Clinical Spectrum and Functional Consequences Associated with Bi-Allelic Pathogenic PNPT1 Variants

 , , , , ,
, , , , ,
Abstract
:1. Introduction
2. Experimental Section
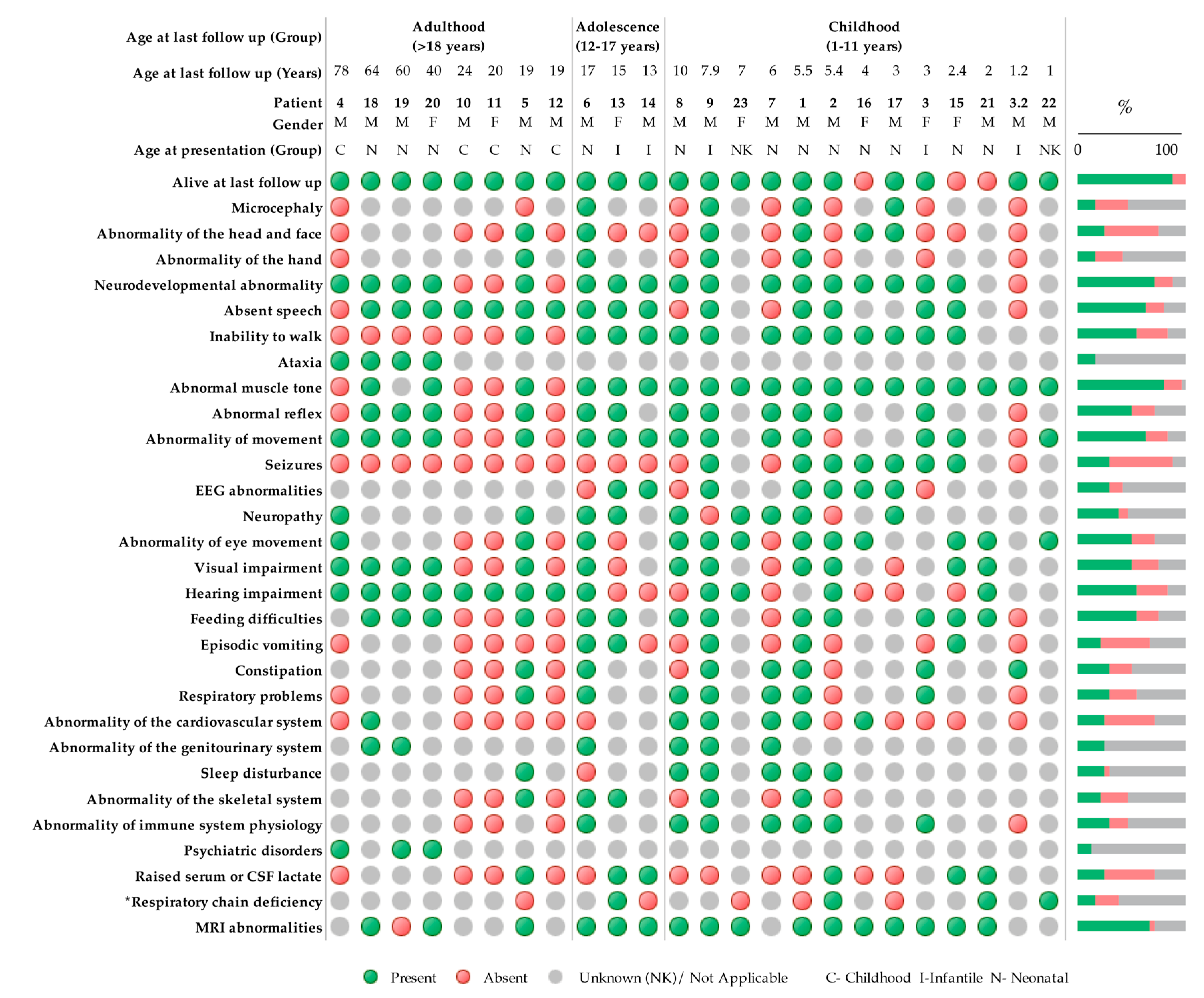
2.1. Patients
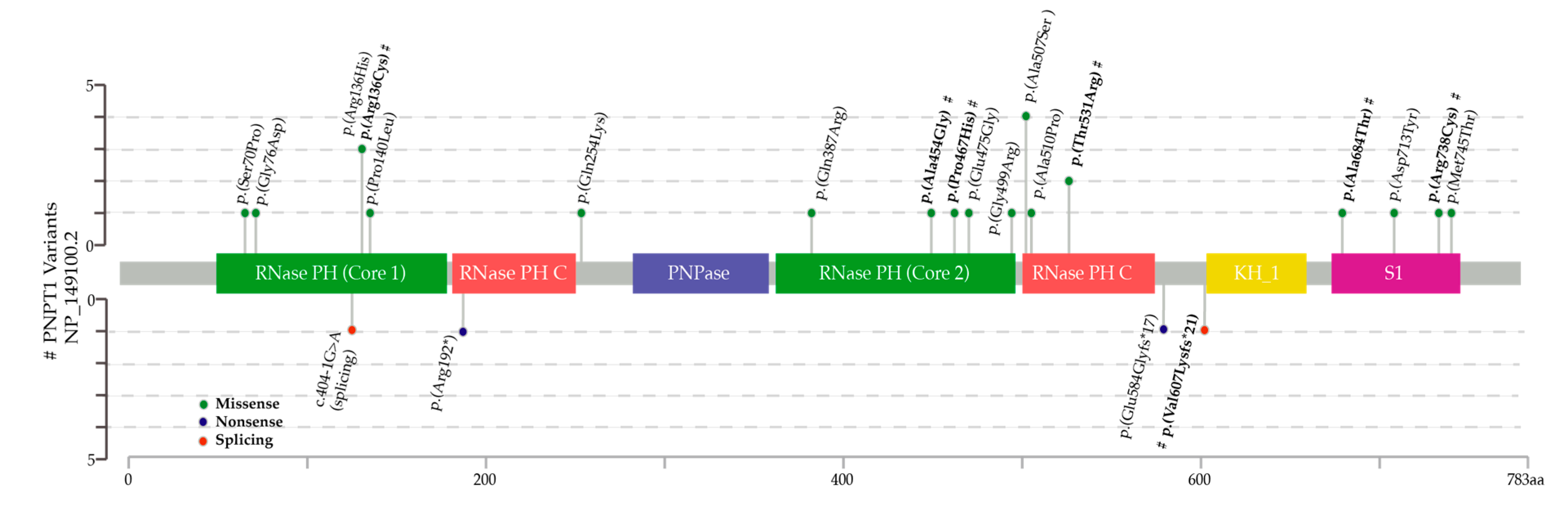
2.2. Next Generation Sequencing (NGS) and in Silico Tools
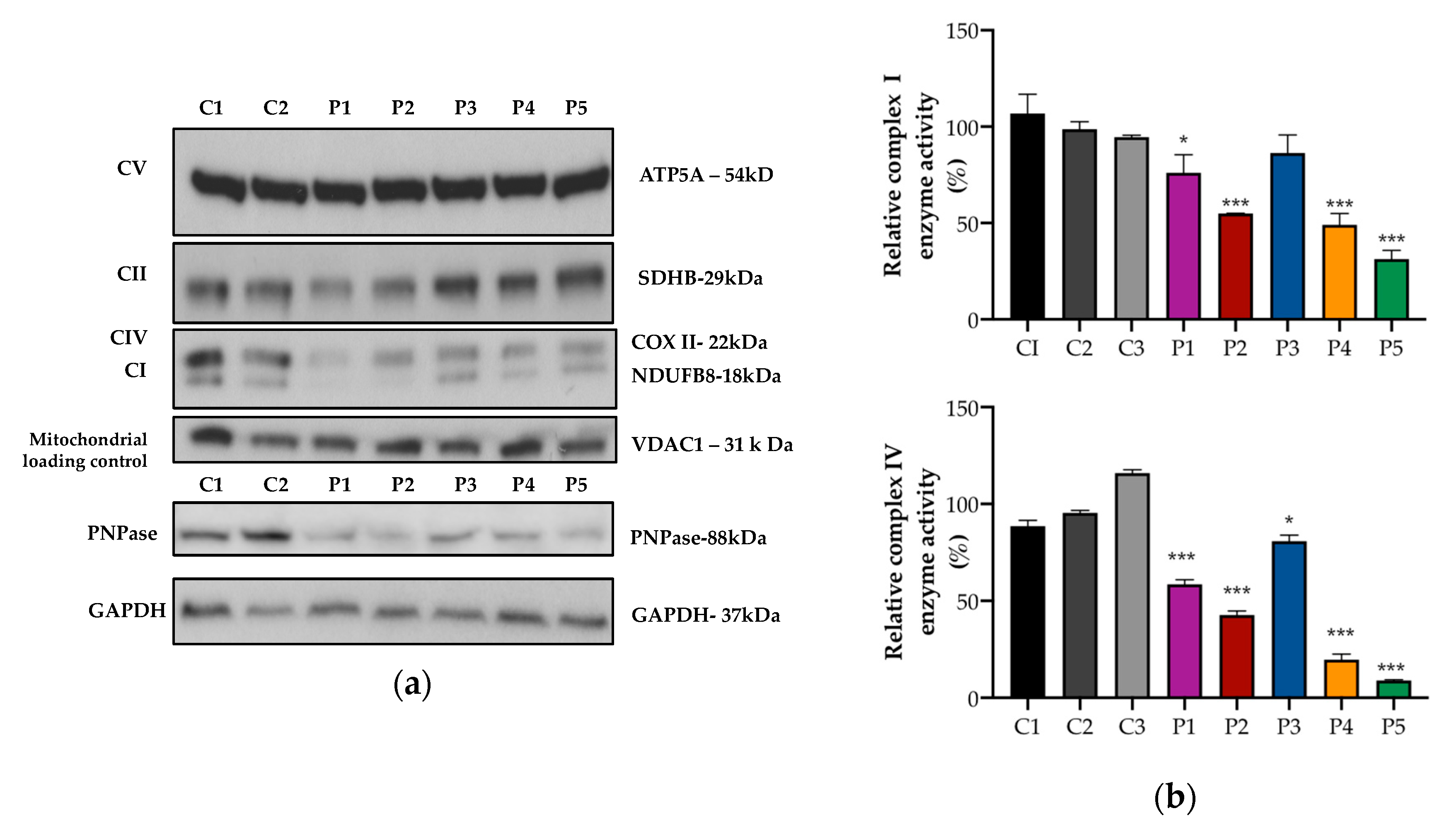
2.3. Western Blotting
2.4. Mitochondrial Oxidative Phosphorylation (OXPHOS) Enzyme Activities
2.5. Fibroblast Culture, RNA Extraction, and Complementary DNA (cDNA) Studies
2.6. RNA Extraction from Blood
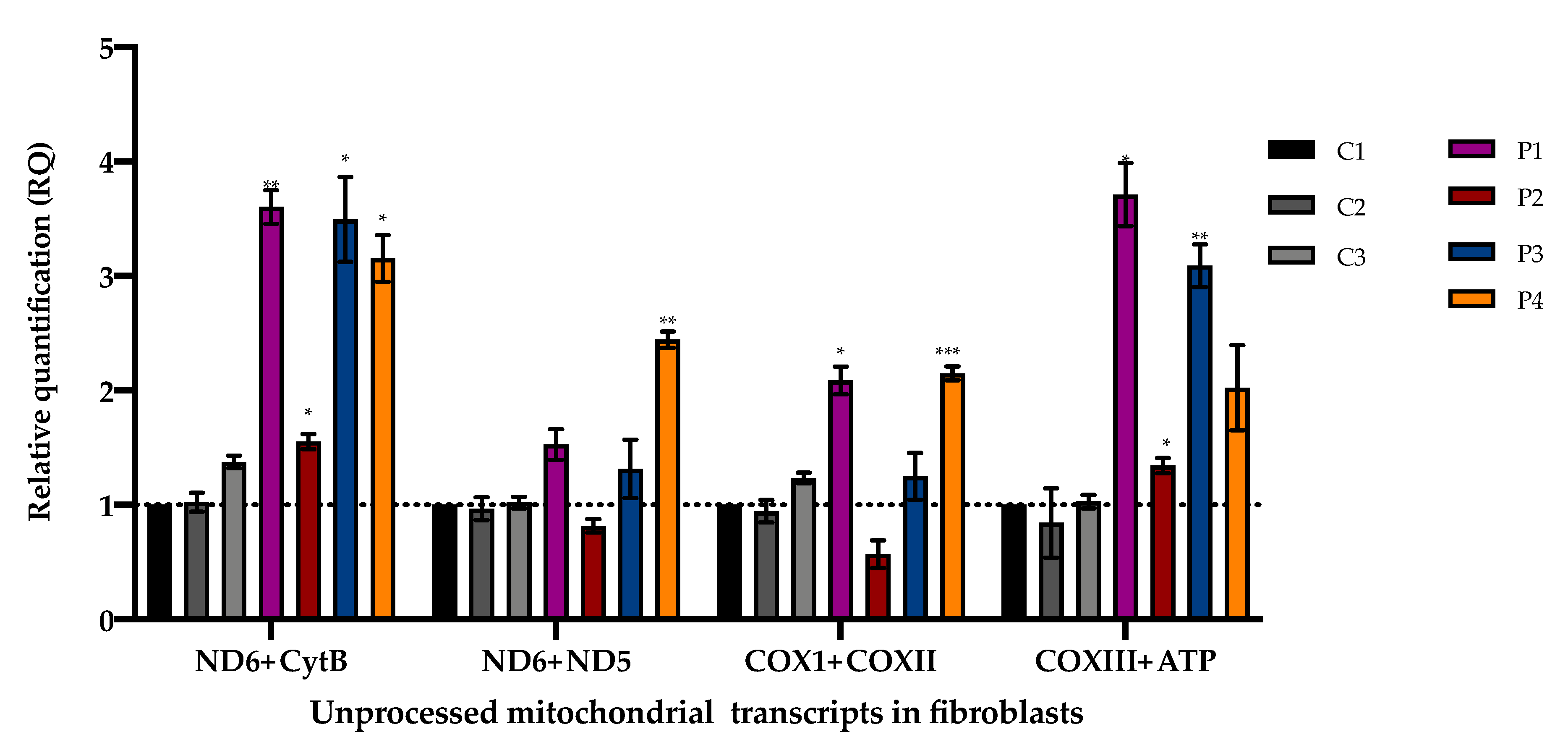
2.7. PCR Quantification of Unprocessed Mitochondrial Transcripts
2.8. Interferon Signature Analysis
3. Results
3.1. cDNA Studies Identify a Splicing Defect in P2
3.2. Mitochondrial OXPHOS Protein Expression and Activity
3.3. Accumulation of Mitochondrial Unprocessed Transcripts
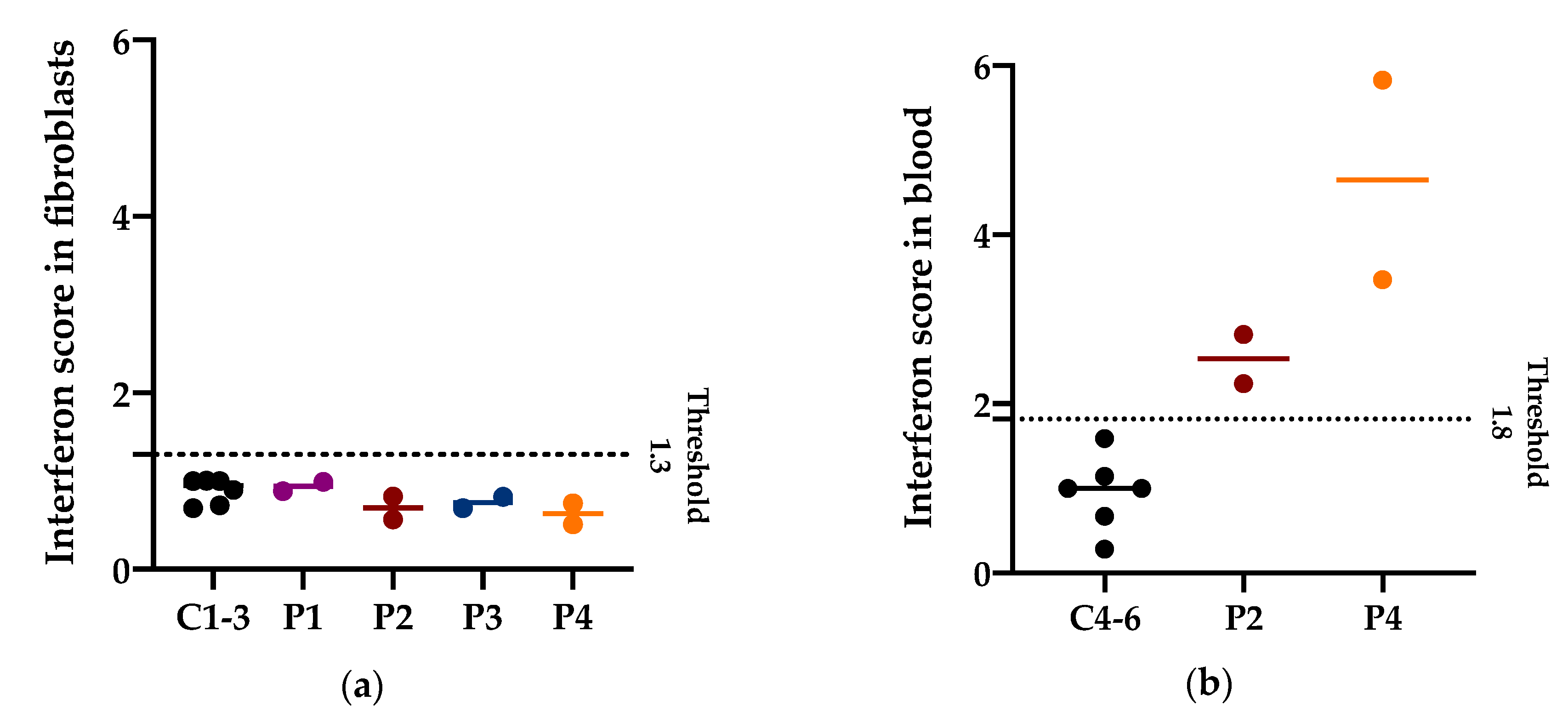
3.4. Interferon Signature in Patients with PNPT1 Variants
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Chen, H.W.; Rainey, R.N.; Balatoni, C.E.; Dawson, D.W.; Troke, J.J.; Wasiak, S.; Hong, J.S.; McBride, H.M.; Koehler, C.M.; Teitell, M.A.; et al. Mammalian polynucleotide phosphorylase is an intermembrane space RNase that maintains mitochondrial homeostasis. Mol. Cell. Biol. 2006, 26, 8475–8487. [Google Scholar] [CrossRef]
- Borowski, L.S.; Dziembowski, A.; Hejnowicz, M.S.; Stepien, P.P.; Szczesny, R.J. Human mitochondrial RNA decay mediated by PNPase-hSuv3 complex takes place in distinct foci. Nucleic Acids Res. 2013, 41, 1223–1240. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Chen, H.W.; Oktay, Y.; Zhang, J.; Allen, E.L.; Smith, G.M.; Fan, K.C.; Hong, J.S.; French, S.W.; McCaffery, J.M.; et al. PNPASE regulates RNA import into mitochondria. Cell 2010, 142, 456–467. [Google Scholar] [CrossRef] [PubMed]
- Golzarroshan, B.; Lin, C.L.; Li, C.L.; Yang, W.Z.; Chu, L.Y.; Agrawal, S.; Yuan, H.S. Crystal structure of dimeric human PNPase reveals why disease-linked mutants suffer from low RNA import and degradation activities. Nucleic Acids Res. 2018, 46, 8630–8640. [Google Scholar] [CrossRef] [PubMed]
- Gammage, P.A.; Moraes, C.T.; Minczuk, M. Mitochondrial Genome Engineering: The Revolution May Not Be CRISPR-Ized. Trends Genet. 2018, 34, 101–110. [Google Scholar] [CrossRef]
- Dhir, A.; Dhir, S.; Borowski, L.S.; Jimenez, L.; Teitell, M.; Rotig, A.; Crow, Y.J.; Rice, G.I.; Duffy, D.; Tamby, C.; et al. Mitochondrial double-stranded RNA triggers antiviral signalling in humans. Nature 2018, 560, 238–242. [Google Scholar] [CrossRef]
- Pajak, A.; Laine, I.; Clemente, P.; El-Fissi, N.; Schober, F.A.; Maffezzini, C.; Calvo-Garrido, J.; Wibom, R.; Filograna, R.; Dhir, A.; et al. Defects of mitochondrial RNA turnover lead to the accumulation of double-stranded RNA in vivo. PLoS Genet. 2019, 15, e1008240. [Google Scholar] [CrossRef]
- Von Ameln, S.; Wang, G.; Boulouiz, R.; Rutherford, M.A.; Smith, G.M.; Li, Y.; Pogoda, H.M.; Nürnberg, G.; Stiller, B.; Volk, A.E.; et al. A mutation in PNPT1, encoding mitochondrial-RNA-import protein PNPase, causes hereditary hearing loss. Am. J. Hum. Genet. 2012, 91, 919–927. [Google Scholar] [CrossRef]
- Matilainen, S.; Carroll, C.J.; Richter, U.; Euro, L.; Pohjanpelto, M.; Paetau, A.; Isohanni, P.; Suomalainen, A. Defective mitochondrial RNA processing due to PNPT1 variants causes Leigh syndrome. Hum. Mol. Genet. 2017, 26, 3352–3361. [Google Scholar] [CrossRef]
- Alodaib, A.; Sobreira, N.; Gold, W.A.; Riley, L.G.; Van Bergen, N.J.; Wilson, M.J.; Bennetts, B.; Thorburn, D.R.; Boehm, C.; Christodoulou, J. Whole-exome sequencing identifies novel variants in PNPT1 causing oxidative phosphorylation defects and severe multisystem disease. Eur. J. Hum. Genet. 2016, 25, 79. [Google Scholar] [CrossRef]
- Vedrenne, V.; Gowher, A.; De Lonlay, P.; Nitschke, P.; Serre, V.; Boddaert, N.; Altuzarra, C.; Mager-Heckel, A.M.; Chretien, F.; Entelis, N.; et al. Mutation in PNPT1, which encodes a polyribonucleotide nucleotidyltransferase, impairs RNA import into mitochondria and causes respiratory-chain deficiency. Am. J. Hum. Genet. 2012, 91, 912–918. [Google Scholar] [CrossRef] [PubMed]
- Sato, R.; Arai-Ichinoi, N.; Kikuchi, A.; Matsuhashi, T.; Numata-Uematsu, Y.; Uematsu, M.; Fujii, Y.; Murayama, K.; Ohtake, A.; Abe, T.; et al. Novel biallelic mutations in the PNPT1 gene encoding a mitochondrial-RNA-import protein PNPase cause delayed myelination. Clin. Genet. 2018, 93, 242–247. [Google Scholar] [CrossRef] [PubMed]
- Eaton, A.; Bernier, F.P.; Goedhart, C.; Caluseriu, O.; Lamont, R.E.; Boycott, K.M.; Parboosingh, J.S.; Innes, A.M. Care4Rare Canada Consortium. Is PNPT1-related hearing loss ever non-syndromic? Whole exome sequencing of adult siblings expands the natural history of PNPT1-related disorders. Am. J. Med. Genet. A 2018, 176, 2487–2493. [Google Scholar] [CrossRef] [PubMed]
- Riley, L.G.; Cowley, M.J.; Gayevskiy, V.; Roscioli, T.; Thorburn, D.R.; Prelog, K.; Bahlo, M.; Sue, C.M.; Balasubramaniam, S.; Christodoulou, J. A SLC39A8 variant causes manganese deficiency, and glycosylation and mitochondrial disorders. J. Inherit. Metab. Dis. 2017, 40, 261–269. [Google Scholar] [CrossRef]
- Adzhubei, I.A.; Schmidt, S.; Peshkin, L.; Ramensky, V.E.; Gerasimova, A.; Bork, P.; Kondrashov, A.S.; Sunyaev, S.R. A method and server for predicting damaging missense mutations. Nat. Methods 2010, 7, 248–249. [Google Scholar] [CrossRef]
- Choi, Y.; Sims, G.E.; Murphy, S.; Miller, J.R.; Chan, A.P. Predicting the functional effect of amino acid substitutions and indels. PLoS ONE 2012, 7, e46688. [Google Scholar] [CrossRef]
- Kircher, M.; Witten, D.M.; Jain, P.; O’Roak, B.J.; Cooper, G.M.; Shendure, J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat. Genet. 2014, 46, 310–315. [Google Scholar] [CrossRef]
- Schwarz, J.M.; Rodelsperger, C.; Schuelke, M.; Seelow, D. MutationTaster evaluates disease-causing potential of sequence alterations. Nat. Methods 2010, 7, 575–576. [Google Scholar] [CrossRef]
- Desmet, F.O.; Hamroun, D.; Lalande, M.; Collod-Beroud, G.; Claustres, M.; Beroud, C. Human Splicing Finder: An online bioinformatics tool to predict splicing signals. Nucleic Acids Res. 2009, 37, e67. [Google Scholar] [CrossRef]
- Punta, M.; Coggill, P.C.; Eberhardt, R.Y.; Mistry, J.; Tate, J.; Boursnell, C.; Pang, N.; Forslund, K.; Ceric, G.; Clements, J.; et al. The Pfam protein families database. Nucleic Acids Res. 2012, 40, D290–D301. [Google Scholar] [CrossRef]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [PubMed]
- Lek, M.; Karczewski, K.J.; Minikel, E.V.; Samocha, K.E.; Banks, E.; Fennell, T.; O’Donnell-Luria, A.H.; Ware, J.S.; Hill, A.J.; Cummings, B.B.; et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 2016, 536, 285–291. [Google Scholar] [CrossRef] [PubMed]
- Frazier, A.E.; Thorburn, D.R. Biochemical analyses of the electron transport chain complexes by spectrophotometry. Methods Mol. Biol. 2012, 837, 49–62. [Google Scholar] [PubMed]
- Calvo, S.E.; Tucker, E.J.; Compton, A.G.; Kirby, D.M.; Crawford, G.; Burtt, N.P.; Rivas, M.; Guiducci, C.; Bruno, D.L.; Goldberger, O.A.; et al. High-throughput, pooled sequencing identifies mutations in NUBPL and FOXRED1 in human complex I deficiency. Nat. Genet. 2010, 42, 851–858. [Google Scholar] [CrossRef]
- Schmittgen, T.D.; Livak, K.J. Analyzing real-time PCR data by the comparative CT method. Nat. Protoc. 2008, 3, 1101–1108. [Google Scholar] [CrossRef]
- Rice, G.I.; Forte, G.M.A.; Szynkiewicz, M.; Chase, D.S.; Aeby, A.; Abdel-Hamid, M.S.; Ackroyd, S.; Allcock, R.; Bailey, K.M.; Balottin, U.; et al. Assessment of interferon-related biomarkers in Aicardi-Goutières syndrome associated with mutations in TREX1, RNASEH2A, RNASEH2B, RNASEH2C, SAMHD1, and ADAR: A case-control study. Lancet Neurol. 2013, 12, 1159–1169. [Google Scholar] [CrossRef]
- Fuchs, S.A.; Schene, I.F.; Kok, G.; Jansen, J.M.; Nikkels, P.G.J.; van Gassen, K.L.I.; Terheggen-Lagro, S.W.J.; van der Crabben, S.N.; Hoeks, S.E.; Niers, L.E.M.; et al. Aminoacyl-tRNA synthetase deficiencies in search of common themes. Genet. Med. 2019, 21, 319–330. [Google Scholar] [CrossRef]
- Robinson, P.N. Deep phenotyping for precision medicine. Hum. Mutat. 2012, 33, 777–780. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| P1 (Fibroblasts) | P2 (Muscle) | |||||
|---|---|---|---|---|---|---|
| Enzyme | Residual Activity (%) | CS Ratio (%) | CII Ratio (%) | Residual Activity (%) | CS Ratio (%) | CII Ratio (%) |
| I | 150 | 107 | 75 | 75 | 73 | 126 |
| II | 194 | 140 | - | 59 | 58 | - |
| III | 266 | 187 | 133 | 69 | 65 | 113 |
| IV | 91 | 67 | 47 | 34 | 34 | 59 |
| CS | 137 | 102 | ||||
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rius, R.; Van Bergen, N.J.; Compton, A.G.; Riley, L.G.; Kava, M.P.; Balasubramaniam, S.; Amor, D.J.; Fanjul-Fernandez, M.; Cowley, M.J.; Fahey, M.C.; et al. Clinical Spectrum and Functional Consequences Associated with Bi-Allelic Pathogenic PNPT1 Variants. J. Clin. Med. 2019, 8, 2020. https://doi.org/10.3390/jcm8112020
Rius R, Van Bergen NJ, Compton AG, Riley LG, Kava MP, Balasubramaniam S, Amor DJ, Fanjul-Fernandez M, Cowley MJ, Fahey MC, et al. Clinical Spectrum and Functional Consequences Associated with Bi-Allelic Pathogenic PNPT1 Variants. Journal of Clinical Medicine. 2019; 8(11):2020. https://doi.org/10.3390/jcm8112020
Chicago/Turabian StyleRius, Rocio, Nicole J. Van Bergen, Alison G. Compton, Lisa G. Riley, Maina P. Kava, Shanti Balasubramaniam, David J. Amor, Miriam Fanjul-Fernandez, Mark J. Cowley, Michael C. Fahey, and et al. 2019. "Clinical Spectrum and Functional Consequences Associated with Bi-Allelic Pathogenic PNPT1 Variants" Journal of Clinical Medicine 8, no. 11: 2020. https://doi.org/10.3390/jcm8112020