
Evidence for Lignocellulose-Decomposing Enzymes in the Genome and Transcriptome of the Aquatic Hyphomycete Clavariopsis aquatica

 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
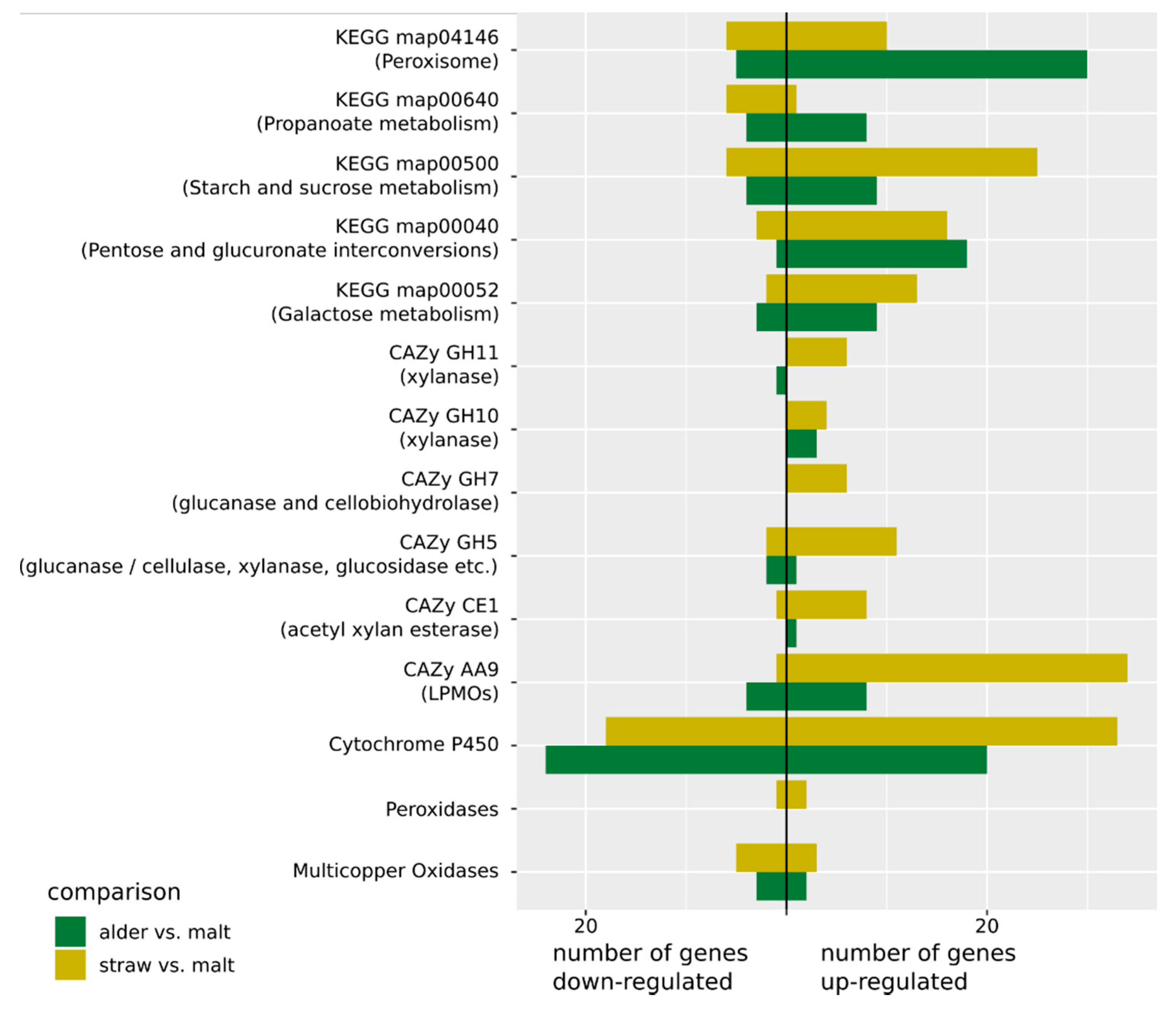
3. Results
3.1. Genome Assembly and Annotation
3.2. RNA-Sequencing and Differential Expression
3.3. Gene and Gene Set Activation
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Krauss, G.-J.; Solé, M.; Krauss, G.; Schlosser, D.; Wesenberg, D.; Bärlocher, F. Fungi in freshwaters: Ecology, physiology and biochemical potential. FEMS Microbiol. Rev. 2011, 35, 620–651. [Google Scholar] [CrossRef]
- Grossart, H.-P.; Wyngaert, S.V.D.; Kagami, M.; Wurzbacher, C.; Cunliffe, M.; Rojas-Jimenez, K. Fungi in aquatic ecosystems. Nat. Rev. Genet. 2019, 17, 339–354. [Google Scholar] [CrossRef] [Green Version]
- Gessner, M.O.; Gulis, K.A.; Kuehn, E.; Chauvet, E.; Suberkopp, K. Fungal decomposers of plant litter in aquatic ecosystems. In Environmental and Microbial Relationships; Springer: Berlin/Heidelberg, Germany, 2007; Volume 4, pp. 301–324. ISBN 978-3-540-71839-0. [Google Scholar]
- Janusz, G.; Pawlik, A.; Sulej, J.; Świderska-Burek, U.; Jarosz-Wilkolazka, A.; Paszczyński, A. Lignin degradation: Microorganisms, enzymes involved, genomes analysis and evolution. FEMS Microbiol. Rev. 2017, 41, 941–962. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bissaro, B.; Várnai, A.; Røhr, Å.K.; Eijsink, V.G.H. Oxidoreductases and reactive oxygen species in conversion of lignocellulosic biomass. Microbiol. Mol. Biol. Rev. 2018, 82, e00029-18. [Google Scholar] [CrossRef] [Green Version]
- Floudas, D.; Bentzer, J.; Ahrén, D.; Johansson, T.; Persson, P.; Tunlid, A. Uncovering the hidden diversity of litter-decomposition mechanisms in mushroom-forming fungi. ISME J. 2020, 14, 2046–2059. [Google Scholar] [CrossRef] [PubMed]
- Liers, C.; Arnstadt, T.; Ullrich, R.; Hofrichter, M. Patterns of lignin degradation and oxidative enzyme secretion by different wood- and litter-colonizing basidiomycetes and ascomycetes grown on beech-wood. FEMS Microbiol. Ecol. 2011, 78, 91–102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riley, R.; Salamov, A.A.; Brown, D.W.; Nagy, L.G.; Floudas, D.; Held, B.W.; Levasseur, A.; Lombard, V.; Morin, E.; Otillar, R.; et al. Extensive sampling of basidiomycete genomes demonstrates inadequacy of the white-rot/brown-rot paradigm for wood decay fungi. Proc. Natl. Acad. Sci. USA 2014, 111, 9923–9928. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, J.D.; Parker, L.A.; Perkins, A.D.; Sonstegard, T.S.; Schroeder, S.; Nicholas, D.D.; Diehl, S.V. Gene expression analysis of copper tolerance and wood decay in the brown rot fungus Fibroporia radiculosa. Appl. Environ. Microbiol. 2012, 79, 1523–1533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Y.; Fan, F.; Zhuo, R.; Ma, F.; Gong, Y.; Wan, X.; Jiang, M.; Zhang, X. Expression of the laccase gene from a white rot fungus in Pichia pastoris can enhance the resistance of this yeast to H2O2-mediated oxidative stress by stimulating the glutathione-based antioxidative system. Appl. Environ. Microbiol. 2012, 78, 5845–5854. [Google Scholar] [CrossRef] [Green Version]
- Iqbal, S.; Webster, J. Aquatic hyphomycete spora of the River Exe and its tributaries. Trans. Br. Mycol. Soc. 1973, 61, 331–346. [Google Scholar] [CrossRef]
- Suberkropp, K.; Klug, M.J. Fungi and bacteria associated with leaves during processing in a woodland stream. Ecology 1976, 57, 707–719. [Google Scholar] [CrossRef]
- Clavariopsis Aquatica De Wild. Available online: https://www.gbif.org/species/8268939 (accessed on 25 August 2021).
- Junghanns, C.; Moeder, M.; Krauss, G.; Martin, C.; Schlosser, D. Degradation of the xenoestrogen nonylphenol by aquatic fungi and their laccases. Microbiology 2005, 151, 45–57. [Google Scholar] [CrossRef] [Green Version]
- Martin, C.; Moeder, M.; Daniel, X.; Krauss, G.; Schlosser, D. Biotransformation of the polycyclic musks HHCB and AHTN and metabolite formation by fungi occurring in freshwater environments. Environ. Sci. Technol. 2007, 41, 5395–5402. [Google Scholar] [CrossRef] [PubMed]
- Harms, H.; Schlosser, D.; Wick, L.Y. Untapped potential: Exploiting fungi in bioremediation of hazardous chemicals. Nat. Rev. Genet. 2011, 9, 177–192. [Google Scholar] [CrossRef] [PubMed]
- Syed, K.; Shale, K.; Pagadala, N.S.; Tuszynski, J. Systematic Identification and evolutionary analysis of catalytically versatile cytochrome P450 monooxygenase families enriched in model basidiomycete fungi. PLoS ONE 2014, 9, e86683. [Google Scholar] [CrossRef]
- Morel, M.; Meux, E.; Mathieu, Y.; Thuillier, A.; Chibani, K.; Harvengt, L.; Jacquot, J.-P.; Gelhaye, E. Xenomic networks variability and adaptation traits in wood decaying fungi. Microb. Biotechnol. 2013, 6, 248–263. [Google Scholar] [CrossRef]
- Hill, R.; Leitch, I.J.; Gaya, E. Targeting ascomycota genomes: What and how big? Fungal Biol. Rev. 2021, 36, 52–59. [Google Scholar] [CrossRef]
- Alemdar, A.; Sain, M. Biocomposites from wheat straw nanofibers: Morphology, thermal and mechanical properties. Compos. Sci. Technol. 2008, 68, 557–565. [Google Scholar] [CrossRef]
- Bjerre, A.B.; Olesen, A.B.; Fernqvist, T.; Plöger, A.; Schmidt, A.S. Pretreatment of wheat straw using combined wet oxidation and alkaline hydrolysis resulting in convertible cellulose and hemicellulose. Biotechnol. Bioeng. 1996, 49, 568–577. [Google Scholar] [CrossRef]
- Chauvet, E. Changes in the chemical composition of alder, poplar and willow leaves during decomposition in a river. Hydrobiologia 1987, 148, 35–44. [Google Scholar] [CrossRef] [Green Version]
- Lecerf, A.; Chauvet, E. Intraspecific variability in leaf traits strongly affects alder leaf decomposition in a stream. Basic Appl. Ecol. 2008, 9, 598–605. [Google Scholar] [CrossRef] [Green Version]
- Schlosser, D.; Höfer, C. Laccase-catalyzed oxidation of Mn2+ in the presence of natural Mn3+ chelators as a novel source of extracellular H2O2 production and its impact on manganese peroxidase. Appl. Environ. Microbiol. 2002, 68, 3514–3521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Solé, M.; Müller, I.; Pecyna, M.J.; Fetzer, I.; Harms, H.; Schlosser, D. Differential regulation by organic compounds and heavy metals of multiple laccase genes in the aquatic hyphomycete Clavariopsis aquatica. Appl. Environ. Microbiol. 2012, 78, 4732–4739. [Google Scholar] [CrossRef] [Green Version]
- Delmas, S.; Pullan, S.T.; Gaddipati, S.; Kokolski, M.; Malla, S.; Blythe, M.J.; Ibbett, R.; Campbell, M.; Liddell, S.; Aboobaker, A.; et al. Uncovering the genome-wide transcriptional responses of the filamentous fungus aspergillus niger to lignocellulose using RNA sequencing. PLoS Genet. 2012, 8, e1002875. [Google Scholar] [CrossRef] [PubMed]
- Bourne, E.C.; Johnston, P.R.; Funke, E.; Monaghan, M.T. Gene expression analysis of litter-associated fungi using RNA-seq. In Methods to Study Litter Decomposition; Springer: Berlin/Heidelberg, Germany, 2020; ISBN 978-3-030-30514-7. [Google Scholar]
- Johnson, M.T.J.; Carpenter, E.J.; Tian, Z.; Bruskiewich, R.; Burris, J.N.; Carrigan, C.T.; Chase, M.W.; Clarke, N.D.; Covshoff, S.; Depamphilis, C.W.; et al. Evaluating methods for isolating total RNA and predicting the success of sequencing phylogenetically diverse plant transcriptomes. PLoS ONE 2012, 7, e50226. [Google Scholar] [CrossRef] [PubMed]
- Crusoe, M.R.; Alameldin, H.; Awad, S.; Bucher, E.; Caldwell, A.; Cartwright, R.; Charbonneau, A.; Constantinides, B.; Edvenson, G.; Fay, S.; et al. The khmer software package: Enabling efficient nucleotide sequence analysis. F1000Research 2015, 4, 900. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, C.T.; Howe, A.; Zhang, Q.; Pyrkosz, A.B.; Brom, T.H. A reference-free algorithm for computational normalization of shotgun sequencing data. arXiv 2012, arXiv:1203.4802. [Google Scholar]
- Zhang, Q.; Pell, J.; Canino-Koning, R.; Howe, A.C.; Brown, C.T. These are not the K-mers you are looking for: Efficient online K-mer counting using a probabilistic data structure. PLoS ONE 2014, 9, e101271. [Google Scholar] [CrossRef] [Green Version]
- Zerbino, D.R.; Birney, E. Velvet: Algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 2008, 18, 821–829. [Google Scholar] [CrossRef] [Green Version]
- Simão, F.A.; Waterhouse, R.M.; Ioannidis, P.; Kriventseva, E.V.; Zdobnov, E.M. Busco: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 2015, 31, 3210–3212. [Google Scholar] [CrossRef] [Green Version]
- Palmer, J.; Stajich, J. Nextgenusfs/Funannotate: Funannotate v1.5.3; Zenodo: Geneva, Switzerland, 2019. [Google Scholar] [CrossRef]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Haas, B.J. Improving the Arabidopsis genome annotation using maximal transcript alignment assemblies. Nucleic Acids Res. 2003, 31, 5654–5666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, P.; Binns, D.; Chang, H.Y.; Fraser, M.; Li, W.; McAnulla, C.; McWilliam, H.; Maslen, J.; Mitchell, A.; Nuka, G.; et al. InterProScan 5: Genome-scale protein function classification. Bioinformatics 2014, 30, 1236–1240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Finn, R.D.; Attwood, T.; Babbitt, P.C.; Bateman, A.; Bork, P.; Bridge, A.; Chang, H.Y.; Dosztányi, Z.; El-Gebali, S.; Fraser, M.; et al. InterPro in 2017—beyond protein family and domain annotations. Nucleic Acids Res. 2017, 45, D190–D199. [Google Scholar] [CrossRef]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [Green Version]
- The Gene Ontology Consortium. Expansion of the gene ontology knowledgebase and resources. Nucleic Acids Res. 2017, 45, D331–D338. [Google Scholar] [CrossRef] [Green Version]
- Lombard, V.; Ramulu, H.G.; Drula, E.; Coutinho, P.M.; Henrissat, B. The carbohydrate-active enzymes database (CAZy) in 2013. Nucleic Acids Res. 2014, 42, D490–D495. [Google Scholar] [CrossRef] [Green Version]
- Yin, Y.; Mao, X.; Yang, J.; Chen, X.; Mao, F.; Xu, Y. dbCAN: A web resource for automated carbohydrate-active enzyme annotation. Nucleic Acids Res. 2012, 40, W445–W451. [Google Scholar] [CrossRef]
- Petersen, T.N.; Brunak, S.; von Heijne, G.; Nielsen, H. SignalP 4.0: Discriminating signal peptides from transmembrane regions. Nat. Methods 2011, 8, 785–786. [Google Scholar] [CrossRef]
- Kanehisa, M.; Sato, Y.; Kawashima, M.; Furumichi, M.; Tanabe, M. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res. 2016, 44, D457–D462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanehisa, M.; Sato, Y.; Morishima, K. BlastKOALA and GhostKOALA: KEGG tools for functional characterization of genome and metagenome sequences. J. Mol. Biol. 2016, 428, 726–731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sirim, D.; Wagner, F.; Wang, L.; Schmid, R.D.; Pleiss, J. The laccase engineering database: A classification and analysis system for laccases and related multicopper oxidases. Database 2011, 2011, bar006. [Google Scholar] [CrossRef] [Green Version]
- Koua, D.; Cerutti, L.; Falquet, L.; Sigrist, C.J.A.; Theiler, G.; Hulo, N.; Dunand, C. PeroxiBase: A database with new tools for peroxidase family classification. Nucleic Acids Res. 2008, 37, D261–D266. [Google Scholar] [CrossRef] [Green Version]
- Fawal, N.; Li, Q.; Savelli, B.; Brette, M.; Passaia, G.; Fabre, M.; Mathé, C.; Dunand, C. PeroxiBase: A database for large-scale evolutionary analysis of peroxidases. Nucleic Acids Res. 2012, 41, D441–D444. [Google Scholar] [CrossRef] [Green Version]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef] [Green Version]
- Köster, J.; Rahmann, S. Snakemake—A scalable bioinformatics workflow engine. Bioinformatics 2012, 28, 2520–2522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bauer, S.; Gagneur, J.; Robinson, P.N. Going bayesian: Model-based gene set analysis of genome-scale data. Nucleic Acids Res. 2010, 38, 3523–3532. [Google Scholar] [CrossRef] [Green Version]
- Bauer, S.; Robinson, P.N.; Gagneur, J. Model-based gene set analysis for bioconductor. Bioinformatics 2011, 27, 1882–1883. [Google Scholar] [CrossRef]
- Grigoriev, I.V.; Nikitin, R.; Haridas, S.; Kuo, A.; Ohm, R.A.; Otillar, R.; Riley, R.; Salamov, A.A.; Zhao, X.; Korzeniewski, F.; et al. MycoCosm portal: Gearing up for 1000 fungal genomes. Nucleic Acids Res. 2014, 42, D699–D704. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.V.S.; Phale, P.S.; Durani, S.; Wangikar, P.P. Combined sequence and structure analysis of the fungal laccase family. Biotechnol. Bioeng. 2003, 83, 386–394. [Google Scholar] [CrossRef]
- Zhang, J.; Siika-Aho, M.; Tenkanen, M.; Viikari, L. The role of acetyl xylan esterase in the solubilization of xylan and enzymatic hydrolysis of wheat straw and giant reed. Biotechnol. Biofuels 2011, 4, 60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giardina, P.; Faraco, V.; Pezzella, C.; Piscitelli, A.; VanHulle, S.; Sannia, G. Laccases: A never-ending story. Cell. Mol. Life Sci. 2010, 67, 369–385. [Google Scholar] [CrossRef] [PubMed]
- Kües, U. Multiple multi-copper oxidase gene families in Basidiomycetes—What for? Curr. Genom. 2011, 12, 72–94. [Google Scholar] [CrossRef] [Green Version]
- Xie, N.; Chapeland-Leclerc, F.; Silar, P.; Ruprich-Robert, G. Systematic gene deletions evidences that laccases are involved in several stages of wood degradation in the filamentous fungus Podospora anserina. Environ. Microbiol. 2013, 16, 141–161. [Google Scholar] [CrossRef]
- Sugano, Y.; Yoshida, T. DyP-type peroxidases: Recent advances and perspectives. Int. J. Mol. Sci. 2021, 22, 5556. [Google Scholar] [CrossRef]
- Mathé, C.; Fawal, N.; Roux, C.; Dunand, C. In Silico definition of new ligninolytic peroxidase sub-classes in fungi and putative relation to fungal life style. Sci. Rep. 2019, 9, 20373. [Google Scholar] [CrossRef] [Green Version]
- Ries, L.; Pullan, S.T.; Delmas, S.; Malla, S.; Blythe, M.J.; Archer, D.B. Genome-wide transcriptional response of Trichoderma reesei to lignocellulose using RNA sequencing and comparison with Aspergillus niger. BMC Genom. 2013, 14, 541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaaje-Kolstad, G.; Forsberg, Z.; Loose, J.S.; Bissaro, B.; Eijsink, V.G. Structural diversity of lytic polysaccharide monooxygenases. Curr. Opin. Struct. Biol. 2017, 44, 67–76. [Google Scholar] [CrossRef]
- Martínez, A.T.; Ruiz-Dueñas, F.J.; Camarero, S.; Serrano, A.; Linde, D.; Lund, H.; Vind, J.; Tovborg, M.; Herold-Majumdar, O.M.; Hofrichter, M.; et al. Oxidoreductases on their way to industrial biotransformations. Biotechnol. Adv. 2017, 35, 815–831. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frommhagen, M.; Sforza, S.; Westphal, A.H.; Visser, J.; Hinz, S.W.; Koetsier, M.J.; Van Berkel, W.J.H.; Gruppen, H.; Kabel, M.A. Discovery of the combined oxidative cleavage of plant xylan and cellulose by a new fungal polysaccharide monooxygenase. Biotechnol. Biofuels 2015, 8, 101. [Google Scholar] [CrossRef] [Green Version]
- Otzen, C.; Bardl, B.; Jacobsen, I.D.; Nett, M.; Brock, M. Candida albicans utilizes a modified β-oxidation pathway for the degradation of toxic propionyl-CoA. J. Biol. Chem. 2014, 289, 8151–8169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bucher, V.; Pointing, S.; Hyde, K.; Reddy, C. Production of wood decay enzymes, loss of mass, and lignin solubilization in wood by diverse tropical freshwater fungi. Microb. Ecol. 2004, 48, 331–337. [Google Scholar] [CrossRef] [PubMed]
- Kerr, J.L.; Baldwin, D.S.; Tobin, M.; Puskar, L.; Kappen, P.; Rees, G.; Silvester, E. High spatial resolution infrared micro-spectroscopy reveals the mechanism of leaf lignin decomposition by aquatic fungi. PLoS ONE 2013, 8, e60857. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
| Sample Code | Culture | Medium | Growth Phase | Number of Reads |
|---|---|---|---|---|
| A3 | liquid | wheat straw | exponential | 50,666,214 |
| A4 | liquid | wheat straw | exponential | 57,490,827 |
| A6 | liquid | wheat straw | stationary | 16,839,738 |
| A7 | liquid | wheat straw | stationary | 17,367,382 |
| A9 | liquid | wheat straw | stationary | 17,584,532 |
| B1 | liquid | alder leaves | exponential | 17,922,153 |
| B3 | liquid | alder leaves | exponential | 16,700,439 |
| B5 | liquid | alder leaves | exponential | 14,857,484 |
| D1 | liquid | malt extract | exponential | 17,796,408 |
| D3 | liquid | malt extract | exponential | 17,863,281 |
| D4 | liquid | malt extract | exponential | 19,469,408 |
| E2 | solid | wheat straw | NA * | 16,859,013 |
| E3 | solid | wheat straw | NA * | 16,904,004 |
| E4 | solid | wheat straw | NA * | 18,861,228 |
| Comparison | Differential Expression | ||
|---|---|---|---|
| Condition 1 | Condition 2 | Upregulated | Downregulated |
| exponential growth on wheat straw in liquid culture | exponential growth on malt extract in liquid culture | 1430 | 1570 |
| exponential growth on alder leaves in liquid culture | exponential growth on malt extract in liquid culture | 1033 | 1462 |
| exponential growth on wheat straw in liquid culture | stationary growth on wheat straw in liquid culture | 1380 | 883 |
| growth on wheat straw in solid culture | exponential growth on wheat straw in liquid culture | 2731 | 2328 |
| growth on wheat straw in solid culture | stationary growth on wheat straw in liquid culture | 2683 | 2478 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Heeger, F.; Bourne, E.C.; Wurzbacher, C.; Funke, E.; Lipzen, A.; He, G.; Ng, V.; Grigoriev, I.V.; Schlosser, D.; Monaghan, M.T. Evidence for Lignocellulose-Decomposing Enzymes in the Genome and Transcriptome of the Aquatic Hyphomycete Clavariopsis aquatica. J. Fungi 2021, 7, 854. https://doi.org/10.3390/jof7100854
Heeger F, Bourne EC, Wurzbacher C, Funke E, Lipzen A, He G, Ng V, Grigoriev IV, Schlosser D, Monaghan MT. Evidence for Lignocellulose-Decomposing Enzymes in the Genome and Transcriptome of the Aquatic Hyphomycete Clavariopsis aquatica. Journal of Fungi. 2021; 7(10):854. https://doi.org/10.3390/jof7100854
Chicago/Turabian StyleHeeger, Felix, Elizabeth C. Bourne, Christian Wurzbacher, Elisabeth Funke, Anna Lipzen, Guifen He, Vivian Ng, Igor V. Grigoriev, Dietmar Schlosser, and Michael T. Monaghan. 2021. "Evidence for Lignocellulose-Decomposing Enzymes in the Genome and Transcriptome of the Aquatic Hyphomycete Clavariopsis aquatica" Journal of Fungi 7, no. 10: 854. https://doi.org/10.3390/jof7100854