
The Sugar Metabolic Model of Aspergillus niger Can Only Be Reliably Transferred to Fungi of Its Phylum
 , , , , , , , and
, , , , , , , and 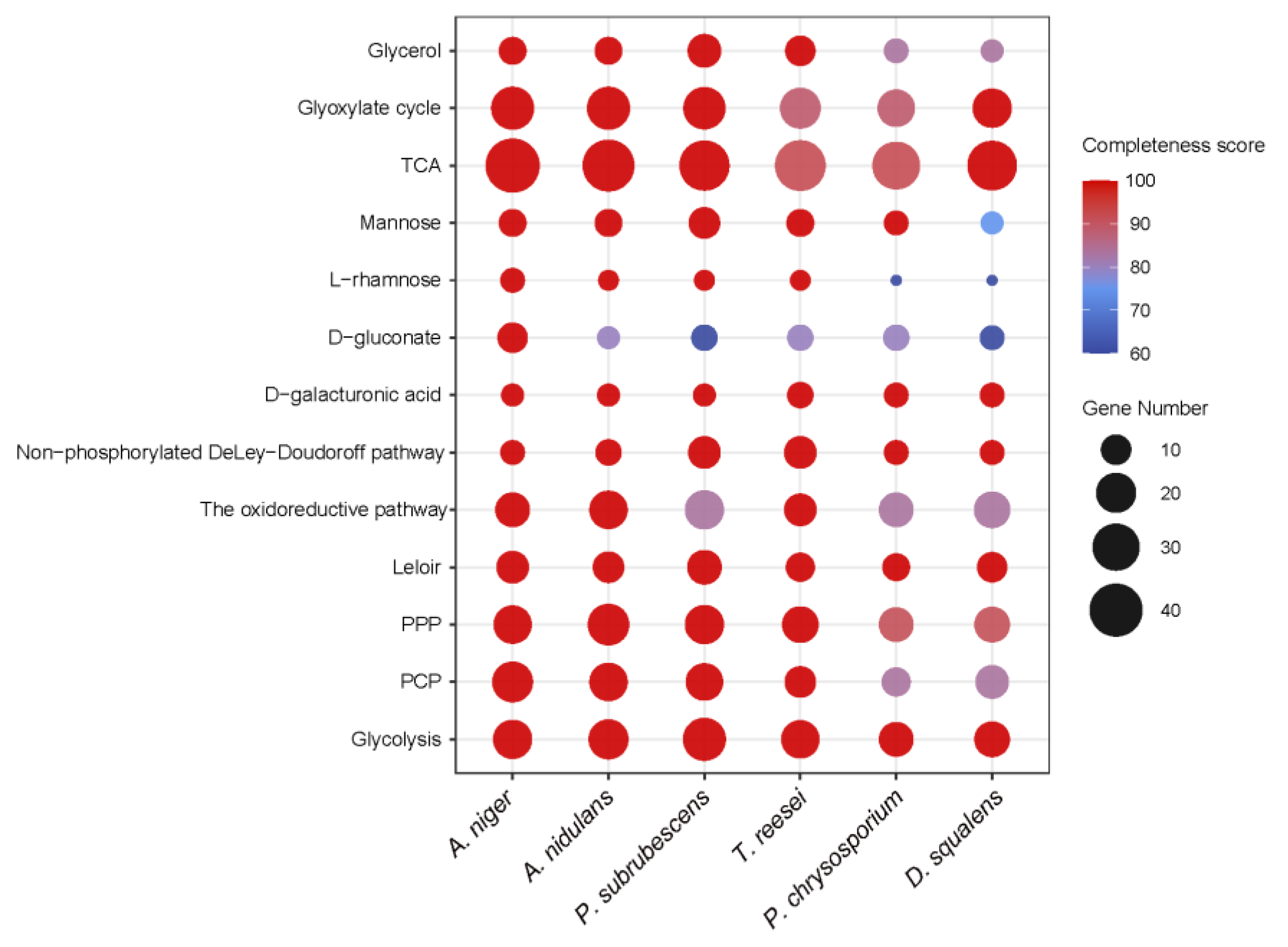{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
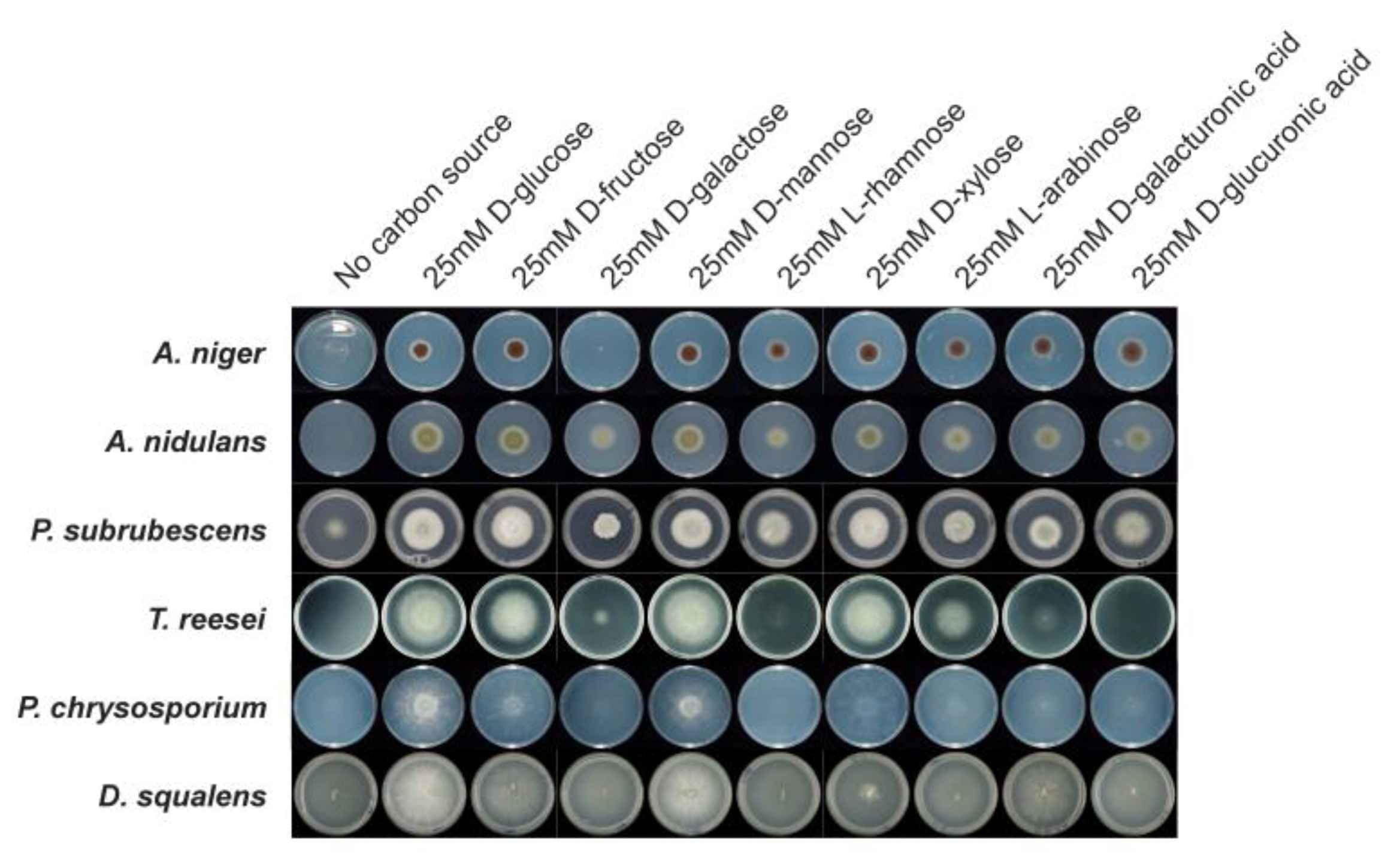{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Fungal Strains
2.2. Identification of Orthologs of Sugar Metabolism Genes across Six Fungal Species
2.3. Data Gathering and Preparation for Pathway Tools
2.4. Transcriptome Sequencing and Analysis
2.5. Proteome Quantitation and Statistical Analysis
2.6. Metabolomics Data Preprocessing and Analysis
2.7. Growth Profiling on Nine Monosaccharides
3. Results
3.1. Identification of Orthologs between A. niger and Five Other Fungi
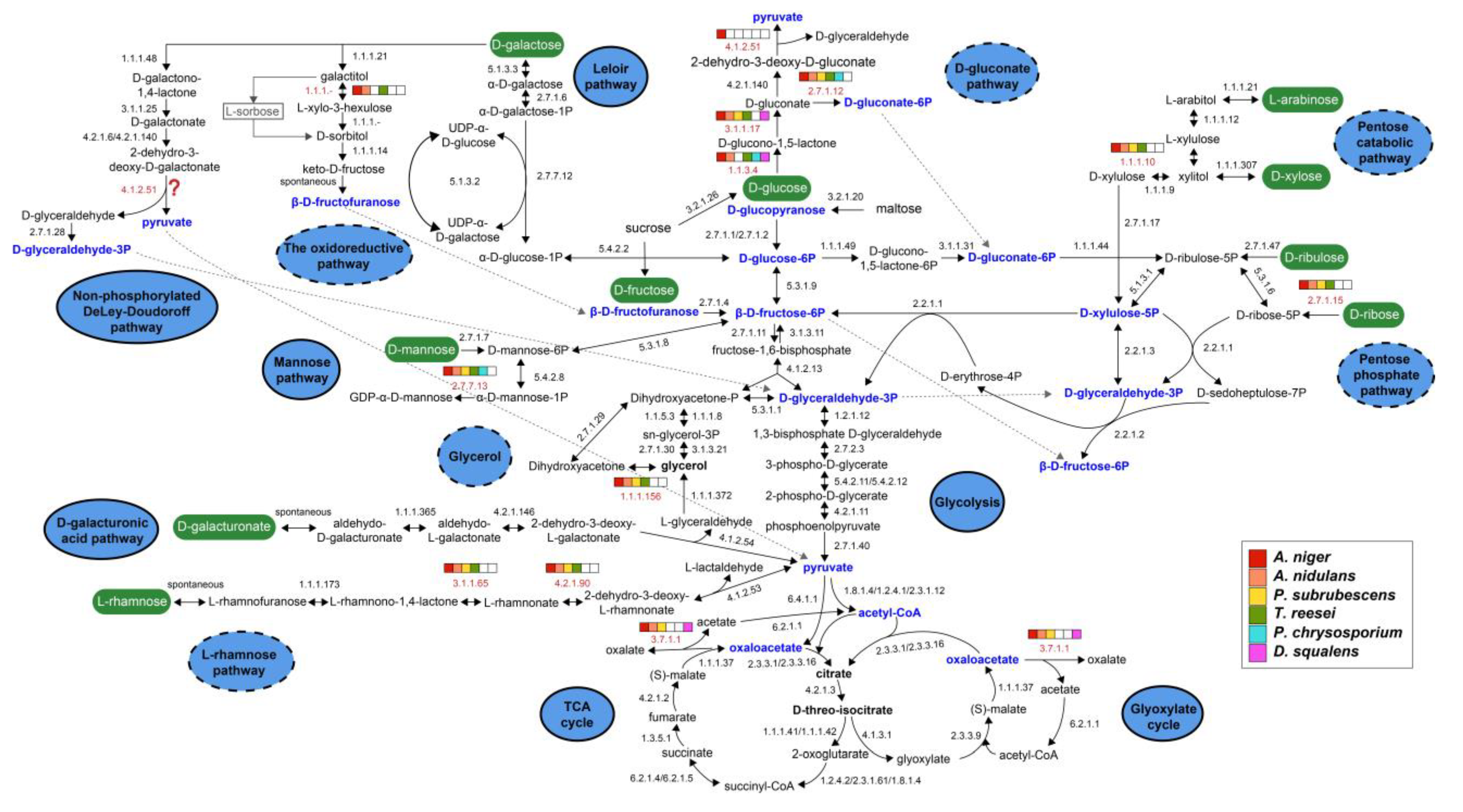
3.2. Generation of Sugar Metabolic Models for the Selected Fungal Species
3.3. Conservation of Sugar Metabolism among Different Fungal Species
3.3.1. Sugar Catabolic Pathways with Strong Conservation
- Glycolysis:
- The tricarboxylic acid (TCA) and glyoxylate cycles:
- D-Galacturonic acid metabolism:
3.3.2. Sugar Catabolism with Moderate Conservation
- Glycerol metabolism:
- D-Mannose metabolism:
- Pentose phosphate pathway (PPP):
- D-Galactose metabolism:
3.3.3. Sugar Catabolism with Low Conservation
- Pentose catabolic pathway (PCP):
- L-Rhamnose metabolism:
- D-Gluconate metabolism:
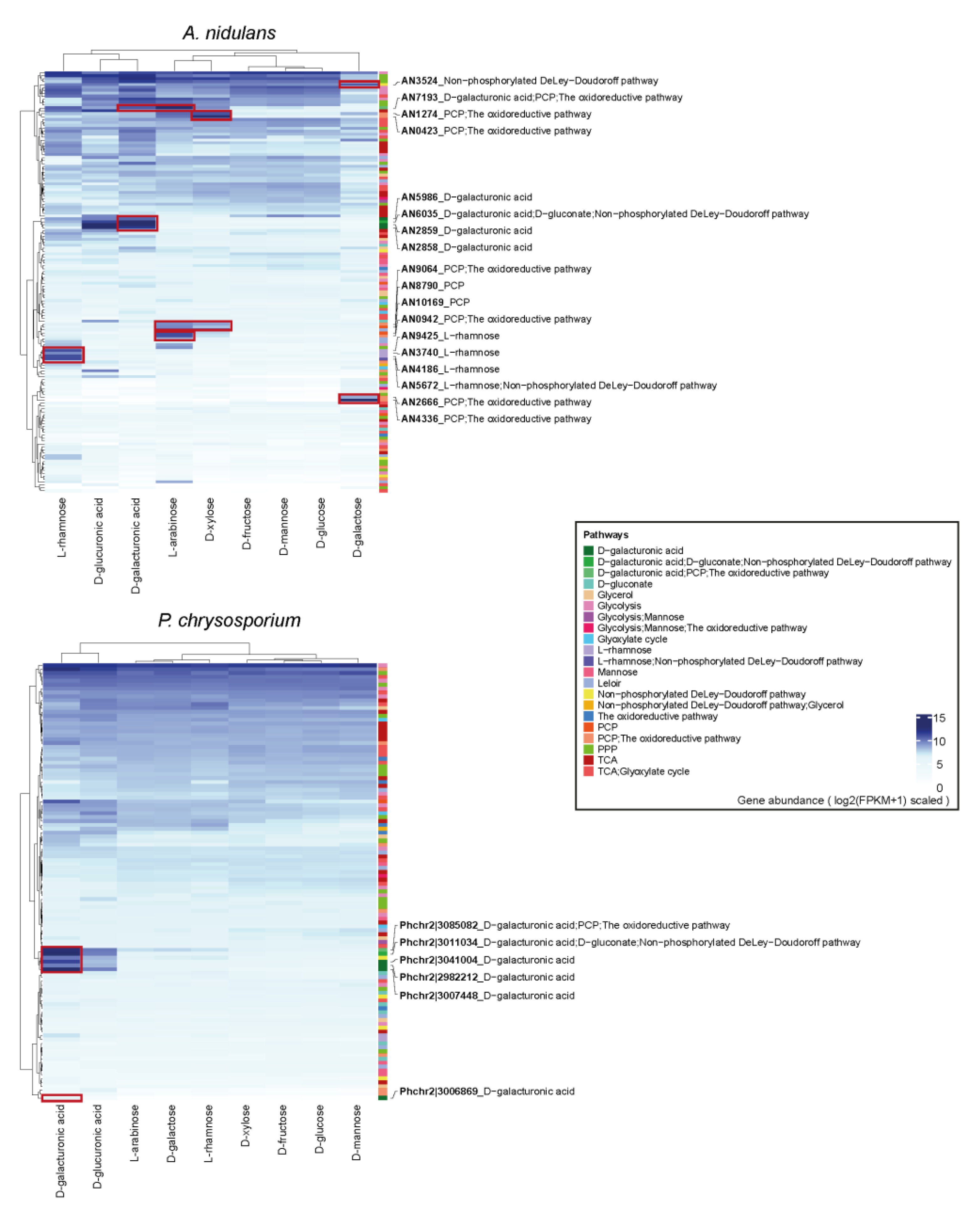
3.4. The Expression Profiling of Sugar Metabolic Genes in Each Fungus
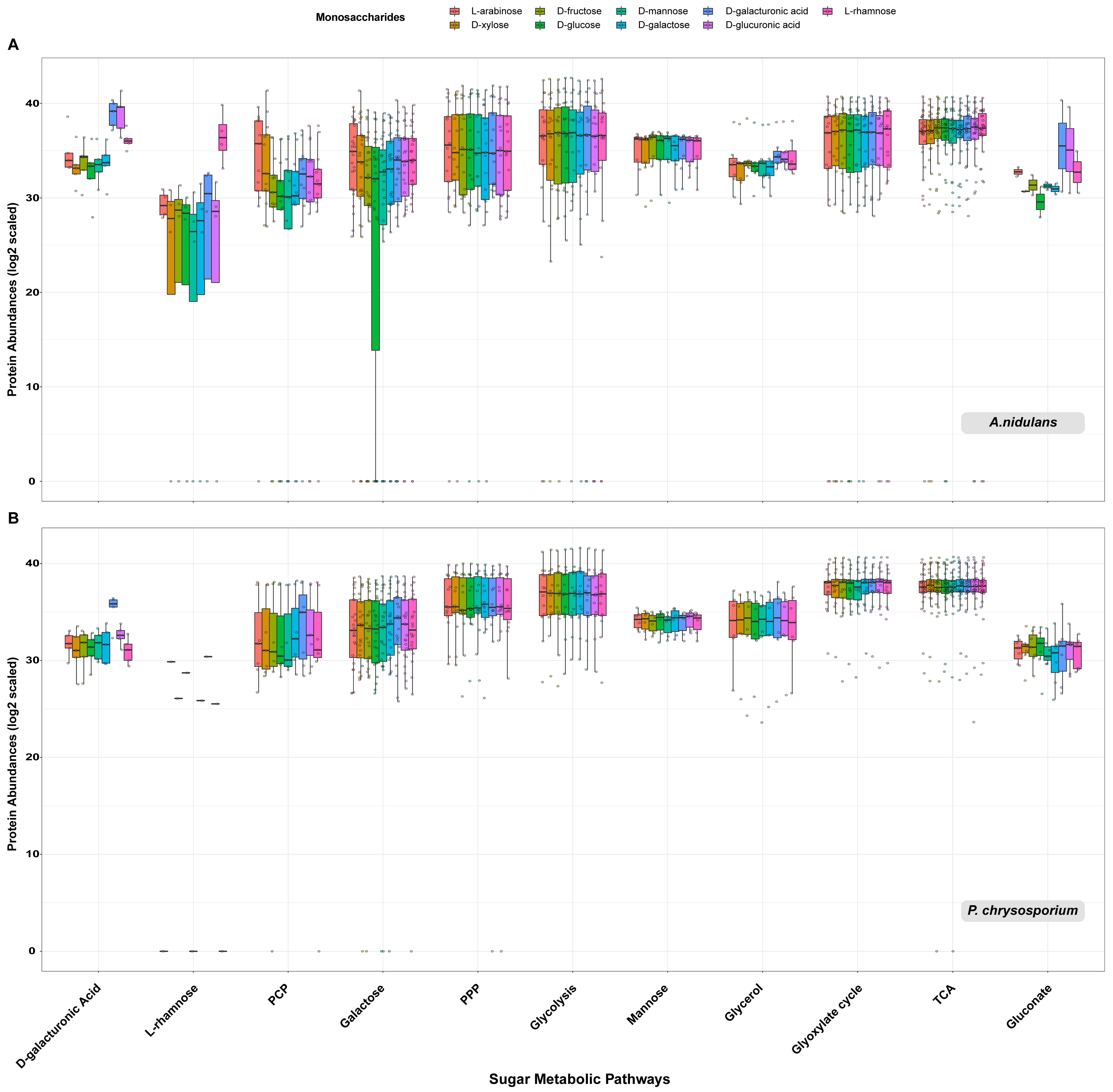
3.5. Proteome Profile of Sugar Metabolism-Related Genes (SMGs) Showed Similar Sugar Inducing Pattern as Transcriptome Data
3.6. Correlation between the Abundance of Metabolites and Gene Expression Levels of Sugar Metabolism-Related Genes (SMGs)
3.7. Comparative Growth Profiles on Different Carbon Sources
4. Discussion
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Guerriero, G.; Hausman, J.F.; Strauss, J.; Ertan, H.; Siddiqui, K.S. Lignocellulosic biomass: Biosynthesis, degradation, and industrial utilization. Eng. Life Sci. 2016, 16, 1–16. [Google Scholar] [CrossRef]
- de Vries, R.P.; Visser, J. Aspergillus enzymes involved in degradation of plant cell wall polysaccharides. Microbiol. Mol. Biol. Rev. 2001, 65, 497–522. [Google Scholar] [CrossRef] [Green Version]
- Culleton, H.; McKie, V.; de Vries, R.P. Physiological and molecular aspects of degradation of plant polysaccharides by fungi: What have we learned from Aspergillus? Biotechnol. J. 2013, 8, 884–894. [Google Scholar] [CrossRef] [PubMed]
- de Vries, R.P.; Mäkelä, M.R. Genomic and postgenomic diversity of fungal plant biomass degradation approaches. Trends Microbiol. 2020, 28, 487–499. [Google Scholar] [CrossRef]
- Khosravi, C.; Benocci, T.; Battaglia, E.; Benoit, I.; de Vries, R.P. Sugar catabolism in Aspergillus and other fungi related to the utilization of plant biomass. Adv. Appl. Microbiol. 2015, 90, 1–28. [Google Scholar] [CrossRef]
- Mäkelä, M.R.; Donofrio, N.; de Vries, R.P. Plant biomass degradation by fungi. Fungal Genet. Biol. 2014, 72, 2–9. [Google Scholar] [CrossRef]
- Panneman, H.; Ruijter, G.J.; Van Den Broeck, H.C.; Driever, E.T.; Visser, J. Cloning and biochemical characterisation of an Aspergillus niger glucokinase: Evidence for the presence of separate glucokinase and hexokinase enzymes. Eur. J. Biochem. 1996, 240, 518–525. [Google Scholar] [CrossRef]
- Panneman, H.; Ruijter, G.J.; van den Broeck, H.C.; Visser, J. Cloning and biochemical characterisation of Aspergillus niger hexokinase: The enzyme is strongly inhibited by physiological concentrations of trehalose 6-phosphate. Eur. J. Biochem. 1998, 258, 223–232. [Google Scholar] [CrossRef]
- Chroumpi, T.; Peng, M.; Aguilar-Pontes, M.V.; Müller, A.; Wang, M.; Yan, J.; Lipzen, A.; Ng, V.; Grigoriev, I.V.; Mäkelä, M.R. Revisiting a ‘simple’fungal metabolic pathway reveals redundancy, complexity and diversity. Microb. Biotechnol. 2021, 14, 2525–2537. [Google Scholar] [CrossRef]
- Aguilar-Pontes, M.V.; Brandl, J.; McDonnell, E.; Strasser, K.; Nguyen, T.; Riley, R.; Mondo, S.; Salamov, A.; Nybo, J.L.; Vesth, T.C. The gold-standard genome of Aspergillus niger NRRL 3 enables a detailed view of the diversity of sugar catabolism in fungi. Stud. Mycol. 2018, 91, 61–78. [Google Scholar] [CrossRef]
- Kubicek, C.P. Regulatory aspects of the tricarboxylic acid cycle in filamentous fungi—A review. Trans. Br. Mycol. Soc 1988, 90, 339–349. [Google Scholar] [CrossRef]
- Strijbis, K.; Distel, B. Intracellular acetyl unit transport in fungal carbon metabolism. Eukaryot. Cell 2010, 9, 1809–1815. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kunze, M.; Pracharoenwattana, I.; Smith, S.M.; Hartig, A. A central role for the peroxisomal membrane in glyoxylate cycle function. Biochim. Biophys. Acta 2006, 1763, 1441–1452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chroumpi, T.; Aguilar-Pontes, M.V.; Peng, M.; Wang, M.; Lipzen, A.; Ng, V.; Grigoriev, I.V.; Mäkelä, M.R.; de Vries, R.P. Identification of a gene encoding the last step of the L-rhamnose catabolic pathway in Aspergillus niger revealed the inducer of the pathway regulator. Microbiol. Res. 2020, 234, 126426. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Stoeckert, C.J.; Roos, D.S. OrthoMCL: Identification of ortholog groups for eukaryotic genomes. Genome Res. 2003, 13, 2178–2189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Emms, D.M.; Kelly, S. OrthoFinder: Phylogenetic orthology inference for comparative genomics. Genome Biol. 2019, 20, 238. [Google Scholar] [CrossRef] [Green Version]
- Grigoriev, I.V.; Nikitin, R.; Haridas, S.; Kuo, A.; Ohm, R.; Otillar, R.; Riley, R.; Salamov, A.; Zhao, X.; Korzeniewski, F. MycoCosm portal: Gearing up for 1000 fungal genomes. Nucleic Acids Res. 2014, 42, D699–D704. [Google Scholar] [CrossRef]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 2015, 12, 59–60. [Google Scholar] [CrossRef]
- Kelly, S.; Maini, P.K. DendroBLAST: Approximate phylogenetic trees in the absence of multiple sequence alignments. PLoS ONE 2013, 8, e58537. [Google Scholar] [CrossRef] [Green Version]
- Karp, P.D.; Paley, S.; Romero, P. The pathway tools software. Bioinformatics 2002, 18, S225–S232. [Google Scholar] [CrossRef] [Green Version]
- Karp, P.D.; Midford, P.E.; Billington, R.; Kothari, A.; Krummenacker, M.; Latendresse, M.; Ong, W.K.; Subhraveti, P.; Caspi, R.; Fulcher, C. Pathway Tools version 23.0 update: Software for pathway/genome informatics and systems biology. Brief. Bioinform. 2021, 22, 109–126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Consortium, G.O. The gene ontology project in 2008. Nucleic Acids Res. 2008, 36, D440–D444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hunter, S.; Apweiler, R.; Attwood, T.K.; Bairoch, A.; Bateman, A.; Binns, D.; Bork, P.; Das, U.; Daugherty, L.; Duquenne, L. InterPro: The integrative protein signature database. Nucleic Acids Res. 2009, 37, D211–D215. [Google Scholar] [CrossRef] [Green Version]
- Petersen, T.N.; Brunak, S.; Von Heijne, G.; Nielsen, H. SignalP 4.0: Discriminating signal peptides from transmembrane regions. Nat. Methods 2011, 8, 785–786. [Google Scholar] [CrossRef]
- Tatusov, R.L.; Fedorova, N.D.; Jackson, J.D.; Jacobs, A.R.; Kiryutin, B.; Koonin, E.V.; Krylov, D.M.; Mazumder, R.; Mekhedov, S.L.; Nikolskaya, A.N. The COG database: An updated version includes eukaryotes. BMC Bioinform. 2003, 4, 41. [Google Scholar] [CrossRef] [Green Version]
- Sonnhammer, E.L.; Eddy, S.R.; Durbin, R. Pfam: A comprehensive database of protein domain families based on seed alignments. Proteins Struct. Funct. Bioinform. 1997, 28, 405–420. [Google Scholar] [CrossRef]
- Mistry, J.; Chuguransky, S.; Williams, L.; Qureshi, M.; Salazar, G.A.; Sonnhammer, E.L.; Tosatto, S.C.; Paladin, L.; Raj, S.; Richardson, L.J. Pfam: The protein families database in 2021. Nucleic Acids Res. 2021, 49, D412–D419. [Google Scholar] [CrossRef]
- Bairoch, A.; Apweiler, R. The SWISS-PROT protein sequence database and its supplement TrEMBL in 2000. Nucleic Acids Res. 2000, 28, 45–48. [Google Scholar] [CrossRef]
- Keseler, I.M.; Mackie, A.; Peralta-Gil, M.; Santos-Zavaleta, A.; Gama-Castro, S.; Bonavides-Martínez, C.; Fulcher, C.; Huerta, A.M.; Kothari, A.; Krummenacker, M. EcoCyc: Fusing model organism databases with systems biology. Nucleic Acids Res. 2013, 41, D605–D612. [Google Scholar] [CrossRef]
- Caspi, R.; Altman, T.; Billington, R.; Dreher, K.; Foerster, H.; Fulcher, C.A.; Holland, T.A.; Keseler, I.M.; Kothari, A.; Kubo, A. The MetaCyc database of metabolic pathways and enzymes and the BioCyc collection of Pathway/Genome Databases. Nucleic Acids Res. 2014, 42, D459–D471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gruben, B.S.; Mäkelä, M.R.; Kowalczyk, J.E.; Zhou, M.; Benoit-Gelber, I.; De Vries, R.P. Expression-based clustering of CAZyme-encoding genes of Aspergillus niger. BMC Genom. 2017, 18, 900. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Casado López, S.; Peng, M.; Issak, T.Y.; Daly, P.; de Vries, R.P.; Mäkelä, M.R. Induction of genes encoding plant cell wall-degrading carbohydrate-active enzymes by lignocellulose-derived monosaccharides and cellobiose in the white-rot fungus Dichomitus squalens. Appl. Environ. Microbiol. 2018, 84, e00403–e00418. [Google Scholar] [CrossRef] [Green Version]
- de Vries, R.P.; Burgers, K.; van de Vondervoort, P.J.; Frisvad, J.C.; Samson, R.A.; Visser, J. A new black Aspergillus species, A. vadensis, is a promising host for homologous and heterologous protein production. Appl. Environ. Microbiol. 2004, 70, 3954–3959. [Google Scholar] [CrossRef] [Green Version]
- Klaubauf, S.; Narang, H.M.; Post, H.; Zhou, M.; Brunner, K.; Mach-Aigner, A.R.; Mach, R.L.; Heck, A.J.; Altelaar, A.M.; de Vries, R.P. Similar is not the same: Differences in the function of the (hemi-) cellulolytic regulator XlnR (Xlr1/Xyr1) in filamentous fungi. Fungal Genet. Biol. 2014, 72, 73–81. [Google Scholar] [CrossRef] [PubMed]
- Eastwood, D.C.; Floudas, D.; Binder, M.; Majcherczyk, A.; Schneider, P.; Aerts, A.; Asiegbu, F.O.; Baker, S.E.; Barry, K.; Bendiksby, M. The plant cell wall–decomposing machinery underlies the functional diversity of forest fungi. Science 2011, 333, 762–765. [Google Scholar] [CrossRef] [Green Version]
- Daly, P.; Peng, M.; Mitchell, H.D.; Kim, Y.M.; Ansong, C.; Brewer, H.; De Gijsel, P.; Lipton, M.S.; Markillie, L.M.; Nicora, C.D. Colonies of the fungus Aspergillus niger are highly differentiated to adapt to local carbon source variation. Environ. Microbiol. 2020, 22, 1154–1166. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.; Gupta, N.; Pevzner, P.A. Spectral probabilities and generating functions of tandem mass spectra: A strike against decoy databases. J. Proteome Res. 2008, 7, 3354–3363. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.; Pevzner, P.A. MS-GF+ makes progress towards a universal database search tool for proteomics. Nat. Commun. 2014, 5, 5277. [Google Scholar] [CrossRef] [Green Version]
- Monroe, M.E.; Shaw, J.L.; Daly, D.S.; Adkins, J.N.; Smith, R.D. MASIC: A software program for fast quantitation and flexible visualization of chromatographic profiles from detected LC–MS (/MS) features. Comput. Biol. Chem. 2008, 32, 215–217. [Google Scholar] [CrossRef] [PubMed]
- Polpitiya, A.D.; Qian, W.-J.; Jaitly, N.; Petyuk, V.A.; Adkins, J.N.; Camp, D.G.; Anderson, G.A.; Smith, R.D. DAnTE: A statistical tool for quantitative analysis of-omics data. Bioinformatics 2008, 24, 1556–1558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chroumpi, T.; Peng, M.; Markillie, L.M.; Mitchell, H.D.; Nicora, C.D.; Hutchinson, C.M.; Paurus, V.; Tolic, N.; Clendinen, C.S.; Orr, G. Re-routing of sugar catabolism provides a better insight into fungal flexibility in using plant biomass-derived monomers as substrates. Front. Bioeng. Biotechnol. 2021, 9, 167. [Google Scholar] [CrossRef] [PubMed]
- Hiller, K.; Hangebrauk, J.; Jäger, C.; Spura, J.; Schreiber, K.; Schomburg, D. MetaboliteDetector: Comprehensive analysis tool for targeted and nontargeted GC/MS based metabolome analysis. Anal. Chem. 2009, 81, 3429–3439. [Google Scholar] [CrossRef] [PubMed]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romano, A.; Conway, T. Evolution of carbohydrate metabolic pathways. Res. Microbiol. 1996, 147, 448–455. [Google Scholar] [CrossRef]
- Ruijter, G.J.; Visser, J. Characterization of Aspergillus niger phosphoglucose isomerase. Use for quantitative determination of erythrose 4-phosphate. Biochimie 1999, 81, 267–272. [Google Scholar] [CrossRef]
- Ruijter, G.; Panneman, H.; Visser, J. Overexpression of phosphofructokinase and pyruvate kinase in citric acid-producing Aspergillus niger. Biochim. Biophys. Acta 1997, 1334, 317–326. [Google Scholar] [CrossRef]
- Pontremoli, S.; Traniello, S.; Luppis, B.; Wood, W. Fructose diphosphatase from rabbit liver I. Purification and properties. J. Biol. Chem. 1965, 240, 3459–3463. [Google Scholar] [CrossRef]
- Jagannathan, V.; Singh, K.; Damodaran, M. Carbohydrate metabolism in citric acid fermentation. 4. Purification and properties of aldolase from Aspergillus niger. Biochem. J. 1956, 63, 94. [Google Scholar] [CrossRef] [Green Version]
- Punt, P.J.; Dingemanse, M.A.; Jacobs-Meijsing, B.J.; Pouwels, P.H.; van den Hondel, C.A. Isolation and characterization of the glyceraldehyde-3-phosphate dehydrogenase gene of Aspergillus nidulans. Gene 1988, 69, 49–57. [Google Scholar] [CrossRef]
- Clements, J.M.; Roberts, C.F. Molecular cloning of the 3-phosphoglycerate kinase (PGK) gene from Aspergillus nidulans. Curr. Genet. 1985, 9, 293–298. [Google Scholar] [CrossRef] [PubMed]
- Machida, M.; Chang, Y.-C.; Manabe, M.; Yasukawa, M.; Kunihiro, S.; Jigami, Y. Molecular cloning of a cDNA encoding enolase from the filamentous fungus, Aspergillus oryzae. Curr. Genet. 1996, 30, 423–431. [Google Scholar] [CrossRef] [PubMed]
- de Graaff, L.; van den Broeck, H.; Visser, J. Isolation and characterization of the Aspergillus niger pyruvate kinase gene. Curr. Genet. 1992, 22, 21–27. [Google Scholar] [CrossRef]
- Khitan, Z.; Kim, D.H. Fructose: A key factor in the development of metabolic syndrome and hypertension. J. Nutr. Metab. 2013, 2013, 682673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jun, H.; Guangye, H.; Daiwen, C. Insights into enzyme secretion by filamentous fungi: Comparative proteome analysis of Trichoderma reesei grown on different carbon sources. J. Proteom. 2013, 89, 191–201. [Google Scholar] [CrossRef]
- Stappler, E.; Dattenböck, C.; Tisch, D.; Schmoll, M. Analysis of light-and carbon-specific transcriptomes implicates a class of G-protein-coupled receptors in cellulose sensing. Msphere 2017, 2, e00089-17. [Google Scholar] [CrossRef] [Green Version]
- Connerton, I.; Fincham, J.; Sandeman, R.; Hynes, M. Comparison and cross-species expression of the acetyl-CoA synthetase genes of the ascomycete fungi, Aspergillus nidulans and Neurospora crassa. Mol. Microbiol. 1990, 4, 451–460. [Google Scholar] [CrossRef]
- Ruijter, G.J.; van de Vondervoort, P.J.; Visser, J. Oxalic acid production by Aspergillus niger: An oxalate-non-producing mutant produces citric acid at pH 5 and in the presence of manganese. Microbiology 1999, 145, 2569–2576. [Google Scholar] [CrossRef] [Green Version]
- Richard, P.; Hilditch, S. D-galacturonic acid catabolism in microorganisms and its biotechnological relevance. Appl. Microbiol. Biotechnol. 2009, 82, 597–604. [Google Scholar] [CrossRef]
- Martens-Uzunova, E.S.; Schaap, P.J. An evolutionary conserved D-galacturonic acid metabolic pathway operates across filamentous fungi capable of pectin degradation. Fungal Genet. Biol. 2008, 45, 1449–1457. [Google Scholar] [CrossRef]
- Hilditch, S. Identification of the Fungal Catabolic D-Galacturonate Pathway; VTT Technical Research Centre of Finland: Espoo, Finland, 2010. [Google Scholar]
- Kuorelahti, S.; Kalkkinen, N.; Penttilä, M.; Londesborough, J.; Richard, P. Identification in the mold Hypocrea jecorina of the first fungal D-galacturonic acid reductase. Biochemistry 2005, 44, 11234–11240. [Google Scholar] [CrossRef] [PubMed]
- Kuorelahti, S.; Jouhten, P.; Maaheimo, H.; Penttilä, M.; Richard, P. L-galactonate dehydratase is part of the fungal path for D-galacturonic acid catabolism. Mol. Microbiol. 2006, 61, 1060–1068. [Google Scholar] [CrossRef]
- Zhang, L.; Thiewes, H.; van Kan, J.A. The D-galacturonic acid catabolic pathway in Botrytis cinerea. Fungal Genet. Biol. 2011, 48, 990–997. [Google Scholar] [CrossRef] [PubMed]
- Alazi, E.; Khosravi, C.; Homan, T.G.; du Pré, S.; Arentshorst, M.; Di Falco, M.; Pham, T.T.; Peng, M.; Aguilar-Pontes, M.V.; Visser, J. The pathway intermediate 2-keto-3-deoxy-L-galactonate mediates the induction of genes involved in D-galacturonic acid utilization in Aspergillus niger. FEBS Lett. 2017, 591, 1408–1418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klein, M.; Swinnen, S.; Thevelein, J.M.; Nevoigt, E. Glycerol metabolism and transport in yeast and fungi: Established knowledge and ambiguities. Environ. Microbiol. 2017, 19, 878–893. [Google Scholar] [CrossRef] [Green Version]
- Nicol, R.; Marchand, K.; Lubitz, W. Bioconversion of crude glycerol by fungi. Appl. Microbiol. Biotechnol. 2012, 93, 1865–1875. [Google Scholar] [CrossRef]
- Hondmann, D.H.; Busink, R.; Witteveen, C.F.; Vlsser, J. Glycerol catabolism in Aspergillus nidulans. Microbiology 1991, 137, 629–636. [Google Scholar] [CrossRef] [Green Version]
- Liepins, J.; Kuorelahti, S.; Penttilä, M.; Richard, P. Enzymes for the NADPH-dependent reduction of dihydroxyacetone and D-glyceraldehyde and L-glyceraldehyde in the mould Hypocrea jecorina. FEBS J. 2006, 273, 4229–4235. [Google Scholar] [CrossRef]
- de Vries, R.P.; Flitter, S.J.; Van De Vondervoort, P.J.; Chaveroche, M.K.; Fontaine, T.; Fillinger, S.; Ruijter, G.J.; D’Enfert, C.; Visser, J. Glycerol dehydrogenase, encoded by gldB is essential for osmotolerance in Aspergillus nidulans. Mol. Microbiol. 2003, 49, 131–141. [Google Scholar] [CrossRef] [Green Version]
- Stryer, L. Biochemistry; WH Freeman: New York, NY, USA, 1995. [Google Scholar]
- Kruger, N.J.; von Schaewen, A. The oxidative pentose phosphate pathway: Structure and organisation. Curr. Opin. Plant Biol. 2003, 6, 236–246. [Google Scholar] [CrossRef]
- Mojzita, D.; Herold, S.; Metz, B.; Seiboth, B.; Richard, P. L-xylo-3-hexulose reductase is the missing link in the oxidoreductive pathway for D-galactose catabolism in filamentous fungi. J. Biol. Chem. 2012, 287, 26010–26018. [Google Scholar] [CrossRef] [Green Version]
- Németh, Z.; Kulcsár, L.; Flipphi, M.; Orosz, A.; Aguilar-Pontes, M.V.; de Vries, R.P.; Karaffa, L.; Fekete, E. L-Arabinose induces D-galactose catabolism via the Leloir pathway in Aspergillus nidulans. Fungal Genet. Biol. 2019, 123, 53–59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pail, M.; Peterbauer, T.; Seiboth, B.; Hametner, C.; Druzhinina, I.; Kubicek, C.P. The metabolic role and evolution of L-arabinitol 4-dehydrogenase of Hypocrea jecorina. Eur. J. Biochem. 2004, 271, 1864–1872. [Google Scholar] [CrossRef] [PubMed]
- Mojzita, D.; Koivistoinen, O.M.; Maaheimo, H.; Penttilä, M.; Ruohonen, L.; Richard, P. Identification of the galactitol dehydrogenase, LadB, that is part of the oxido-reductive D-galactose catabolic pathway in Aspergillus niger. Fungal Genet. Biol. 2012, 49, 152–159. [Google Scholar] [CrossRef] [PubMed]
- Seiboth, B.; Metz, B. Fungal arabinan and L-arabinose metabolism. Appl. Microbiol. Biotechnol. 2011, 89, 1665–1673. [Google Scholar] [CrossRef] [Green Version]
- Fekete, E.; Karaffa, L.; Sándor, E.; Bányai, I.; Seiboth, B.; Gyémánt, G.; Sepsi, A.; Szentirmai, A.; Kubicek, C.P. The alternative D-galactose degrading pathway of Aspergillus nidulans proceeds via L-sorbose. Arch. Microbiol. 2004, 181, 35–44. [Google Scholar] [CrossRef]
- Orosz, A.; Fekete, E.; Flipphi, M.; Karaffa, L. Metabolism of D-galactose is dispensable for the induction of the beta-galactosidase (bgaD) and lactose permease (lacpA) genes in Aspergillus nidulans. FEMS Microbiol. Lett. 2014, 359, 19–25. [Google Scholar] [CrossRef] [Green Version]
- Kowalczyk, J.E.; Gruben, B.S.; Battaglia, E.; Wiebenga, A.; Majoor, E.; de Vries, R.P. Genetic interaction of Aspergillus nidulans galR, xlnR and araR in regulating D-galactose and L-arabinose release and catabolism gene expression. PLoS ONE 2015, 10, e0143200. [Google Scholar] [CrossRef]
- Flipphi, M.; Sun, J.; Robellet, X.; Karaffa, L.; Fekete, E.; Zeng, A.-P.; Kubicek, C.P. Biodiversity and evolution of primary carbon metabolism in Aspergillus nidulans and other Aspergillus spp. Fungal Genet. Biol. 2009, 46, S19–S44. [Google Scholar] [CrossRef]
- Kulcsár, L.; Flipphi, M.; Jónás, Á.; Sándor, E.; Fekete, E.; Karaffa, L. Identification of a mutarotase gene involved in D-galactose utilization in Aspergillus nidulans. FEMS Microbiol. Lett. 2017, 364, fnx202. [Google Scholar] [CrossRef]
- Mojzita, D.; Vuoristo, K.; Koivistoinen, O.M.; Penttilä, M.; Richard, P. The ‘true’L-xylulose reductase of filamentous fungi identified in Aspergillus niger. FEBS Lett. 2010, 584, 3540–3544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seiboth, B.; Gamauf, C.; Pail, M.; Hartl, L.; Kubicek, C.P. The D-xylose reductase of Hypocrea jecorina is the major aldose reductase in pentose and D-galactose catabolism and necessary for β-galactosidase and cellulase induction by lactose. Mol. Microbiol. 2007, 66, 890–900. [Google Scholar] [CrossRef] [PubMed]
- Khosravi, C.; Kun, R.S.; Visser, J.; Aguilar-Pontes, M.V.; de Vries, R.P.; Battaglia, E. In vivo functional analysis of L-rhamnose metabolic pathway in Aspergillus niger: A tool to identify the potential inducer of RhaR. BMC Microbiol. 2017, 17, 214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koivistoinen, O.M.; Arvas, M.; Headman, J.R.; Andberg, M.; Penttilä, M.; Jeffries, T.W.; Richard, P. Characterisation of the gene cluster for L-rhamnose catabolism in the yeast Scheffersomyces (Pichia) stipitis. Gene 2012, 492, 177–185. [Google Scholar] [CrossRef] [PubMed]
- MacCabe, A.P.; Ninou, E.I.; Pardo, E.; Orejas, M. Catabolism of L-rhamnose in A. nidulans proceeds via the non-phosphorylated pathway and is glucose repressed by a CreA-independent mechanism. Microb. Cell Factories 2020, 19, 188. [Google Scholar] [CrossRef]
- Shindia, A.; EI-Sherbeny, G.; EI-Esawy, A.; Sheriff, Y. Production of gluconic acid by some local fungi. Mycobiology 2006, 34, 22–29. [Google Scholar] [CrossRef] [Green Version]
- Peng, M.; Aguilar-Pontes, M.V.; de Vries, R.P.; Mäkelä, M.R. In silico analysis of putative sugar transporter genes in Aspergillus niger using phylogeny and comparative transcriptomics. Front. Microbiol. 2018, 9, 1045. [Google Scholar] [CrossRef] [Green Version]
- Hayer, K.; Stratford, M.; Archer, D.B. Structural features of sugars that trigger or support conidial germination in the filamentous fungus Aspergillus niger. Appl. Environ. Microbiol. 2013, 79, 6924–6931. [Google Scholar] [CrossRef] [Green Version]
- Hayer, K.; Stratford, M.; Archer, D.B. Germination of Aspergillus niger conidia is triggered by nitrogen compounds related to L-amino acids. Appl. Environ. Microbiol. 2014, 80, 6046–6053. [Google Scholar] [CrossRef] [Green Version]
- Fekete, E.; de Vries, R.P.; Seiboth, B.; vanKuyk, P.A.; Sándor, E.; Fekete, É.; Metz, B.; Kubicek, C.P.; Karaffa, L. D-Galactose uptake is nonfunctional in the conidiospores of Aspergillus niger. FEMS Microbiol. Lett. 2012, 329, 198–203. [Google Scholar] [CrossRef]
- Moore, B.M.; Wang, P.; Fan, P.; Leong, B.; Schenck, C.A.; Lloyd, J.P.; Lehti-Shiu, M.D.; Last, R.L.; Pichersky, E.; Shiu, S.-H. Robust predictions of specialized metabolism genes through machine learning. Proc. Natl. Acad. Sci. USA 2019, 116, 2344–2353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamanishi, Y.; Vert, J.-P.; Kanehisa, M. Supervised enzyme network inference from the integration of genomic data and chemical information. Bioinformatics 2005, 21, i468–i477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, M.; de Vries, R.P. Machine learning prediction of novel pectinolytic enzymes in Aspergillus niger through integrating heterogeneous (post-) genomics data. Microb. Genom. 2021, 7, 000674. [Google Scholar] [CrossRef] [PubMed]
- Todd, R.B.; Zhou, M.; Ohm, R.A.; Leeggangers, H.A.; Visser, L.; De Vries, R.P. Prevalence of transcription factors in ascomycete and basidiomycete fungi. BMC Genom. 2014, 15, 214. [Google Scholar] [CrossRef] [Green Version]
- Benocci, T.; Aguilar-Pontes, M.V.; Zhou, M.; Seiboth, B.; de Vries, R.P. Regulators of plant biomass degradation in ascomycetous fungi. Biotechnol. Biofuels 2017, 10, 152. [Google Scholar] [CrossRef] [Green Version]
- Vesth, T.C.; Nybo, J.L.; Theobald, S.; Frisvad, J.C.; Larsen, T.O.; Nielsen, K.F.; Hoof, J.B.; Brandl, J.; Salamov, A.; Riley, R.; et al. Investigation of inter-and intraspecies variation through genome sequencing of Aspergillus section Nigri. Nat. Genet. 2018, 50, 1688–1695. [Google Scholar] [CrossRef] [Green Version]
- Arnaud, M.B.; Cerqueira, G.C.; Inglis, D.O.; Skrzypek, M.S.; Binkley, J.; Chibucos, M.C.; Crabtree, J.; Howarth, C.; Orvis, J.; Shah, P.; et al. The Aspergillus Genome Database (AspGD): Recent developments in comprehensive multispecies curation, comparative genomics and community resources. Nucleic Acids Res. 2012, 40, D653–D659. [Google Scholar] [CrossRef]
- Galagan, J.E.; Calvo, S.E.; Cuomo, C.; Ma, L.J.; Wortman, J.R.; Batzoglou, S.; Lee, S.I.; Baştürkmen, M.; Spevak, C.C.; Clutterbuck, J.; et al. Sequencing of Aspergillus nidulans and comparative analysis with A. fumigatus and A. oryzae. Nature 2005, 438, 1105–1115. [Google Scholar] [CrossRef] [Green Version]
- Peng, M.; Dilokpimol, A.; Mäkelä, M.R.; Hildén, K.; Bervoets, S.; Riley, R.; Grigoriev, I.V.; Hainaut, M.; Henrissat, B.; de Vries, R.P.; et al. The draft genome sequence of the ascomycete fungus Penicillium subrubescens reveals a highly enriched content of plant biomass related CAZymes compared to related fungi. J. Biotechnol. 2017, 246, 1–3. [Google Scholar] [CrossRef]
- Li, W.-C.; Huang, C.-H.; Chen, C.-L.; Chuang, Y.-C.; Tung, S.-Y.; Wang, T.-F. Trichoderma reesei complete genome sequence, repeat-induced point mutation, and partitioning of CAZyme gene clusters. Biotechnol. Biofuels. 2017, 10, 170. [Google Scholar] [CrossRef]
- Martinez, D.; Berka, R.M.; Henrissat, B.; Saloheimo, M.; Arvas, M.; Baker, S.E.; Chapman, J.; Chertkov, O.; Coutinho, P.M.; Cullen, D.; et al. Genome sequencing and analysis of the biomass-degrading fungus Trichoderma reesei (syn. Hypocrea jecorina). Nat. Biotechnol. 2008, 26, 553–560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohm, R.A.; Riley, R.; Salamov, A.; Min, B.; Choi, I.-G.; Grigoriev, I.V. Genomics of wood-degrading fungi. Fungal Genet. Biol. 2014, 72, 82–90. [Google Scholar] [CrossRef] [PubMed]
- Casado López, S.; Peng, M.; Daly, P.; Andreopoulos, B.; Pangilinan, J.; Lipzen, A.; Riley, R.; Ahrendt, S.; Ng, V.; Barry, K.; et al. Draft genome sequences of three monokaryotic isolates of the white-rot basidiomycete fungus Dichomitus squalens. Microbiol. Resour. Announc. 2019, 8, e00264-19. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, J.; Chroumpi, T.; Garrigues, S.; Kun, R.S.; Meng, J.; Salazar-Cerezo, S.; Aguilar-Pontes, M.V.; Zhang, Y.; Tejomurthula, S.; Lipzen, A.; et al. The Sugar Metabolic Model of Aspergillus niger Can Only Be Reliably Transferred to Fungi of Its Phylum. J. Fungi 2022, 8, 1315. https://doi.org/10.3390/jof8121315
Li J, Chroumpi T, Garrigues S, Kun RS, Meng J, Salazar-Cerezo S, Aguilar-Pontes MV, Zhang Y, Tejomurthula S, Lipzen A, et al. The Sugar Metabolic Model of Aspergillus niger Can Only Be Reliably Transferred to Fungi of Its Phylum. Journal of Fungi. 2022; 8(12):1315. https://doi.org/10.3390/jof8121315
Chicago/Turabian StyleLi, Jiajia, Tania Chroumpi, Sandra Garrigues, Roland S. Kun, Jiali Meng, Sonia Salazar-Cerezo, Maria Victoria Aguilar-Pontes, Yu Zhang, Sravanthi Tejomurthula, Anna Lipzen, and et al. 2022. "The Sugar Metabolic Model of Aspergillus niger Can Only Be Reliably Transferred to Fungi of Its Phylum" Journal of Fungi 8, no. 12: 1315. https://doi.org/10.3390/jof8121315