oncol. 2020, 21, 271–282. [Google Scholar] [CrossRef]
- Al-Salama, Z.T.; Keam, S.J. Entrectinib: First Global Approval. Drugs 2019, 79, 1477–1483. [Google Scholar] [CrossRef]
- Scott, L.J. Larotrectinib: First Global Approval. Drugs 2019, 79, 201–206. [Google Scholar] [CrossRef]
- Drilon, A.; Siena, S.; Ou, S.-H.I.; Patel, M.; Ahn, M.J.; Lee, J.; Bauer, T.M.; Farago, A.F.; Wheler, J.J.; Liu, S.V.; et al. Safety and Antitumor Activity of the Multitargeted Pan-TRK, ROS1, and ALK Inhibitor Entrectinib: Combined Results from Two Phase I Trials (ALKA-372-001 and STARTRK-1). Cancer Discov. 2017, 7, 400–409. [Google Scholar] [CrossRef] [Green Version]
- Maekawa, T.; Ashihara, E.; Kimura, S. The Bcr-Abl Tyrosine Kinase Inhibitor Imatinib and Promising New Agents against Philadelphia Chromosome-Positive Leukemias. Int. J. Clin. Oncol. 2007, 12, 327–340. [Google Scholar] [CrossRef]
- Wozniak, A.; Rutkowski, P.; Schöffski, P.; Ray-Coquard, I.; Hostein, I.; Schildhaus, H.; Le Cesne, A.; Bylina, E.; Limon, J.; Blay, J.; et al. Tumor Genotype Is an Independent Prognostic Factor in Primary Gastrointestinal Stromal Tumors of Gastric Origin: A European Multicenter Analysis Based on ConticaGIST. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2014, 20, 6105–6116. [Google Scholar] [CrossRef] [Green Version]
- Blanke, C.D.; Rankin, C.; Demetri, G.D.; Ryan, C.W.; von Mehren, M.; Benjamin, R.S.; Raymond, A.K.; Bramwell, V.H.C.; Baker, L.H.; Maki, R.G.; et al. Phase III Randomized, Intergroup Trial Assessing Imatinib Mesylate at Two Dose Levels in Patients with Unresectable or Metastatic Gastrointestinal Stromal Tumors Expressing the Kit Receptor Tyrosine Kinase: S0033. J. Clin. Oncol. 2008, 26, 626–632. [Google Scholar] [CrossRef]
- Verweij, J.; Casali, P.G.; Zalcberg, J.; LeCesne, A.; Reichardt, P.; Blay, J.-Y.; Issels, R.; van Oosterom, A.; Hogendoorn, P.C.W.; Van Glabbeke, M.; et al. Progression-Free Survival in Gastrointestinal Stromal Tumours with High-Dose Imatinib: Randomised Trial. Lancet 2004, 364, 1127–1134. [Google Scholar] [CrossRef]
- Mazzocca, A.; Napolitano, A.; Silletta, M.; Spalato Ceruso, M.; Santini, D.; Tonini, G.; Vincenzi, B. New Frontiers in the Medical Management of Gastrointestinal Stromal Tumours. Ther. Adv. Med. Oncol. 2019, 11, 1758835919841946. [Google Scholar] [CrossRef] [Green Version]
- Bamboat, Z.M.; Dematteo, R.P. Updates on the Management of Gastrointestinal Stromal Tumors. Surg. Oncol. Clin. N. Am. 2012, 21, 301–316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reid T Reintroduction of Imatinib in GIST. J. Gastrointest. Cancer 2013, 44, 385–392. [CrossRef] [PubMed] [Green Version]
- Li, G.Z.; Raut, C.P. Targeted Therapy and Personalized Medicine in Gastrointestinal Stromal Tumors: Drug Resistance, Mechanisms, and Treatment Strategies. Onco Targets Ther. 2019, 12, 5123–5133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heinrich, M.C.; Corless, C.L.; Blanke, C.D.; Demetri, G.D.; Joensuu, H.; Roberts, P.J.; Eisenberg, B.L.; von Mehren, M.; Fletcher, C.D.M.; Sandau, K.; et al. Molecular Correlates of Imatinib Resistance in Gastrointestinal Stromal Tumors. J. Clin. Oncol. 2006, 24, 4764–4774. [Google Scholar] [CrossRef]
- Liegl, B.; Kepten, I.; Le, C.; Zhu, M.; Demetri, G.D.; Heinrich, M.C.; Fletcher, C.D.M.; Corless, C.L.; Fletcher, J.A. Heterogeneity of Kinase Inhibitor Resistance Mechanisms in GIST. J. Pathol. 2008, 216, 64–74. [Google Scholar] [CrossRef] [Green Version]
- Kinoshita, K.; Isozaki, K.; Hirota, S.; Nishida, T.; Chen, H.; Nakahara, M.; Nagasawa, Y.; Ohashi, A.; Shinomura, Y.; Kitamura, Y.; et al. C-Kit Gene Mutation at Exon 17 or 13 Is Very Rare in Sporadic Gastrointestinal Stromal Tumors. J. Gastroenterol. Hepatol. 2003, 18, 147–151. [Google Scholar] [CrossRef]
- Antonescu, C.R.; Besmer, P.; Guo, T.; Arkun, K.; Hom, G.; Koryotowski, B.; Leversha, M.A.; Jeffrey, P.D.; Desantis, D.; Singer, S.; et al. Acquired Resistance to Imatinib in Gastrointestinal Stromal Tumor Occurs Through Secondary Gene Mutation. Clin. Cancer Res. 2005, 11, 4182–4190. [Google Scholar] [CrossRef] [Green Version]
- Heinrich, M.C.; Maki, R.G.; Corless, C.L.; Antonescu, C.R.; Harlow, A.; Griffith, D.; Town, A.; McKinley, A.; Ou, W.-B.; Fletcher, J.A.; et al. Primary and Secondary Kinase Genotypes Correlate with the Biological and Clinical Activity of Sunitinib in Imatinib-Resistant Gastrointestinal Stromal Tumor. J. Clin. Oncol. 2008, 26, 5352–5359. [Google Scholar] [CrossRef] [Green Version]
- Antonescu, C.R.; DeMatteo, R.P. CCR 20th Anniversary Commentary: A Genetic Mechanism of Imatinib Resistance in Gastrointestinal Stromal Tumor-Where Are We a Decade Later? Clin. Cancer Res. 2015, 21, 3363–3365. [Google Scholar] [CrossRef] [Green Version]
- Tamborini, E.; Pricl, S.; Negri, T.; Lagonigro, M.S.; Miselli, F.; Greco, A.; Gronchi, A.; Casali, P.G.; Ferrone, M.; Fermeglia, M.; et al. Functional Analyses and Molecular Modeling of Two C-Kit Mutations Responsible for Imatinib Secondary Resistance in GIST Patients. Oncogene 2006, 25, 6140–6146. [Google Scholar] [CrossRef] [Green Version]
- Abrams, T.J.; Lee, L.B.; Murray, L.J.; Pryer, N.K.; Cherrington, J.M. SU11248 Inhibits KIT and Platelet-Derived Growth Factor Receptor Beta in Preclinical Models of Human Small Cell Lung Cancer. Mol. Cancer Ther. 2003, 2, 471–478. [Google Scholar]
- Mendel, D.; Laird, A.; Xin, X.; Louie, S.; Christensen, J.; Li, G.; Schreck, R.; Abrams, T.; Ngai, T.; Lee, L.; et al. In Vivo Antitumor Activity of SU11248, a Novel Tyrosine Kinase Inhibitor Targeting Vascular Endothelial Growth Factor and Platelet-Derived Growth Factor Receptors: Determination of a Pharmacokinetic/Pharmacodynamic Relationship. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2003, 9, 327–337. [Google Scholar]
- Mulet-Margalef, N.; Garcia-Del-Muro, X. Sunitinib in the Treatment of Gastrointestinal Stromal Tumor: Patient Selection and Perspectives. Onco Targets Ther. 2016, 9, 7573–7582. [Google Scholar] [CrossRef] [Green Version]
- Gajiwala, K.S.; Wu, J.C.; Christensen, J.; Deshmukh, G.D.; Diehl, W.; DiNitto, J.P.; English, J.M.; Greig, M.J.; He, Y.-A.; Jacques, S.L.; et al. KIT Kinase Mutants Show Unique Mechanisms of Drug Resistance to Imatinib and Sunitinib in Gastrointestinal Stromal Tumor Patients. Proc. Natl. Acad. Sci. USA 2009, 106, 1542–1547. [Google Scholar] [CrossRef] [Green Version]
- Demetri, G.D.; van Oosterom, A.T.; Garrett, C.R.; Blackstein, M.E.; Shah, M.H.; Verweij, J.; McArthur, G.; Judson, I.R.; Heinrich, M.C.; Morgan, J.A.; et al. Efficacy and Safety of Sunitinib in Patients with Advanced Gastrointestinal Stromal Tumour after Failure of Imatinib: A Randomised Controlled Trial. Lancet 2006, 368, 1329–1338. [Google Scholar] [CrossRef]
- George, S.; Blay, J.Y.; Casali, P.G.; Le Cesne, A.; Stephenson, P.; Deprimo, S.E.; Harmon, C.S.; Law, C.N.J.; Morgan, J.A.; Ray-Coquard, I.; et al. Clinical Evaluation of Continuous Daily Dosing of Sunitinib Malate in Patients with Advanced Gastrointestinal Stromal Tumour after Imatinib Failure. Eur. J. Cancer 2009, 45, 1959–1968. [Google Scholar] [CrossRef]
- Rutkowski, P.; Bylina, E.; Klimczak, A.; Switaj, T.; Falkowski, S.; Kroc, J.; Lugowska, I.; Brzeskwiniewicz, M.; Melerowicz, W.; Osuch, C.; et al. The Outcome and Predictive Factors of Sunitinib Therapy in Advanced Gastrointestinal Stromal Tumors (GIST) after Imatinib Failure—One Institution Study. BMC Cancer 2012, 12, 107. [Google Scholar] [CrossRef] [Green Version]
- Guo, T.; Hajdu, M.; Agaram, N.P.; Shinoda, H.; Veach, D.; Clarkson, B.D.; Maki, R.G.; Singer, S.; Dematteo, R.P.; Besmer, P.; et al. Mechanisms of Sunitinib Resistance in Gastrointestinal Stromal Tumors Harboring KITAY502-3ins Mutation: An in Vitro Mutagenesis Screen for Drug Resistance. Clin. Cancer Res. 2009, 15, 6862–6870. [Google Scholar] [CrossRef] [Green Version]
- Judson, I.R. Prognosis, Imatinib Dose, and Benefit of Sunitinib in GIST: Knowing the Genotype. J. Clin. Oncol. 2008, 26, 5322–5325. [Google Scholar] [CrossRef]
- Nishida, T.; Takahashi, T.; Nishitani, A.; Doi, T.; Shirao, K.; Komatsu, Y.; Nakajima, K.; Hirota, S. Japanese Study Group on GIST Sunitinib-Resistant Gastrointestinal Stromal Tumors Harbor Cis-Mutations in the Activation Loop of the KIT Gene. Int. J. Clin. Oncol. 2009, 14, 143–149. [Google Scholar] [CrossRef]
- Joensuu, H.; De Braud, F.; Grignagni, G.; De Pas, T.; Spitalieri, G.; Coco, P.; Spreafico, C.; Boselli, S.; Toffalorio, F.; Bono, P.; et al. Vatalanib for Metastatic Gastrointestinal Stromal Tumour (GIST) Resistant to Imatinib: Final Results of a Phase II Study. Br. J. Cancer 2011, 104, 1686–1690. [Google Scholar] [CrossRef] [PubMed]
- Call, J.W.; Wang, Y.; Montoya, D.; Scherzer, N.J.; Heinrich, M.C. Survival in Advanced GIST Has Improved over Time and Correlates with Increased Access to Post-Imatinib Tyrosine Kinase Inhibitors: Results from Life Raft Group Registry. Clin. Sarcoma Res. 2019, 9, 4. [Google Scholar] [CrossRef] [PubMed]
- Wilhelm, S.M.; Dumas, J.; Adnane, L.; Lynch, M.; Carter, C.A.; Schütz, G.; Thierauch, K.-H.; Zopf, D. Regorafenib (BAY 73-4506): A New Oral Multikinase Inhibitor of Angiogenic, Stromal and Oncogenic Receptor Tyrosine Kinases with Potent Preclinical Antitumor Activity. Int. J. Cancer 2011, 129, 245–255. [Google Scholar] [CrossRef] [PubMed]
- Mross, K.; Frost, A.; Steinbild, S.; Hedbom, S.; Büchert, M.; Fasol, U.; Unger, C.; Krätzschmar, J.; Heinig, R.; Boix, O.; et al. A Phase I Dose-Escalation Study of Regorafenib (BAY 73-4506), an Inhibitor of Oncogenic, Angiogenic, and Stromal Kinases, in Patients with Advanced Solid Tumors. Clin. Cancer Res. 2012, 18, 2658–2667. [Google Scholar] [CrossRef] [Green Version]
- Ferraro, D.; Zalcberg, J. Regorafenib in Gastrointestinal Stromal Tumors: Clinical Evidence and Place in Therapy. Ther. Adv. Med. Oncol. 2014, 6, 222–228. [Google Scholar] [CrossRef] [Green Version]
- Chamberlain, F.; Farag, S.; Williams-Sharkey, C.; Collingwood, C.; Chen, L.; Mansukhani, S.; Engelmann, B.; Al-Muderis, O.; Chauhan, D.; Thway, K.; et al. Toxicity Management of Regorafenib in Patients with Gastro-Intestinal Stromal Tumour (GIST) in a Tertiary Cancer Centre. Clin. Sarcoma Res. 2020, 10, 1. [Google Scholar] [CrossRef]
- Takahashi, T.; Nishida, T.; Kudo, T.; Boku, N.; Honma, Y.; Komatsu, Y.; Nakatsumi, H.; Matsumoto, K.; Onoe, T.; Oki, E.; et al. Regorafenib as Second-Line Therapy for Imatinib-Resistant Gastrointestinal Stromal Tumor (GIST). JCO 2020, 38, 823. [Google Scholar] [CrossRef]
- Demetri, G.D.; Reichardt, P.; Kang, Y.-K.; Blay, J.-Y.; Rutkowski, P.; Gelderblom, H.; Hohenberger, P.; Leahy, M.; von Mehren, M.; Joensuu, H.; et al. Efficacy and Safety of Regorafenib for Advanced Gastrointestinal Stromal Tumours after Failure of Imatinib and Sunitinib (GRID): An International, Multicentre, Randomised, Placebo-Controlled, Phase 3 Trial. Lancet 2013, 381, 295–302. [Google Scholar] [CrossRef] [Green Version]
- Komatsu, Y.; Doi, T.; Sawaki, A.; Kanda, T.; Yamada, Y.; Kuss, I.; Demetri, G.D.; Nishida, T. Regorafenib for Advanced Gastrointestinal Stromal Tumors Following Imatinib and Sunitinib Treatment: A Subgroup Analysis Evaluating Japanese Patients in the Phase III GRID Trial. Int. J. Clin. Oncol. 2015, 20, 905–912. [Google Scholar] [CrossRef] [Green Version]
- Abbaspour Babaei, M.; Kamalidehghan, B.; Saleem, M.; Huri, H.Z.; Ahmadipour, F. Receptor Tyrosine Kinase (c-Kit) Inhibitors: A Potential Therapeutic Target in Cancer Cells. Drug Des. Devel. Ther. 2016, 10, 2443–2459. [Google Scholar] [CrossRef] [Green Version]
- Montemurro, M.; Schöffski, P.; Reichardt, P.; Gelderblom, H.; Schütte, J.; Hartmann, J.T.; von Moos, R.; Seddon, B.; Joensuu, H.; Wendtner, C.M.; et al. Nilotinib in the Treatment of Advanced Gastrointestinal Stromal Tumours Resistant to Both Imatinib and Sunitinib. Eur. J. Cancer 2009, 45, 2293–2297. [Google Scholar] [CrossRef] [PubMed]
- Sawaki, A.; Nishida, T.; Doi, T.; Yamada, Y.; Komatsu, Y.; Kanda, T.; Kakeji, Y.; Onozawa, Y.; Yamasaki, M.; Ohtsu, A. Phase 2 Study of Nilotinib as Third-Line Therapy for Patients with Gastrointestinal Stromal Tumor. Cancer 2011, 117, 4633–4641. [Google Scholar] [CrossRef]
- Reichardt, P.; Blay, J.-Y.; Gelderblom, H.; Schlemmer, M.; Demetri, G.D.; Bui-Nguyen, B.; McArthur, G.A.; Yazji, S.; Hsu, Y.; Galetic, I.; et al. Phase III Study of Nilotinib versus Best Supportive Care with or without a TKI in Patients with Gastrointestinal Stromal Tumors Resistant to or Intolerant of Imatinib and Sunitinib. Ann. Oncol. 2012, 23, 1680–1687. [Google Scholar] [CrossRef] [PubMed]
- Casali, P.G.; Joensuu, H.; Martin Broto, J.; Garcia del Muro, X.; Blay, J.; May, C.; Pustowka, A.; Reichardt, P. Preliminary Data of Nilotinib in the First-Line Treatment of Patients with Metastatic or Unresectable Gastrointestinal Stromal Tumors (GIST). JCO 2010, 28, TPS332. [Google Scholar] [CrossRef]
- Mir, O.; Cropet, C.; Toulmonde, M.; Cesne, A.L.; Molimard, M.; Bompas, E.; Cassier, P.; Ray-Coquard, I.; Rios, M.; Adenis, A.; et al. Pazopanib plus Best Supportive Care versus Best Supportive Care Alone in Advanced Gastrointestinal Stromal Tumours Resistant to Imatinib and Sunitinib (PAZOGIST): A Randomised, Multicentre, Open-Label Phase 2 Trial. Lancet Oncol. 2016, 17, 632–641. [Google Scholar] [CrossRef]
- Ramaswamy, A.; Pande, N.; Shetty, O.; Shetty, N.; Gupta, S.; Ostwal, V. Pazopanib in Metastatic Multiply Treated Progressive Gastrointestinal Stromal Tumors: Feasible and Efficacious. J. Gastrointest. Oncol. 2016, 7, 638–643. [Google Scholar] [CrossRef] [Green Version]
- Eriksson, M.; Reichardt, P.; Joensuu, H.; Krarup-Hansen, A.; Hagberg, O.; Hohenberger, P.; Hagberg, H.; Hansson, L.; Foukakis, T.; Pulkkanen, K.; et al. Benefit of Pazopanib in Advanced Gastrointestinal Stromal Tumours: Results from a Phase II Trial (SSG XXI, PAGIST). ESMO Open 2021, 6, 100217. [Google Scholar] [CrossRef]
- Park, S.H.; Ryu, M.H.; Ryoo, B.Y.; Im, S.A.; Kwon, H.C.; Lee, S.S.; Park, S.R.; Kang, B.Y.; Kang, Y.K. Sorafenib in Patients with Metastatic Gastrointestinal Stromal Tumors Who Failed Two or More Prior Tyrosine Kinase Inhibitors: A Phase II Study of Korean Gastrointestinal Stromal Tumors Study Group. Investig. New Drugs 2012, 30, 2377–2383. [Google Scholar] [CrossRef]
- Montemurro, M.; Gelderblom, H.; Bitz, U.; Schütte, J.; Blay, J.Y.; Joensuu, H.; Trent, J.; Bauer, S.; Rutkowski, P.; Duffaud, F.; et al. Sorafenib as Third- or Fourth-Line Treatment of Advanced Gastrointestinal Stromal Tumour and Pretreatment Including Both Imatinib and Sunitinib, and Nilotinib: A Retrospective Analysis. Eur. J. Cancer 2013, 49, 1027–1031. [Google Scholar] [CrossRef]
- Brinch, C.; Dehnfeld, M.; Hogdall, E.; Poulsen, T.S.; Toxvaerd, A.; Al-Farra, G.; Bergenfeldt, M.; Krarup-Hansen, A. Outstanding Response to Sorafenib in a Patient with Metastatic Gastrointestinal Stromal Tumour. Case Rep. Oncol. 2021, 14, 1567–1573. [Google Scholar] [CrossRef]
- Montemurro, M.; Cioffi, A.; Dômont, J.; Rutkowski, P.; Roth, A.D.; von Moos, R.; Inauen, R.; Toulmonde, M.; Burkhard, R.O.; Knuesli, C.; et al. Long-Term Outcome of Dasatinib First-Line Treatment in Gastrointestinal Stromal Tumor: A Multicenter, 2-Stage Phase 2 Trial (Swiss Group for Clinical Cancer Research 56/07). Cancer 2018, 124, 1449–1454. [Google Scholar] [CrossRef]
- Schuetze, S.M.; Bolejack, V.; Thomas, D.G.; von Mehren, M.; Patel, S.; Samuels, B.; Choy, E.; D’Amato, G.; Staddon, A.P.; Ganjoo, K.N.; et al. Association of Dasatinib with Progression-Free Survival Among Patients with Advanced Gastrointestinal Stromal Tumors Resistant to Imatinib. JAMA Oncol. 2018, 4, 814–820. [Google Scholar] [CrossRef]
- Li, J.; Zhou, Y.; Zhang, X.; Wu, X.; Zhou, Y.; Liu, X.; Zhang, B.; Wu, X.; Lin, S. Safety and Efficacy of Dasatinib in Patients with Advanced Gastrointestinal Stromal Tumors Refractory to Imatinib and Sunitinib: A Single Arm, Multicenters, Phase 2 Trial. JCO 2019, 37, 138. [Google Scholar] [CrossRef]
- Smith, B.D.; Hood, M.M.; Wise, S.C.; Kaufman, M.D.; Lu, W.-P.; Rutkoski, T.; Flynn, D.L.; Heinrich, M.C. Abstract 2690: DCC-2618 Is a Potent Inhibitor of Wild-Type and Mutant KIT, Including Refractory Exon 17 D816 KIT Mutations, and Exhibits Efficacy in Refractory GIST and AML Xenograft Models. Cancer Res. 2015, 75, 2690. [Google Scholar] [CrossRef]
- Smith, B.D.; Kaufman, M.D.; Lu, W.-P.; Gupta, A.; Leary, C.B.; Wise, S.C.; Rutkoski, T.J.; Ahn, Y.M.; Al-Ani, G.; Bulfer, S.L.; et al. Ripretinib (DCC-2618) Is a Switch Control Kinase Inhibitor of a Broad Spectrum of Oncogenic and Drug-Resistant KIT and PDGFRA Variants. Cancer Cell 2019, 35, 738–751.e9. [Google Scholar] [CrossRef]
- George, S.; Heinrich, M.; Chi, P.; Razak, A.; von Mehren, M.; Gordon, M.; Ganjoo, K.N.; Somaiah, N.; Trent, J.C.; Ahnert, J.R.; et al. Initial Results of Phase I Study of DCC-2618, a Broad-Spectrum KIT and PDGFRa Inhibitor, in Patients (Pts) with Gastrointestinal Stromal Tumor (GIST) by Number of Prior Regimens. Ann. Oncol. 2018, 29, viii576–viii577. [Google Scholar] [CrossRef]
- Blay, J.-Y.; Serrano, C.; Heinrich, M.C.; Zalcberg, J.; Bauer, S.; Gelderblom, H.; Schöffski, P.; Jones, R.L.; Attia, S.; D’Amato, G.; et al. Ripretinib in Patients with Advanced Gastrointestinal Stromal Tumours (INVICTUS): A Double-Blind, Randomised, Placebo-Controlled, Phase 3 Trial. Lancet Oncol. 2020, 21, 923–934. [Google Scholar] [CrossRef]
- Heinrich, M.C.; Jones, R.L.; Gelderblom, H.; George, S.; Schöffski, P.; von Mehren, M.; Zalcberg, J.R.; Kang, Y.-K.; Abdul Razak, A.R.; Trent, J.C.; et al. INTRIGUE: A Phase III, Randomized, Open-Label Study to Evaluate the Efficacy and Safety of Ripretinib versus Sunitinib in Patients with Advanced Gastrointestinal Stromal Tumor Previously Treated with Imatinib. JCO 2022, 40, 359881. [Google Scholar] [CrossRef]
- Liu, B.; Kou, Y. Fourth-Line Rescue Treatment Ripretinib of Advanced Small Intestine Gastrointestinal Stromal Tumors Who Achieved Partial Response: A Case Report. J. Gastrointest. Oncol. 2022, 13, 1505–1513. [Google Scholar] [CrossRef]
- Li, J.; Cai, S.; Zhou, Y.; Zhang, J.; Zhou, Y.; Cao, H.; Wu, X.; Deng, Y.; Huang, Z.; Dong, J.; et al. Efficacy and Safety of Ripretinib in Chinese Patients with Advanced Gastrointestinal Stromal Tumors as a Fourth- or Later-Line Therapy: A Multicenter, Single-Arm, Open-Label Phase II Study. Clin. Cancer Res. 2022, 28, 3425–3432. [Google Scholar] [CrossRef]
- Xiao, X.; Yuan, W.; Wang, C.; Song, H. A Systematic Review and Network Meta-Analysis of the Efficacy and Safety of Third-Line and over Third-Line Therapy after Imatinib and TKI Resistance in Advanced Gastrointestinal Stromal Tumor. Front. Pharmacol. 2022, 13, 978885. [Google Scholar] [CrossRef] [PubMed]
- Gebreyohannes, Y.K.; Burton, E.A.; Wozniak, A.; Matusow, B.; Habets, G.; Wellens, J.; Cornillie, J.; Lin, J.; Nespi, M.; Wu, G.; et al. PLX9486 Shows Anti-Tumor Efficacy in Patient-Derived, Tyrosine Kinase Inhibitor-Resistant KIT-Mutant Xenograft Models of Gastrointestinal Stromal Tumors. Clin. Exp. Med. 2019, 19, 201–210. [Google Scholar] [CrossRef] [PubMed]
- Wagner, A.J.; Tap, W.D.; Shields, A.F.; Patnaik, A.; Chugh, R.; Tinoco, G.; Michelson, G.; Alcantar, O.; Pelayo, M.; Zhang, C.; et al. A Phase I Pharmacokinetic (PK) and Pharmacodynamic (PD) Study of PLX9486 Alone and in Combination (Combo) with the KIT Inhibitors Pexidartinib (Pexi) or Sunitinib (Su) in Patients (Pts) with Advanced Solid Tumors and Gastrointestinal Stromal Tumor (GIST). JCO 2018, 36, 11509. [Google Scholar] [CrossRef]
- Wagner, A.J.; Severson, P.L.; Shields, A.F.; Patnaik, A.; Chugh, R.; Tinoco, G.; Wu, G.; Nespi, M.; Lin, J.; Zhang, Y.; et al. Association of Combination of Conformation-Specific KIT Inhibitors with Clinical Benefit in Patients with Refractory Gastrointestinal Stromal Tumors: A Phase 1b/2a Nonrandomized Clinical Trial. JAMA Oncol. 2021, 7, 1343–1350. [Google Scholar] [CrossRef] [PubMed]
- Markowitz, J.N.; Fancher, K.M. Cabozantinib: A Multitargeted Oral Tyrosine Kinase Inhibitor. Pharmacotherapy 2018, 38, 357–369. [Google Scholar] [CrossRef]
- Gebreyohannes, Y.K.; Schöffski, P.; Van Looy, T.; Wellens, J.; Vreys, L.; Cornillie, J.; Vanleeuw, U.; Aftab, D.T.; Debiec-Rychter, M.; Sciot, R.; et al. Cabozantinib Is Active against Human Gastrointestinal Stromal Tumor Xenografts Carrying Different KIT Mutations. Mol. Cancer Ther. 2016, 15, 2845–2852. [Google Scholar] [CrossRef] [Green Version]
- Lu, T.; Chen, C.; Wang, A.; Jiang, Z.; Qi, Z.; Hu, Z.; Hu, C.; Liu, F.; Wang, W.; Wu, H.; et al. Repurposing Cabozantinib to GISTs: Overcoming Multiple Imatinib-Resistant CKIT Mutations Including Gatekeeper and Activation Loop Mutants in GISTs Preclinical Models. Cancer Lett. 2019, 447, 105–114. [Google Scholar] [CrossRef]
- Schöffski, P.; Mir, O.; Kasper, B.; Papai, Z.; Blay, J.-Y.; Italiano, A.; Benson, C.; Kopeckova, K.; Ali, N.; Dileo, P.; et al. Activity and Safety of the Multi-Target Tyrosine Kinase Inhibitor Cabozantinib in Patients with Metastatic Gastrointestinal Stromal Tumour after Treatment with Imatinib and Sunitinib: European Organisation for Research and Treatment of Cancer Phase II Trial 1317 “CaboGIST”. Eur. J. Cancer 2020, 134, 62–74. [Google Scholar] [CrossRef]
- Evans, E.K.; Hodous, B.L.; Gardino, A.K.; Davis, A.; Zhu, J.; Shutes, A.; Kim, J.L.; Wilson, K.J.; Wilson, D.; Zhang, Y.; et al. Abstract 791: BLU-285, the First Selective Inhibitor of PDGFRα D842V and KIT Exon 17 Mutants. Cancer Res. 2015, 75, 791. [Google Scholar] [CrossRef]
- Heinrich, M.C.; Jones, R.L.; von Mehren, M.; Schöffski, P.; Serrano, C.; Kang, Y.-K.; Cassier, P.A.; Mir, O.; Eskens, F.; Tap, W.D.; et al. Avapritinib in Advanced PDGFRA D842V-Mutant Gastrointestinal Stromal Tumour (NAVIGATOR): A Multicentre, Open-Label, Phase 1 Trial. Lancet Oncol. 2020, 21, 935–946. [Google Scholar] [CrossRef]
- Jones, R.L.; Serrano, C.; von Mehren, M.; George, S.; Heinrich, M.C.; Kang, Y.-K.; Schöffski, P.; Cassier, P.A.; Mir, O.; Chawla, S.P.; et al. Avapritinib in Unresectable or Metastatic PDGFRA D842V-Mutant Gastrointestinal Stromal Tumours: Long-Term Efficacy and Safety Data from the NAVIGATOR Phase I Trial. Eur. J. Cancer 2021, 145, 132–142. [Google Scholar] [CrossRef]
- George, S.; Jones, R.L.; Bauer, S.; Kang, Y.-K.; Schöffski, P.; Eskens, F.; Mir, O.; Cassier, P.A.; Serrano, C.; Tap, W.D.; et al. Avapritinib in Patients with Advanced Gastrointestinal Stromal Tumors Following at Least Three Prior Lines of Therapy. Oncologist 2021, 26, e639–e649. [Google Scholar] [CrossRef]
- Kang, Y.-K.; George, S.; Jones, R.L.; Rutkowski, P.; Shen, L.; Mir, O.; Patel, S.; Zhou, Y.; von Mehren, M.; Hohenberger, P.; et al. Avapritinib Versus Regorafenib in Locally Advanced Unresectable or Metastatic GI Stromal Tumor: A Randomized, Open-Label Phase III Study. J. Clin. Oncol. 2021, 39, 3128–3139. [Google Scholar] [CrossRef]
- von Mehren, M.; Heinrich, M.C.; Shi, H.; Iannazzo, S.; Mankoski, R.; Dimitrijević, S.; Hoehn, G.; Chiroli, S.; George, S. Clinical Efficacy Comparison of Avapritinib with Other Tyrosine Kinase Inhibitors in Gastrointestinal Stromal Tumors with PDGFRA D842V Mutation: A Retrospective Analysis of Clinical Trial and Real-World Data. BMC Cancer 2021, 21, 291. [Google Scholar] [CrossRef]
- Mu, J.; Sun, X.; Zhao, Z.; Sun, H.; Sun, P. BRD9 Inhibition Promotes PUMA-Dependent Apoptosis and Augments the Effect of Imatinib in Gastrointestinal Stromal Tumors. Cell Death Dis. 2021, 12, 962. [Google Scholar] [CrossRef]
- Volckmar, A.-L.; Sültmann, H.; Riediger, A.; Fioretos, T.; Schirmacher, P.; Endris, V.; Stenzinger, A.; Dietz, S. A Field Guide for Cancer Diagnostics Using Cell-Free DNA: From Principles to Practice and Clinical Applications. Genes Chromosomes Cancer 2018, 57, 123–139. [Google Scholar] [CrossRef]
- Kang, G.; Sohn, B.S.; Pyo, J.-S.; Kim, J.Y.; Lee, B.; Kim, K.-M. Detecting Primary KIT Mutations in Presurgical Plasma of Patients with Gastrointestinal Stromal Tumor. Mol. Diagn. Ther. 2016, 20, 347–351. [Google Scholar] [CrossRef]
- Wada, N.; Kurokawa, Y.; Takahashi, T.; Hamakawa, T.; Hirota, S.; Naka, T.; Miyazaki, Y.; Makino, T.; Yamasaki, M.; Nakajima, K.; et al. Detecting Secondary C-KIT Mutations in the Peripheral Blood of Patients with Imatinib-Resistant Gastrointestinal Stromal Tumor. Oncology 2016, 90, 112–117. [Google Scholar] [CrossRef]
- Deprimo, S.; Huang, X.; Blackstein, M.; Garrett, C.; Harmon, C.; Schöffski, P.; Shah, M.; Verweij, J.; Baum, C.; Demetri, G. Circulating Levels of Soluble KIT Serve as a Biomarker for Clinical Outcome in Gastrointestinal Stromal Tumor Patients Receiving Sunitinib Following Imatinib Failure. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2009, 15, 5869–5877. [Google Scholar] [CrossRef] [Green Version]
- Maier, J.; Lange, T.; Kerle, I.; Specht, K.; Bruegel, M.; Wickenhauser, C.; Jost, P.; Niederwieser, D.; Peschel, C.; Duyster, J.; et al. Detection of Mutant Free Circulating Tumor DNA in the Plasma of Patients with Gastrointestinal Stromal Tumor Harboring Activating Mutations of CKIT or PDGFRA. Clin. Cancer Res. 2013, 19, 4854–4867. [Google Scholar] [CrossRef] [Green Version]
- Jilg, S.; Rassner, M.; Maier, J.; Waldeck, S.; Kehl, V.; Follo, M.; Philipp, U.; Sauter, A.; Specht, K.; Mitschke, J.; et al. Circulating CKIT and PDGFRA DNA Indicates Disease Activity in Gastrointestinal Stromal Tumor (GIST). Int. J. Cancer 2019, 145, 2292–2303. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, Y.; Fujisawa, T.; Taniguchi, H.; Bando, H.; Okamoto, W.; Tsuchihara, K.; Yoshino, T.; Ohtsu, A. SCRUM-Japan GI-SCREEN and MONSTAR-SCREEN: Path to the Realization of Biomarker-Guided Precision Oncology in Advanced Solid Tumors. Cancer Sci. 2021, 112, 4425–4432. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, Y.; Taniguchi, H.; Ikeda, M.; Bando, H.; Kato, K.; Morizane, C.; Esaki, T.; Komatsu, Y.; Kawamoto, Y.; Takahashi, N.; et al. Clinical Utility of Circulating Tumor DNA Sequencing in Advanced Gastrointestinal Cancer: SCRUM-Japan GI-SCREEN and GOZILA Studies. Nat. Med. 2020, 26, 1859–1864. [Google Scholar] [CrossRef] [PubMed]
- Ko, T.K.; Lee, E.; Ng, C.C.-Y.; Yang, V.S.; Farid, M.; Teh, B.T.; Chan, J.Y.; Somasundaram, N. Circulating Tumor DNA Mutations in Progressive Gastrointestinal Stromal Tumors Identify Biomarkers of Treatment Resistance and Uncover Potential Therapeutic Strategies. Front. Oncol. 2022, 12, 840843. [Google Scholar] [CrossRef]
- Lasota, J.; Felisiak-Golabek, A.; Wasag, B.; Kowalik, A.; Zięba, S.; Chłopek, M.; Wang, Z.-F.; Coates, T.; Kopczynski, J.; Gozdz, S.; et al. Frequency and Clinicopathologic Profile of PIK3CA Mutant GISTs: Molecular Genetic Study of 529 Cases. Mod. Pathol. 2016, 29, 275–282. [Google Scholar] [CrossRef] [Green Version]
- Floris, G.; Wozniak, A.; Sciot, R.; Li, H.; Friedman, L.; Van Looy, T.; Wellens, J.; Vermaelen, P.; Deroose, C.M.; Fletcher, J.A.; et al. A Potent Combination of the Novel PI3K Inhibitor, GDC-0941, with Imatinib in Gastrointestinal Stromal Tumor Xenografts: Long-Lasting Responses after Treatment Withdrawal. Clin. Cancer Res. 2013, 19, 620–630. [Google Scholar] [CrossRef] [Green Version]
- Gupta, A.; Ma, S.; Che, K.; Pobbati, A.V.; Rubin, B.P. Inhibition of PI3K and MAPK Pathways along with KIT Inhibitors as a Strategy to Overcome Drug Resistance in Gastrointestinal Stromal Tumors. PLoS ONE 2021, 16, e0252689. [Google Scholar] [CrossRef]
- Pantaleo, M.A.; Heinrich, M.C.; Italiano, A.; Valverde, C.; Schöffski, P.; Grignani, G.; Reyners, A.K.L.; Bauer, S.; Reichardt, P.; Stark, D.; et al. A Multicenter, Dose-Finding, Phase 1b Study of Imatinib in Combination with Alpelisib as Third-Line Treatment in Patients with Advanced Gastrointestinal Stromal Tumor. BMC Cancer 2022, 22, 511. [Google Scholar] [CrossRef]


| Mutations | Molecular GIST Subtype | Imatinib | Sunitinib | Regorafenib | Ripretinib | Avapritinib |
|---|---|---|---|---|---|---|
| KIT | Exon 9 | S | ||||
| Exon 11 | S | |||||
| Exon 13 (V654A) | R | S | R | R | ||
| Exon 14 (T670I) | R | S | S | S | ||
| Exon 17 (D816V, D820E, and N822K) | R | R | S/R | S/R | ||
| Exon 18 (A829P) | R | R | S | S | ||
| PDGFRA | Exon 12 | S | S | S | S | S |
| Exon 13 | R | R | R | R | ||
| Exon 14 | R | R | R | R | R | |
| Exon 15 | R | R | R | R | R | |
| Exon 18 (D842V) | R | R | R | R | S | |
| Exon 18 (Non-D842V) | S | S | S | S | S |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Masucci, M.T.; Motti, M.L.; Minopoli, M.; Di Carluccio, G.; Carriero, M.V. Emerging Targeted Therapeutic Strategies to Overcome Imatinib Resistance of Gastrointestinal Stromal Tumors. Int. J. Mol. Sci. 2023, 24, 6026. https://doi.org/10.3390/ijms24076026
Masucci MT, Motti ML, Minopoli M, Di Carluccio G, Carriero MV. Emerging Targeted Therapeutic Strategies to Overcome Imatinib Resistance of Gastrointestinal Stromal Tumors. International Journal of Molecular Sciences. 2023; 24(7):6026. https://doi.org/10.3390/ijms24076026
Chicago/Turabian StyleMasucci, Maria Teresa, Maria Letizia Motti, Michele Minopoli, Gioconda Di Carluccio, and Maria Vincenza Carriero. 2023. "Emerging Targeted Therapeutic Strategies to Overcome Imatinib Resistance of Gastrointestinal Stromal Tumors" International Journal of Molecular Sciences 24, no. 7: 6026. https://doi.org/10.3390/ijms24076026