
Clinical Impact of Physical Activity and Cough on Disease Progression in Fibrotic Interstitial Lung Disease
 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Design and Cohort
2.2. Physical Activity
2.3. Cough
2.4. Questionnaires Assessing Health-Related Quality of Life
2.5. Statistical Analysis
3. Results
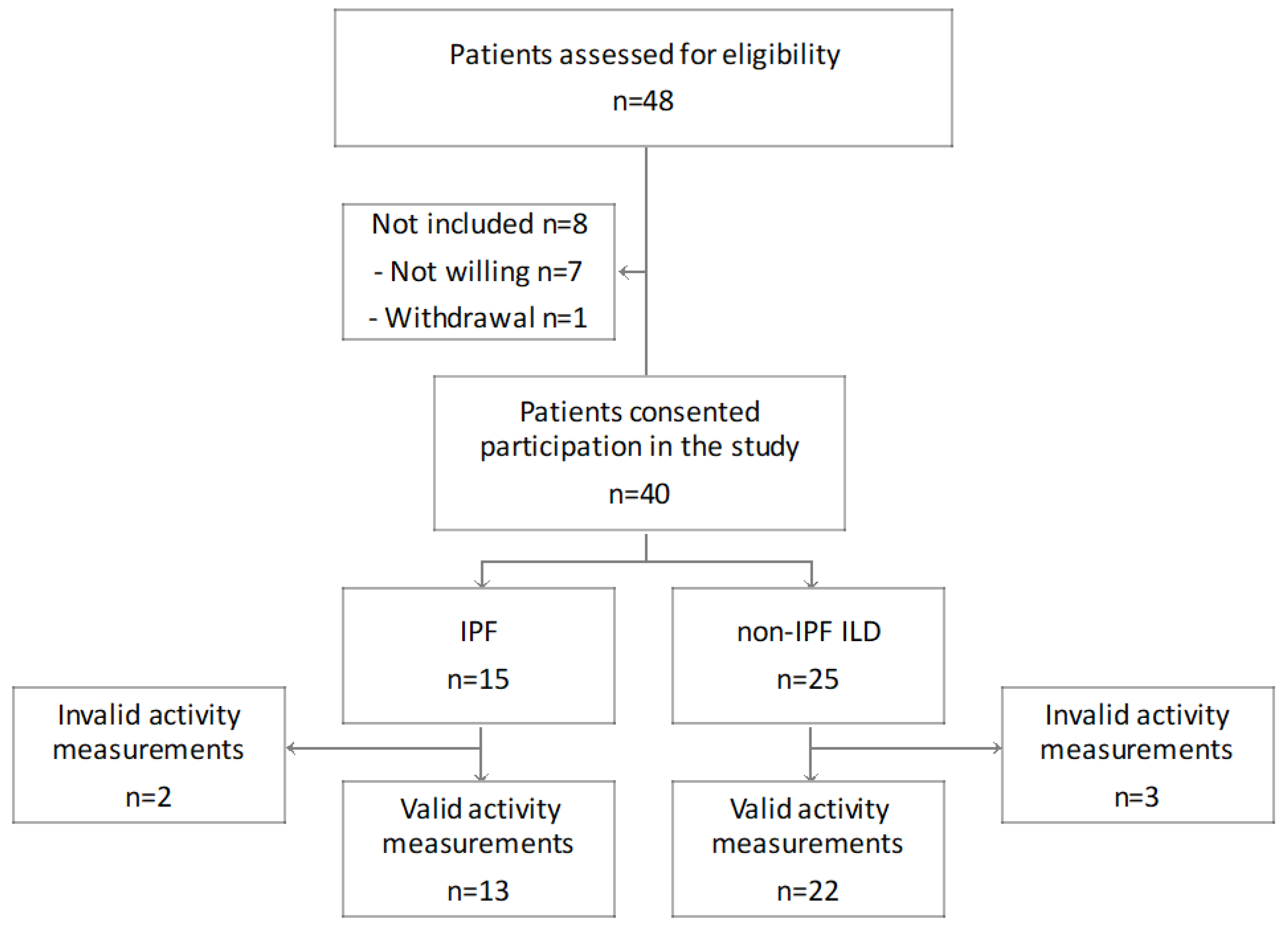
3.1. Study Cohort
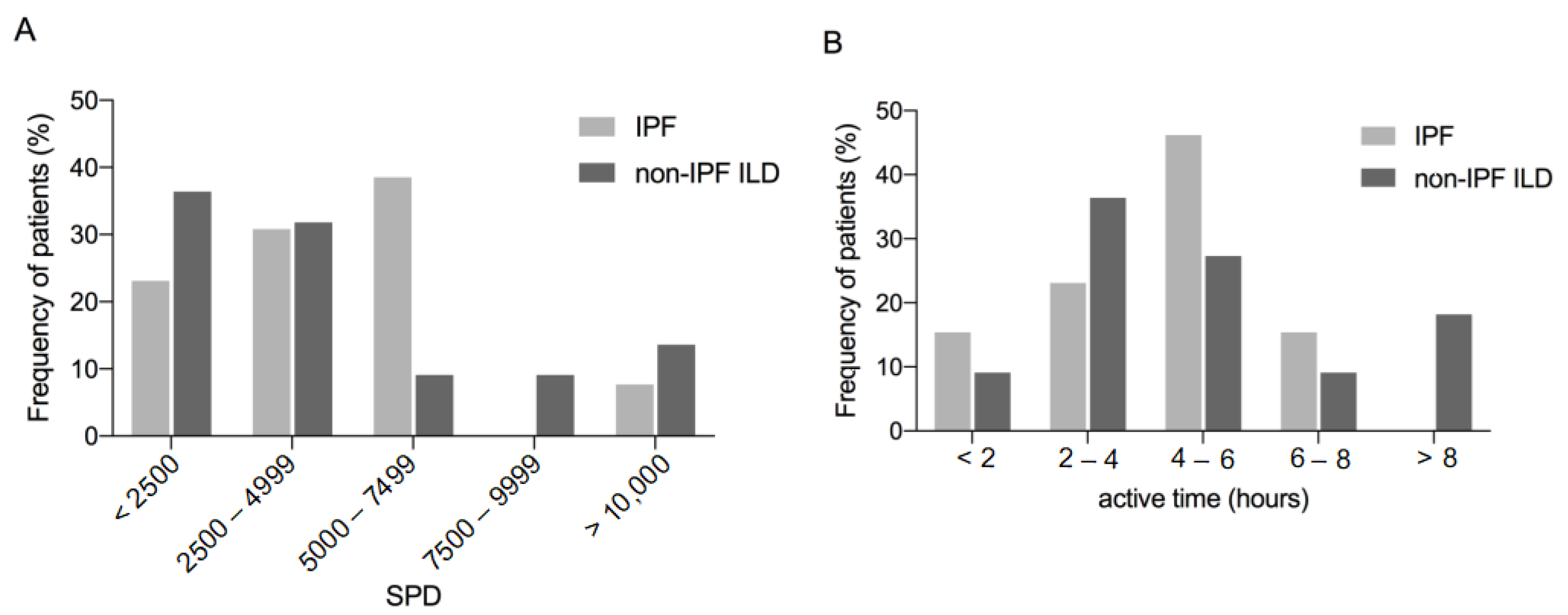
3.2. Physical Activity
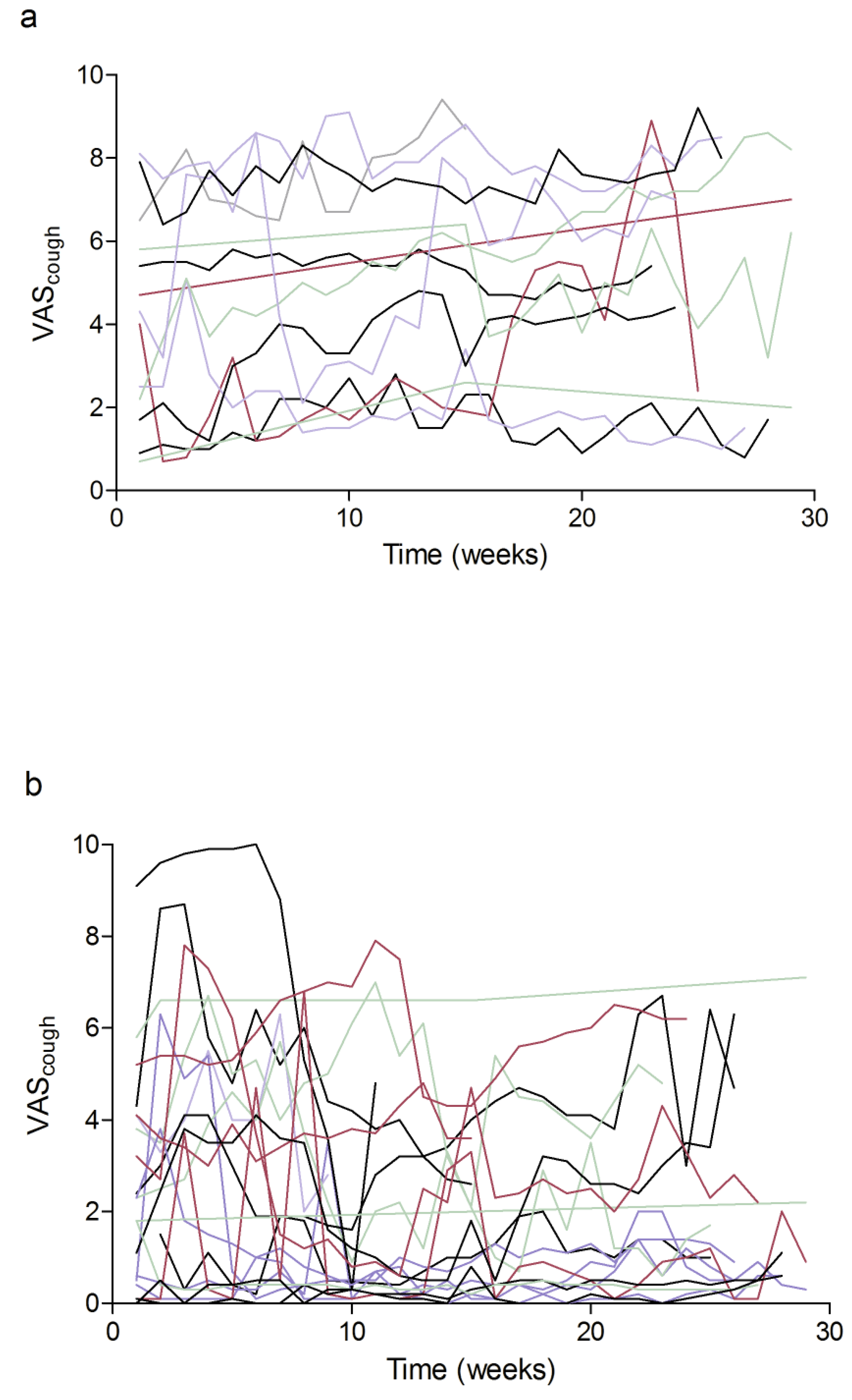
3.3. Cough
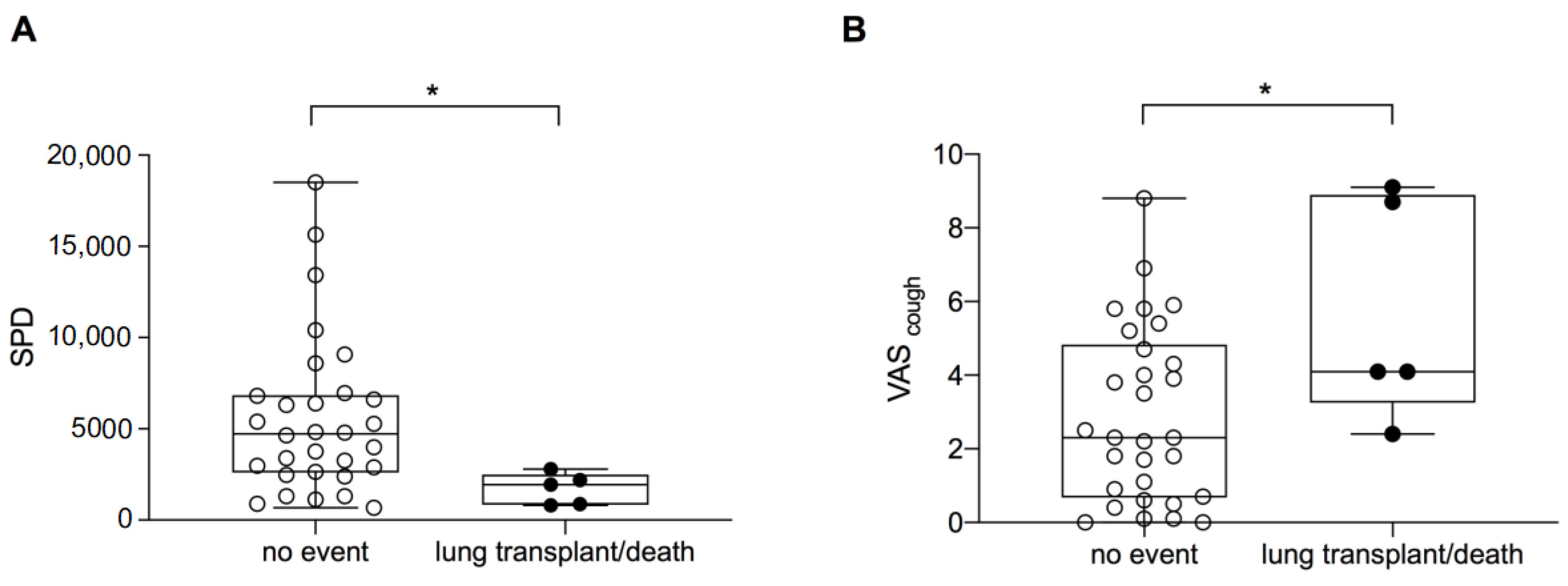
3.4. Transplant-Free Survival
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Glaspole, I.N.; Chapman, S.A.; Cooper, W.A.; Ellis, S.J.; Goh, N.S.; Hopkins, P.M.; Macansh, S.; Mahar, A.; Moodley, Y.P.; Paul, E.; et al. Health-related quality of life in idiopathic pulmonary fibrosis: Data from the Australian IPF Registry. Respirology 2017, 22, 950–956. [Google Scholar] [CrossRef] [PubMed]
- Bahmer, T.; Kirsten, A.-M.; Waschki, B.; Rabe, K.F.; Magnussen, H.; Kirsten, D.; Gramm, M.; Hummler, S.; Brunnemer, E.; Kreuter, M.; et al. Prognosis and longitudinal changes of physical activity in idiopathic pulmonary fibrosis. BMC Pulm. Med. 2017, 17, 104. [Google Scholar] [CrossRef] [PubMed]
- Wallaert, B.; Monge, E.; Le Rouzic, O.; Wémeau-Stervinou, L.; Salleron, J.; Grosbois, J.-M. Physical Activity in Daily Life of Patients With Fibrotic Idiopathic Interstitial Pneumonia. Chest 2013, 144, 1652–1658. [Google Scholar] [CrossRef] [PubMed]
- Nakayama, M.; Bando, M.; Araki, K.; Sekine, T.; Kurosaki, F.; Sawata, T.; Nakazawa, S.; Mato, N.; Yamasawa, H.; Sugiyama, Y. Physical activity in patients with idiopathic pulmonary fibrosis. Respirology 2015, 20, 640–646. [Google Scholar] [CrossRef]
- Wickerson, L.; Mathur, S.; Helm, D.; Singer, L.; Brooks, D. Physical Activity Profile of Lung Transplant Candidates With Interstitial Lung Disease. J. Cardiopulm. Rehabil. Prev. 2013, 33, 106–112. [Google Scholar] [CrossRef]
- Giacomini, M.; DeJean, D.; Simeonov, D.; Smith, A. Experiences of living and dying with COPD: A systematic review and synthesis of the qualitative empirical literature. Ont. Health Technol. Assess. Ser. 2012, 12, 1–47. [Google Scholar]
- Hur, S.A.; Guler, S.A.; Khalil, N.; Camp, P.G.; Guenette, J.A.; Ryerson, C.J. Impact of Psychological Deficits and Pain on Physical Activity of Patients with Interstitial Lung Disease. Lung 2019, 197, 415–425. [Google Scholar] [CrossRef]
- Cheng, J.Z.; Wilcox, P.G.; Glaspole, I.; Corte, T.J.; Murphy, D.; Hague, C.J.; Ryerson, C.J. Cough is less common and less severe in systemic sclerosis-associated interstitial lung disease compared to other fibrotic interstitial lung diseases. Respirology 2017, 22, 1592–1597. [Google Scholar] [CrossRef]
- Key, A.L.; Holt, K.; Hamilton, A.; Smith, J.; Earis, J. Objective cough frequency in Idiopathic Pulmonary Fibrosis. Cough 2010, 6, 4. [Google Scholar]
- Ryerson, C.J.; Abbritti, M.; Ley, B.; Elicker, B.M.; Jones, K.D.; Collard, H.R. Cough predicts prognosis in idiopathic pulmonary fibrosis. Respirology 2011, 16, 969–975. [Google Scholar] [CrossRef]
- Birring, S.S.; Kavanagh, J.E.; Irwin, R.S.; Keogh, K.A.; Lim, K.G.; Ryu, J.H.; Adams, T.M.; Altman, K.W.; Azoulay, E.; Barker, A.F.; et al. Treatment of Interstitial Lung Disease Associated Cough: CHEST Guideline and Expert Panel Report. Chest 2018, 154, 904–917. [Google Scholar] [CrossRef] [PubMed]
- Raghu, G.; Remy-Jardin, M.; Myers, J.L.; Richeldi, L.; Ryerson, C.J.; Lederer, D.J.; Behr, J.; Cottin, V.; Danoff, S.K.; Morell, F.; et al. Diagnosis of Idiopathic Pulmonary Fibrosis. An Official ATS/ERS/JRS/ALAT Clinical Practice Guideline. Am. J. Respir. Crit. Care Med. 2018, 198, e44–e68. [Google Scholar] [CrossRef] [PubMed]
- Travis, W.D.; Costabel, U.; Hansell, D.M.; King, T.E., Jr.; Lynch, D.A.; Nicholson, A.G.; Ryerson, C.J.; Ryu, J.H.; Selman, M.; Wells, A.U.; et al. An official American Thoracic Society/European Respiratory Society statement: Update of the international multidisciplinary classification of the idiopathic interstitial pneumonias. Am. J. Respir. Crit. Care Med. 2013, 188, 733–748. [Google Scholar] [CrossRef] [PubMed]
- Sennekamp, J.; Muller-Wening, D.; Amthor, M.; Baur, X.; Bergmann, K.C.; Costabel, U.; Kirsten, D.; Koschel, D.; Kroidl, R.; Liebetrau, G.; et al. Guidelines for diagnosing extrinsic allergic alveolitis (hypersensitivity pneumonitis) (German Extrinsic Allergic Alveolitis Study Group). Pneumologie 2007, 61, 52–56. [Google Scholar] [CrossRef] [PubMed]
- Morice, A.H.; Fontana, G.A.; Belvisi, M.G.; Birring, S.S.; Chung, K.F.; Dicpinigaitis, P.V.; Kastelik, J.A.; McGarvey, L.P.; Smith, J.; Tatar, M.; et al. ERS guidelines on the assessment of cough. Eur. Respir. J. 2007, 29, 1256–1276. [Google Scholar] [CrossRef]
- Nguyen, A.M.; Bacci, E.D.; Vernon, M.; Birring, S.S.; La Rosa, C.; Muccino, D.; Schelfhout, J. Validation of a visual analog scale for assessing cough severity in patients with chronic cough. Ther. Adv. Respir. Dis. 2021, 15, 17534666211049743. [Google Scholar]
- Jones, P.W.; Quirk, F.H.; Baveystock, C.M. The St George’s Respiratory Questionnaire. Respir. Med. 1991, 85 (Suppl. SB), 25–31. [Google Scholar] [CrossRef]
- Peng, S.; Li, Z.; Kang, J.; Hou, X. Cross-sectional and longitudinal construct validity of the Saint George’s Respiratory Questionnaire in patients with IPF. Respirology 2008, 13, 871–879. [Google Scholar] [CrossRef]
- Swigris, J.J.; Brown, K.K.; Behr, J.; du Bois, R.M.; King, T.E.; Raghu, G.; Wamboldt, F.S. The SF-36 and SGRQ: Validity and first look at minimum important differences in IPF. Respir. Med. 2010, 104, 296–304. [Google Scholar] [CrossRef]
- Patel, A.S.; Siegert, R.J.; Brignall, K.; Gordon, P.; Steer, S.; Desai, S.R.; Maher, T.M.; Renzoni, E.A.; Wells, A.U.; Higginson, I.J.; et al. The development and validation of the King’s Brief Interstitial Lung Disease (K-BILD) health status questionnaire. Thorax 2012, 67, 804–810. [Google Scholar] [CrossRef]
- Szentes, B.L.; Kreuter, M.; Bahmer, T.; Birring, S.S.; Claussen, M.; Waelscher, J.; Leidl, R.; Schwarzkopf, L. Quality of life assessment in interstitial lung diseases:a comparison of the disease-specific K-BILD with the generic EQ-5D-5L. Respir. Res. 2018, 19, 101. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.W.; Quirk, F.H.; Baveystock, C.M.; Littlejohns, P. A Self-complete Measure of Health Status for Chronic Airflow Limitation: The St. George’s Respiratory Questionnaire. Am. Rev. Respir. Dis. 1992, 145, 1321–1327. [Google Scholar] [CrossRef] [PubMed]
- Kreuter, M.; Birring, S.S.; Wijsenbeek, M.; Wapenaar, M.; Oltmanns, U.; Costabel, U.; Bonella, F. German Validation of the “King’s Brief Interstitial Lung Disease (K-Bild) Health Status Questionnaire”. Pneumologie 2016, 70, 742–746. [Google Scholar] [PubMed]
- Bahmer, T.; Kirsten, A.-M.; Waschki, B.; Rabe, K.F.; Magnussen, H.; Kirsten, D.; Gramm, M.; Hummler, S.; Brunnemer, E.; Kreuter, M.; et al. Clinical Correlates of Reduced Physical Activity in Idiopathic Pulmonary Fibrosis. Respiration 2016, 91, 497–502. [Google Scholar] [CrossRef] [PubMed]
- Sinha, A.; Lee, K.K.; Rafferty, G.; Yousaf, N.; Pavord, I.; Galloway, J.; Birring, S.S. Predictors of objective cough frequency in pulmonary sarcoidosis. Eur. Respir. J. 2016, 47, 1461–1471. [Google Scholar] [CrossRef] [PubMed]
- Tashkin, D.P.; Elashoff, R.; Clements, P.J.; Goldin, J.; Roth, M.D.; Furst, D.E.; Arriola, E.; Silver, R.; Strange, C.; Bolster, M.; et al. Cyclophosphamide versus placebo in scleroderma lung disease. N. Engl. J. Med. 2006, 354, 2655–2666. [Google Scholar] [CrossRef]
- Tashkin, D.P.; Roth, M.D.; Clements, P.J.; Furst, D.E.; Khanna, D.; Kleerup, E.C.; Goldin, J.; Arriola, E.; Volkmann, E.R.; Kafaja, S.; et al. Mycophenolate mofetil versus oral cyclophosphamide in scleroderma-related interstitial lung disease (SLS II): A randomised controlled, double-blind, parallel group trial. Lancet Respir. Med. 2016, 4, 708–719. [Google Scholar] [CrossRef]
- Sinha, A.; Patel, A.S.; Siegert, R.J.; Bajwah, S.; Maher, T.M.; Renzoni, E.; Wells, A.U.; Higginson, I.J.; Birring, S.S. The King’s Brief Interstitial Lung Disease (KBILD) questionnaire: An updated minimal clinically important difference. BMJ Open Respir. Res. 2019, 6, e000363. [Google Scholar] [CrossRef]
- Prior, T.S.; Hoyer, N.; Shaker, S.B.; Davidsen, J.R.; Yorke, J.; Hilberg, O.; Bendstrup, E. Validation of the IPF-specific version of St. George’s Respiratory Questionnaire. Respir. Res. 2019, 20, 199. [Google Scholar] [CrossRef]
- Henriksen, A.; Mikalsen, M.H.; Woldaregay, A.Z.; Muzny, M.; Hartvigsen, G.; Hopstock, L.A.; Grimsgaard, S. Using Fitness Trackers and Smartwatches to Measure Physical Activity in Research: Analysis of Consumer Wrist-Worn Wearables. J. Med Internet Res. 2018, 20, e110. [Google Scholar] [CrossRef]
- Raghu, G.; Remy-Jardin, M.; Richeldi, L.; Thomson, C.C.; Inoue, Y.; Johkoh, T.; Kreuter, M.; Lynch, D.A.; Maher, T.M.; Martinez, F.J.; et al. Idiopathic Pulmonary Fibrosis (an Update) and Progressive Pulmonary Fibrosis in Adults: An Official ATS/ERS/JRS/ALAT Clinical Practice Guideline. Am. J. Respir. Crit. Care Med. 2022, 205, e18–e47. [Google Scholar] [CrossRef] [PubMed]
- Behr, J.; Bonella, F.; Frye, B.C.; Gunther, A.; Hagmeyer, L.; Henes, J.; Klemm, P.; Koschel, D.; Kreuter, M.; Leuschner, G.; et al. Pharmacological treatment of idiopathic pulmonary fibrosis (update) and progressive pulmonary fibrosis-S2k Guideline of the German Respiratory Society. Pneumologie 2023, 77, e1. [Google Scholar] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| All (n = 35) | IPF (n = 13) | Non-IPF ILD (n = 22) | p-Value | |
|---|---|---|---|---|
| Age (years) | 61.8 ± 10.8 | 65.3 ± 7.2 | 59.8 ± 12.1 | 0.145 |
| Sex (male), n (%) | 20 (57.1) | 10 (76.9) | 10 (45.5) | 0.069 |
| BMI (kg/m2) | 27.7 ± 4.2 | 27.0 ± 4.7 | 28.1 ± 4.0 | 0.471 |
| Smoking status | ||||
| Non-smokers | 16 (45.7) | 3 (23.1) | 13 (59.1) | 0.08 |
| Ex-smoker | 19 (54.3) | 10 (76.9) | 9 (40.9) | 0.08 |
| Current smokers | 0 | 0 | 0 | 1.0 |
| Smoking history (py) | 21.4 ± 13.4 | 23.5 ± 11.3 | 19.0 ± 15.7 | 0.479 |
| Lung function | ||||
| FVC, % predicted | 65.3 ± 21.7 | 68.5 ± 18.7 | 63.4 ± 23.5 | 0.507 |
| FVC (L) | 2.4 ± 0.8 | 2.7 ± 0.8 | 2.2 ± 0.8 | 0.135 |
| TLC, % predicted | 70.5 ± 20.4 | 70.5 ± 19.1 | 70.5 ± 21.6 | 0.991 |
| TLC (L) | 4.2 ± 1.1 | 4.6 ± 1.2 | 4.0 ± 1.0 | 0.210 |
| DLCO, % predicted a | 42.8 ± 16.4 | 38.9 ± 11.5 | 45.4 ± 19.1 | 0.427 |
| 6MWD (m) b | 381.3 ± 125.7 | 431.2 ± 89.5 | 353.9 ± 136.0 | 0.102 |
| KBILD | 52.4 ± 11.3 | 48.2 ± 2.6 | 53.1 ± 12.1 | 0.022 |
| SGRQ | 49.2 ± 18.7 | 51.1 ± 9.8 | 48.9 ± 20.0 | 0.193 |
| Comorbidity | All (n = 35) | IPF (n = 13) | Non-IPF ILD (n = 22) | p-Value |
|---|---|---|---|---|
| Arterial hypertension, n (%) | 16 (45.7) | 6 (46.2) | 10 (45.5) | 0.968 |
| Diabetes mellitus, n (%) | 4 (11.4) | 2 (15.4) | 2 (9.1) | 0.572 |
| Hyperlipoproteinemia, n (%) | 3 (8.6) | 2 (15.4) | 1 (4.5) | 0.268 |
| Coronary artery disease, n (%) | 8 (22.9) | 1 (7.7) | 7 (31.8) | 0.101 |
| History of revascularization, n (%) | 5 (14.3) | 1 (7.7) | 4 (18.2) | 0.392 |
| Osteoporosis, n (%) | 3 (8.6) | 1 (7.7) | 2 (9.1) | 0.886 |
| Obstructive sleep apnea syndrome, n (%) | 4 (8.7) | 2 (15.4) | 2 (9.1) | 0.572 |
| Gastroesophageal reflux disease, n (%) | 8 (22.9) | 3 (23.1) | 5 (22.7) | 0.981 |
| Atrial fibrillation, n (%) | 1 (4.3) | 0 (0) | 1 (4.5) | 0.435 |
| Pulmonary hypertension, n (%) a | 9 (25.7) | 3 (23.1) | 6 (27.3) | 0.926 |
| All | IPF | Non-IPF-ILD | ||||
|---|---|---|---|---|---|---|
| r | p-Value | r | p-Value | r | p-Value | |
| FVC, % predicted | 0.495 | 0.003 | 0.706 | 0.007 | 0.464 | 0.03 |
| DLCO, % predicted | 0.323 | 0.165 | 0.440 | 0.276 | 0.282 | 0.375 |
| 6MWD, m | 0.537 | 0.002 | 0.695 | 0.018 | 0.518 | 0.019 |
| BMI (kg/m2) | −0.050 | 0.776 | −0.185 | 0.545 | −0.019 | 0.934 |
| KBILD | 0.353 | 0.038 | 0.067 | 0.827 | 0.405 | 0.062 |
| SGRQ | −0.56 | 0.755 | 0.414 | 0.160 | −0.131 | 0.571 |
| All | IPF | Non-IPF-ILD | ||||
|---|---|---|---|---|---|---|
| r | p-Value | r | p-Value | r | p-Value | |
| FVC, % predicted | −0.222 | 0.119 | 0.107 | 0.725 | −0.478 | 0.025 |
| DLCO, % predicted | −0.466 | 0.038 | −0.148 | 0.727 | −0.640 | 0.025 |
| 6MWD, m | −0.367 | 0.042 | 0.144 | 0.738 | −0.773 | <0.01 |
| SPD | −0.227 | 0.189 | 0.086 | 0.779 | −0.350 | 0.110 |
| Active time | −0.207 | 0.232 | −0.090 | 0.771 | −0.221 | 0.324 |
| KBILD | −0.539 | 0.001 | −0.616 | 0.025 | −0.437 | 0.042 |
| SGRQ | 0.195 | 0.269 | −0.063 | 0.838 | 0.204 | 0.374 |
| Hazard Ratio | 95%–CI | p-Value | |
|---|---|---|---|
| Baseline FVC, % predicted | 0.973 | 0.941–1.007 | 0.119 |
| IPF | 1.708 | 0.569–5.125 | 0.340 |
| VAScough | 1.387 | 1.081–1.781 | 0.010 |
| SPD (per 1000 SPD) | 0.606 | 0.412–0.892 | 0.011 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Veit, T.; Barnikel, M.; Kneidinger, N.; Munker, D.; Arnold, P.; Barton, J.; Crispin, A.; Milger, K.; Behr, J.; Neurohr, C.; et al. Clinical Impact of Physical Activity and Cough on Disease Progression in Fibrotic Interstitial Lung Disease. J. Clin. Med. 2023, 12, 3787. https://doi.org/10.3390/jcm12113787
Veit T, Barnikel M, Kneidinger N, Munker D, Arnold P, Barton J, Crispin A, Milger K, Behr J, Neurohr C, et al. Clinical Impact of Physical Activity and Cough on Disease Progression in Fibrotic Interstitial Lung Disease. Journal of Clinical Medicine. 2023; 12(11):3787. https://doi.org/10.3390/jcm12113787
Chicago/Turabian StyleVeit, Tobias, Michaela Barnikel, Nikolaus Kneidinger, Dieter Munker, Paola Arnold, Jürgen Barton, Alexander Crispin, Katrin Milger, Jürgen Behr, Claus Neurohr, and et al. 2023. "Clinical Impact of Physical Activity and Cough on Disease Progression in Fibrotic Interstitial Lung Disease" Journal of Clinical Medicine 12, no. 11: 3787. https://doi.org/10.3390/jcm12113787