Evaluation of the Predictive Ability, Environmental Regulation and Pharmacogenetics Utility of a BMI-Predisposing Genetic Risk Score during Childhood and Puberty

 , , , , , , , ,
, , , , , , , ,  and
and
Abstract
:1. Introduction
2. Objectives
- (1)
- To demonstrate how a pGRS can quantify inherited susceptibility to obesity and its cardio-metabolic comorbidities in children.
- (2)
- To evaluate the effects of genetic predisposition for obesity during childhood and how they evolve when entering puberty.
- (3)
- To describe the plausible modulatory role of environmental factors over inherited genetic susceptibility in children.
- (4)
- To investigate the utility of the pGRS for the pharmacological management of obesity in children.
3. Materials and Methods
3.1. Study Design
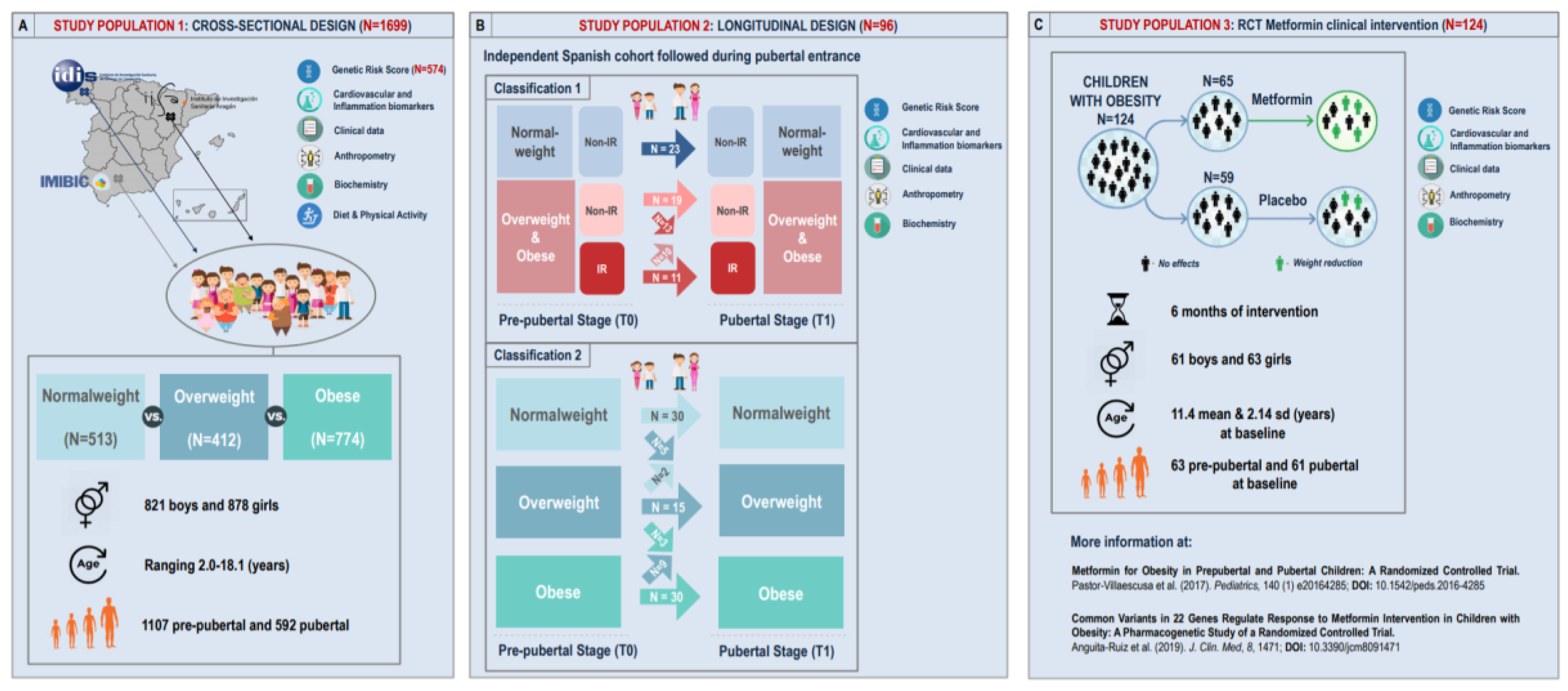
3.1.1. Study Population 1: Cross-Sectional Approach
3.1.2. Study Population 2: Longitudinal Approach
3.1.3. Study Population 3: RCT Metformin Clinical Intervention
3.2. Ethics Statement
3.3. DNA Extraction, Genotyping and pGRS Construction
3.4. Phenotypic Measurements and Lifestyle Factors
3.5. Statistical Analysis
3.5.1. General Descriptive Analysis
3.5.2. Association between the pGRS and Obesity Outcomes and Evaluation of the pGRS Predictive Ability
3.5.3. Identification of Gene × Environment Modulatory Effects
3.5.4. pGRS-Drug Interaction
4. Results
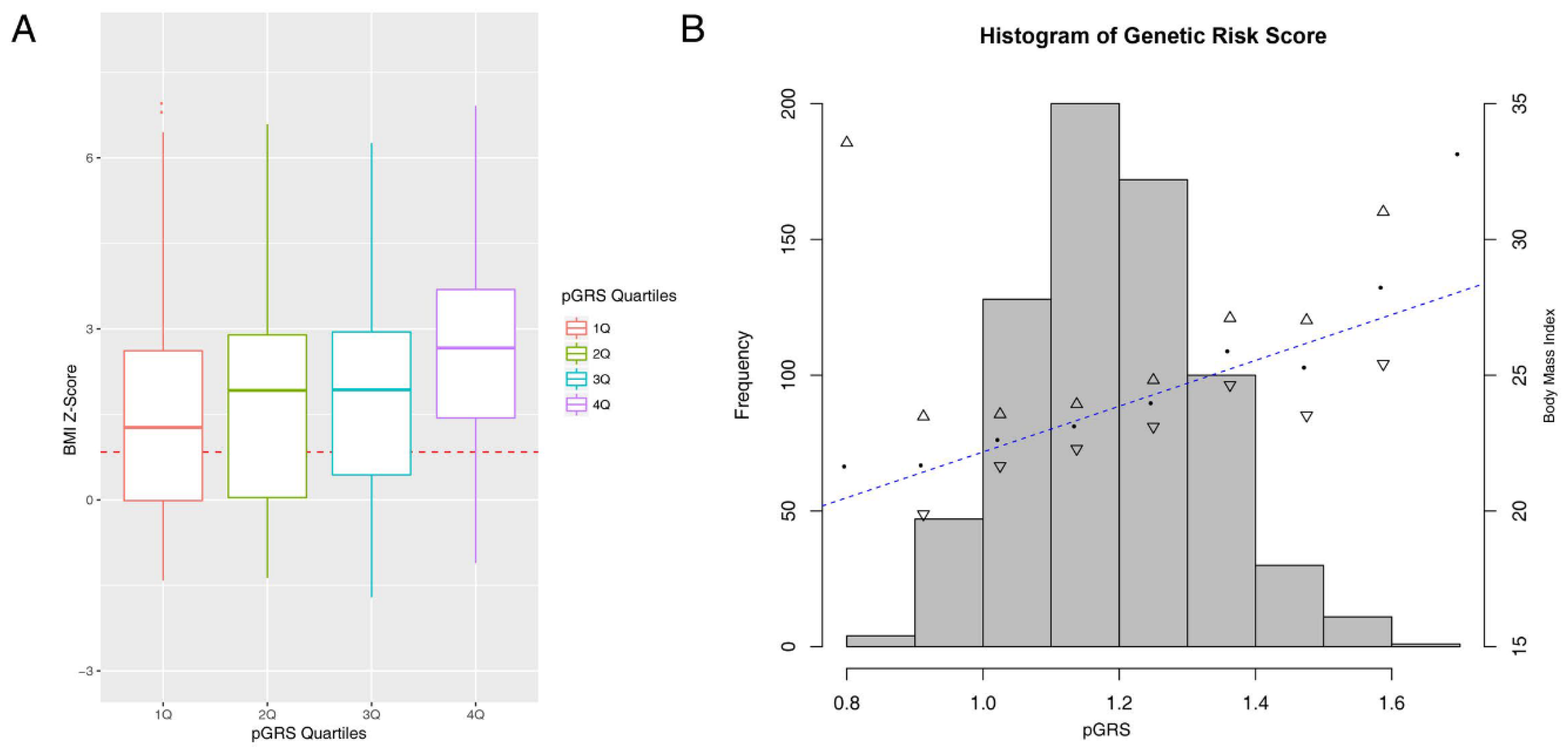
4.1. The pGRS Associates with BMI Z-Score and Performs Well in the Identification of High-Risk Children
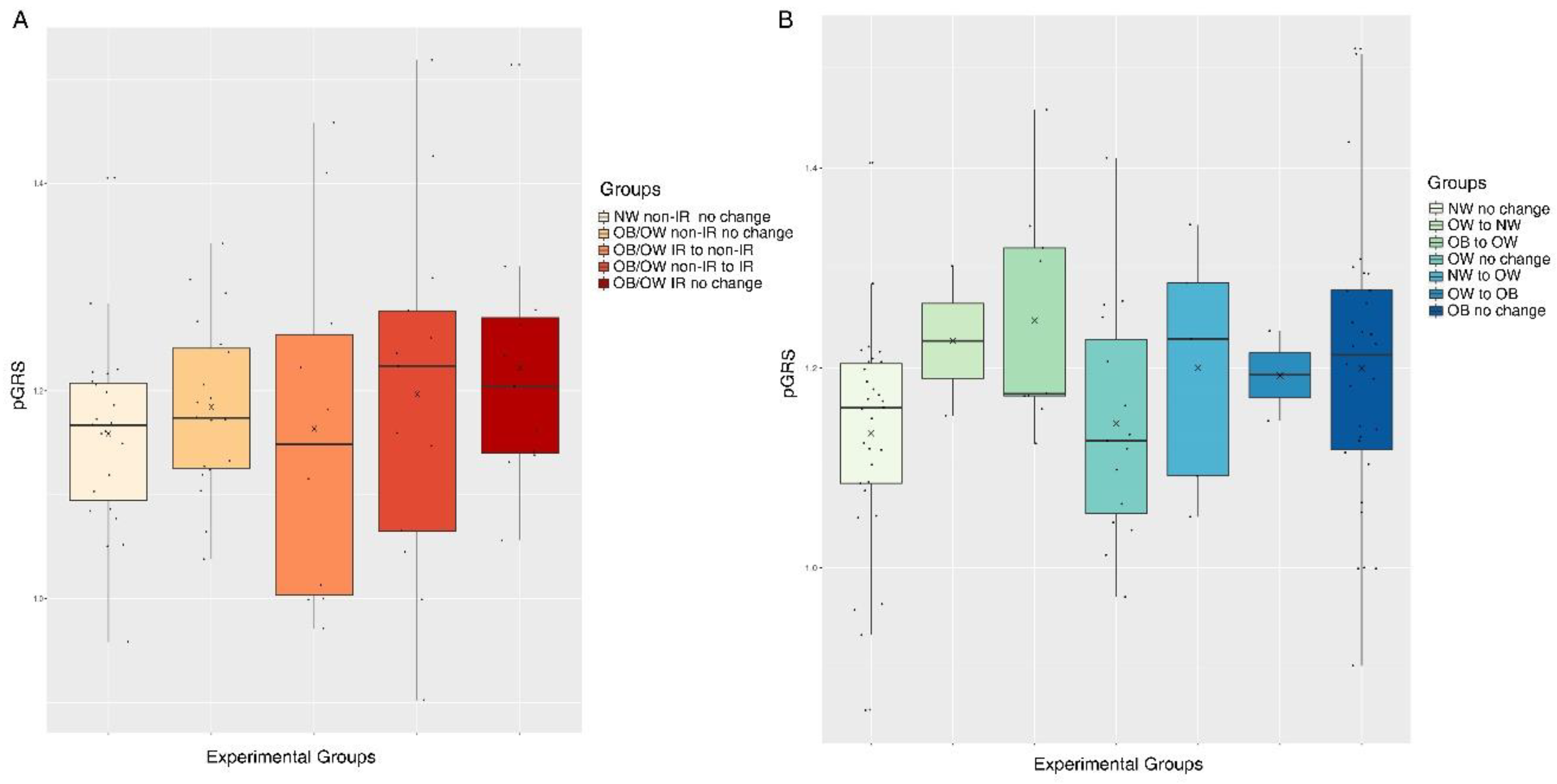
4.2. The pGRS is Associated with Longitudinal Trajectories for Obesity and IR in Children Undergoing Puberty
4.3. The pGRS Does not Correlate with Increased Cardio-Metabolic Alterations in Children and Adolescents
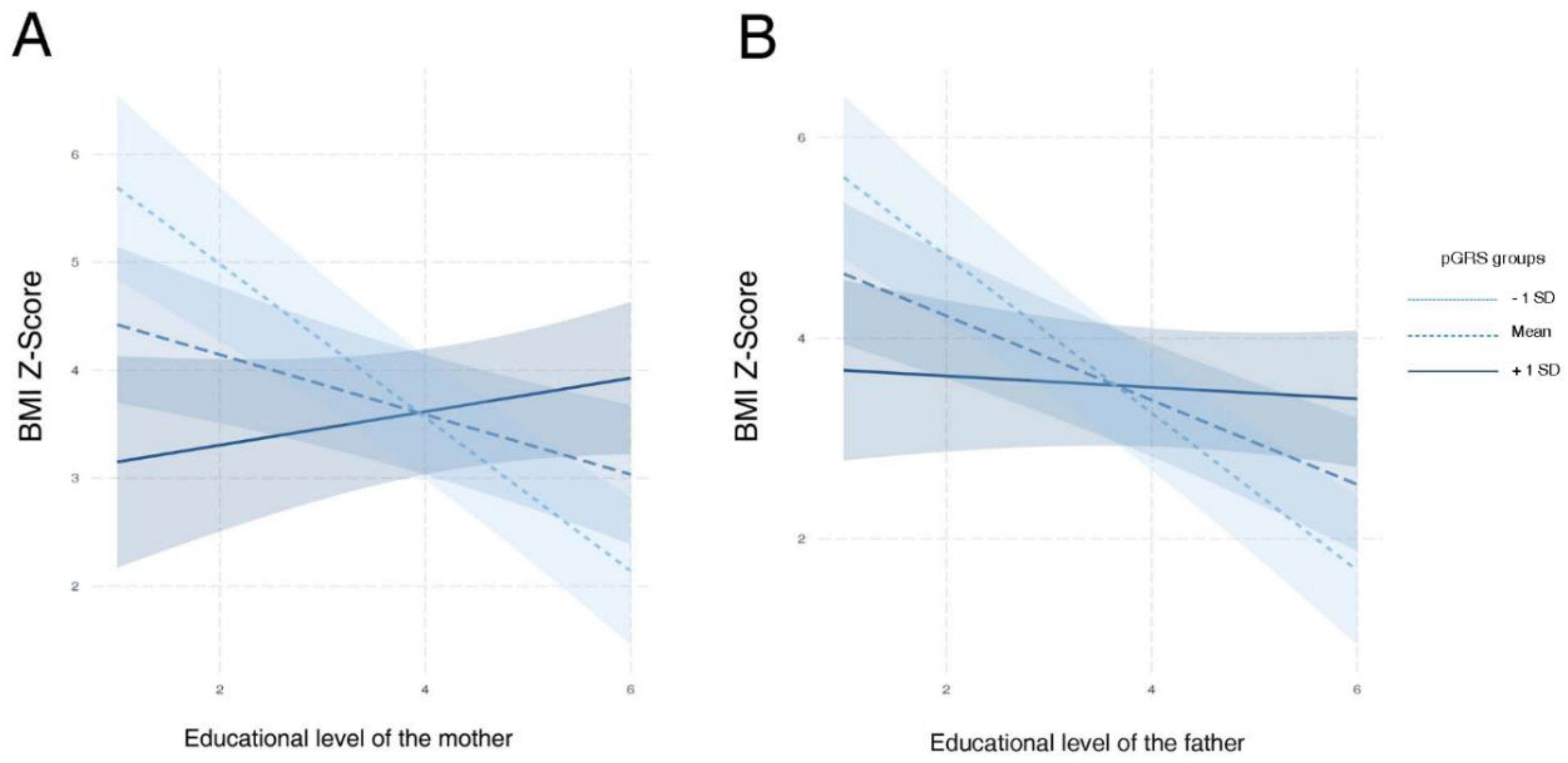
4.4. Lifestyle Factors Interact with the Inherited Genetic Susceptibility to Obesity in Children
4.5. The pGRS is not Helpful for the Pharmacogenetics Management of Obesity in Children
5. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- GBD 2015 Obesity Collaborators; Afshin, A.; Forouzanfar, M.H.; Reitsma, M.B.; Sur, P.; Estep, K.; Lee, A.; Marczak, L.; Mokdad, A.H.; Moradi-Lakeh, M.; et al. Health Effects of Overweight and Obesity in 195 Countries over 25 Years. N. Engl. J. Med. 2017, 377, 13–27. [Google Scholar] [CrossRef] [PubMed]
- Jones, R.E.; Jewell, J.; Saksena, R.; Ramos Salas, X.; Breda, J. Overweight and Obesity in Children under 5 Years: Surveillance Opportunities and Challenges for the WHO European Region. Front. Public Health 2017, 5, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Locke, A.E.; Kahali, B.; Berndt, S.I.; Justice, A.E.; Pers, T.H.; Day, F.R.; Powell, C.; Vedantam, S.; Buchkovich, M.L.; Yang, J.; et al. Genetic studies of body mass index yield new insights for obesity biology. Nature 2015, 518, 197–206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belsky, D.W.; Moffitt, T.E.; Houts, R.; Bennett, G.G.; Biddle, A.K.; Blumenthal, J.A.; Evans, J.P.; Harrington, H.L.; Sugden, K.; Williams, B.; et al. Polygenic risk, rapid childhood growth, and the development of obesity: Evidence from a 4-decade longitudinal study. Arch. Pediatr. Adolesc. Med. 2012, 166, 515–521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elks, C.E.; Loos, R.J.F.; Hardy, R.; Wills, A.K.; Wong, A.; Wareham, N.J.; Kuh, D.; Ong, K.K. Adult obesity susceptibility variants are associated with greater childhood weight gain and a faster tempo of growth: The 1946 British Birth Cohort Study. Am. J. Clin. Nutr. 2012, 95, 1150–1156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Felix, J.F.; Bradfield, J.P.; Monnereau, C.; van der Valk, R.J.P.; Stergiakouli, E.; Chesi, A.; Gaillard, R.; Feenstra, B.; Thiering, E.; Kreiner-Møller, E.; et al. Genome-wide association analysis identifies three new susceptibility loci for childhood body mass index. Hum. Mol. Genet. 2016, 25, 389–403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mäkelä, J.; Lagström, H.; Pitkänen, N.; Kuulasmaa, T.; Kaljonen, A.; Laakso, M.; Niinikoski, H. Genetic risk clustering increases children’s body weight at 2 years of age - the STEPS Study. Pediatr. Obes. 2016, 11, 459–467. [Google Scholar] [CrossRef] [PubMed]
- Zandoná, M.R.; Sangalli, C.N.; Campagnolo, P.D.B.; Vitolo, M.R.; Almeida, S.; Mattevi, V.S. Validation of obesity susceptibility loci identified by genome-wide association studies in early childhood in South Brazilian children. Pediatr. Obes. 2017, 12, 85–92. [Google Scholar] [CrossRef] [PubMed]
- Tekola-Ayele, F.; Lee, A.; Workalemahu, T.; Sánchez-Pozos, K. Shared genetic underpinnings of childhood obesity and adult cardiometabolic diseases. Hum. Genom. 2019, 13, 17. [Google Scholar] [CrossRef] [Green Version]
- Moon, J.Y.; Wang, T.; Sofer, T.; North, K.E.; Isasi, C.R.; Cai, J.; Gellman, M.D.; Moncrieft, A.E.; Sotres-Alvarez, D.; Argos, M.; et al. Objectively measured physical activity, sedentary behavior, and genetic predisposition to obesity in U.S. Hispanics/Latinos: Results from the hispanic community health study/study of Latinos (HCHS/SOL). Diabetes 2017, 66, 3001–3012. [Google Scholar] [CrossRef] [Green Version]
- Mead, E.; Brown, T.; Rees, K.; Azevedo, L.B.; Whittaker, V.; Jones, D.; Olajide, J.; Mainardi, G.M.; Corpeleijn, E.; O’Malley, C.; et al. Diet, physical activity and behavioural interventions for the treatment of overweight or obese children from the age of 6 to 11 years. Cochrane Database Syst. Rev. 2017, 2017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, J.; Gong, C.; Wan, Y.; Xu, Y.; Tao, F.; Sun, Y. Polygenic risk, adherence to a healthy lifestyle, and childhood obesity. Pediatr. Obes. 2019, 14. [Google Scholar] [CrossRef] [PubMed]
- Manolio, T.A.; Collins, F.S.; Cox, N.J.; Goldstein, D.B.; Hindorff, L.A.; Hunter, D.J.; McCarthy, M.I.; Ramos, E.M.; Cardon, L.R.; Chakravarti, A.; et al. Finding the missing heritability of complex diseases. Nature 2009, 461, 747–753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schrodi, S.J.; Mukherjee, S.; Shan, Y.; Tromp, G.; Sninsky, J.J.; Callear, A.P.; Carter, T.C.; Ye, Z.; Haines, J.L.; Brilliant, M.H.; et al. Genetic-based prediction of disease traits: Prediction is very difficult, especially about the future. Front. Genet. 2014, 5, 1–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khera, A.V.; Chaffin, M.; Aragam, K.G.; Haas, M.E.; Roselli, C.; Choi, S.H.; Natarajan, P.; Lander, E.S.; Lubitz, S.A.; Ellinor, P.T.; et al. Genome-wide polygenic scores for common diseases identify individuals with risk equivalent to monogenic mutations. Nat. Genet. 2018, 50, 1219–1224. [Google Scholar] [CrossRef]
- Torkamani, A.; Wineinger, N.E.; Topol, E.J. The personal and clinical utility of polygenic risk scores. Nat. Rev. Genet. 2018, 19, 581–590. [Google Scholar] [CrossRef]
- Chatterjee, N.; Shi, J.; García-Closas, M. Developing and evaluating polygenic risk prediction models for stratified disease prevention. Nat. Rev. Genet. 2016, 17, 392–406. [Google Scholar] [CrossRef]
- Khera, A.V.; Chaffin, M.; Wade, K.H.; Zahid, S.; Brancale, J.; Xia, R.; Distefano, M.; Senol-Cosar, O.; Haas, M.E.; Bick, A.; et al. Polygenic Prediction of Weight and Obesity Trajectories from Birth to Adulthood. Cell 2019, 177, 587–596.e9. [Google Scholar] [CrossRef] [Green Version]
- Hohenadel, M.G.; Baier, L.J.; Piaggi, P.; Muller, Y.L.; Hanson, R.L.; Krakoff, J.; Thearle, M.S. The impact of genetic variants on BMI increase during childhood versus adulthood. Int. J. Obes. 2016, 40, 1301–1309. [Google Scholar] [CrossRef]
- Choh, A.C.; Lee, M.; Kent, J.W.; Diego, V.P.; Johnson, W.; Curran, J.E.; Dyer, T.D.; Bellis, C.; Blangero, J.; Siervogel, R.M.; et al. Gene-by-age effects on BMI from birth to adulthood: The fels longitudinal study. Obesity 2014, 22, 875–881. [Google Scholar] [CrossRef]
- Song, M.; Zheng, Y.; Qi, L.; Hu, F.B.; Chan, A.T.; Giovannucci, E.L. Associations between genetic variants associated with body mass index and trajectories of body fatness across the life course: A longitudinal analysis. Int. J. Epidemiol. 2018, 47, 506–515. [Google Scholar] [CrossRef] [PubMed]
- Torkamani, A.; Topol, E. Polygenic Risk Scores Expand to Obesity. Cell 2019, 177, 518–520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hannon, T.S.; Janosky, J.; Arslanian, S.A. Longitudinal study of physiologic insulin resistance and metabolic changes of puberty. Pediatr. Res. 2006, 60, 759–763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reinehr, T.; Roth, C.L. Is there a causal relationship between obesity and puberty? Lancet Child Adolesc. Health 2019, 3, 44–54. [Google Scholar] [CrossRef]
- Mega, J.L.; Stitziel, N.O.; Smith, J.G.; Chasman, D.I.; Caulfield, M.; Devlin, J.J.; Nordio, F.; Hyde, C.; Cannon, C.P.; Sacks, F.; et al. Genetic risk, coronary heart disease events, and the clinical benefit of statin therapy: An analysis of primary and secondary prevention trials. Lancet 2015, 385, 2264–2271. [Google Scholar] [CrossRef] [Green Version]
- Olza, J.; Aguilera, C.M.; Gil-Campos, M.; Leis, R.; Bueno, G.; Martínez-Jiménez, M.D.; Valle, M.; Canẽte, R.; Tojo, R.; Moreno, L.A.; et al. Myeloperoxidase is an early biomarker of inflammation and cardiovascular risk in prepubertal obese children. Diabetes Care 2012, 35, 2373–2376. [Google Scholar] [CrossRef] [Green Version]
- Anguita-Ruiz, A.; Plaza-Diaz, J.; Ruiz-Ojeda, F.J.; Ruperez, A.I.; Leis, R.; Bueno, G.; Gil-Campos, M.; Vazquez-Cobela, R.; Canete, R.; Moreno, L.A.; et al. X chromosome genetic data in a Spanish children cohort, dataset description and analysis pipeline. Sci. Data 2019, 6, 130. [Google Scholar] [CrossRef]
- Cole, T.J.; Bellizzi, M.C.; Flegal, K.M.; Dietz, W.H. Establishing a standard definition for child overweight and obesity worldwide: International survey. BMJ 2000, 320, 1240–1243. [Google Scholar] [CrossRef] [Green Version]
- Anguita-Ruiz, A.; Mendez-Gutierrez, A.; Ruperez, A.I.; Leis, R.; Bueno, G.; Gil-Campos, M.; Tofe, I.; Gomez-Llorente, C.; Moreno, L.A.; Gil, Á.; et al. The protein S100A4 as a novel marker of insulin resistance in prepubertal and pubertal children with obesity. Metabolism 2020, 105. [Google Scholar] [CrossRef]
- Tanner, J.M.; Whitehouse, R.H. Clinical longitudinal standards for height, weight, height velocity, weight velocity, and stages of puberty. Arch. Dis. Child. 1976, 51, 170–179. [Google Scholar] [CrossRef] [Green Version]
- Tang, Q.; Li, X.; Song, P.; Xu, L. Optimal cut-off values for the homeostasis model assessment of insulin resistance (HOMA-IR) and pre-diabetes screening: Developments in research and prospects for the future. Drug Discov. Ther. 2015, 9, 380–385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Andrade, M.I.S.; Oliveira, J.S.; Leal, V.S.; da Lima, N.M.S.; Costa, E.C.; de Aquino, N.B.; de Lira, P.I.C. Identification of cutoff points for Homeostatic Model Assessment for Insulin Resistance index in adolescents: Systematic review. Rev. Paul. Pediatr. 2016, 34, 234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rupérez, A.I.; Olza, J.; Gil-Campos, M.; Leis, R.; Bueno, G.; Aguilera, C.M.; Gil, A.; Moreno, L.A. Cardiovascular risk biomarkers and metabolically unhealthy status in prepubertal children: Comparison of definitions. Nutr. Metab. Cardiovasc. Dis. 2018, 28, 524–530. [Google Scholar] [CrossRef] [PubMed]
- Pastor-Villaescusa, B.; Caballero-Villarraso, J.; Cañete, M.D.; Hoyos, R.; Maldonado, J.; Bueno, G.; Leis, R.; Gil, Á.; Cañete, R.; Aguilera, C.M. Evaluation of differential effects of metformin treatment in obese children according to pubertal stage and genetic variations: Study protocol for a randomized controlled trial. Trials 2016, 17, 323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pastor-Villaescusa, B.; Cañete, M.D.; Caballero-Villarraso, J.; Hoyos, R.; Latorre, M.; Vázquez-Cobela, R.; Plaza-Díaz, J.; Maldonado, J.; Bueno, G.; Leis, R.; et al. Metformin for Obesity in Prepubertal and Pubertal Children: A Randomized Controlled Trial. Pediatrics 2017, 140, e20164285. [Google Scholar] [CrossRef] [Green Version]
- Anguita-Ruiz, A.; Pastor-Villaescusa, B.; Leis, R.; Bueno, G.; Hoyos, R.; Vázquez-Cobela, R.; Latorre-Millán, M.; Cañete, M.D.; Caballero-Villarraso, J.; Gil, Á.; et al. Common Variants in 22 Genes Regulate Response to Metformin Intervention in Children with Obesity: A Pharmacogenetic Study of a Randomized Controlled Trial. J. Clin. Med. 2019, 8, 1471. [Google Scholar] [CrossRef] [Green Version]
- Sobradillo, B.; Aguirre, A.; Aresti, U.; Bilbao, A.; Fernández-Ramos, C.; Lizárraga, A.; Lorenzo, H.; Madariaga, L.; Rica, I.; Ruiz, I.; et al. Curvas y Tablas de Crecimiento (Estudios Longitudinal y Transversal); Fundación Faustino Orbegozo Eizaguirre: Madrid, Spain, 2004; ISBN 84-607-9967-0. [Google Scholar]
- Wray, N.R.; Yang, J.; Goddard, M.E.; Visscher, P.M. The Genetic Interpretation of Area under the ROC Curve in Genomic Profiling. PLoS Genet. 2010, 6, e1000864. [Google Scholar] [CrossRef] [Green Version]
- Cook, N.R. Use and Misuse of the Receiver Operating Characteristic Curve in Risk Prediction. Circulation 2007, 115, 928–935. [Google Scholar] [CrossRef] [Green Version]
- Keller, M.C. Gene × Environment Interaction Studies Have Not Properly Controlled for Potential Confounders: The Problem and the (Simple) Solution. Biol. Psychiatry 2014, 75, 18–24. [Google Scholar] [CrossRef] [Green Version]
- Loos, R.J.F.; Yeo, G.S.H. The bigger picture of FTO - The first GWAS-identified obesity gene. Nat. Rev. Endocrinol. 2014, 10, 51–61. [Google Scholar] [CrossRef]
- Apalasamy, Y.D.; Mohamed, Z. Obesity and genomics: Role of technology in unraveling the complex genetic architecture of obesity. Hum. Genet. 2015, 134, 361–374. [Google Scholar] [CrossRef] [PubMed]
- Durbin, R.M.; Altshuler, D.L.; Durbin, R.M.; Abecasis, G.R.; Bentley, D.R.; Chakravarti, A.; Clark, A.G.; Collins, F.S.; De La Vega, F.M.; Donnelly, P.; et al. A map of human genome variation from population-scale sequencing. Nature 2010, 467, 1061–1073. [Google Scholar] [CrossRef] [Green Version]
- Butler, É.M.; Derraik, J.G.B.; Taylor, R.W.; Cutfield, W.S. Prediction Models for Early Childhood Obesity: Applicability and Existing Issues. Horm. Res. Paediatr. 2018, 90, 358–367. [Google Scholar] [CrossRef] [PubMed]
- Goran, M.I.; Shaibi, G.Q.; Weigensberg, M.J.; Davis, J.N.; Cruz, M.L. Deterioration of insulin sensitivity and beta-cell function in overweight Hispanic children during pubertal transition: A longitudinal assessment. Int. J. Pediatr. Obes. 2006, 1, 139–145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reinehr, T.; Wabitsch, M.; Kleber, M.; de Sousa, G.; Denzer, C.; Toschke, A.M. Parental diabetes, pubertal stage, and extreme obesity are the main risk factors for prediabetes in children and adolescents: A simple risk score to identify children at risk for prediabetes. Pediatr. Diabetes 2009, 10, 395–400. [Google Scholar] [CrossRef] [PubMed]
- McCrory, C.; Leahy, S.; Ribeiro, A.I.; Fraga, S.; Barros, H.; Avendano, M.; Vineis, P.; Layte, R.; Alenius, H.; Baglietto, L.; et al. Maternal educational inequalities in measured body mass index trajectories in three European countries. Paediatr. Perinat. Epidemiol. 2019, 33, 226–237. [Google Scholar] [CrossRef]
- Adler, N.E.; Boyce, T.; Chesney, M.A.; Cohen, S.; Folkman, S.; Kahn, R.L.; Syme, S.L. Socioeconomic Status and Health: The Challenge of the Gradient. Am. Psychol. 1994, 49, 15–24. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Whole Population | Training Set | Test Set | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Predictors | AUC [95% CI] | n | n Normal-Weight | n Obese | n | n Normal-Weight | n Obese | n | n Normal-Weight | n Obese |
| Tanner, Origin, Sex and Age | 0.66 [0.61–0.72] | 1285 | 512 | 773 | 901 | 359 | 542 | 384 | 153 | 231 |
| pGRS | 0.72 [0.63–0.80] | 443 | 187 | 256 | 311 | 131 | 180 | 132 | 56 | 76 |
| Obesity Family History | 0.70 [0.63–0.77] | 686 | 232 | 454 | 481 | 163 | 318 | 205 | 69 | 136 |
| Maternal Smoking | 0.49 [0.43–0.55] | 632 | 218 | 414 | 443 | 153 | 290 | 189 | 65 | 124 |
| Gestational Diabetes | 0.49 [0.45–0.54] | 620 | 214 | 406 | 435 | 150 | 285 | 185 | 64 | 121 |
| Birthweight | 0.60 [0.51–0.69] | 610 | 211 | 399 | 428 | 148 | 280 | 182 | 63 | 119 |
| Gestational Weight Gain | 0.54 [0.45–0.62] | 569 | 206 | 363 | 400 | 145 | 255 | 169 | 61 | 108 |
| Parents BMI | 0.76 [0.68–0.84] | 530 | 199 | 331 | 372 | 140 | 232 | 158 | 59 | 99 |
| Type of Breastfeeding | 0.55 [0.45–0.64 ] | 555 | 193 | 362 | 390 | 136 | 254 | 165 | 57 | 108 |
| Tanner, Origin, Sex, Age, pGRS, Obesity Family History, Birthweight, and Parents BMI | 0.81 [0.7–0.93] | 176 | 78 | 98 | 124 | 55 | 69 | 52 | 23 | 29 |
| Measurement | Beta | SE | CI.LO | CI.HI | T-Value | p-Value | FDR |
|---|---|---|---|---|---|---|---|
| APO B/LDLc Ratio | −0.12 | 0.05 | −0.21 | −0.02 | −2.29 | 0.02 | 0.60 |
| Triglycerides (mg/dL) | −17.69 | 9.17 | −35.67 | 0.28 | −1.93 | 0.05 | 0.68 |
| APO B (mg/dL) | −10.94 | 6.49 | −23.66 | 1.78 | −1.69 | 0.09 | 0.68 |
| APO A/APO B | 0.40 | 0.25 | −0.09 | 0.88 | 1.61 | 0.11 | 0.68 |
| WC/Height Ratio | 0.03 | 0.02 | −0.01 | 0.06 | 1.51 | 0.13 | 0.68 |
| WC (cm) | 3.75 | 2.52 | -1.19 | 8.70 | 1.49 | 0.14 | 0.68 |
| Adiponectin/Leptin Ratio | 0.50 | 0.36 | -0.21 | 1.22 | 1.38 | 0.17 | 0.68 |
| MCP1 (ng/L) | −21.94 | 16.22 | −53.73 | 9.84 | −1.35 | 0.18 | 0.68 |
| aPAI (ug/L) | −3.56 | 2.90 | −9.24 | 2.12 | −1.23 | 0.22 | 0.69 |
| IL8 (ng/L) | −0.60 | 0.54 | −1.66 | 0.45 | −1.12 | 0.26 | 0.69 |
| QUICKI | 0.01 | 0.01 | −0.01 | 0.03 | 0.96 | 0.34 | 0.69 |
| DBP (mmHg) | 3.11 | 3.35 | −3.46 | 9.68 | 0.93 | 0.35 | 0.69 |
| Adiponectin (mg/L) | −3.09 | 3.34 | −9.63 | 3.45 | −0.93 | 0.35 | 0.69 |
| IL6 (ng/L) | 2.32 | 2.57 | −2.71 | 7.35 | 0.90 | 0.37 | 0.69 |
| HC (cm) | 1.96 | 2.19 | −2.32 | 6.25 | 0.90 | 0.37 | 0.69 |
| HOMA-IR index | −0.37 | 0.42 | −1.19 | 0.46 | −0.87 | 0.38 | 0.69 |
| WC/HC Ratio | 0.02 | 0.03 | −0.03 | 0.08 | 0.85 | 0.39 | 0.69 |
| Total cholesterol (mg/dL) | −5.95 | 9.48 | −24.53 | 12.63 | −0.63 | 0.53 | 0.88 |
| hsCRP (mg/L) | 0.42 | 0.81 | −1.16 | 2.01 | 0.52 | 0.60 | 0.89 |
| HDLc/LDLc Ratio | −0.05 | 0.09 | −0.22 | 0.13 | −0.49 | 0.62 | 0.89 |
| Glucose (mg/dL) | −1.14 | 2.38 | −5.81 | 3.53 | −0.48 | 0.63 | 0.89 |
| Resistin (ug/L) | 1.28 | 2.88 | −4.37 | 6.93 | 0.44 | 0.66 | 0.89 |
| SBP (mmHg) | −1.74 | 4.17 | −9.92 | 6.44 | −0.42 | 0.68 | 0.89 |
| LDLc (mg/dL) | −2.43 | 8.57 | −19.21 | 14.36 | −0.28 | 0.78 | 0.89 |
| TNF (ng/L) | 0.15 | 0.55 | −0.93 | 1.24 | 0.28 | 0.78 | 0.89 |
| Leptin (ug/L) | 0.77 | 2.86 | −4.84 | 6.37 | 0.27 | 0.79 | 0.89 |
| APO A (mg/dL) | −2.35 | 9.74 | −21.44 | 16.75 | −0.24 | 0.81 | 0.89 |
| MPO (ug/L) | −2.17 | 10.09 | −21.95 | 17.60 | −0.22 | 0.83 | 0.89 |
| HDLc (mg/dL) | −0.50 | 4.22 | −8.76 | 7.77 | −0.12 | 0.91 | 0.94 |
| MMP9 (ug/L) | −0.05 | 20.57 | −40.38 | 40.27 | 0.00 | 1.00 | 1.00 |
| Measurement (Delta T1–T0) | Beta | SE | CI.LO | CI.HI | T-Value | p-Value | FDR |
|---|---|---|---|---|---|---|---|
| APO B (mg/dL) | 57.44 | 21.63 | 15.05 | 99.82 | 2.66 | 0.02 | 0.29 |
| HDLc (mg/dL) | −18.91 | 8.98 | −36.51 | −1.32 | −2.11 | 0.03 | 0.33 |
| Triglycerides (mg/dL) | 45.38 | 31.58 | −16.51 | 107.27 | 1.44 | 0.15 | 0.78 |
| APO A (mg/dL) | −35.22 | 30.97 | −95.92 | 25.48 | −1.14 | 0.26 | 0.78 |
| DBP (mmHg) | −13.34 | 12.98 | −38.79 | 12.10 | −1.03 | 0.31 | 0.78 |
| Insulin (mU/L) | 8.94 | 8.89 | −8.49 | 26.37 | 1.00 | 0.32 | 0.78 |
| SBP (mmHg) | 15.57 | 16.77 | −17.29 | 48.43 | 0.93 | 0.36 | 0.78 |
| HOMA-IR index | 1.88 | 2.07 | −2.17 | 5.94 | 0.91 | 0.37 | 0.78 |
| HDLc/LDLc Ratio | −0.29 | 0.43 | −1.14 | 0.56 | −0.67 | 0.51 | 0.86 |
| Total cholesterol (mg/dL) | −13.63 | 24.38 | −61.42 | 34.16 | −0.56 | 0.58 | 0.86 |
| Glucose (mg/dL) | −3.80 | 8.72 | −20.89 | 13.29 | −0.44 | 0.66 | 0.86 |
| WC/HC | −0.03 | 0.08 | −0.19 | 0.12 | −0.41 | 0.68 | 0.86 |
| WC (cm) | −3.11 | 9.17 | −21.09 | 14.87 | −0.34 | 0.74 | 0.86 |
| LDLc (mg/dL) | −5.94 | 18.41 | −42.02 | 30.15 | −0.32 | 0.75 | 0.86 |
| QUICKI | 0.01 | 0.04 | −0.06 | 0.09 | 0.31 | 0.75 | 0.86 |
| Lifestyle Factor | Beta | SE | CI.LO | CI.HI | T-Value | p-Value | FDR |
|---|---|---|---|---|---|---|---|
| Educational level of the mother | 3.41 | 0.95 | 1.56 | 5.27 | 3.60 | 3.86 × 10−4 | 0.02 |
| Educational level of the father | 2.93 | 0.86 | 1.24 | 4.62 | 3.40 | 7.92 × 10−4 | 0.02 |
| How many minutes per week do you spend exercising at a sport program? | 0.02 | 0.01 | 0.00 | 0.04 | 2.13 | 0.03 | 0.47 |
| Presence of AH in father or mother | −4.01 | 2.01 | −7.95 | −0.08 | −2.00 | 0.05 | 0.56 |
| Mother BMI | −0.22 | 0.11 | −0.44 | 0.00 | −1.93 | 0.06 | 0.56 |
| How long does it take to get to the school on walk? | −0.24 | 0.14 | −0.52 | 0.04 | −1.66 | 0.10 | 0.58 |
| How often do you eat fruit while watching TV? | 1.73 | 1.05 | −0.34 | 3.80 | 1.64 | 0.10 | 0.58 |
| How often do you eat snacks while watching TV? | 1.80 | 1.14 | −0.44 | 4.03 | 1.58 | 0.12 | 0.58 |
| How many hours do you spend doing home activities? | −2.55 | 1.62 | −5.73 | 0.63 | −1.57 | 0.12 | 0.58 |
| How much time do you play videogames in a day during weekend? | 1.03 | 0.70 | −0.35 | 2.41 | 1.46 | 0.15 | 0.58 |
| How many hours each day do you spend doing vigorous efforts like training activity? | 2.27 | 1.55 | −0.77 | 5.32 | 1.46 | 0.15 | 0.58 |
| Presence of hypercholesterolemia in father or mother | 1.78 | 1.24 | −0.64 | 4.21 | 1.44 | 0.15 | 0.58 |
| Presence of heart stroke in father or mother | −9.74 | 6.86 | −23.19 | 3.70 | −1.42 | 0.16 | 0.58 |
| Presence of vascular problems in father or mother | −39.77 | 29.84 | −98.26 | 18.72 | −1.33 | 0.18 | 0.59 |
| How many hours do you spend exercising in a sport club? | 0.04 | 0.03 | −0.02 | 0.10 | 1.30 | 0.19 | 0.59 |
| How often do you eat salted potatoes while watching TV? | 2.11 | 1.65 | −1.12 | 5.34 | 1.28 | 0.20 | 0.59 |
| Presence of diabetes in father or mother | 11.59 | 9.96 | −7.94 | 31.12 | 1.16 | 0.25 | 0.64 |
| How often do you eat nuts while watching TV? | 2.21 | 1.95 | −1.62 | 6.04 | 1.13 | 0.26 | 0.64 |
| How many days per week do you spend walking with vigorous efforts? | 0.63 | 0.56 | −0.47 | 1.73 | 1.13 | 0.26 | 0.64 |
| How many hours each day do you spend practicing activities that do not require physical activity (e.g., reading) | 0.41 | 0.44 | −0.44 | 1.27 | 0.95 | 0.34 | 0.77 |
| How much time do you play videogames in a day during the week? | −0.79 | 0.85 | −2.46 | 0.87 | −0.93 | 0.35 | 0.77 |
| How many hours do you usually sleep every day during the week? | 0.68 | 0.80 | −0.89 | 2.26 | 0.85 | 0.40 | 0.77 |
| How many hours do you spend doing physical activity in family? | 1.21 | 1.42 | −1.58 | 4.00 | 0.85 | 0.40 | 0.77 |
| How often do you eat candies while watching TV? | 1.50 | 1.77 | −1.96 | 4.96 | 0.85 | 0.40 | 0.77 |
| How many hours do you usually sleep every day during the weekends? | −0.55 | 0.67 | −1.85 | 0.76 | −0.82 | 0.41 | 0.77 |
| Diagnosed hypertriglyceridemia in father or mother | 1.06 | 1.34 | −1.57 | 3.69 | 0.79 | 0.43 | 0.77 |
| How often do you eat sweets while watching TV? | 1.37 | 1.78 | −2.12 | 4.86 | 0.77 | 0.44 | 0.77 |
| How much time per weekend do you usually use the smartphone? | 0.69 | 0.95 | −1.17 | 2.55 | 0.73 | 0.47 | 0.79 |
| Do you usually eat in front of the TV? | 0.77 | 1.15 | −1.49 | 3.02 | 0.67 | 0.51 | 0.83 |
| How many hours per week do you spend on physical education during school hours? | −2.01 | 3.48 | −8.83 | 4.82 | −0.58 | 0.57 | 0.85 |
| How often do you eat fruits while playing video games? | −2.07 | 3.73 | −9.38 | 5.23 | −0.56 | 0.58 | 0.85 |
| Presence of obesity in the father or mother | −0.45 | 0.82 | −2.07 | 1.17 | −0.54 | 0.59 | 0.85 |
| How much time during the week do you usually watch TV? | 0.39 | 0.75 | −1.07 | 1.85 | 0.52 | 0.60 | 0.85 |
| How often do you eat fruits while surfing internet? | 1.81 | 3.88 | −5.80 | 9.43 | 0.47 | 0.64 | 0.88 |
| How much time in per weekend do you usually use internet | 0.25 | 0.63 | −0.98 | 1.48 | 0.40 | 0.69 | 0.92 |
| How many hours a day do you spend walking with vigorous efforts? | −0.35 | 1.00 | −2.32 | 1.62 | −0.35 | 0.73 | 0.92 |
| How often do you eat snacks while surfing the internet? | 1.26 | 3.77 | −6.13 | 8.65 | 0.33 | 0.74 | 0.92 |
| How many days per week do you spend doing home activities? | 0.20 | 0.64 | −1.05 | 1.44 | 0.31 | 0.75 | 0.92 |
| How many days per week do you spend doing physical activity in family? | −0.26 | 0.91 | −2.05 | 1.53 | −0.29 | 0.78 | 0.92 |
| How many hours each day you spend walking quite a lot without vigorous efforts? | 0.17 | 0.63 | −1.07 | 1.40 | 0.26 | 0.79 | 0.92 |
| Father BMI | −0.04 | 0.17 | −0.38 | 0.30 | −0.25 | 0.80 | 0.92 |
| How often do you eat snacks while playing videogames? | −0.73 | 3.99 | −8.56 | 7.09 | −0.18 | 0.85 | 0.94 |
| How much time during the weekend do you spend watching TV and DVD? | 0.14 | 0.79 | −1.41 | 1.70 | 0.18 | 0.86 | 0.94 |
| How many days per week do you exercise in a sport club? | 0.07 | 0.69 | −1.28 | 1.43 | 0.11 | 0.92 | 0.98 |
| How many hours do you spend doing homework outside of school hours? | 0.05 | 0.99 | −1.88 | 1.99 | 0.05 | 0.96 | 0.99 |
| How much time per day do you use the internet during the week? | −0.02 | 0.71 | −1.41 | 1.37 | −0.03 | 0.98 | 0.99 |
| How many days do you spend doing vigorous efforts like training activity? | −0.01 | 0.62 | −1.22 | 1.19 | −0.02 | 0.99 | 0.99 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Anguita-Ruiz, A.; González-Gil, E.M.; Rupérez, A.I.; Llorente-Cantarero, F.J.; Pastor-Villaescusa, B.; Alcalá-Fdez, J.; Moreno, L.A.; Gil, Á.; Gil-Campos, M.; Bueno, G.; et al. Evaluation of the Predictive Ability, Environmental Regulation and Pharmacogenetics Utility of a BMI-Predisposing Genetic Risk Score during Childhood and Puberty. J. Clin. Med. 2020, 9, 1705. https://doi.org/10.3390/jcm9061705
Anguita-Ruiz A, González-Gil EM, Rupérez AI, Llorente-Cantarero FJ, Pastor-Villaescusa B, Alcalá-Fdez J, Moreno LA, Gil Á, Gil-Campos M, Bueno G, et al. Evaluation of the Predictive Ability, Environmental Regulation and Pharmacogenetics Utility of a BMI-Predisposing Genetic Risk Score during Childhood and Puberty. Journal of Clinical Medicine. 2020; 9(6):1705. https://doi.org/10.3390/jcm9061705
Chicago/Turabian StyleAnguita-Ruiz, Augusto, Esther M. González-Gil, Azahara I. Rupérez, Francisco Jesús Llorente-Cantarero, Belén Pastor-Villaescusa, Jesús Alcalá-Fdez, Luis A. Moreno, Ángel Gil, Mercedes Gil-Campos, Gloria Bueno, and et al. 2020. "Evaluation of the Predictive Ability, Environmental Regulation and Pharmacogenetics Utility of a BMI-Predisposing Genetic Risk Score during Childhood and Puberty" Journal of Clinical Medicine 9, no. 6: 1705. https://doi.org/10.3390/jcm9061705