
Pathogenesis of IgA Nephropathy: Current Understanding and Implications for Development of Disease-Specific Treatment

 and
and 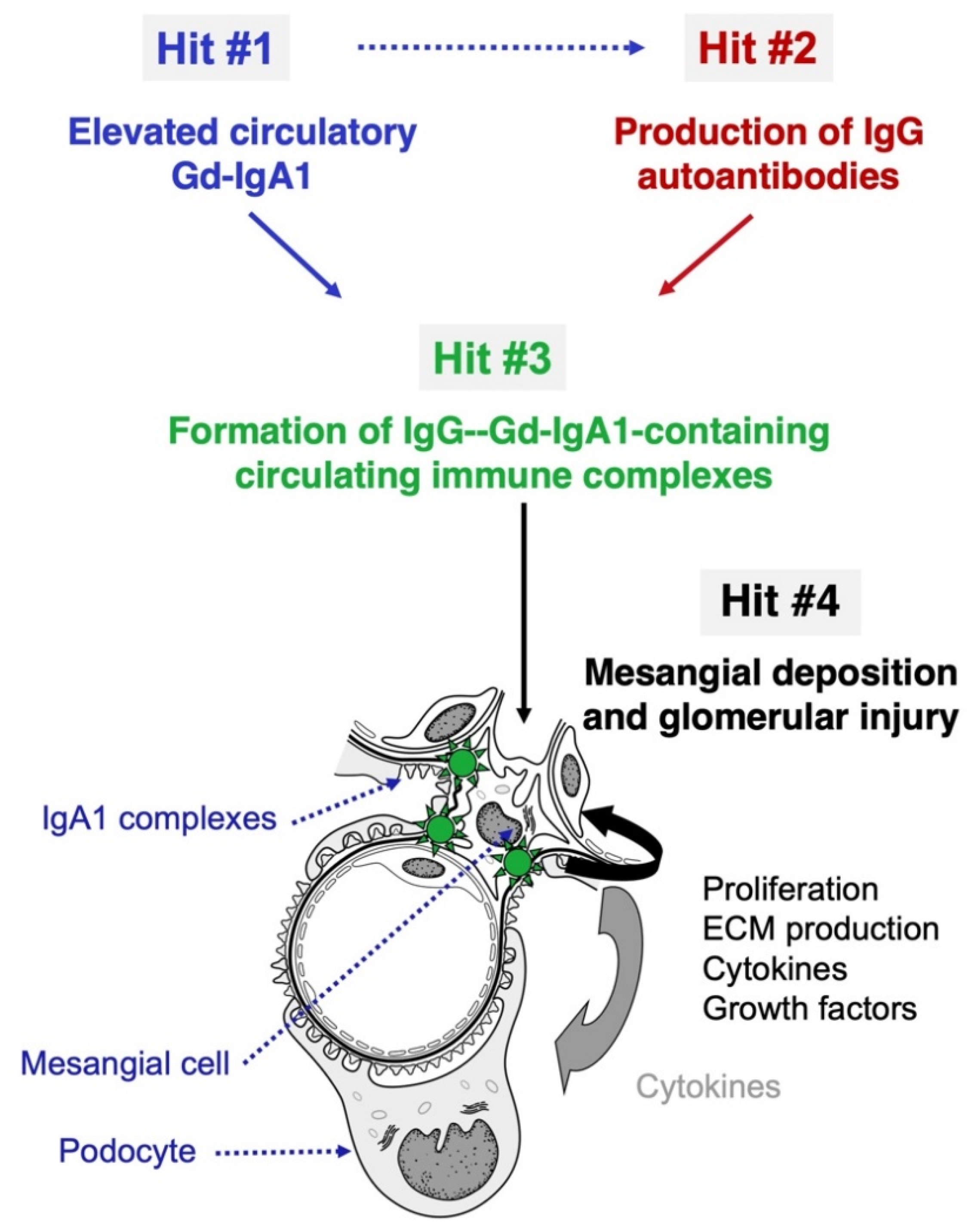{kind=link}
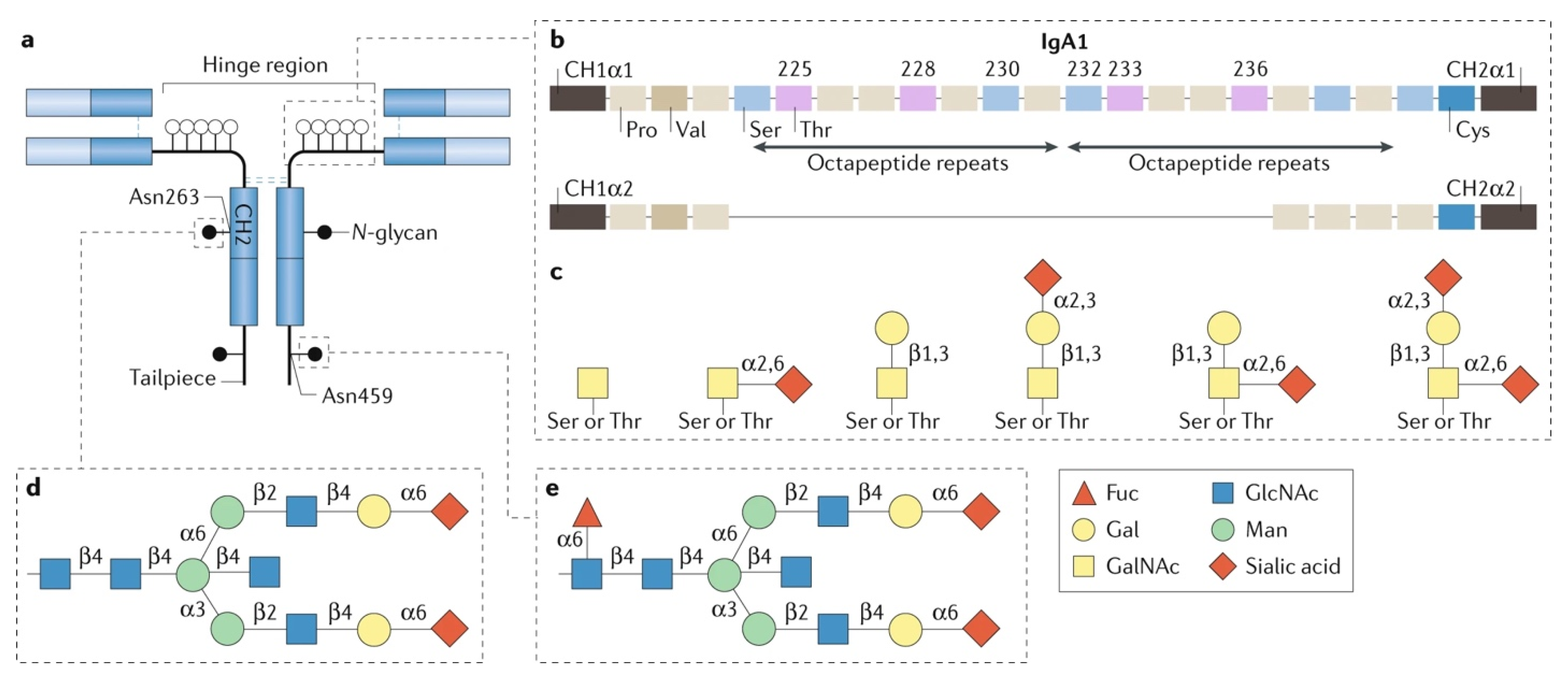{kind=link}
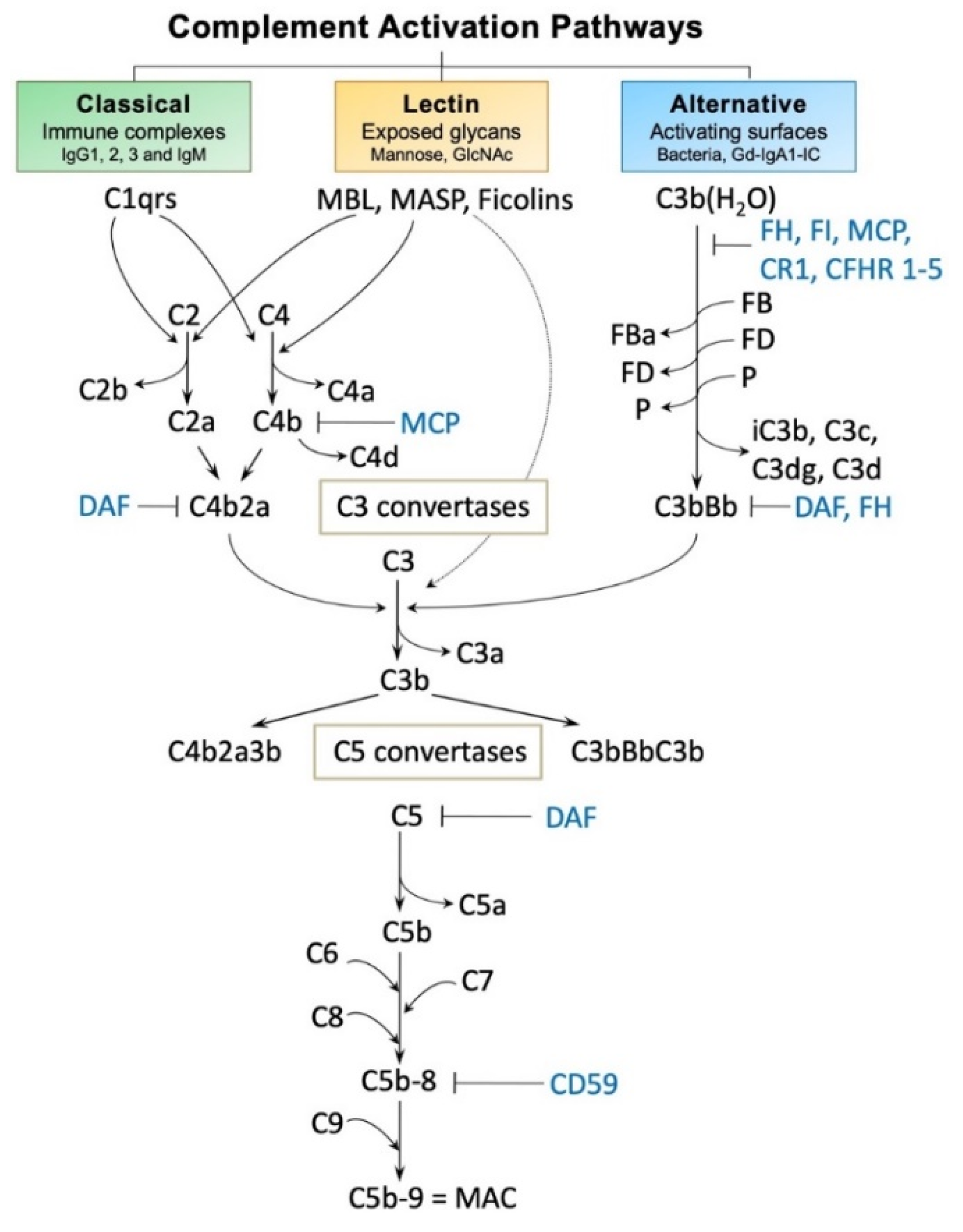{kind=link}
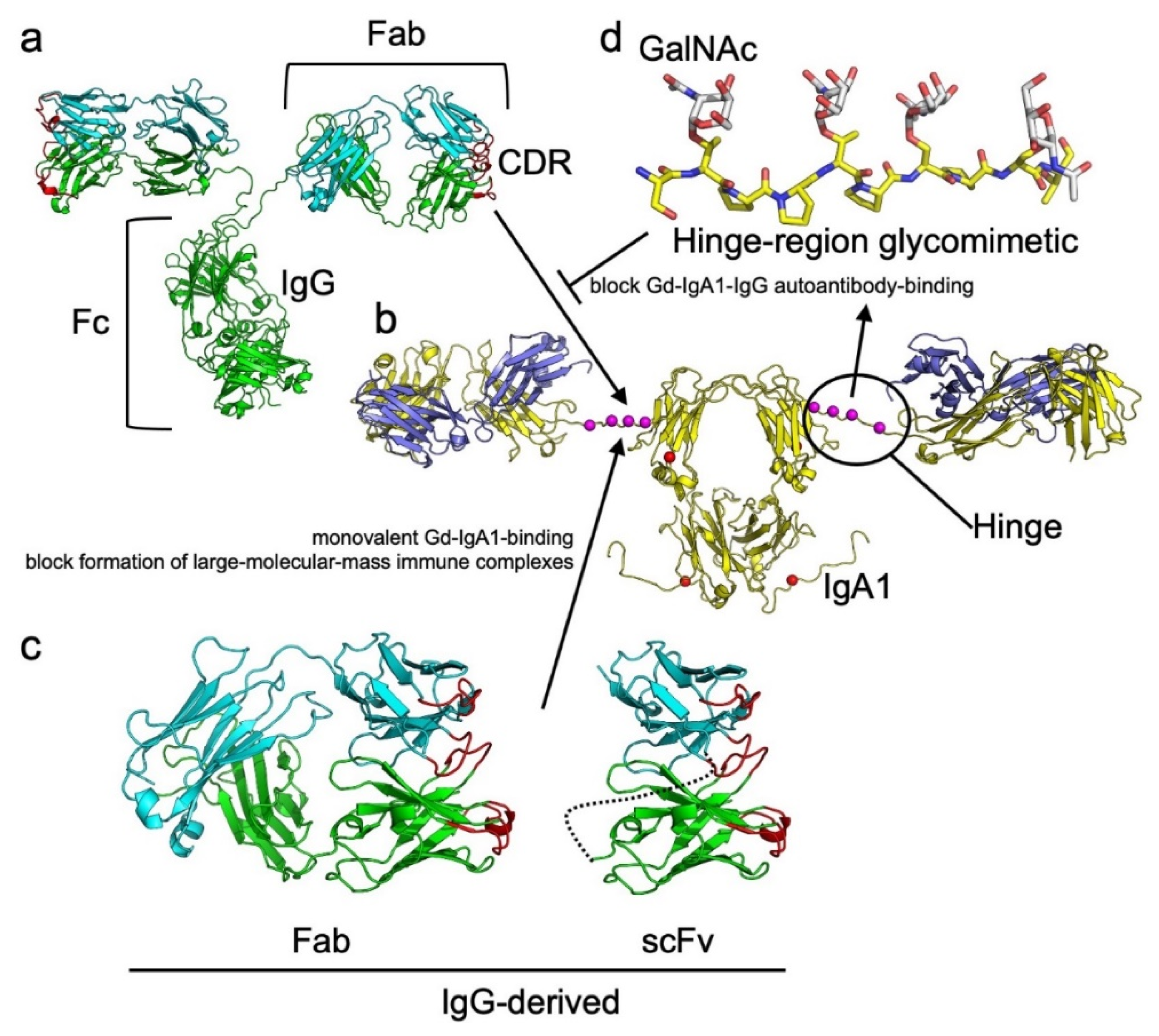{kind=link}
Abstract
:1. Introduction
2. Galactose-Deficient IgA1 (Hit #1)
3. Autoantibodies (Hit #2)
4. Other Types of Autoantibodies Targeting Aberrantly Glycosylated Proteins
5. Circulating Immune Complexes (Hit #3)
6. Deposition of Circulating Immune Complexes and Renal Injury (Hit #4)
7. IgA Nephropathy—Disease-Specific Treatment Approaches
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- D’Amico, G. The commonest glomerulonephritis in the world: IgA nephropathy. Q. J. Med. 1987, 64, 709–727. [Google Scholar]
- Berger, J.; Hinglais, N. Intercapillary deposits of IgA-IgG. J. Urol. Nephrol. 1968, 74, 694–695. [Google Scholar]
- Conley, M.E.; Cooper, M.D.; Michael, A.F. Selective deposition of immunoglobulin A1 in immunoglobulin A nephropathy, anaphylactoid purpura nephritis, and systemic lupus erythematosus. J. Clin. Investig. 1980, 66, 1432–1436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allen, A.C.; Bailey, E.M.; Brenchley, P.E.; Buck, K.S.; Barratt, J.; Feehally, J. Mesangial IgA1 in IgA nephropathy exhibits aberrant O-glycosylation: Observations in three patients. Kidney Int. 2001, 60, 969–973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hiki, Y.; Odani, H.; Takahashi, M.; Yasuda, Y.; Nishimoto, A.; Iwase, H.; Shinzato, T.; Kobayashi, Y.; Maeda, K. Mass spectrometry proves under-O-glycosylation of glomerular IgA1 in IgA nephropathy. Kidney Int. 2001, 59, 1077–1085. [Google Scholar] [CrossRef] [Green Version]
- Jennette, J.C. The immunohistology of IgA nephropathy. Am. J. Kidney Dis. 1988, 12, 348–352. [Google Scholar] [CrossRef]
- Wyatt, R.J.; Julian, B.A. IgA nephropathy. N. Engl. J. Med. 2013, 368, 2402–2414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roberts, I.S. Pathology of IgA nephropathy. Nat. Rev. Nephrol. 2014, 10, 445–454. [Google Scholar] [CrossRef]
- D’Amico, G.; Colasanti, G.; Barbiano di Belgioioso, G.; Fellin, G.; Ragni, A.; Egidi, F.; Radaelli, L.; Fogazzi, G.; Ponticelli, C.; Minetti, L. Long-term follow-up of IgA mesangial nephropathy: Clinico-histological study in 374 patients. Semin. Nephrol. 1987, 7, 355–358. [Google Scholar]
- D’Amico, G. Natural history of idiopathic IgA nephropathy and factors predictive of disease outcome. Semin. Nephrol. 2004, 24, 179–196. [Google Scholar] [CrossRef]
- Barratt, J.; Feehally, J. IgA nephropathy. J. Am. Soc. Nephrol. 2005, 16, 2088–2097. [Google Scholar] [CrossRef] [PubMed]
- Reich, H.N.; Troyanov, S.; Scholey, J.W.; Cattran, D.C.; Registry, T.G. Remission of proteinuria improves prognosis in IgA nephropathy. J. Am. Soc. Nephrol. 2007, 18, 3177–3183. [Google Scholar] [CrossRef] [PubMed]
- Hastings, M.C.; Bursac, Z.; Julian, B.A.; Villa Baca, E.; Featherston, J.; Woodford, S.Y.; Bailey, L.; Wyatt, R.J. Life expectancy for patients from the southeastern United States with IgA nephropathy. Kidney Int. Rep. 2018, 3, 99–104. [Google Scholar] [CrossRef] [Green Version]
- Woo, K.T.; Lau, Y.K.; Chan, C.M.; Wong, K.S. Angiotensin-converting enzyme inhibitor versus angiotensin 2 receptor antagonist therapy and the influence of angiotensin-converting enzyme gene polymorphism in IgA nephritis. Ann. Acad. Med. Singap. 2008, 37, 372–376. [Google Scholar]
- Yamagata, K.; Iseki, K.; Nitta, K.; Imai, H.; Iino, Y.; Matsuo, S.; Makino, H.; Hishida, A. Chronic kidney disease perspectives in Japan and the importance of urinalysis screening. Clin. Exp. Nephrol. 2008, 12, 1–8. [Google Scholar] [CrossRef]
- Wyatt, R.J.; Julian, B.A.; Baehler, R.W.; Stafford, C.C.; McMorrow, R.G.; Ferguson, T.; Jackson, E.; Woodford, S.Y.; Miller, P.M.; Kritchevsky, S. Epidemiology of IgA nephropathy in central and eastern Kentucky for the period 1975 through 1994. Central Kentucky Region of the Southeastern United States IgA Nephropathy DATABANK Project. J. Am. Soc. Nephrol. 1998, 9, 853–858. [Google Scholar] [CrossRef]
- Schena, F.P. A retrospective analysis of the natural history of primary IgA nephropathy worldwide. Am. J. Med. 1990, 89, 209–215. [Google Scholar] [CrossRef]
- Wyatt, R.J.; Kritchevsky, S.B.; Woodford, S.Y.; Miller, P.M.; Roy, S.; Holland, N.H.; Jackson, E.; Bishof, N.A. IgA nephropathy: Long-term prognosis for pediatric patients. J. Pediatr. 1995, 127, 913–919. [Google Scholar] [CrossRef]
- Geddes, C.C.; Rauta, V.; Gronhagen-Riska, C.; Bartosik, L.P.; Jardine, A.G.; Ibels, L.S.; Pei, Y.; Cattran, D.C. A tricontinental view of IgA nephropathy. Nephrol. Dial. Transplant. 2003, 18, 1541–1548. [Google Scholar] [CrossRef]
- Shen, A.Y.; Brar, S.S.; Khan, S.S.; Kujubu, D.A. Association of race, heart failure and chronic kidney disease. Future Cardiol. 2006, 2, 441–454. [Google Scholar] [CrossRef] [PubMed]
- Varis, J.; Rantala, I.; Pasternack, A.; Oksa, H.; Jäntti, M.; Paunu, E.S.; Pirhonen, R. Immunoglobulin and complement deposition in glomeruli of 756 subjects who had committed suicide or met with a violent death. J. Clin. Pathol. 1993, 46, 607–610. [Google Scholar] [CrossRef] [Green Version]
- Sinniah, R. Occurrence of mesangial IgA and IgM deposits in a control necropsy population. J. Clin. Pathol. 1983, 36, 276–279. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, K.; Honda, K.; Tanabe, K.; Toma, H.; Nihei, H.; Yamaguchi, Y. Incidence of latent mesangial IgA deposition in renal allograft donors in Japan. Kidney Int. 2003, 63, 2286–2294. [Google Scholar] [CrossRef] [Green Version]
- Nakazawa, S.; Imamura, R.; Kawamura, M.; Kato, T.; Abe, T.; Namba, T.; Iwatani, H.; Yamanaka, K.; Uemura, M.; Kishikawa, H.; et al. Difference in IgA1 O-glycosylation between IgA deposition donors and IgA nephropathy recipients. Biochem. Biophys. Res. Commun. 2019, 508, 1106–1112. [Google Scholar] [CrossRef]
- Berger, J. Recurrence of IgA nephropathy in renal allografts. Am. J. Kidney Dis. 1988, 12, 371–372. [Google Scholar] [CrossRef]
- Floege, J. Recurrent IgA nephropathy after renal transplantation. Semin. Nephrol. 2004, 24, 287–291. [Google Scholar] [CrossRef]
- Chandrakantan, A.; Ratanapanichkich, P.; Said, M.; Barker, C.V.; Julian, B.A. Recurrent IgA nephropathy after renal transplantation despite immunosuppressive regimens with mycophenolate mofetil. Nephrol. Dial. Transplant. 2005, 20, 1214–1221. [Google Scholar] [CrossRef] [Green Version]
- Silva, F.G.; Chander, P.; Pirani, C.L.; Hardy, M.A. Disappearance of glomerular mesangial IgA deposits after renal allograft transplantation. Transplantation 1982, 33, 241–246. [Google Scholar] [PubMed]
- Gharavi, A.G.; Moldoveanu, Z.; Wyatt, R.J.; Barker, C.V.; Woodford, S.Y.; Lifton, R.P.; Mestecky, J.; Novak, J.; Julian, B.A. Aberrant IgA1 glycosylation is inherited in familial and sporadic IgA nephropathy. J. Am. Soc. Nephrol. 2008, 19, 1008–1014. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, H.; Fan, R.; Zhang, Z.; Brown, R.; Hall, S.; Julian, B.A.; Chatham, W.W.; Suzuki, Y.; Wyatt, R.J.; Moldoveanu, Z.; et al. Aberrantly glycosylated IgA1 in IgA nephropathy patients is recognized by IgG antibodies with restricted heterogeneity. J. Clin. Investig. 2009, 119, 1668–1677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzuki, H.; Kiryluk, K.; Novak, J.; Moldoveanu, Z.; Herr, A.B.; Renfrow, M.B.; Wyatt, R.J.; Scolari, F.; Mestecky, J.; Gharavi, A.G.; et al. The pathophysiology of IgA nephropathy. J. Am. Soc. Nephrol. 2011, 22, 1795–1803. [Google Scholar] [CrossRef] [Green Version]
- Berthoux, F.; Suzuki, H.; Thibaudin, L.; Yanagawa, H.; Maillard, N.; Mariat, C.; Tomino, Y.; Julian, B.A.; Novak, J. Autoantibodies targeting galactose-deficient IgA1 associate with progression of IgA nephropathy. J. Am. Soc. Nephrol. 2012, 23, 1579–1587. [Google Scholar] [CrossRef] [Green Version]
- Zhao, N.; Hou, P.; Lv, J.; Moldoveanu, Z.; Li, Y.; Kiryluk, K.; Gharavi, A.G.; Novak, J.; Zhang, H. The level of galactose-deficient IgA1 in the sera of patients with IgA nephropathy is associated with disease progression. Kidney Int. 2012, 82, 790–796. [Google Scholar] [CrossRef] [Green Version]
- Maixnerova, D.; Ling, C.; Hall, S.; Reily, C.; Brown, R.; Neprasova, M.; Suchanek, M.; Honsova, E.; Zima, T.; Novak, J.; et al. Galactose-deficient IgA1 and the corresponding IgG autoantibodies predict IgA nephropathy progression. PLoS ONE 2019, 14, e0212254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aucouturier, P.; Monteiro, R.C.; Noël, L.H.; Preud’homme, J.L.; Lesavre, P. Glomerular and serum immunoglobulin G subclasses in IgA nephropathy. Clin. Immunol. Immunopathol. 1989, 51, 338–347. [Google Scholar] [CrossRef]
- Placzek, W.J.; Yanagawa, H.; Makita, Y.; Renfrow, M.B.; Julian, B.A.; Rizk, D.V.; Suzuki, Y.; Novak, J.; Suzuki, H. Serum galactose-deficient-IgA1 and IgG autoantibodies correlate in patients with IgA nephropathy. PLoS ONE 2018, 13, e0190967. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berthelot, L.; Robert, T.; Vuiblet, V.; Tabary, T.; Braconnier, A.; Dramé, M.; Toupance, O.; Rieu, P.; Monteiro, R.C.; Touré, F. Recurrent IgA nephropathy is predicted by altered glycosylated IgA, autoantibodies and soluble CD89 complexes. Kidney Int. 2015, 88, 815–822. [Google Scholar] [CrossRef] [Green Version]
- Maixnerova, D.; Reily, C.; Bian, Q.; Neprasova, M.; Novak, J.; Tesar, V. Markers for the progression of IgA nephropathy. J. Nephrol. 2016, 29, 535–541. [Google Scholar] [CrossRef] [Green Version]
- Berthoux, F.; Suzuki, H.; Mohey, H.; Maillard, N.; Mariat, C.; Novak, J.; Julian, B.A. Prognostic value of serum biomarkers of autoimmunity for recurrence of IgA nephropathy after kidney transplantation. J. Am. Soc. Nephrol. 2017, 28, 1943–1950. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nieuwhof, C.; Kruytzer, M.; Frederiks, P.; van Breda Vriesman, P.J. Chronicity index and mesangial IgG deposition are risk factors for hypertension and renal failure in early IgA nephropathy. Am. J. Kidney Dis. 1998, 31, 962–970. [Google Scholar] [CrossRef]
- Wada, Y.; Ogata, H.; Takeshige, Y.; Takeshima, A.; Yoshida, N.; Yamamoto, M.; Ito, H.; Kinugasa, E. Clinical significance of IgG deposition in the glomerular mesangial area in patients with IgA nephropathy. Clin. Exp. Nephrol. 2013, 17, 73–82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shin, D.H.; Lim, B.J.; Han, I.M.; Han, S.G.; Kwon, Y.E.; Park, K.S.; Lee, M.J.; Oh, H.J.; Park, J.T.; Han, S.H.; et al. Glomerular IgG deposition predicts renal outcome in patients with IgA nephropathy. Mod. Pathol. 2016, 29, 743–752. [Google Scholar] [CrossRef] [Green Version]
- Reily, C.; Stewart, T.J.; Renfrow, M.B.; Novak, J. Glycosylation in health and disease. Nat. Rev. Nephrol. 2019, 15, 346–366. [Google Scholar] [CrossRef] [PubMed]
- Novak, J.; Julian, B.A.; Mestecky, J.; Renfrow, M.B. Glycosylation of IgA1 and pathogenesis of IgA nephropathy. Semin. Immunopathol. 2012, 34, 365–382. [Google Scholar] [CrossRef] [PubMed]
- Bellur, S.S.; Troyanov, S.; Cook, H.T.; Roberts, I.S.; Working Group of the International IgA Nephropathy Network and the Renal Pathology Society. Immunostaining findings in IgA nephropathy: Correlation with histology and clinical outcome in the Oxford classification patient cohort. Nephrol. Dial. Transplant. 2011, 26, 2533–2536. [Google Scholar] [CrossRef] [Green Version]
- Rizk, D.V.; Saha, M.K.; Hall, S.; Novak, L.; Brown, R.; Huang, Z.Q.; Fatima, H.; Julian, B.A.; Novak, J. Glomerular immunodeposits of patients with IgA nephropathy are enriched for IgG autoantibodies specific for galactose-deficient IgA1. J. Am. Soc. Nephrol. 2019, 30, 2017–2026. [Google Scholar] [CrossRef] [PubMed]
- Moldoveanu, Z.; Suzuki, H.; Reily, C.; Satake, K.; Novak, L.; Xu, N.; Huang, Z.Q.; Knoppova, B.; Khan, A.; Hall, S.; et al. Experimental evidence of pathogenic role of IgG autoantibodies in IgA nephropathy. J. Autoimmun. 2021, 118, 102593. [Google Scholar] [CrossRef]
- Julian, B.A.; Quiggins, P.A.; Thompson, J.S.; Woodford, S.Y.; Gleason, K.; Wyatt, R.J. Familial IgA nephropathy. Evidence of an inherited mechanism of disease. N. Engl. J. Med. 1985, 312, 202–208. [Google Scholar] [CrossRef]
- Yeo, S.C.; Goh, S.M.; Barratt, J. Is immunoglobulin A nephropathy different in different ethnic populations? Nephrology (Carlton) 2019, 24, 885–895. [Google Scholar] [CrossRef] [Green Version]
- Gharavi, A.G.; Kiryluk, K.; Choi, M.; Li, Y.; Hou, P.; Xie, J.; Sanna-Cherchi, S.; Men, C.J.; Julian, B.A.; Wyatt, R.J.; et al. Genome-wide association study identifies susceptibility loci for IgA nephropathy. Nat. Genet. 2011, 43, 321–327. [Google Scholar] [CrossRef]
- Kiryluk, K.; Li, Y.; Sanna-Cherchi, S.; Rohanizadegan, M.; Suzuki, H.; Eitner, F.; Snyder, H.J.; Choi, M.; Hou, P.; Scolari, F.; et al. Geographic differences in genetic susceptibility to IgA nephropathy: GWAS replication study and geospatial risk analysis. PLoS Genet. 2012, 8, e1002765. [Google Scholar] [CrossRef]
- Kiryluk, K.; Novak, J.; Gharavi, A.G. Pathogenesis of immunoglobulin A nephropathy: Recent insight from genetic studies. Annu. Rev. Med. 2013, 64, 339–356. [Google Scholar] [CrossRef] [Green Version]
- Kiryluk, K.; Novak, J. The genetics and immunobiology of IgA nephropathy. J. Clin. Investig. 2014, 124, 2325–2332. [Google Scholar] [CrossRef] [Green Version]
- Kiryluk, K.; Li, Y.; Scolari, F.; Sanna-Cherchi, S.; Choi, M.; Verbitsky, M.; Fasel, D.; Lata, S.; Prakash, S.; Shapiro, S.; et al. Discovery of new risk loci for IgA nephropathy implicates genes involved in immunity against intestinal pathogens. Nat. Genet. 2014, 46, 1187–1196. [Google Scholar] [CrossRef]
- Li, M.; Wang, L.; Shi, D.C.; Foo, J.N.; Zhong, Z.; Khor, C.C.; Lanzani, C.; Citterio, L.; Salvi, E.; Yin, P.R.; et al. Genome-wide meta-analysis identifies three novel susceptibility loci and reveals ethnic heterogeneity of genetic susceptibility for IgA nephropathy. J. Am. Soc. Nephrol. 2020, 31, 2949–2963. [Google Scholar] [CrossRef]
- Gale, D.P.; Molyneux, K.; Wimbury, D.; Higgins, P.; Levine, A.P.; Caplin, B.; Ferlin, A.; Yin, P.; Nelson, C.P.; Stanescu, H.; et al. Galactosylation of IgA1 is associated with common variation in C1GALT1. J. Am. Soc. Nephrol. 2017, 28, 2158–2166. [Google Scholar] [CrossRef] [Green Version]
- Kiryluk, K.; Li, Y.; Moldoveanu, Z.; Suzuki, H.; Reily, C.; Hou, P.; Xie, J.; Mladkova, N.; Prakash, S.; Fischman, C.; et al. GWAS for serum galactose-deficient IgA1 implicates critical genes of the O-glycosylation pathway. PLoS Genet. 2017, 13, e1006609. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.N.; Zhou, X.J.; Chen, P.; Yu, G.Z.; Zhang, X.; Hou, P.; Liu, L.J.; Shi, S.F.; Lv, J.C.; Zhang, H. Interaction between GALNT12 and C1GALT1 associates with galactose-deficient IgA1 and IgA nephropathy. J. Am. Soc. Nephrol. 2021, 32, 545–552. [Google Scholar] [CrossRef] [PubMed]
- Maillard, N.; Wyatt, R.J.; Julian, B.A.; Kiryluk, K.; Gharavi, A.; Fremeaux-Bacchi, V.; Novak, J. Current understanding of the role of complement in IgA nephropathy. J. Am. Soc. Nephrol. 2015, 26, 1503–1512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woof, J.M.; Russell, M.W. Structure and function relationships in IgA. Mucosal Immunol. 2011, 4, 590–597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brandtzaeg, P.; Johansen, F.E. Mucosal B cells: Phenotypic characteristics, transcriptional regulation, and homing properties. Immunol. Rev. 2005, 206, 32–63. [Google Scholar] [CrossRef]
- Kaetzel, C.S. The polymeric immunoglobulin receptor: Bridging innate and adaptive immune responses at mucosal surfaces. Immunol. Rev. 2005, 206, 83–99. [Google Scholar] [CrossRef]
- Rojas, R.; Apodaca, G. Immunoglobulin transport across polarized epithelial cells. Nat. Rev. Mol. Cell Biol. 2002, 3, 944–955. [Google Scholar] [CrossRef]
- Woof, J.M.; Mestecky, J. Mucosal immunoglobulins. Immunol. Rev. 2005, 206, 64–82. [Google Scholar] [CrossRef]
- Franc, V.; Řehulka, P.; Raus, M.; Stulík, J.; Novak, J.; Renfrow, M.B.; Šebela, M. Elucidating heterogeneity of IgA1 hinge-region O-glycosylation by use of MALDI-TOF/TOF mass spectrometry: Role of cysteine alkylation during sample processing. J. Proteom. 2013, 92, 299–312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frangione, B.; Wolfenstein-Todel, C. Partial duplication in the "hinge" region of IgA 1 myeloma proteins. Proc. Natl. Acad. Sci. USA 1972, 69, 3673–3676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohyama, Y.; Renfrow, M.B.; Novak, J.; Takahashi, K. Aberrantly glycosylated IgA1 in IgA nephropathy: What we know and what we don’t know. J. Clin. Med. 2021, 10, 3467. [Google Scholar] [CrossRef] [PubMed]
- Renfrow, M.B.; Cooper, H.J.; Tomana, M.; Kulhavy, R.; Hiki, Y.; Toma, K.; Emmett, M.R.; Mestecky, J.; Marshall, A.G.; Novak, J. Determination of aberrant O-glycosylation in the IgA1 hinge region by electron capture dissociation fourier transform-ion cyclotron resonance mass spectrometry. J. Biol. Chem. 2005, 280, 19136–19145. [Google Scholar] [CrossRef] [Green Version]
- Tarelli, E.; Smith, A.C.; Hendry, B.M.; Challacombe, S.J.; Pouria, S. Human serum IgA1 is substituted with up to six O-glycans as shown by matrix assisted laser desorption ionisation time-of-flight mass spectrometry. Carbohydr. Res. 2004, 339, 2329–2335. [Google Scholar] [CrossRef]
- Ju, T.; Cummings, R.D. Protein glycosylation: Chaperone mutation in Tn syndrome. Nature 2005, 437, 1252. [Google Scholar] [CrossRef] [PubMed]
- Qin, W.; Zhou, Q.; Yang, L.C.; Li, Z.; Su, B.H.; Luo, H.; Fan, J.M. Peripheral B lymphocyte beta1,3-galactosyltransferase and chaperone expression in immunoglobulin A nephropathy. J. Intern. Med. 2005, 258, 467–477. [Google Scholar] [CrossRef]
- Field, M.C.; Dwek, R.A.; Edge, C.J.; Rademacher, T.W. O-linked oligosaccharides from human serum immunoglobulin A1. Biochem. Soc. Trans. 1989, 17, 1034–1035. [Google Scholar] [CrossRef] [PubMed]
- Tomana, M.; Niedermeier, W.; Mestecky, J.; Skvaril, F. The differences in carbohydrate composition between the subclasses of IgA immunoglobulins. Immunochemistry 1976, 13, 325–328. [Google Scholar] [CrossRef]
- Reily, C.; Ueda, H.; Huang, Z.Q.; Mestecky, J.; Julian, B.A.; Willey, C.D.; Novak, J. Cellular signaling and production of galactose-deficient IgA1 in IgA nephropathy, an autoimmune disease. J. Immunol. Res. 2014, 2014, 197548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzuki, H.; Raska, M.; Yamada, K.; Moldoveanu, Z.; Julian, B.A.; Wyatt, R.J.; Tomino, Y.; Gharavi, A.G.; Novak, J. Cytokines alter IgA1 O-glycosylation by dysregulating C1GalT1 and ST6GalNAc-II enzymes. J. Biol. Chem. 2014, 289, 5330–5339. [Google Scholar] [CrossRef] [Green Version]
- Mestecky, J.; Tomana, M.; Crowley-Nowick, P.A.; Moldoveanu, Z.; Julian, B.A.; Jackson, S. Defective galactosylation and clearance of IgA1 molecules as a possible etiopathogenic factor in IgA nephropathy. Contrib. Nephrol. 1993, 104, 172–182. [Google Scholar] [CrossRef]
- Mestecky, J.; Tomana, M.; Moldoveanu, Z.; Julian, B.A.; Suzuki, H.; Matousovic, K.; Renfrow, M.B.; Novak, L.; Wyatt, R.J.; Novak, J. Role of aberrant glycosylation of IgA1 molecules in the pathogenesis of IgA nephropathy. Kidney Blood Press. Res. 2008, 31, 29–37. [Google Scholar] [CrossRef] [Green Version]
- Hastings, M.C.; Moldoveanu, Z.; Julian, B.A.; Novak, J.; Sanders, J.T.; McGlothan, K.R.; Gharavi, A.G.; Wyatt, R.J. Galactose-deficient IgA1 in African Americans with IgA nephropathy: Serum levels and heritability. Clin. J. Am. Soc. Nephrol. 2010, 5, 2069–2074. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, H.; Moldoveanu, Z.; Hall, S.; Brown, R.; Vu, H.L.; Novak, L.; Julian, B.A.; Tomana, M.; Wyatt, R.J.; Edberg, J.C.; et al. IgA1-secreting cell lines from patients with IgA nephropathy produce aberrantly glycosylated IgA1. J. Clin. Investig. 2008, 118, 629–639. [Google Scholar] [CrossRef] [Green Version]
- Barratt, J.; Smith, A.C.; Molyneux, K.; Feehally, J. Immunopathogenesis of IgAN. Semin. Immunopathol. 2007, 29, 427–443. [Google Scholar] [CrossRef]
- Novak, J.; Julian, B.A.; Tomana, M.; Mestecky, J. Progress in molecular and genetic studies of IgA nephropathy. J. Clin. Immunol. 2001, 21, 310–327. [Google Scholar] [CrossRef]
- Xing, Y.; Li, L.; Zhang, Y.; Wang, F.; He, D.; Liu, Y.; Jia, J.; Yan, T.; Lin, S. C1GALT1 expression is associated with galactosylation of IgA1 in peripheral B lymphocyte in immunoglobulin a nephropathy. BMC Nephrol. 2020, 21, 18. [Google Scholar] [CrossRef] [PubMed]
- Yamada, K.; Huang, Z.Q.; Raska, M.; Reily, C.; Anderson, J.C.; Suzuki, H.; Ueda, H.; Moldoveanu, Z.; Kiryluk, K.; Suzuki, Y.; et al. Inhibition of STAT3 signaling reduces IgA1 autoantigen production in IgA nephropathy. Kidney Int. Rep. 2017, 2, 1194–1207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamada, K.; Huang, Z.Q.; Raska, M.; Reily, C.; Anderson, J.C.; Suzuki, H.; Kiryluk, K.; Gharavi, A.G.; Julian, B.A.; Willey, C.D.; et al. Leukemia inhibitory factor signaling enhances production of galactose-deficient IgA1 in IgA nephropathy. Kidney Dis. 2020, 6, 168–180. [Google Scholar] [CrossRef] [PubMed]
- Kiryluk, K.; Moldoveanu, Z.; Sanders, J.T.; Eison, T.M.; Suzuki, H.; Julian, B.A.; Novak, J.; Gharavi, A.G.; Wyatt, R.J. Aberrant glycosylation of IgA1 is inherited in both pediatric IgA nephropathy and Henoch-Schönlein purpura nephritis. Kidney Int. 2011, 80, 79–87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, Y.X.; He, L.Y.; Chen, X.; Peng, X.F.; Ye, M.Y.; Zhao, Y.J.; Yan, W.Z.; Liu, C.; Shao, J.; Peng, Y.M. Potential diagnostic biomarkers for IgA nephropathy: A comparative study pre- and post-tonsillectomy. Int. Urol. Nephrol. 2016, 48, 1855–1861. [Google Scholar] [CrossRef]
- Hirano, K.; Matsuzaki, K.; Yasuda, T.; Nishikawa, M.; Yasuda, Y.; Koike, K.; Maruyama, S.; Yokoo, T.; Matsuo, S.; Kawamura, T.; et al. Association between tonsillectomy and outcomes in patients with immunoglobulin A nephropathy. JAMA Netw. Open 2019, 2, e194772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Enya, T.; Miyazaki, K.; Miyazawa, T.; Oshima, R.; Morimoto, Y.; Okada, M.; Takemura, T.; Sugimoto, K. Early tonsillectomy for severe immunoglobulin A nephropathy significantly reduces proteinuria. Pediatr. Int. 2020, 62, 1054–1057. [Google Scholar] [CrossRef]
- Kawabe, M.; Yamamoto, I.; Yamakawa, T.; Katsumata, H.; Isaka, N.; Katsuma, A.; Nakada, Y.; Kobayashi, A.; Koike, K.; Ueda, H.; et al. Association between galactose-deficient IgA1 derived from the tonsils and recurrence of IgA nephropathy in patients who underwent kidney transplantation. Front. Immunol. 2020, 11, 2068. [Google Scholar] [CrossRef]
- Aratani, S.; Matsunobu, T.; Shimizu, A.; Okubo, K.; Kashiwagi, T.; Sakai, Y. Tonsillectomy combined with steroid pulse therapy prevents the progression of chronic kidney disease in patients with immunoglobulin A (IgA) nephropathy in a single Japanese institution. Cureus 2021, 13, e15736. [Google Scholar] [CrossRef]
- Nakata, J.; Suzuki, Y.; Suzuki, H.; Sato, D.; Kano, T.; Yanagawa, H.; Matsuzaki, K.; Horikoshi, S.; Novak, J.; Tomino, Y. Changes in nephritogenic serum galactose-deficient IgA1 in IgA nephropathy following tonsillectomy and steroid therapy. PLoS ONE 2014, 9, e89707. [Google Scholar] [CrossRef]
- Kawamura, T.; Yoshimura, M.; Miyazaki, Y.; Okamoto, H.; Kimura, K.; Hirano, K.; Matsushima, M.; Utsunomiya, Y.; Ogura, M.; Yokoo, T.; et al. A multicenter randomized controlled trial of tonsillectomy combined with steroid pulse therapy in patients with immunoglobulin A nephropathy. Nephrol. Dial. Transplant. 2014, 29, 1546–1553. [Google Scholar] [CrossRef] [PubMed]
- Feehally, J.; Coppo, R.; Troyanov, S.; Bellur, S.S.; Cattran, D.; Cook, T.; Roberts, I.S.; Verhave, J.C.; Camilla, R.; Vergano, L.; et al. Tonsillectomy in a European cohort of 1,147 patients with IgA nephropathy. Nephron 2016, 132, 15–24. [Google Scholar] [CrossRef]
- Kim, M.J.; Schaub, S.; Molyneux, K.; Koller, M.T.; Stampf, S.; Barratt, J. Effect of immunosuppressive drugs on the changes of serum galactose-deficient IgA1 in patients with IgA nephropathy. PLoS ONE 2016, 11, e0166830. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kosztyu, P.; Hill, M.; Jemelkova, J.; Czernekova, L.; Kafkova, L.R.; Hruby, M.; Matousovic, K.; Vondrak, K.; Zadrazil, J.; Sterzl, I.; et al. Glucocorticoids reduce aberrant O-glycosylation of IgA1 in IgA nephropathy patients. Kidney Blood Press. Res. 2018, 43, 350–359. [Google Scholar] [CrossRef]
- Rauen, T.; Fitzner, C.; Eitner, F.; Sommerer, C.; Zeier, M.; Otte, B.; Panzer, U.; Peters, H.; Benck, U.; Mertens, P.R.; et al. Effects of two immunosuppressive treatment protocols for IgA nephropathy. J. Am. Soc. Nephrol. 2018, 29, 317–325. [Google Scholar] [CrossRef] [PubMed]
- Fellström, B.C.; Barratt, J.; Cook, H.; Coppo, R.; Feehally, J.; de Fijter, J.W.; Floege, J.; Hetzel, G.; Jardine, A.G.; Locatelli, F.; et al. Targeted-release budesonide versus placebo in patients with IgA nephropathy (NEFIGAN): A double-blind, randomised, placebo-controlled phase 2b trial. Lancet 2017, 389, 2117–2127. [Google Scholar] [CrossRef] [Green Version]
- Coppo, R. Biomarkers and targeted new therapies for IgA nephropathy. Pediatr. Nephrol. 2017, 32, 725–731. [Google Scholar] [CrossRef]
- Coppo, R.; Mariat, C. Systemic corticosteroids and mucosal-associated lymphoid tissue-targeted therapy in immunoglobulin A nephropathy: Insight from the NEFIGAN study. Nephrol. Dial. Transplant. 2020, 35, 1291–1294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomana, M.; Kulhavy, R.; Mestecky, J. Receptor-mediated binding and uptake of immunoglobulin A by human liver. Gastroenterology 1988, 94, 762–770. [Google Scholar] [CrossRef]
- Mestecky, J.; Moldoveanu, Z.; Tomana, M.; Epps, J.M.; Thorpe, S.R.; Phillips, J.O.; Kulhavy, R. The role of the liver in catabolism of mouse and human IgA. Immunol. Investig. 1989, 18, 313–324. [Google Scholar] [CrossRef] [PubMed]
- Baenziger, J.U.; Maynard, Y. Human hepatic lectin. Physiochemical properties and specificity. J. Biol. Chem. 1980, 255, 4607–4613. [Google Scholar] [CrossRef]
- Tomana, M.; Phillips, J.O.; Kulhavy, R.; Mestecky, J. Carbohydrate-mediated clearance of secretory IgA from the circulation. Mol. Immunol. 1985, 22, 887–892. [Google Scholar] [CrossRef]
- Basset, C.; Devauchelle, V.; Durand, V.; Jamin, C.; Pennec, Y.L.; Youinou, P.; Dueymes, M. Glycosylation of immunoglobulin A influences its receptor binding. Scand. J. Immunol. 1999, 50, 572–579. [Google Scholar] [CrossRef] [PubMed]
- Park, E.I.; Mi, Y.; Unverzagt, C.; Gabius, H.J.; Baenziger, J.U. The asialoglycoprotein receptor clears glycoconjugates terminating with sialic acid alpha 2,6GalNAc. Proc. Natl. Acad. Sci. USA 2005, 102, 17125–17129. [Google Scholar] [CrossRef] [Green Version]
- Steirer, L.M.; Park, E.I.; Townsend, R.R.; Baenziger, J.U. The asialoglycoprotein receptor regulates levels of plasma glycoproteins terminating with sialic acid alpha2,6-galactose. J. Biol. Chem. 2009, 284, 3777–3783. [Google Scholar] [CrossRef] [Green Version]
- Roccatello, D.; Picciotto, G.; Torchio, M.; Ropolo, R.; Ferro, M.; Franceschini, R.; Quattrocchio, G.; Cacace, G.; Coppo, R.; Sena, L.M. Removal systems of immunoglobulin A and immunoglobulin A containing complexes in IgA nephropathy and cirrhosis patients. The role of asialoglycoprotein receptors. Lab. Investig. 1993, 69, 714–723. [Google Scholar] [PubMed]
- Tomana, M.; Matousovic, K.; Julian, B.A.; Radl, J.; Konecny, K.; Mestecky, J. Galactose-deficient IgA1 in sera of IgA nephropathy patients is present in complexes with IgG. Kidney Int. 1997, 52, 509–516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzuki, H.; Moldoveanu, Z.; Hall, S.; Brown, R.; Julian, B.A.; Wyatt, R.J.; Tomana, M.; Tomino, Y.; Novak, J.; Mestecky, J. IgA nephropathy: Characterization of IgG antibodies specific for galactose-deficient IgA1. Contrib. Nephrol. 2007, 157, 129–133. [Google Scholar] [CrossRef]
- Cisar, J.O.; Sandberg, A.L.; Reddy, G.P.; Abeygunawardana, C.; Bush, C.A. Structural and antigenic types of cell wall polysaccharides from viridans group streptococci with receptors for oral actinomyces and streptococcal lectins. Infect. Immun. 1997, 65, 5035–5041. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, D.C.; Spear, P.G. O-linked oligosaccharides are acquired by herpes simplex virus glycoproteins in the Golgi apparatus. Cell 1983, 32, 987–997. [Google Scholar] [CrossRef]
- Kieff, E.; Dambaugh, T.; Heller, M.; King, W.; Cheung, A.; van Santen, V.; Hummel, M.; Beisel, C.; Fennewald, S.; Hennessy, K.; et al. The biology and chemistry of Epstein-Barr virus. J. Infect. Dis. 1982, 146, 506–517. [Google Scholar] [CrossRef] [PubMed]
- Wertz, G.W.; Krieger, M.; Ball, L.A. Structure and cell surface maturation of the attachment glycoprotein of human respiratory syncytial virus in a cell line deficient in O glycosylation. J. Virol. 1989, 63, 4767–4776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yanagawa, H.; Suzuki, H.; Suzuki, Y.; Kiryluk, K.; Gharavi, A.G.; Matsuoka, K.; Makita, Y.; Julian, B.A.; Novak, J.; Tomino, Y. A panel of serum biomarkers differentiates IgA nephropathy from other renal diseases. PLoS ONE 2014, 9, e98081. [Google Scholar] [CrossRef] [Green Version]
- Hamel, K.M.; Liarski, V.M.; Clark, M.R. Germinal center B-cells. Autoimmunity 2012, 45, 333–347. [Google Scholar] [CrossRef]
- Huang, Z.Q.; Raska, M.; Stewart, T.J.; Reily, C.; King, R.G.; Crossman, D.K.; Crowley, M.R.; Hargett, A.; Zhang, Z.; Suzuki, H.; et al. Somatic mutations modulate autoantibodies against galactose-deficient IgA1 in IgA nephropathy. J. Am. Soc. Nephrol. 2016, 27, 3278–3284. [Google Scholar] [CrossRef]
- Tomana, M.; Novak, J.; Julian, B.A.; Matousovic, K.; Konecny, K.; Mestecky, J. Circulating immune complexes in IgA nephropathy consist of IgA1 with galactose-deficient hinge region and antiglycan antibodies. J. Clin. Investig. 1999, 104, 73–81. [Google Scholar] [CrossRef] [Green Version]
- Springer, G.F.; Tegtmeyer, H. Origin of anti-Thomsen-Friedenreich (T) and Tn agglutinins in man and in White Leghorn chicks. Br. J. Haematol. 1981, 47, 453–460. [Google Scholar] [CrossRef]
- Mestecky, J.; Novak, J.; Moldoveanu, Z.; Raska, M. IgA nephropathy enigma. Clin. Immunol. 2016, 172, 72–77. [Google Scholar] [CrossRef] [Green Version]
- Raška, M.; Zadražil, J.; Horynová, M.S.; Kafková, L.R.; Vráblíková, A.; Matoušovic, K.; Novak, J.; Městecký, J. IgA nephropathy-research-generated questions. Vnitrni Lekarstvi 2016, 62 (Suppl. 6), 67–77. [Google Scholar]
- Ito, S.; Misaki, T.; Naka, S.; Wato, K.; Nagasawa, Y.; Nomura, R.; Otsugu, M.; Matsumoto-Nakano, M.; Nakano, K.; Kumagai, H.; et al. Specific strains of Streptococcus mutans, a pathogen of dental caries, in the tonsils, are associated with IgA nephropathy. Sci. Rep. 2019, 9, 20130. [Google Scholar] [CrossRef]
- Yamaguchi, H.; Goto, S.; Takahashi, N.; Tsuchida, M.; Watanabe, H.; Yamamoto, S.; Kaneko, Y.; Higashi, K.; Mori, H.; Nakamura, Y.; et al. Aberrant mucosal immunoreaction to tonsillar microbiota in immunoglobulin A nephropathy. Nephrol. Dial. Transplant. 2021, 36, 75–86. [Google Scholar] [CrossRef]
- Muthana, S.M.; Gildersleeve, J.C. Factors affecting anti-glycan IgG and IgM repertoires in human serum. Sci. Rep. 2016, 6, 19509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huflejt, M.E.; Vuskovic, M.; Vasiliu, D.; Xu, H.; Obukhova, P.; Shilova, N.; Tuzikov, A.; Galanina, O.; Arun, B.; Lu, K.; et al. Anti-carbohydrate antibodies of normal sera: Findings, surprises and challenges. Mol. Immunol. 2009, 46, 3037–3049. [Google Scholar] [CrossRef] [PubMed]
- New, J.S.; Dizon, B.L.P.; Fucile, C.F.; Rosenberg, A.F.; Kearney, J.F.; King, R.G. Neonatal exposure to commensal-bacteria-derived antigens directs polysaccharide-specific B-1 B cell repertoire development. Immunity 2020, 53, 172–186 e176. [Google Scholar] [CrossRef] [PubMed]
- Stuchlová Horynová, M.; Raška, M.; Clausen, H.; Novak, J. Aberrant O-glycosylation and anti-glycan antibodies in an autoimmune disease IgA nephropathy and breast adenocarcinoma. Cell Mol. Life Sci. 2013, 70, 829–839. [Google Scholar] [CrossRef] [Green Version]
- Springer, G.F.; Taylor, C.R.; Howard, D.R.; Tegtmeyer, H.; Desai, P.R.; Murthy, S.M.; Felder, B.; Scanlon, E.F. Tn, a carcinoma-associated antigen, reacts with anti-Tn of normal human sera. Cancer 1985, 55, 561–569. [Google Scholar] [CrossRef]
- Dobrochaeva, K.; Khasbiullina, N.; Shilova, N.; Antipova, N.; Obukhova, P.; Ovchinnikova, T.; Galanina, O.; Blixt, O.; Kunz, H.; Filatov, A.; et al. Specificity of human natural antibodies referred to as anti-Tn. Mol. Immunol. 2020, 120, 74–82. [Google Scholar] [CrossRef]
- Zlocowski, N.; Grupe, V.; Garay, Y.C.; Nores, G.A.; Lardone, R.D.; Irazoqui, F.J. Purified human anti-Tn and anti-T antibodies specifically recognize carcinoma tissues. Sci. Rep. 2019, 9, 8097. [Google Scholar] [CrossRef] [Green Version]
- Gendler, S.J.; Lancaster, C.A.; Taylor-Papadimitriou, J.; Duhig, T.; Peat, N.; Burchell, J.; Pemberton, L.; Lalani, E.N.; Wilson, D. Molecular cloning and expression of human tumor-associated polymorphic epithelial mucin. J. Biol. Chem. 1990, 265, 15286–15293. [Google Scholar] [CrossRef]
- Blixt, O.; Clo, E.; Nudelman, A.S.; Sorensen, K.K.; Clausen, T.; Wandall, H.H.; Livingston, P.O.; Clausen, H.; Jensen, K.J. A high-throughput O-glycopeptide discovery platform for seromic profiling. J. Proteome Res. 2010, 9, 5250–5261. [Google Scholar] [CrossRef]
- Karsten, U.; Serttas, N.; Paulsen, H.; Danielczyk, A.; Goletz, S. Binding patterns of DTR-specific antibodies reveal a glycosylation-conditioned tumor-specific epitope of the epithelial mucin (MUC1). Glycobiology 2004, 14, 681–692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fiedler, W.; DeDosso, S.; Cresta, S.; Weidmann, J.; Tessari, A.; Salzberg, M.; Dietrich, B.; Baumeister, H.; Goletz, S.; Gianni, L.; et al. A phase I study of PankoMab-GEX, a humanised glyco-optimised monoclonal antibody to a novel tumour-specific MUC1 glycopeptide epitope in patients with advanced carcinomas. Eur. J. Cancer 2016, 63, 55–63. [Google Scholar] [CrossRef] [Green Version]
- Wu, G.; Maharjan, S.; Kim, D.; Kim, J.N.; Park, B.K.; Koh, H.; Moon, K.; Lee, Y.; Kwon, H.J. A novel monoclonal antibody targets mucin1 and attenuates growth in pancreatic cancer model. Int. J. Mol. Sci. 2018, 19, 2004. [Google Scholar] [CrossRef] [Green Version]
- Danielczyk, A.; Stahn, R.; Faulstich, D.; Loffler, A.; Marten, A.; Karsten, U.; Goletz, S. PankoMab: A potent new generation anti-tumour MUC1 antibody. Cancer Immunol. Immunother. 2006, 55, 1337–1347. [Google Scholar] [CrossRef] [PubMed]
- Doi, M.; Yokoyama, A.; Kondo, K.; Ohnishi, H.; Ishikawa, N.; Hattori, N.; Kohno, N. Anti-tumor effect of the anti-KL-6/MUC1 monoclonal antibody through exposure of surface molecules by MUC1 capping. Cancer Sci. 2006, 97, 420–429. [Google Scholar] [CrossRef] [PubMed]
- de Bono, J.S.; Rha, S.Y.; Stephenson, J.; Schultes, B.C.; Monroe, P.; Eckhardt, G.S.; Hammond, L.A.; Whiteside, T.L.; Nicodemus, C.F.; Cermak, J.M.; et al. Phase I trial of a murine antibody to MUC1 in patients with metastatic cancer: Evidence for the activation of humoral and cellular antitumor immunity. Ann. Oncol. 2004, 15, 1825–1833. [Google Scholar] [CrossRef]
- Berlyn, K.A.; Schultes, B.; Leveugle, B.; Noujaim, A.A.; Alexander, R.B.; Mann, D.L. Generation of CD4(+) and CD8(+) T lymphocyte responses by dendritic cells armed with PSA/anti-PSA (antigen/antibody) complexes. Clin. Immunol. 2001, 101, 276–283. [Google Scholar] [CrossRef] [PubMed]
- Gordon, A.N.; Schultes, B.C.; Gallion, H.; Edwards, R.; Whiteside, T.L.; Cermak, J.M.; Nicodemus, C.F. CA125- and tumor-specific T-cell responses correlate with prolonged survival in oregovomab-treated recurrent ovarian cancer patients. Gynecol. Oncol. 2004, 94, 340–351. [Google Scholar] [CrossRef]
- Iwase, H.; Yokozeki, Y.; Hiki, Y.; Tanaka, A.; Kokubo, T.; Sano, T.; Ishii-Karakasa, I.; Hisatani, K.; Kobayashi, Y.; Hotta, K. Human serum immunoglobulin G3 subclass bound preferentially to asialo-, agalactoimmunoglobulin A1/Sepharose. Biochem. Biophys. Res. Commun. 1999, 264, 424–429. [Google Scholar] [CrossRef]
- Kokubo, T.; Hiki, Y.; Iwase, H.; Tanaka, A.; Nishikido, J.; Hotta, K.; Kobayashi, Y. Exposed peptide core of IgA1 hinge region in IgA nephropathy. Nephrol. Dial. Transplant. 1999, 14, 81–85. [Google Scholar] [CrossRef] [PubMed]
- Cederholm, B.; Wieslander, J.; Bygren, P.; Heinegård, D. Circulating complexes containing IgA and fibronectin in patients with primary IgA nephropathy. Proc. Natl. Acad. Sci. USA 1988, 85, 4865–4868. [Google Scholar] [CrossRef] [Green Version]
- Jennette, J.C.; Wieslander, J.; Tuttle, R.; Falk, R.J. Serum IgA-fibronectin aggregates in patients with IgA nephropathy and Henoch-Schönlein purpura: Diagnostic value and pathogenic implications. The Glomerular Disease Collaborative Network. Am. J. Kidney Dis. 1991, 18, 466–471. [Google Scholar] [CrossRef]
- Nakamura, I.; Iwase, H.; Ohba, Y.; Hiki, Y.; Katsumata, T.; Kobayashi, Y. Quantitative analysis of IgA1 binding protein prepared from human serum by hypoglycosylated IgA1/Sepharose affinity chromatography. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2002, 776, 101–106. [Google Scholar] [CrossRef]
- Novak, J.; Tomana, M.; Matousovic, K.; Brown, R.; Hall, S.; Novak, L.; Julian, B.A.; Wyatt, R.J.; Mestecky, J. IgA1-containing immune complexes in IgA nephropathy differentially affect proliferation of mesangial cells. Kidney Int. 2005, 67, 504–513. [Google Scholar] [CrossRef] [Green Version]
- Novak, J.; Raskova Kafkova, L.; Suzuki, H.; Tomana, M.; Matousovic, K.; Brown, R.; Hall, S.; Sanders, J.T.; Eison, T.M.; Moldoveanu, Z.; et al. IgA1 immune complexes from pediatric patients with IgA nephropathy activate cultured human mesangial cells. Nephrol. Dial. Transplant. 2011, 26, 3451–3457. [Google Scholar] [CrossRef] [Green Version]
- Yanagihara, T.; Brown, R.; Hall, S.; Moldoveanu, Z.; Goepfert, A.; Tomana, M.; Julian, B.A.; Mestecky, J.; Novak, J. In vitro-generated immune complexes containing galactose-deficient IgA1 stimulate proliferation of mesangial cells. Results Immunol. 2012, 2, 166–172. [Google Scholar] [CrossRef] [Green Version]
- Czerkinsky, C.; Koopman, W.J.; Jackson, S.; Collins, J.E.; Crago, S.S.; Schrohenloher, R.E.; Julian, B.A.; Galla, J.H.; Mestecky, J. Circulating immune complexes and immunoglobulin A rheumatoid factor in patients with mesangial immunoglobulin A nephropathies. J. Clin. Investig. 1986, 77, 1931–1938. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maillard, N.; Boerma, L.; Hall, S.; Huang, Z.Q.; Mrug, M.; Moldoveanu, Z.; Julian, B.A.; Renfrow, M.B.; Novak, J. Proteomic analysis of engineered IgA1-IgG immune complexes reveals association with activated complement C3. J. Am. Soc. Nephrol. 2013, 24, 490A. [Google Scholar]
- Wyatt, R.J.; Kanayama, Y.; Julian, B.A.; Negoro, N.; Sugimoto, S.; Hudson, E.C.; Curd, J.G. Complement activation in IgA nephropathy. Kidney Int. 1987, 31, 1019–1023. [Google Scholar] [CrossRef] [Green Version]
- Rizk, D.V.; Maillard, N.; Julian, B.A.; Knoppova, B.; Green, T.J.; Novak, J.; Wyatt, R.J. The emerging role of complement proteins as a target for therapy of IgA nephropathy. Front. Immunol. 2019, 10, 504. [Google Scholar] [CrossRef] [PubMed]
- Medjeral-Thomas, N.R.; Cook, H.T.; Pickering, M.C. Complement activation in IgA nephropathy. Semin. Immunopathol. 2021, 31, 1019–1023. [Google Scholar] [CrossRef]
- Xu, B.; Zhu, L.; Wang, Q.; Zhao, Y.; Jia, M.; Shi, S.; Liu, L.; Lv, J.; Lai, W.; Ji, J.; et al. Mass spectrometry-based screening identifies circulating immunoglobulinA-α1-microglobulin complex as potential biomarker in immunoglobulin A nephropathy. Nephrol. Dial. Transplant. 2021, 36, 782–792. [Google Scholar] [CrossRef] [PubMed]
- Coppo, R.; Basolo, B.; Martina, G.; Rollino, C.; De Marchi, M.; Giacchino, F.; Mazzucco, G.; Messina, M.; Piccoli, G. Circulating immune complexes containing IgA, IgG and IgM in patients with primary IgA nephropathy and with Henoch-Schoenlein nephritis. Correlation with clinical and histologic signs of activity. Clin. Nephrol. 1982, 18, 230–239. [Google Scholar]
- Schena, F.P.; Pastore, A.; Ludovico, N.; Sinico, R.A.; Benuzzi, S.; Montinaro, V. Increased serum levels of IgA1-IgG immune complexes and anti-F(ab’)2 antibodies in patients with primary IgA nephropathy. Clin. Exp. Immunol. 1989, 77, 15–20. [Google Scholar]
- Kemper, C.; Pangburn, M.K.; Fishelson, Z. Complement nomenclature 2014. Mol. Immunol. 2014, 61, 56–58. [Google Scholar] [CrossRef]
- Floege, J.; Daha, M.R. IgA nephropathy: New insights into the role of complement. Kidney Int. 2018, 94, 16–18. [Google Scholar] [CrossRef]
- Novak, J.; Rizk, D.; Takahashi, K.; Zhang, X.; Bian, Q.; Ueda, H.; Ueda, Y.; Reily, C.; Lai, L.Y.; Hao, C.; et al. New Insights into the Pathogenesis of IgA Nephropathy. Kidney Dis. 2015, 1, 8–18. [Google Scholar] [CrossRef]
- Knoppova, B.; Reily, C.; Maillard, N.; Rizk, D.V.; Moldoveanu, Z.; Mestecky, J.; Raska, M.; Renfrow, M.B.; Julian, B.A.; Novak, J. The origin and activities of IgA1-containing immune complexes in IgA nephropathy. Front. Immunol. 2016, 7, 117. [Google Scholar] [CrossRef] [Green Version]
- Chen, A.; Chen, W.P.; Sheu, L.F.; Lin, C.Y. Pathogenesis of IgA nephropathy: In vitro activation of human mesangial cells by IgA immune complex leads to cytokine secretion. J. Pathol. 1994, 173, 119–126. [Google Scholar] [CrossRef]
- Coppo, R.; Amore, A.; Cirina, P.; Messina, M.; Basolo, B.; Segoloni, G.; Berthoux, F.; Boulahrouz, R.; Egido, J.; Alcazar, R. Characteristics of IgA and macromolecular IgA in sera from IgA nephropathy transplanted patients with and without IgAN recurrence. Contrib. Nephrol. 1995, 111, 85–92. [Google Scholar] [CrossRef] [PubMed]
- Amore, A.; Cirina, P.; Conti, G.; Brusa, P.; Peruzzi, L.; Coppo, R. Glycosylation of circulating IgA in patients with IgA nephropathy modulates proliferation and apoptosis of mesangial cells. J. Am. Soc. Nephrol. 2001, 12, 1862–1871. [Google Scholar] [CrossRef]
- Leung, J.C.; Tsang, A.W.; Chan, L.Y.; Tang, S.C.; Lam, M.F.; Lai, K.N. Size-dependent binding of IgA to HepG2, U937, and human mesangial cells. J. Lab. Clin. Med. 2002, 140, 398–406. [Google Scholar] [CrossRef] [PubMed]
- Novak, J.; Vu, H.L.; Novak, L.; Julian, B.A.; Mestecky, J.; Tomana, M. Interactions of human mesangial cells with IgA and IgA-containing immune complexes. Kidney Int. 2002, 62, 465–475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leung, J.C.; Tang, S.C.; Chan, L.Y.; Tsang, A.W.; Lan, H.Y.; Lai, K.N. Polymeric IgA increases the synthesis of macrophage migration inhibitory factor by human mesangial cells in IgA nephropathy. Nephrol. Dial. Transplant. 2003, 18, 36–45. [Google Scholar] [CrossRef] [Green Version]
- Moura, I.C.; Arcos-Fajardo, M.; Sadaka, C.; Leroy, V.; Benhamou, M.; Novak, J.; Vrtovsnik, F.; Haddad, E.; Chintalacharuvu, K.R.; Monteiro, R.C. Glycosylation and size of IgA1 are essential for interaction with mesangial transferrin receptor in IgA nephropathy. J. Am. Soc. Nephrol. 2004, 15, 622–634. [Google Scholar] [CrossRef] [Green Version]
- Leung, J.C.; Tang, S.C.; Chan, L.Y.; Chan, W.L.; Lai, K.N. Synthesis of TNF-alpha by mesangial cells cultured with polymeric anionic IgA--role of MAPK and NF-kappaB. Nephrol. Dial. Transplant. 2008, 23, 72–81. [Google Scholar] [CrossRef] [Green Version]
- Lai, K.N.; Leung, J.C.; Chan, L.Y.; Saleem, M.A.; Mathieson, P.W.; Tam, K.Y.; Xiao, J.; Lai, F.M.; Tang, S.C. Podocyte injury induced by mesangial-derived cytokines in IgA nephropathy. Nephrol. Dial. Transplant. 2009, 24, 62–72. [Google Scholar] [CrossRef] [Green Version]
- Tam, K.Y.; Leung, J.C.K.; Chan, L.Y.Y.; Lam, M.F.; Tang, S.C.W.; Lai, K.N. Macromolecular IgA1 taken from patients with familial IgA nephropathy or their asymptomatic relatives have higher reactivity to mesangial cells in vitro. Kidney Int. 2009, 75, 1330–1339. [Google Scholar] [CrossRef] [Green Version]
- Coppo, R.; Fonsato, V.; Balegno, S.; Ricotti, E.; Loiacono, E.; Camilla, R.; Peruzzi, L.; Amore, A.; Bussolati, B.; Camussi, G. Aberrantly glycosylated IgA1 induces mesangial cells to produce platelet-activating factor that mediates nephrin loss in cultured podocytes. Kidney Int. 2010, 77, 417–427. [Google Scholar] [CrossRef] [Green Version]
- Tamouza, H.; Chemouny, J.M.; Raskova Kafkova, L.; Berthelot, L.; Flamant, M.; Demion, M.; Mesnard, L.; Paubelle, E.; Walker, F.; Julian, B.A.; et al. The IgA1 immune complex-mediated activation of the MAPK/ERK kinase pathway in mesangial cells is associated with glomerular damage in IgA nephropathy. Kidney Int. 2012, 82, 1284–1296. [Google Scholar] [CrossRef] [Green Version]
- Novak, J.; Moldoveanu, Z.; Julian, B.A.; Raska, M.; Wyatt, R.J.; Suzuki, Y.; Tomino, Y.; Gharavi, A.G.; Mestecky, J.; Suzuki, H. Aberrant glycosylation of IgA1 and anti-glycan antibodies in IgA nephropathy: Role of mucosal immune system. Adv. Otorhinolaryngol. 2011, 72, 60–63. [Google Scholar] [CrossRef]
- Novak, J.; Barratt, J.; Julian, B.A.; Renfrow, M.B. Aberrant glycosylation of the IgA1 molecule in IgA nephropathy. Semin. Nephrol. 2018, 38, 461–476. [Google Scholar] [CrossRef]
- Moura, I.C.; Arcos-Fajardo, M.; Gdoura, A.; Leroy, V.; Sadaka, C.; Mahlaoui, N.; Lepelletier, Y.; Vrtovsnik, F.; Haddad, E.; Benhamou, M.; et al. Engagement of transferrin receptor by polymeric IgA1: Evidence for a positive feedback loop involving increased receptor expression and mesangial cell proliferation in IgA nephropathy. J. Am. Soc. Nephrol. 2005, 16, 2667–2676. [Google Scholar] [CrossRef]
- Moura, I.C.; Centelles, M.N.; Arcos-Fajardo, M.; Malheiros, D.M.; Collawn, J.F.; Cooper, M.D.; Monteiro, R.C. Identification of the transferrin receptor as a novel immunoglobulin (Ig)A1 receptor and its enhanced expression on mesangial cells in IgA nephropathy. J. Exp. Med. 2001, 194, 417–425. [Google Scholar] [CrossRef]
- Tamouza, H.; Vende, F.; Tiwari, M.; Arcos-Fajardo, M.; Vrtovsnik, F.; Benhamou, M.; Monteiro, R.C.; Moura, I.C. Transferrin receptor engagement by polymeric IgA1 induces receptor expression and mesangial cell proliferation: Role in IgA nephropathy. Contrib. Nephrol. 2007, 157, 144–147. [Google Scholar] [CrossRef] [PubMed]
- Kaneko, Y.; Otsuka, T.; Tsuchida, Y.; Gejyo, F.; Narita, I. Integrin α1/β1 and α2/β1 as a receptor for IgA1 in human glomerular mesangial cells in IgA nephropathy. Int. Immunol. 2012, 24, 219–232. [Google Scholar] [CrossRef] [Green Version]
- Molyneux, K.; Wimbury, D.; Pawluczyk, I.; Muto, M.; Bhachu, J.; Mertens, P.R.; Feehally, J.; Barratt, J. β1,4-galactosyltransferase 1 is a novel receptor for IgA in human mesangial cells. Kidney Int. 2017, 92, 1458–1468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Launay, P.; Grossetête, B.; Arcos-Fajardo, M.; Gaudin, E.; Torres, S.P.; Beaudoin, L.; Patey-Mariaud de Serre, N.; Lehuen, A.; Monteiro, R.C. Fcalpha receptor (CD89) mediates the development of immunoglobulin A (IgA) nephropathy (Berger’s disease). Evidence for pathogenic soluble receptor-Iga complexes in patients and CD89 transgenic mice. J. Exp. Med. 2000, 191, 1999–2009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hansen, I.S.; Baeten, D.L.P.; den Dunnen, J. The inflammatory function of human IgA. Cell Mol. Life Sci. 2019, 76, 1041–1055. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heineke, M.H.; van Egmond, M. Immunoglobulin A: Magic bullet or Trojan horse? Eur. J. Clin. Investig. 2017, 47, 184–192. [Google Scholar] [CrossRef] [Green Version]
- Breedveld, A.; van Egmond, M. IgA and FcαRI: Pathological roles and therapeutic opportunities. Front. Immunol. 2019, 10, 553. [Google Scholar] [CrossRef]
- Cheung, C.K.; Rajasekaran, A.; Barratt, J.; Rizk, D.V. An update on the current state of management and clinical trials for IgA nephropathy. J. Clin. Med. 2021, 10, 2493. [Google Scholar] [CrossRef]
- Harris, L.J.; Larson, S.B.; Hasel, K.W.; McPherson, A. Refined structure of an intact IgG2a monoclonal antibody. Biochemistry 1997, 36, 1581–1597. [Google Scholar] [CrossRef]
- Harris, L.J.; Skaletsky, E.; McPherson, A. Crystallographic structure of an intact IgG1 monoclonal antibody. J. Mol. Biol. 1998, 275, 861–872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saphire, E.O.; Parren, P.W.; Pantophlet, R.; Zwick, M.B.; Morris, G.M.; Rudd, P.M.; Dwek, R.A.; Stanfield, R.L.; Burton, D.R.; Wilson, I.A. Crystal structure of a neutralizing human IgG against HIV-1: A template for vaccine design. Science 2001, 293, 1155–1159. [Google Scholar] [CrossRef] [PubMed]
- Boehm, M.K.; Woof, J.M.; Kerr, M.A.; Perkins, S.J. The Fab and Fc fragments of IgA1 exhibit a different arrangement from that in IgG: A study by X-ray and neutron solution scattering and homology modelling. J. Mol. Biol. 1999, 286, 1421–1447. [Google Scholar] [CrossRef] [PubMed]
- Almogren, A.; Furtado, P.B.; Sun, Z.; Perkins, S.J.; Kerr, M.A. Purification, properties and extended solution structure of the complex formed between human immunoglobulin A1 and human serum albumin by scattering and ultracentrifugation. J. Mol. Biol. 2006, 356, 413–431. [Google Scholar] [CrossRef] [PubMed]
- Bonner, A.; Furtado, P.B.; Almogren, A.; Kerr, M.A.; Perkins, S.J. Implications of the near-planar solution structure of human myeloma dimeric IgA1 for mucosal immunity and IgA nephropathy. J. Immunol. 2008, 180, 1008–1018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonner, A.; Almogren, A.; Furtado, P.B.; Kerr, M.A.; Perkins, S.J. Location of secretory component on the Fc edge of dimeric IgA1 reveals insight into the role of secretory IgA1 in mucosal immunity. Mucosal Immunol. 2009, 2, 74–84. [Google Scholar] [CrossRef]
- Woods Group. GLYCAM Web. Complex. Carbohydrate Research Center, University of Georgia, Athens, Georgia, 2005–2021. Available online: http://glycam.org (accessed on 8 August 2021).
- Krieger, E.; Joo, K.; Lee, J.; Lee, J.; Raman, S.; Thompson, J.; Tyka, M.; Baker, D.; Karplus, K. Improving physical realism, stereochemistry, and side-chain accuracy in homology modeling: Four approaches that performed well in CASP8. Proteins 2009, 77 (Suppl. 9), 114–122. Available online: http://www.yasara.org (accessed on 20 August 2021). [CrossRef] [Green Version]
- DeLano, W.L. The PyMOL Molecular Graphics System; Version 1.7.1.1; Schrödinger, LLC.: New York, NY, USA, 2002; Available online: http://www.pymol.org (accessed on 17 November 2019).
- Wilson, I.A.; Stanfield, R.L. 50 Years of structural immunology. J. Biol. Chem. 2021, 296, 100745. [Google Scholar] [CrossRef]
- Sarma, V.R.; Silverton, E.W.; Davies, D.R.; Terry, W.D. The three-dimensional structure at 6 A resolution of a human gamma Gl immunoglobulin molecule. J. Biol. Chem. 1971, 246, 3753–3759. [Google Scholar] [CrossRef]
- Poljak, R.J.; Amzel, L.M.; Avey, H.P.; Becka, L.N. Structure of Fab’ New at 6 A resolution. Nat. New Biol. 1972, 235, 137–140. [Google Scholar] [CrossRef] [PubMed]
- Amzel, L.M.; Poljak, R.J.; Saul, F.; Varga, J.M.; Richards, F.F. The three dimensional structure of a combining region-ligand complex of immunoglobulin NEW at 3.5-A resolution. Proc. Natl. Acad. Sci. USA 1974, 71, 1427–1430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deisenhofer, J. Crystallographic refinement and atomic models of a human Fc fragment and its complex with fragment B of protein A from Staphylococcus aureus at 2.9- and 2.8-A resolution. Biochemistry 1981, 20, 2361–2370. [Google Scholar] [CrossRef] [PubMed]
- Silverton, E.W.; Navia, M.A.; Davies, D.R. Three-dimensional structure of an intact human immunoglobulin. Proc. Natl. Acad. Sci. USA 1977, 74, 5140–5144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [Green Version]
- Dunbar, J.; Krawczyk, K.; Leem, J.; Baker, T.; Fuchs, A.; Georges, G.; Shi, J.; Deane, C.M. SAbDab: The structural antibody database. Nucleic Acids Res. 2014, 42, D1140–D1146. [Google Scholar] [CrossRef]
- Kuhlbrandt, W. Biochemistry. The resolution revolution. Science 2014, 343, 1443–1444. [Google Scholar] [CrossRef]
- Callaway, E. The revolution will not be crystallized: A new method sweeps through structural biology. Nature 2015, 525, 172–174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, N.; Arthur, C.P.; Ciferri, C.; Matsumoto, M.L. Structure of the secretory immunoglobulin A core. Science 2020, 367, 1008–1014. [Google Scholar] [CrossRef] [PubMed]
- Kumar Bharathkar, S.; Parker, B.W.; Malyutin, A.G.; Haloi, N.; Huey-Tubman, K.E.; Tajkhorshid, E.; Stadtmueller, B.M. The structures of secretory and dimeric immunoglobulin A. eLife 2020, 9, e56098. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wang, G.; Li, Y.; Zhu, Q.; Shen, H.; Gao, N.; Xiao, J. Structural insights into secretory immunoglobulin A and its interaction with a pneumococcal adhesin. Cell Res. 2020, 30, 602–609. [Google Scholar] [CrossRef]
- Wang, Z.; Rahkola, J.; Redzic, J.S.; Chi, Y.C.; Tran, N.; Holyoak, T.; Zheng, H.; Janoff, E.; Eisenmesser, E. Mechanism and inhibition of Streptococcus pneumoniae IgA1 protease. Nat. Commun. 2020, 11, 6063. [Google Scholar] [CrossRef] [PubMed]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [Green Version]
- Shin, W.H.; Christoffer, C.W.; Wang, J.; Kihara, D. PL-PatchSurfer2: Improved local surface matching-based virtual screening method that is tolerant to target and ligand structure variation. J. Chem. Inf. Model. 2016, 56, 1676–1691. [Google Scholar] [CrossRef] [Green Version]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Knoppova, B.; Reily, C.; King, R.G.; Julian, B.A.; Novak, J.; Green, T.J. Pathogenesis of IgA Nephropathy: Current Understanding and Implications for Development of Disease-Specific Treatment. J. Clin. Med. 2021, 10, 4501. https://doi.org/10.3390/jcm10194501
Knoppova B, Reily C, King RG, Julian BA, Novak J, Green TJ. Pathogenesis of IgA Nephropathy: Current Understanding and Implications for Development of Disease-Specific Treatment. Journal of Clinical Medicine. 2021; 10(19):4501. https://doi.org/10.3390/jcm10194501
Chicago/Turabian StyleKnoppova, Barbora, Colin Reily, R. Glenn King, Bruce A. Julian, Jan Novak, and Todd J. Green. 2021. "Pathogenesis of IgA Nephropathy: Current Understanding and Implications for Development of Disease-Specific Treatment" Journal of Clinical Medicine 10, no. 19: 4501. https://doi.org/10.3390/jcm10194501