
Capsid Assembly Modulators as Antiviral Agents against HBV: Molecular Mechanisms and Clinical Perspectives

 , , , , and
, , , , and
Abstract
:1. Introduction
2. HBV Core Protein: A Multifunctional Protein Essential for HBV Life Cycle
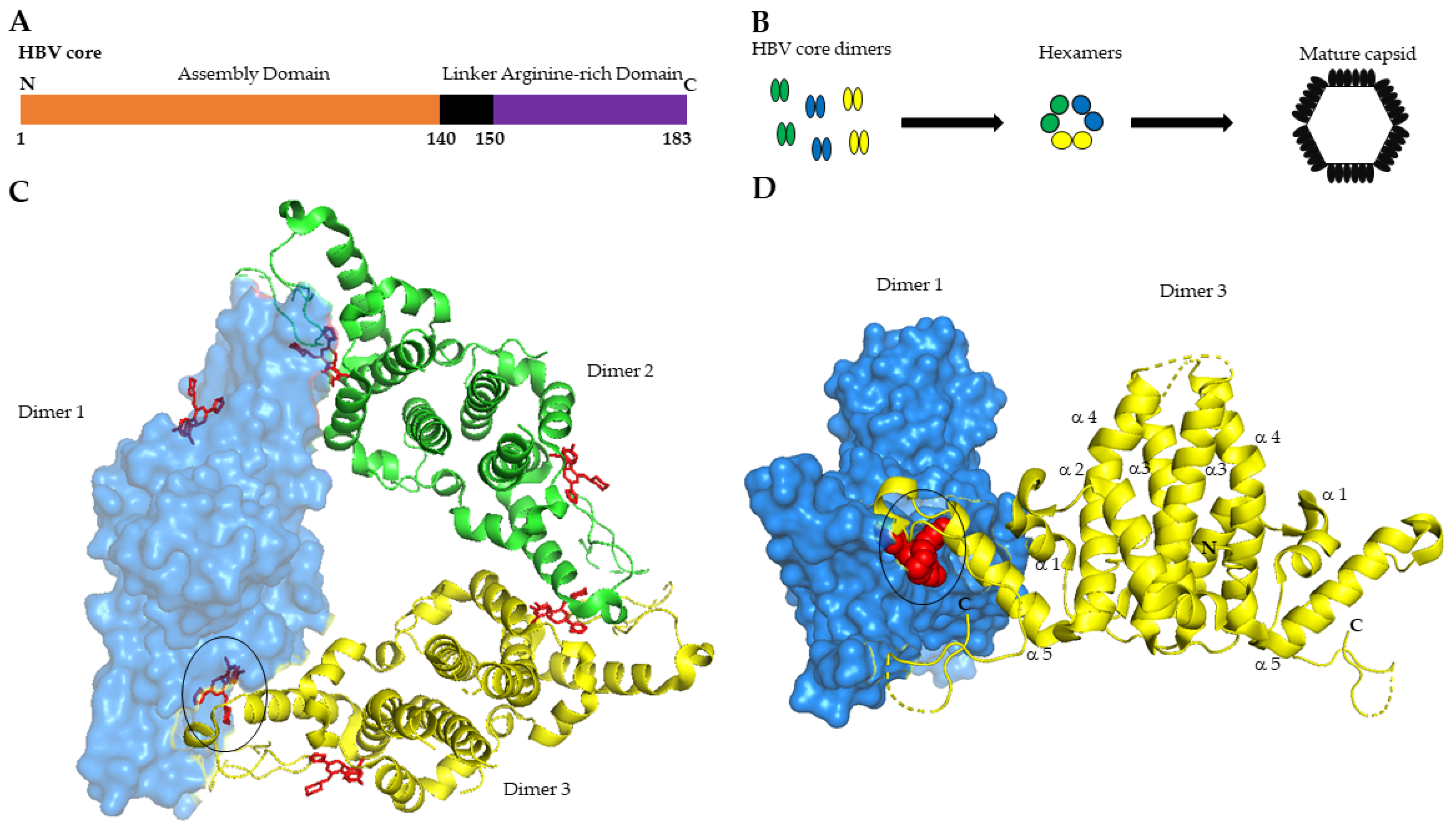
2.1. Core Protein Is the Building Block of HBV Nucleocapsid

2.2. Core Is Essential for the HBV Life Cycle
2.3. Core Interacts with Host Factors for Its Functions
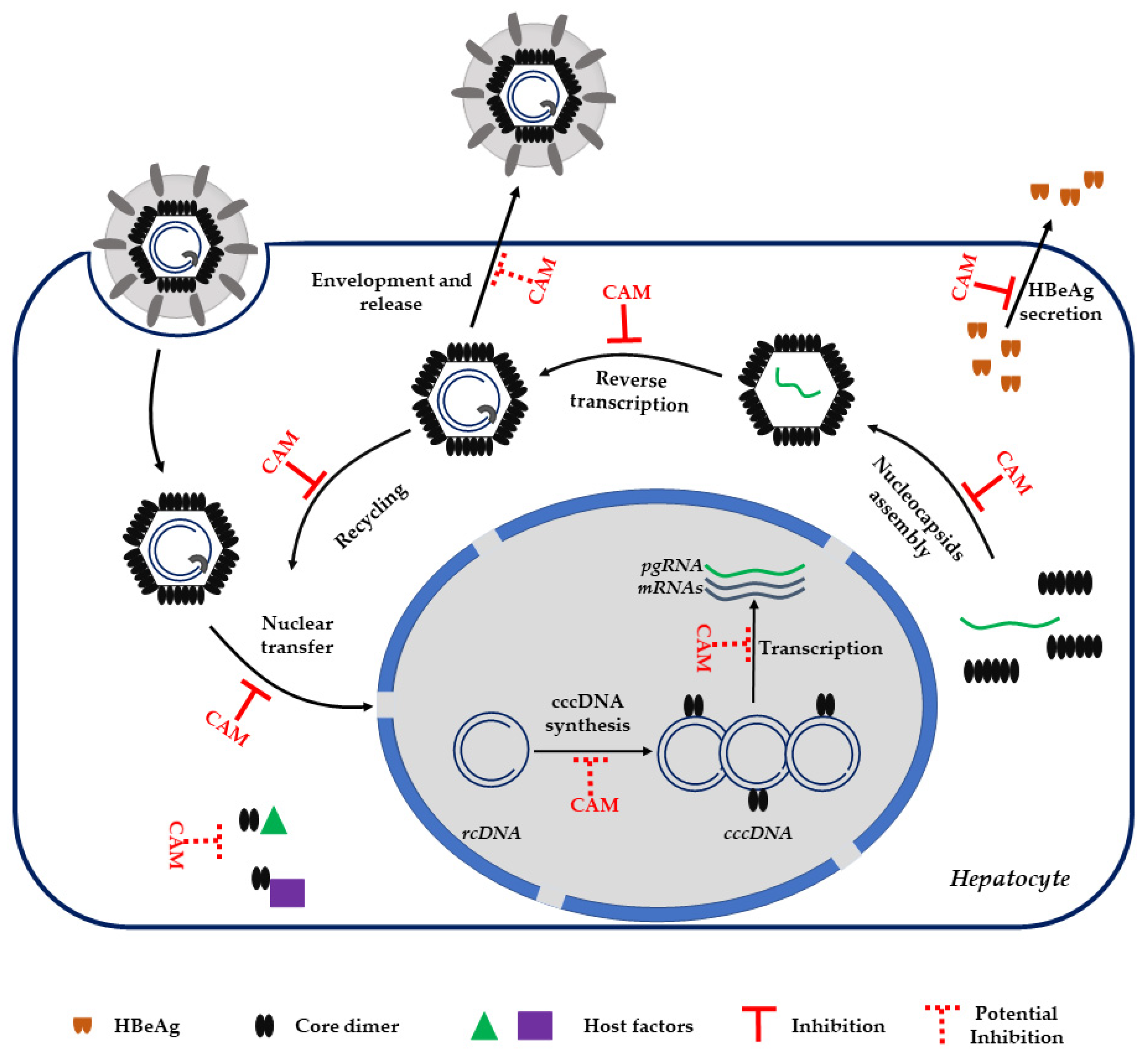
3. Capsid Assembly Modulators
3.1. CAM Chemotypes
3.2. CAM MoA
3.3. CAM and HBeAg
4. CAM Molecules in Preclinical and Clinical Development

{kind=link}
{kind=link}
| CAMs | Clinical Phase | Post-Treatment Reduction of Viral Parameters * |
|---|---|---|
| NVR 3–778 (Novira, Janssen Pharmaceutica) | Discontinued | DNA 1.97 log10 IU/mL, RNA 2.09 log10 copies/mL |
| ABI-H0731 (Assembly Bioscience) | Phase IIA | DNA 2.8 log10 IU/mL, RNA 2.0 log10 copies/mL |
| RO7049389 (Roche) | Phase II | DNA 3.3 log10 IU/mL, RNA 2.77 log10 IU/mL |
| JNJ-56136379 (Janssen) | Phase II | DNA < LLOQ, RNA < LLOQ |
| AB-506 (Arbutus) | Discontinued | NA |
| ABI-H2158 (Assembly Bioscience) | Discontinued | NA |
| ALG-000184 (ALIGOS therapeutics) | Phase I | DNA 3.8 log10 IU/mL, RNA 1.9 log10 IU/mL |
| GLS4JHS (Jilin University) | Phase I/II | DNA 2.13 log10 IU/mL, RNA 1.78 log10 IU/mL |
| EDP-514 (Enanta) | Phase I | NA |
| GLP-26 (Emory University) [77] | Preclinical | NA |
| ABI-H3733 (Assembly Bioscience) | Phase I | NA |
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Cooke, G.S.; Andrieux-Meyer, I.; Applegate, T.L.; Atun, R.; Burry, J.R.; Cheinquer, H.; Dusheiko, G.; Feld, J.J.; Gore, C.; Griswold, M.G.; et al. Accelerating the elimination of viral hepatitis: A Lancet Gastroenterology & Hepatology Commission. Lancet Gastroenterol. Hepatol. 2019, 4, 135–184. [Google Scholar] [CrossRef] [Green Version]
- Trepo, C.; Chan, H.L.; Lok, A. Hepatitis B virus infection. Lancet 2014, 384, 2053–2063. [Google Scholar] [CrossRef]
- Ligat, G.; Verrier, E.R.; Nassal, M.; Baumert, T.F. Hepatitis B virus-host interactions and novel targets for viral cure. Curr. Opin. Virol. 2021, 49, 41–51. [Google Scholar] [CrossRef] [PubMed]
- Beck, J.; Nassal, M. Hepatitis B virus replication. World J. Gastroenterol. 2007, 13, 48–64. [Google Scholar] [CrossRef] [Green Version]
- Verrier, E.R.; Colpitts, C.C.; Bach, C.; Heydmann, L.; Weiss, A.; Renaud, M.; Durand, S.C.; Habersetzer, F.; Durantel, D.; Abou-Jaoude, G.; et al. A targeted functional RNA interference screen uncovers glypican 5 as an entry factor for hepatitis B and D viruses. Hepatology 2016, 63, 35–48. [Google Scholar] [CrossRef] [PubMed]
- Yan, H.; Zhong, G.; Xu, G.; He, W.; Jing, Z.; Gao, Z.; Huang, Y.; Qi, Y.; Peng, B.; Wang, H.; et al. Sodium taurocholate cotransporting polypeptide is a functional receptor for human hepatitis B and D virus. Elife 2012, 1, e00049. [Google Scholar] [CrossRef]
- Ni, Y.; Lempp, F.A.; Mehrle, S.; Nkongolo, S.; Kaufman, C.; Falth, M.; Stindt, J.; Koniger, C.; Nassal, M.; Kubitz, R.; et al. Hepatitis B and D viruses exploit sodium taurocholate co-transporting polypeptide for species-specific entry into hepatocytes. Gastroenterology 2014, 146, 1070–1083. [Google Scholar] [CrossRef] [PubMed]
- Newbold, J.E.; Xin, H.; Tencza, M.; Sherman, G.; Dean, J.; Bowden, S.; Locarnini, S. The covalently closed duplex form of the hepadnavirus genome exists in situ as a heterogeneous population of viral minichromosomes. J. Virol. 1995, 69, 3350–3357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nassal, M. The arginine-rich domain of the hepatitis B virus core protein is required for pregenome encapsidation and productive viral positive-strand DNA synthesis but not for virus assembly. J. Virol. 1992, 66, 4107–4116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seifer, M.; Standring, D.N. Assembly and antigenicity of hepatitis B virus core particles. Intervirology 1995, 38, 47–62. [Google Scholar] [CrossRef]
- Wingfield, P.T.; Stahl, S.J.; Williams, R.W.; Steven, A.C. Hepatitis core antigen produced in Escherichia coli: Subunit composition, conformational analysis, and in vitro capsid assembly. Biochemistry 1995, 34, 4919–4932. [Google Scholar] [CrossRef] [PubMed]
- Wynne, S.A.; Crowther, R.A.; Leslie, A.G. The crystal structure of the human hepatitis B virus capsid. Mol. Cell. 1999, 3, 771–780. [Google Scholar] [CrossRef]
- Bottcher, B.; Wynne, S.A.; Crowther, R.A. Determination of the fold of the core protein of hepatitis B virus by electron cryomicroscopy. Nature 1997, 386, 88–91. [Google Scholar] [CrossRef]
- Conway, J.F.; Cheng, N.; Zlotnick, A.; Wingfield, P.T.; Stahl, S.J.; Steven, A.C. Visualization of a 4-helix bundle in the hepatitis B virus capsid by cryo-electron microscopy. Nature 1997, 386, 91–94. [Google Scholar] [CrossRef] [PubMed]
- Selzer, L.; Zlotnick, A. Assembly and Release of Hepatitis B Virus. Cold Spring Harb. Perspect. Med. 2015, 5, a021394. [Google Scholar] [CrossRef] [Green Version]
- Zlotnick, A. Theoretical aspects of virus capsid assembly. J. Mol. Recognit. 2005, 18, 479–490. [Google Scholar] [CrossRef] [PubMed]
- Zlotnick, A.; Venkatakrishnan, B.; Tan, Z.; Lewellyn, E.; Turner, W.; Francis, S. Core protein: A pleiotropic keystone in the HBV lifecycle. Antivir. Res. 2015, 121, 82–93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, Y.; Cheng, J.; Hu, Z.; Ban, H.; Wu, S.; Hwang, N.; Kulp, J.; Li, Y.; Du, Y.; Chang, J.; et al. Identification of hepatitis B virus core protein residues critical for capsid assembly, pgRNA encapsidation and resistance to capsid assembly modulators. Antivir. Res. 2021, 191, 105080. [Google Scholar] [CrossRef]
- Klumpp, K.; Lam, A.M.; Lukacs, C.; Vogel, R.; Ren, S.; Espiritu, C.; Baydo, R.; Atkins, K.; Abendroth, J.; Liao, G.; et al. High-resolution crystal structure of a hepatitis B virus replication inhibitor bound to the viral core protein. Proc. Natl. Acad. Sci. USA 2015, 112, 15196–15201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kann, M.; Bischof, A.; Gerlich, W.H. In vitro model for the nuclear transport of the hepadnavirus genome. J. Virol. 1997, 71, 1310–1316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blondot, M.L.; Bruss, V.; Kann, M. Intracellular transport and egress of hepatitis B virus. J. Hepatol. 2016, 64, S49–S59. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, A.; Schwarz, A.; Foss, M.; Zhou, L.; Rabe, B.; Hoellenriegel, J.; Stoeber, M.; Pante, N.; Kann, M. Nucleoporin 153 arrests the nuclear import of hepatitis B virus capsids in the nuclear basket. PLoS Pathog. 2010, 6, e1000741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.C.; Huang, E.Y.; Su, P.Y.; Wu, S.Y.; Yang, C.C.; Lin, Y.S.; Chang, W.C.; Shih, C. Nuclear export and import of human hepatitis B virus capsid protein and particles. PLoS Pathog. 2010, 6, e1001162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Selzer, L.; Kant, R.; Wang, J.C.Y.; Bothner, B.; Zlotnick, A. Hepatitis B Virus Core Protein Phosphorylation Sites Affect Capsid Stability and Transient Exposure of the C-terminal Domain. J. Biol. Chem. 2015, 290, 28584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, C.; Wang, J.C.; Zlotnick, A. A kinase chaperones hepatitis B virus capsid assembly and captures capsid dynamics in vitro. PLoS Pathog. 2011, 7, e1002388. [Google Scholar] [CrossRef]
- Lucifora, J.; Pastor, F.; Charles, E.; Pons, C.; Auclair, H.; Fusil, F.; Rivoire, M.; Cosset, F.L.; Durantel, D.; Salvetti, A. Evidence for long-term association of virion-delivered HBV core protein with cccDNA independently of viral protein production. JHEP Rep. 2021, 3, 100330. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.H.; Li, Y.N.; Zhao, J.R.; Zhang, J.; Yan, Z. HBc binds to the CpG islands of HBV cccDNA and promotes an epigenetic permissive state. Epigenetics 2011, 6, 720–726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bock, C.T.; Schwinn, S.; Locarnini, S.; Fyfe, J.; Manns, M.P.; Trautwein, C.; Zentgraf, H. Structural organization of the hepatitis B virus minichromosome. J. Mol. Biol. 2001, 307, 183–196. [Google Scholar] [CrossRef]
- Chong, C.K.; Cheng, C.Y.S.; Tsoi, S.Y.J.; Huang, F.Y.; Liu, F.; Seto, W.K.; Lai, C.L.; Yuen, M.F.; Wong, D.K. Role of hepatitis B core protein in HBV transcription and recruitment of histone acetyltransferases to cccDNA minichromosome. Antivir. Res. 2017, 144, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Lewellyn, E.B.; Loeb, D.D. The arginine clusters of the carboxy-terminal domain of the core protein of hepatitis B virus make pleiotropic contributions to genome replication. J. Virol. 2011, 85, 1298–1309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zlotnick, A.; Cheng, N.; Stahl, S.J.; Conway, J.F.; Steven, A.C.; Wingfield, P.T. Localization of the C terminus of the assembly domain of hepatitis B virus capsid protein: Implications for morphogenesis and organization of encapsidated RNA. Proc. Natl. Acad. Sci. USA 1997, 94, 9556–9561. [Google Scholar] [CrossRef] [Green Version]
- Chu, T.H.; Liou, A.T.; Su, P.Y.; Wu, H.N.; Shih, C. Nucleic acid chaperone activity associated with the arginine-rich domain of human hepatitis B virus core protein. J. Virol. 2014, 88, 2530–2543. [Google Scholar] [CrossRef] [Green Version]
- Diab, A.; Foca, A.; Zoulim, F.; Durantel, D.; Andrisani, O. The diverse functions of the hepatitis B core/capsid protein (HBc) in the viral life cycle: Implications for the development of HBc-targeting antivirals. Antivir. Res. 2018, 149, 211–220. [Google Scholar] [CrossRef]
- Shim, H.Y.; Quan, X.; Yi, Y.S.; Jung, G. Heat shock protein 90 facilitates formation of the HBV capsid via interacting with the HBV core protein dimers. Virology 2011, 410, 161–169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, C.C.; Huang, E.Y.; Li, H.C.; Su, P.Y.; Shih, C. Nuclear export of human hepatitis B virus core protein and pregenomic RNA depends on the cellular NXF1-p15 machinery. PLoS ONE 2014, 9, e106683. [Google Scholar] [CrossRef]
- Hu, Z.; Ban, H.; Zheng, H.; Liu, M.; Chang, J.; Guo, J.T. Protein phosphatase 1 catalyzes HBV core protein dephosphorylation and is co-packaged with viral pregenomic RNA into nucleocapsids. PLoS Pathog. 2020, 16, e1008669. [Google Scholar] [CrossRef] [PubMed]
- Chabrolles, H.; Auclair, H.; Vegna, S.; Lahlali, T.; Pons, C.; Michelet, M.; Coute, Y.; Belmudes, L.; Chadeuf, G.; Kim, Y.; et al. Hepatitis B virus Core protein nuclear interactome identifies SRSF10 as a host RNA-binding protein restricting HBV RNA production. PLoS Pathog. 2020, 16, e1008593. [Google Scholar] [CrossRef] [PubMed]
- Sohn, S.Y.; Kim, S.B.; Kim, J.; Ahn, B.Y. Negative regulation of hepatitis B virus replication by cellular Hsp40/DnaJ proteins through destabilization of viral core and X proteins. J. Gen. Virol. 2006, 87, 1883–1891. [Google Scholar] [CrossRef] [PubMed]
- Qian, G.; Jin, F.; Chang, L.; Yang, Y.; Peng, H.; Duan, C. NIRF, a novel ubiquitin ligase, interacts with hepatitis B virus core protein and promotes its degradation. Biotechnol. Lett. 2012, 34, 29–36. [Google Scholar] [CrossRef] [PubMed]
- Lucifora, J.; Xia, Y.; Reisinger, F.; Zhang, K.; Stadler, D.; Cheng, X.; Sprinzl, M.F.; Koppensteiner, H.; Makowska, Z.; Volz, T.; et al. Specific and nonhepatotoxic degradation of nuclear hepatitis B virus cccDNA. Science 2014, 343, 1221–1228. [Google Scholar] [CrossRef]
- Nijampatnam, B.; Liotta, D.C. Recent advances in the development of HBV capsid assembly modulators. Curr. Opin. Chem. Biol. 2019, 50, 73–79. [Google Scholar] [CrossRef] [PubMed]
- Stray, S.J.; Zlotnick, A. BAY 41-4109 has multiple effects on Hepatitis B virus capsid assembly. J. Mol. Recognit. 2006, 19, 542–548. [Google Scholar] [CrossRef] [PubMed]
- Deres, K.; Schroder, C.H.; Paessens, A.; Goldmann, S.; Hacker, H.J.; Weber, O.; Kramer, T.; Niewohner, U.; Pleiss, U.; Stoltefuss, J.; et al. Inhibition of hepatitis B virus replication by drug-induced depletion of nucleocapsids. Science 2003, 299, 893–896. [Google Scholar] [CrossRef]
- Debing, Y.; Buh Kum, D.; Sanchez, A.A.; Vanrusselt, H.; Liu, C.; Deval, J.; Stoycheva, A.; Zhang, Q.; Mukherjee, S.; Misner, D.; et al. ALG-005398 Is a Potent Non-HAP Class I HBV Capsid Assembly Modulator that Strongly Reduces HBsAg Levels In Vivo. Hepatology 2021, 74, 502A–503A. [Google Scholar]
- Delaney, W.E.t.; Edwards, R.; Colledge, D.; Shaw, T.; Furman, P.; Painter, G.; Locarnini, S. Phenylpropenamide derivatives AT-61 and AT-130 inhibit replication of wild-type and lamivudine-resistant strains of hepatitis B virus in vitro. Antimicrob. Agents Chemother. 2002, 46, 3057–3060. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, Y.H.; Cha, H.M.; Hwang, J.Y.; Park, S.Y.; Vishakantegowda, A.G.; Imran, A.; Lee, J.Y.; Yi, Y.S.; Jun, S.; Kim, G.H.; et al. Sulfamoylbenzamide-based Capsid Assembly Modulators for Selective Inhibition of Hepatitis B Viral Replication. ACS Med. Chem. Lett. 2021, 12, 242–248. [Google Scholar] [CrossRef] [PubMed]
- Katen, S.P.; Tan, Z.; Chirapu, S.R.; Finn, M.G.; Zlotnick, A. Assembly-directed antivirals differentially bind quasiequivalent pockets to modify hepatitis B virus capsid tertiary and quaternary structure. Structure 2013, 21, 1406–1416. [Google Scholar] [CrossRef] [Green Version]
- Weber, O.; Schlemmer, K.H.; Hartmann, E.; Hagelschuer, I.; Paessens, A.; Graef, E.; Deres, K.; Goldmann, S.; Niewoehner, U.; Stoltefuss, J.; et al. Inhibition of human hepatitis B virus (HBV) by a novel non-nucleosidic compound in a transgenic mouse model. Antivir. Res. 2002, 54, 69–78. [Google Scholar] [CrossRef]
- Brezillon, N.; Brunelle, M.N.; Massinet, H.; Giang, E.; Lamant, C.; DaSilva, L.; Berissi, S.; Belghiti, J.; Hannoun, L.; Puerstinger, G.; et al. Antiviral activity of Bay 41-4109 on hepatitis B virus in humanized Alb-uPA/SCID mice. PLoS ONE 2011, 6, e25096. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lahlali, T.; Berke, J.M.; Vergauwen, K.; Foca, A.; Vandyck, K.; Pauwels, F.; Zoulim, F.; Durantel, D. Novel Potent Capsid Assembly Modulators Regulate Multiple Steps of the Hepatitis B Virus Life Cycle. Antimicrob. Agents Chemother. 2018, 62, e00835-18. [Google Scholar] [CrossRef] [Green Version]
- Berke, J.M.; Dehertogh, P.; Vergauwen, K.; Van Damme, E.; Mostmans, W.; Vandyck, K.; Pauwels, F. Capsid Assembly Modulators Have a Dual Mechanism of Action in Primary Human Hepatocytes Infected with Hepatitis B Virus. Antimicrob. Agents Chemother. 2017, 61, e00560-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, F.; Zhao, Q.; Sheraz, M.; Cheng, J.; Qi, Y.; Su, Q.; Cuconati, A.; Wei, L.; Du, Y.; Li, W.; et al. HBV core protein allosteric modulators differentially alter cccDNA biosynthesis from de novo infection and intracellular amplification pathways. PLoS Pathog. 2017, 13, e1006658. [Google Scholar] [CrossRef]
- Wang, J.; Sheng, Q.; Ding, Y.; Chen, R.; Sun, X.; Chen, X.; Dou, X.; Lu, F. HBV RNA virion-like particles produced under nucleos(t)ide analogues treatment are mainly replication-deficient. J. Hepatol. 2018, 68, 847–849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jansen, L.; Kootstra, N.A.; van Dort, K.A.; Takkenberg, R.B.; Reesink, H.W.; Zaaijer, H.L. Hepatitis B Virus Pregenomic RNA Is Present in Virions in Plasma and Is Associated With a Response to Pegylated Interferon Alfa-2a and Nucleos(t)ide Analogues. J. Infect. Dis. 2016, 213, 224–232. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Shen, T.; Huang, X.; Kumar, G.R.; Chen, X.; Zeng, Z.; Zhang, R.; Chen, R.; Li, T.; Zhang, T.; et al. Serum hepatitis B virus RNA is encapsidated pregenome RNA that may be associated with persistence of viral infection and rebound. J. Hepatol. 2016, 65, 700–710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berke, J.M.; Dehertogh, P.; Vergauwen, K.; Mostmans, W.; Vandyck, K.; Raboisson, P.; Pauwels, F. Antiviral Properties and Mechanism of Action Studies of the Hepatitis B Virus Capsid Assembly Modulator JNJ-56136379. Antimicrob. Agents Chemother. 2020, 64, e02439-19. [Google Scholar] [CrossRef] [PubMed]
- Schlicksup, C.J.; Wang, J.C.; Francis, S.; Venkatakrishnan, B.; Turner, W.W.; VanNieuwenhze, M.; Zlotnick, A. Hepatitis B virus core protein allosteric modulators can distort and disrupt intact capsids. Elife 2018, 7, e31473. [Google Scholar] [CrossRef] [PubMed]
- Ito, K.; Kim, K.-H.; Lok, A.S.-F.; Tong, S. Characterization of genotype-specific carboxyl-terminal cleavage sites of hepatitis B virus e antigen precursor and identification of furin as the candidate enzyme. J. Virol. 2009, 83, 3507–3517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DiMattia, M.A.; Watts, N.R.; Stahl, S.J.; Grimes, J.M.; Steven, A.C.; Stuart, D.I.; Wingfield, P.T. Antigenic switching of hepatitis B virus by alternative dimerization of the capsid protein. Structure 2013, 21, 133–142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jegaskanda, S.; Ahn, S.H.; Skinner, N.; Thompson, A.J.; Ngyuen, T.; Holmes, J.; De Rose, R.; Navis, M.; Winnall, W.R.; Kramski, M.; et al. Downregulation of interleukin-18-mediated cell signaling and interferon gamma expression by the hepatitis B virus e antigen. J. Virol. 2014, 88, 10412–10420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lang, T.; Lo, C.; Skinner, N.; Locarnini, S.; Visvanathan, K.; Mansell, A. The hepatitis B e antigen (HBeAg) targets and suppresses activation of the toll-like receptor signaling pathway. J. Hepatol. 2011, 55, 762–769. [Google Scholar] [CrossRef]
- Chen, M.T.; Billaud, J.N.; Sallberg, M.; Guidotti, L.G.; Chisari, F.V.; Jones, J.; Hughes, J.; Milich, D.R. A function of the hepatitis B virus precore protein is to regulate the immune response to the core antigen. Proc. Natl. Acad. Sci. USA 2004, 101, 14913–14918. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fried, M.W.; Piratvisuth, T.; Lau, G.K.; Marcellin, P.; Chow, W.C.; Cooksley, G.; Luo, K.X.; Paik, S.W.; Liaw, Y.F.; Button, P.; et al. HBeAg and hepatitis B virus DNA as outcome predictors during therapy with peginterferon alfa-2a for HBeAg-positive chronic hepatitis B. Hepatology 2008, 47, 428–434. [Google Scholar] [CrossRef] [PubMed]
- Mitra, B.; Wang, J.; Kim, E.S.; Mao, R.; Dong, M.; Liu, Y.; Zhang, J.; Guo, H. Hepatitis B Virus Precore Protein p22 Inhibits Alpha Interferon Signaling by Blocking STAT Nuclear Translocation. J. Virol. 2019, 93, e00196-19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, Z.; Wu, D.; Hu, H.; Zeng, J.; Yu, X.; Xu, Z.; Zhou, Z.; Zhou, X.; Yang, G.; Young, J.A.T.; et al. Direct Inhibition of Hepatitis B e Antigen by Core Protein Allosteric Modulator. Hepatology 2019, 70, 11–24. [Google Scholar] [CrossRef] [Green Version]
- Cornberg, M.; Lok, A.S.; Terrault, N.A.; Zoulim, F.; Faculty, E.-A.H.T.E.C. Guidance for design and endpoints of clinical trials in chronic hepatitis B—Report from the 2019 EASL-AASLD HBV Treatment Endpoints Conference. Hepatology 2019, 71, 1070–1092. [Google Scholar] [CrossRef] [PubMed]
- Klumpp, K.; Shimada, T.; Allweiss, L.; Volz, T.; Lutgehetmann, M.; Hartman, G.; Flores, O.A.; Lam, A.M.; Dandri, M. Efficacy of NVR 3-778, Alone and In Combination With Pegylated Interferon, vs Entecavir In uPA/SCID Mice With Humanized Livers and HBV Infection. Gastroenterology 2018, 154, 652–662.e658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lam, A.M.; Espiritu, C.; Vogel, R.; Ren, S.; Lau, V.; Kelly, M.; Kuduk, S.D.; Hartman, G.D.; Flores, O.A.; Klumpp, K. Preclinical Characterization of NVR 3-778, a First-in-Class Capsid Assembly Modulator against Hepatitis B Virus. Antimicrob. Agents Chemother. 2019, 63, e01734-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Q.; Cai, D.; Yan, R.; Li, L.; Zong, Y.; Guo, L.; Mercier, A.; Zhou, Y.; Tang, A.; Henne, K.; et al. Preclinical Profile and Characterization of the Hepatitis B Virus Core Protein Inhibitor ABI-H0731. Antimicrob. Agents Chemother. 2020, 64, e01463-20. [Google Scholar] [CrossRef] [PubMed]
- Yuen, M.F.; Agarwal, K.; Gane, E.J.; Schwabe, C.; Ahn, S.H.; Kim, D.J.; Lim, Y.S.; Cheng, W.; Sievert, W.; Visvanathan, K.; et al. Safety, pharmacokinetics, and antiviral effects of ABI-H0731, a hepatitis B virus core inhibitor: A randomised, placebo-controlled phase 1 trial. Lancet Gastroenterol. Hepatol. 2020, 5, 152–166. [Google Scholar] [CrossRef]
- Zhou, X.; Zhou, Y.; Tian, X.; Shen, F.; Yang, G.; Zhu, W.; Ottaviani, G.; Xie, J.; Shen, H.; Young, J.; et al. In vitro and in vivo antiviral characterization of RO7049389, a novel small molecule capsid assembly modulator for the treatment of chronic hepatitis B. J. Hepatol. 2018, 68, S770. [Google Scholar] [CrossRef]
- Yuen, M.F.; Zhou, X.; Gane, E.; Schwabe, C.; Tanwandee, T.; Feng, S.; Jin, Y.; Triyatni, M.; Lemenuel-Diot, A.; Cosson, V.; et al. Safety, pharmacokinetics, and antiviral activity of RO7049389, a core protein allosteric modulator, in patients with chronic hepatitis B virus infection: A multicentre, randomised, placebo-controlled, phase 1 trial. Lancet Gastroenterol. Hepatol. 2021, 6, 723–732. [Google Scholar] [CrossRef]
- Vandenbossche, J.; Jessner, W.; van den Boer, M.; Biewenga, J.; Berke, J.M.; Talloen, W.; De Zwart, L.; Snoeys, J.; Yogaratnam, J. Pharmacokinetics, Safety and Tolerability of JNJ-56136379, a Novel Hepatitis B Virus Capsid Assembly Modulator, in Healthy Subjects. Adv. Ther. 2019, 36, 2450–2462. [Google Scholar] [CrossRef]
- Zoulim, F.; Lenz, O.; Vandenbossche, J.J.; Talloen, W.; Verbinnen, T.; Moscalu, I.; Streinu-Cercel, A.; Bourgeois, S.; Buti, M.; Crespo, J.; et al. JNJ-56136379, an HBV Capsid Assembly Modulator, Is Well-Tolerated and Has Antiviral Activity in a Phase 1 Study of Patients With Chronic Infection. Gastroenterology 2020, 159, 521–533.e529. [Google Scholar] [CrossRef]
- Zhang, H.; Wang, F.; Zhu, X.; Chen, Y.; Chen, H.; Li, X.; Wu, M.; Li, C.; Liu, J.; Zhang, Y.; et al. Antiviral Activity and Pharmacokinetics of the Hepatitis B Virus (HBV) Capsid Assembly Modulator GLS4 in Patients With Chronic HBV Infection. Clin. Infect. Dis. 2021, 73, 175–182. [Google Scholar] [CrossRef]
- Amblard, F.; Boucle, S.; Bassit, L.; Cox, B.; Sari, O.; Tao, S.; Chen, Z.; Ozturk, T.; Verma, K.; Russell, O.; et al. Novel Hepatitis B Virus Capsid Assembly Modulator Induces Potent Antiviral Responses In Vitro and in Humanized Mice. Antimicrob. Agents Chemother. 2020, 64, e01701-19. [Google Scholar] [CrossRef] [PubMed]
- Hurwitz, S.J.; McBrearty, N.; Arzumanyan, A.; Bichenkov, E.; Tao, S.; Bassit, L.; Chen, Z.; Kohler, J.J.; Amblard, F.; Feitelson, M.A.; et al. Studies on the Efficacy, Potential Cardiotoxicity and Monkey Pharmacokinetics of GLP-26 as a Potent Hepatitis B Virus Capsid Assembly Modulator. Viruses 2021, 13, 114. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Jekle, A.; Serebryany, V.; Welch, M.; Liu, J.; Vendeville, S.; Debing, Y.; Kum, D.B.; Ren, S.; Liu, C.; et al. Best-In-Class Preclinical Characteristics of Alg-000184, A Prodrug of The Capsid Assembly Modulator Alg-001075 for the Treatment of Chronic Hepatitis B. Hepatology 2020, 72, 503A. [Google Scholar] [CrossRef]
- Gane, E.J.; Yuen, M.; Jucov, A.; Le, K.; Westland, C.; Schwabe, C.; Agarwal, K.; Zhang, Q.L.; Blatt, L.M.; Chanda, S.; et al. Safety, Pharmacokinetics (PK), and Antiviral Activity of the Capsid Assembly Modulator (CAM) ALG-000184 in Subjects with Chronic Hepatitis B (CHB). Hepatology 2021, 74, 516A–517A. [Google Scholar]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Taverniti, V.; Ligat, G.; Debing, Y.; Kum, D.B.; Baumert, T.F.; Verrier, E.R. Capsid Assembly Modulators as Antiviral Agents against HBV: Molecular Mechanisms and Clinical Perspectives. J. Clin. Med. 2022, 11, 1349. https://doi.org/10.3390/jcm11051349
Taverniti V, Ligat G, Debing Y, Kum DB, Baumert TF, Verrier ER. Capsid Assembly Modulators as Antiviral Agents against HBV: Molecular Mechanisms and Clinical Perspectives. Journal of Clinical Medicine. 2022; 11(5):1349. https://doi.org/10.3390/jcm11051349
Chicago/Turabian StyleTaverniti, Valerio, Gaëtan Ligat, Yannick Debing, Dieudonne Buh Kum, Thomas F. Baumert, and Eloi R. Verrier. 2022. "Capsid Assembly Modulators as Antiviral Agents against HBV: Molecular Mechanisms and Clinical Perspectives" Journal of Clinical Medicine 11, no. 5: 1349. https://doi.org/10.3390/jcm11051349