
No Difference in Penetrance between Truncating and Missense/Aberrant Splicing Pathogenic Variants in MLH1 and MSH2: A Prospective Lynch Syndrome Database Study

 , , , , , , , , , , , , ,
, , , , , , , , , , , , ,  , , , , , , , , , , , , , , , , , , , and
, , , , , , , , , , , , , , , , , , , and
Abstract
:1. Introduction
2. Methods
2.1. The Prospective Lynch Syndrome Database (PLSD) Design
2.2. MMR Gene Variant Categorization
2.3. Cancer Risk by Gene and Type of Genetic Variant
2.4. Ethics Statement
3. Results
3.1. Characterization of Path_MLH1 and Path_MSH2 Genetic Variants
3.2. Cumulative Cancer Incidence by Gene and Type of Genetic Variant
4. Discussion and Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Dominguez-Valentin, M.; Sampson, J.R.; Seppälä, T.T.; Broeke, S.W.T.; Be, J.-P.P.; Nakken, S.; Engel, C.; Aretz, S.; Jenkins, M.; Sunde, L.; et al. Cancer risks by gene, age, and gender in 6350 carriers of pathogenic mismatch repair variants: Findings from the Prospective Lynch Syndrome Database. Genet. Med. 2020, 22, 15–25. [Google Scholar] [CrossRef] [Green Version]
- Møller, P.; Seppälä, T.; Bernstein, I.; Holinski-Feder, E.; Sala, P.; Evans, D.G.; Lindblom, A.; Macrae, F.; Blanco, I.; Sijmons, R.; et al. Cancer incidence and survival in Lynch syndrome patients receiving colonoscopic and gynaecological surveillance: First report from the prospective Lynch syndrome database. Gut 2017, 66, 464–472. [Google Scholar] [CrossRef]
- Møller, P.; Seppälä, T.; Bernstein, I.; Holinski-Feder, E.; Sala, P.; Evans, D.G.; Lindblom, A.; Macrae, F.; Blanco, I.; Sijmons, R.; et al. Incidence of and survival after subsequent cancers in carriers of pathogenic MMR variants with previous cancer: A report from the prospective Lynch syndrome database. Gut 2017, 66, 1657–1664. [Google Scholar] [CrossRef]
- Møller, P.; Seppälä, T.; Bernstein, I.; Holinski-Feder, E.; Sala, P.; Evans, D.G.; Lindblom, A.; Macrae, F.; Blanco, I.; Sijmons, R.H.; et al. Cancer risk and survival in path_MMR carriers by gene and gender up to 75 years of age: A report from the Prospective Lynch Syndrome Database. Gut 2018, 67, 1306–1316. [Google Scholar] [CrossRef] [Green Version]
- Lynch, H.T.; Snyder, C.L.; Shaw, T.G.; Heinen, C.D.; Hitchins, M.P. Milestones of Lynch syndrome: 1895–2015. Nat. Rev. Cancer 2015, 15, 181–194. [Google Scholar] [CrossRef] [PubMed]
- Ryan, N.; Morris, J.; Green, K.; Lalloo, F.; Woodward, E.R.; Hill, J.; Crosbie, E.J.; Evans, D.G. Association of Mismatch Repair Mutation with Age at Cancer Onset in Lynch Syndrome. JAMA Oncol. 2017, 3, 1702–1706. [Google Scholar] [CrossRef]
- Seppälä, T.T.; Dominguez-Valentin, M.; Sampson, J.R.; Møller, P. Prospective observational data informs understanding and future management of Lynch syndrome: Insights from the Prospective Lynch Syndrome Database (PLSD). Fam. Cancer 2021, 20, 35–39. [Google Scholar] [CrossRef] [PubMed]
- Wijnen, J.T.; Brohet, R.M.; van Eijk, R.; Jagmohan–Changur, S.; Middeldorp, A.; Tops, C.M.; Van Puijenbroek, M.; Ausems, M.G.E.M.; García, E.G.; Hes, F.J.; et al. Chromosome 8q23.3 and 11q23.1 Variants Modify Colorectal Cancer Risk in Lynch Syndrome. Gastroenterology 2009, 136, 131–137. [Google Scholar] [CrossRef]
- Talseth-Palmer, B.A.; Wijnen, J.T.; Brenne, I.S.; Jagmohan-Changur, S.; Barker, D.; Ashton, K.A.; Tops, C.M.; Evans, T.-J.; McPhillips, M.; Groombridge, C.; et al. Combined analysis of three lynch syndrome cohorts confirms the modifying effects of 8q23.3 and 11q23.1 in MLH1 mutation carriers. Int. J. Cancer 2012, 132, 1556–1564. [Google Scholar] [CrossRef] [PubMed]
- Ten Broeke, S.W.; Van Der Klift, H.M.; Tops, C.M.; Aretz, S.; Bernstein, I.; Buchanan, D.D.; De La Chapelle, A.; Capella, G.; Clendenning, M.; Engel, C.; et al. Cancer Risks for PMS2-Associated Lynch Syndrome. J. Clin. Oncol. 2018, 36, 2961–2968. [Google Scholar] [CrossRef] [Green Version]
- Peltomäki, P.; Olkinuora, A.; Nieminen, T.T. Updates in the field of hereditary nonpolyposis colorectal cancer. Expert Rev. Gastroenterol. Hepatol. 2020, 14, 707–720. [Google Scholar] [CrossRef]
- Peltomäki, P. Update on Lynch syndrome genomics. Fam. Cancer 2016, 15, 385–393. [Google Scholar] [CrossRef] [Green Version]
- Cravo, M.; Afonso, A.; Lage, P.; Albuquerque, C.; Maia, L.; Lacerda, C.; Fidalgo, P.; Chaves, P.; Cruz, C.; Nobre-Leitão, C. Pathogenicity of missense and splice site mutations in hMSH2 and hMLH1 mismatch repair genes: Implications for genetic testing. Gut 2002, 50, 405–412. [Google Scholar] [CrossRef]
- Liu, B.; Parsons, R.E.; Hamilton, S.R.; Petersen, G.M.; Lynch, H.T.; Watson, P.; Markowitz, S.; Willson, J.K.V.; Green, J.; de la Chapelle, A.; et al. hMSH2 mutations in hereditary nonpolyposis colorectal cancer kindreds. Cancer Res. 1994, 54, 4590–4594. [Google Scholar] [PubMed]
- Papadopoulos, N.; Nicolaides, N.C.; Wei, Y.F.; Ruben, S.M.; Carter, K.C.; Rosen, C.A.; Haseltine, W.A.; Fleischmann, R.D.; Fraser, C.; Adams, M.D.; et al. Mutation of a mutL homolog in hereditary colon cancer. Science 1994, 263, 1625–1629. [Google Scholar] [CrossRef] [PubMed]
- Peltomaki, P.; Vasen, H. Mutations predisposing to hereditary nonpolyposis colorectal cancer: Database and results of a collaborative study. The International Collaborative Group on Hereditary Nonpolyposis Colorectal Cancer. Gastroenterology 1997, 113, 1146–1158. [Google Scholar] [CrossRef]
- Li, L.; Hamel, N.; Baker, K.; McGuffin, M.J.; Couillard, M.; Gologan, A.; Marcus, V.A.; Chodirker, B.; Chudley, A.; Stefanovici, C.; et al. A homozygousPMS2founder mutation with an attenuated constitutional mismatch repair deficiency phenotype. J. Med. Genet. 2015, 52, 348–352. [Google Scholar] [CrossRef] [PubMed]
- Auclair, J.; Busine, M.P.; Navarro, C.; Ruano, E.; Montmain, G.; Desseigne, F.; Saurin, J.C.; Lasset, C.; Bonadona, V.; Giraud, S.; et al. Systematic mRNA analysis for the effect ofMLH1 andMSH2 missense and silent mutations on aberrant splicing. Hum. Mutat. 2006, 27, 145–154. [Google Scholar] [CrossRef]
- Soukarieh, O.; Gaildrat, P.; Hamieh, M.; Drouet, A.; Baert-Desurmont, S.; Frébourg, T.; Tosi, M.; Martins, A. Exonic Splicing Mutations Are More Prevalent than Currently Estimated and Can Be Predicted by Using In Silico Tools. PLoS Genet. 2016, 12, e1005756. [Google Scholar] [CrossRef] [Green Version]
- Thompson, B.A.; Walters, R.; Parsons, M.T.; Dumenil, T.; Drost, M.; Tiersma, Y.; Lindor, N.M.; Tavtigian, S.V.; De Wind, N.; Spurdle, A.B.; et al. Contribution of mRNA Splicing to Mismatch Repair Gene Sequence Variant Interpretation. Front. Genet. 2020, 11, 798. [Google Scholar] [CrossRef]
- Maccaroni, E.; Bracci, R.; Giampieri, R.; Bianchi, F.; Belvederesi, L.; Brugiati, C.; Pagliaretta, S.; Del Prete, M.; Scartozzi, M.; Cascinu, S. Prognostic impact of mismatch repair genes germline defects in colorectal cancer patients: Are all mutations equal? Oncotarget 2015, 6, 38737–38748. [Google Scholar] [CrossRef] [Green Version]
- Møller, P. The Prospective Lynch Syndrome Database reports enable evidence-based personal precision health care. Hered. Cancer Clin. Pract. 2020, 18, 6–7. [Google Scholar] [CrossRef]
- Thompson, B.A.; Spurdle, A.B.; Plazzer, J.-P.; Greenblatt, M.S.; Akagi, K.; Al-Mulla, F.; Bapat, B.; Bernstein, I.; Capellá, G.; den Dunnen, J.T.; et al. Application of a 5-tiered scheme for standardized classification of 2360 unique mismatch repair gene variants in the InSiGHT locus-specific database. Nat. Genet. 2014, 46, 107–115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seppälä, T.; Pylvänäinen, K.; Evans, D.G.; Järvinen, H.; Renkonen-Sinisalo, L.; Bernstein, I.; Holinski-Feder, E.; Sala, P.; Lindblom, A.; Macrae, F.; et al. Colorectal cancer incidence in path_MLH1 carriers subjected to different follow-up protocols: A Prospective Lynch Syndrome Database report. Hered. Cancer Clin. Pract. 2017, 15, 18. [Google Scholar] [CrossRef] [Green Version]
- Seppälä, T.T.; Ahadova, A.; Dominguez-Valentin, M.; Macrae, F.; Evans, D.G.; Therkildsen, C.; Sampson, J.; Scott, R.; Burn, J.; Möslein, G.; et al. Lack of association between screening interval and cancer stage in Lynch syndrome may be accounted for by over-diagnosis; a prospective Lynch syndrome database report. Hered. Cancer Clin. Pract. 2019, 17, 8. [Google Scholar] [CrossRef]
- Adzhubei, I.; Jordan, D.; Sunyaev, S.R. Predicting Functional Effect of Human Missense Mutations Using PolyPhen-2. Curr. Protoc. Hum. Genet. 2013, 76, 7.20.1–7.20.41. [Google Scholar] [CrossRef] [Green Version]
- Ng, P.C.; Henikoff, S. SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res. 2003, 31, 3812–3814. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moghadasi, S.; Meeks, H.D.; Vreeswijk, M.P.; Janssen, L.A.; Borg, Å.; Ehrencrona, H.; Paulsson-Karlsson, Y.; Wappenschmidt, B.; Engel, C.; Gehrig, A.; et al. The BRCA1 c. 5096G>A p.Arg1699Gln (R1699Q) intermediate risk variant: Breast and ovarian cancer risk estimation and recommendations for clinical management from the ENIGMA consortium. J. Med. Genet. 2017, 55, 15–20. [Google Scholar] [CrossRef]
- Spurdle, A.B.; Whiley, P.J.; Thompson, B.; Feng, B.; Healey, S.; Brown, M.; Pettigrew, C.; Van Asperen, C.J.; Ausems, M.G.E.M.; Kattentidt-Mouravieva, A.A.; et al. BRCA1 R1699Q variant displaying ambiguous functional abrogation confers intermediate breast and ovarian cancer risk. J. Med. Genet. 2012, 49, 525–532. [Google Scholar] [CrossRef] [PubMed]
- Drost, M.; Ms, Y.T.; Thompson, B.A.; Frederiksen, J.H.; Keijzers, G.; Glubb, D.; Kathe, S.; Osinga, J.; Westers, H.; Pappas, L.; et al. A functional assay–based procedure to classify mismatch repair gene variants in Lynch syndrome. Genet. Med. 2019, 21, 1486–1496. [Google Scholar] [CrossRef]
- Ten Broeke, S.W.; Brohet, R.M.; Tops, C.M.; van der Klift, H.M.; Velthuizen, M.E.; Bernstein, I.; Munar, G.C.; Garcia, E.G.; Hoogerbrugge, N.; Letteboer, T.G.W.; et al. Lynch syndrome caused by germline PMS2 mutations: Delineating the cancer risk. J. Clin. Oncol. 2015, 33, 319–325. [Google Scholar] [CrossRef] [PubMed]
- Suerink, M.; Rodríguez-Girondo, M.; Van Der Klift, H.M.; Colas, C.; Brugieres, L.; Lavoine, N.; Jongmans, M.; Munar, G.C.; Evans, D.G.; Farrell, M.P.; et al. An alternative approach to establishing unbiased colorectal cancer risk estimation in Lynch syndrome. Genet. Med. 2019, 21, 2706–2712. [Google Scholar] [CrossRef] [PubMed]
- Suerink, M.; Van Der Klift, H.M.; Broeke, S.W.T.; Dekkers, O.M.; Bernstein, I.; Munar, G.C.; Garcia, E.G.; Hoogerbrugge, N.; Letteboer, T.G.W.; Menko, F.H.; et al. The effect of genotypes and parent of origin on cancer risk and age of cancer development in PMS2 mutation carriers. Genet. Med. 2015, 18, 405–409. [Google Scholar] [CrossRef]
- Ballhausen, A.; Przybilla, M.J.; Jendrusch, M.; Haupt, S.; Pfaffendorf, E.; Seidler, F.; Witt, J.; Sanchez, A.H.; Urban, K.; Draxlbauer, M.; et al. The shared frameshift mutation landscape of microsatellite-unstable cancers suggests immunoediting during tumor evolution. Nat. Commun. 2020, 11, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Ahadova, A.; Doeberitz, M.V.K.; Bläker, H.; Kloor, M. CTNNB1-mutant colorectal carcinomas with immediate invasive growth: A model of interval cancers in Lynch syndrome. Fam. Cancer 2016, 15, 579–586. [Google Scholar] [CrossRef] [PubMed]
- Ahadova, A.; Seppälä, T.; Engel, C.; Gallon, R.; Burn, J.; Holinski-Feder, E.; Steinke-Lange, V.; Möslein, G.; Nielsen, M.; Broeke, S.T.; et al. The “unnatural” history of colorectal cancer in Lynch syndrome: Lessons from colonoscopy surveillance. Int. J. Cancer 2021, 148, 800–811. [Google Scholar] [CrossRef]



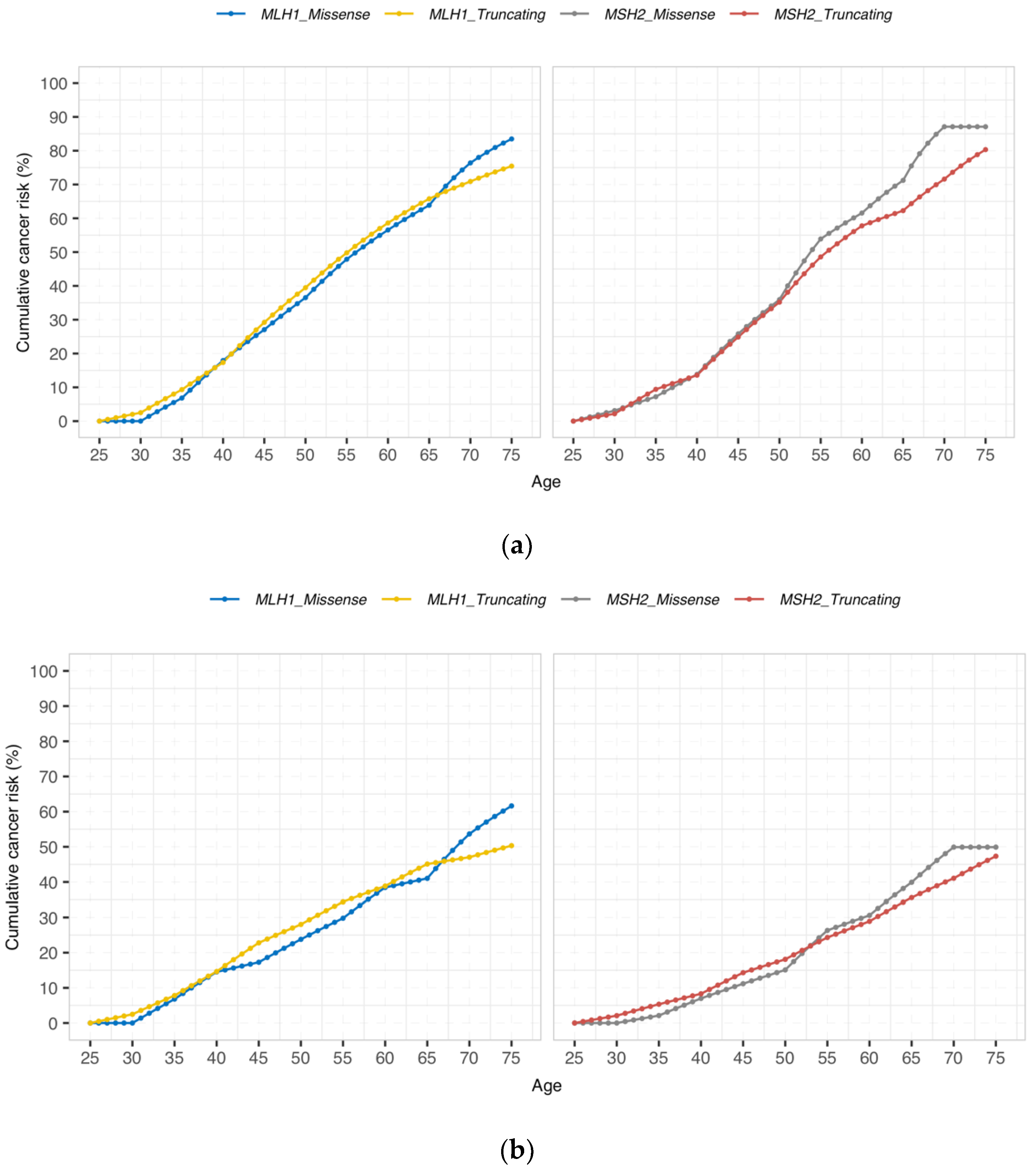{kind=link}
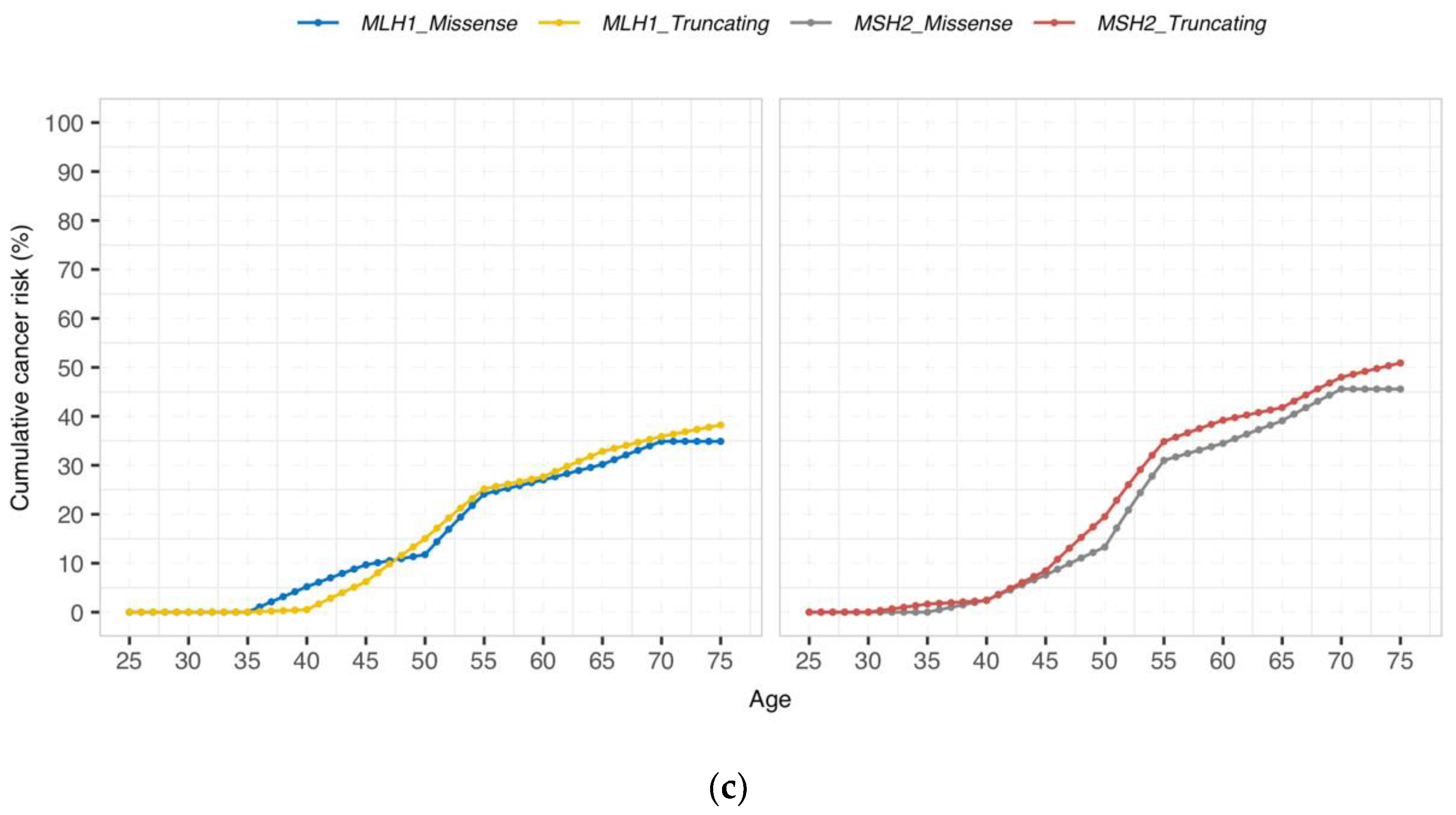{kind=link}
| Categorization Group | Variant Type | Gene | Number of Carriers | Sum of the FUP Years | Mean of the FUP Years | 95% CI |
|---|---|---|---|---|---|---|
| Missense or aberrant splicing | Aberrant Splicing | MLH1 | 233 | 1829 | 7.8 | (7.1–8.5) |
| Aberrant Splicing | MSH2 | 350 | 2778 | 7.9 | (7.4–8.4) | |
| Missense | MLH1 | 345 | 2715 | 7.9 | (7.4–8.4) | |
| Missense | MSH2 | 117 | 883 | 7.5 | (6.7–8.3) | |
| Total | 1045 | 8205 | 7.9 | (7.6–8.2) | ||
| Truncating | Canonical Splicing | MLH1 | 501 | 4709 | 9.4 | (8.9–9.9) |
| Canonical Splicing | MSH2 | 185 | 1635 | 8.8 | (8.0–9.6) | |
| Exon Deletion | MLH1 | 688 | 7643 | 11.1 | (10.6–11.6) | |
| Exon Deletion | MSH2 | 579 | 4207 | 7.3 | (6.9–7.7) | |
| Nonsense | MLH1 | 324 | 2880 | 8.9 | (8.3–9.5) | |
| Nonsense | MSH2 | 608 | 4929 | 8.1 | (7.7–8.5) | |
| Frameshift | MLH1 | 482 | 3722 | 7.7 | (7.3–8.1) | |
| Frameshift | MSH2 | 563 | 4416 | 7.8 | (7.4–8.2) | |
| Total | 3930 | 34,141 | 8.7 | (8.5–8.9) | ||
| Others | Exon Duplication | MLH1 | 1 | 1 | 1 | (1.0–1.0) |
| Exon Duplication | MSH2 | 16 | 71 | 4.4 | (2.7–6.1) | |
| Inframe Indel | MLH1 | 85 | 790 | 9.3 | (8.3–10.3) | |
| Inframe Indel | MSH2 | 93 | 811 | 8.7 | (7.7–9.7) | |
| Initiation Codon | MLH1 | 8 | 36 | 4.5 | (1.5–7.5) | |
| Intronic | MSH2 | 3 | 25 | 8.3 | (2.1–14.5) | |
| Undefined | MLH1 | 18 | 249 | 13.8 | (10.7–16.9) | |
| Total | 224 | 1983 |
| Cumulative Incidences (95% CI) | |||||
|---|---|---|---|---|---|
| Age | MLH1 Missense/Aberrant Splicing | MLH1 Truncating | MSH2 Missense/Aberrant Splicing | MSH2 Truncating | |
| Any cancer | 30 | 0 (0–0) | 2.5 (0.3–4.7) | 3.1 (0–9.1) | 2.2 (0–4.6) |
| 40 | 17.9 (9.3–26.5) | 17.3 (13.1–21.6) | 13.8 (4.1–23.5) | 13.6 (8.9–18.2) | |
| 50 | 36.5 (26.6–46.5) | 39.5 (34.5–44.5) | 36.0 (23.9–48.0) | 35.2 (29.3–41.1) | |
| 60 | 56.6 (44.6–68.4) | 58.6 (53.4–63.9) | 61.6 (49.5–73.6) | 57.8 (51.4–64.1) | |
| 70 | 76.4 (63.6–89.2) | 71.0 (65.1–76.7) | 87.1 (75.6–98.6) | 71.6 (64.4–78.8) | |
| 75 | 83.5 (71.4–95.6) | 75.4 (69.1–81.8) | 87.1 (75.6–98.6) | 80.3 (73.3–87.4) | |
| Colorectal cancer | 30 | 0 (0–0) | 2.5 (0.3–4.6) | 0 (0–0) | 2.1 (0–4.4) |
| 40 | 14.5 (6.5–22.6) | 14.6 (10.6–18.6) | 7.0 (0.3–13.6) | 8.3 (4.5–12.1) | |
| 50 | 23.8 (14.6–33.0) | 28.0 (23.3–32.7) | 15.1 (6.4–23.8) | 18.1 (13.3–22.9) | |
| 60 | 38.4 (26.5–50.4) | 38.9 (33.7–44.0) | 30.6 (19.7–41.5) | 28.9 (23.3–34.5) | |
| 70 | 53.7 (39.0–68.3) | 47.0 (41.2–52.8) | 49.9 (36.4–63.4) | 41.1 (34.2–48.0) | |
| 75 | 61.6 (45.9–77.4) | 50.3 (43.8–56.8) | 49.9 (36.4–63.4) | 47.3 (39.6–55.1) | |
| Endometrial cancer | 30 | 0 (0–0) | 0 (0–0) | 0 (0–0) | 0 (0–0) |
| 40 | 5.2 (0–10.9) | 0.5 (0–1.5) | 2.5 (0–7.2) | 2.4 (0–5.0) | |
| 50 | 11.8 (3.5–20.0) | 15.0 (10.1–19.9) | 13.3 (2.4–24.2) | 19.5 (12.9–26.1) | |
| 60 | 27.0 (13.9–40.1) | 27.7 (21.0–34.3) | 34.5 (17.6–51.3) | 39.2 (30.3–48.1) | |
| 70 | 34.9 (19.2–50.6) | 35.9 (27.6–44.2) | 45.6 (25.6–65.6) | 48.0 (37.4–58.5) | |
| 75 | 34.9 (19.2–50.6) | 38.2 (29.0–47.4) | 45.6 (25.6–65.6) | 50.9 (39.5–62.3) | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dominguez-Valentin, M.; Plazzer, J.-P.; Sampson, J.R.; Engel, C.; Aretz, S.; Jenkins, M.A.; Sunde, L.; Bernstein, I.; Capella, G.; Balaguer, F.; et al. No Difference in Penetrance between Truncating and Missense/Aberrant Splicing Pathogenic Variants in MLH1 and MSH2: A Prospective Lynch Syndrome Database Study. J. Clin. Med. 2021, 10, 2856. https://doi.org/10.3390/jcm10132856
Dominguez-Valentin M, Plazzer J-P, Sampson JR, Engel C, Aretz S, Jenkins MA, Sunde L, Bernstein I, Capella G, Balaguer F, et al. No Difference in Penetrance between Truncating and Missense/Aberrant Splicing Pathogenic Variants in MLH1 and MSH2: A Prospective Lynch Syndrome Database Study. Journal of Clinical Medicine. 2021; 10(13):2856. https://doi.org/10.3390/jcm10132856
Chicago/Turabian StyleDominguez-Valentin, Mev, John-Paul Plazzer, Julian R. Sampson, Christoph Engel, Stefan Aretz, Mark A. Jenkins, Lone Sunde, Inge Bernstein, Gabriel Capella, Francesc Balaguer, and et al. 2021. "No Difference in Penetrance between Truncating and Missense/Aberrant Splicing Pathogenic Variants in MLH1 and MSH2: A Prospective Lynch Syndrome Database Study" Journal of Clinical Medicine 10, no. 13: 2856. https://doi.org/10.3390/jcm10132856