
Expression of Saccharomyces cerevisiae RER2 Gene Encoding Cis-Prenyltransferase in Trichoderma atroviride Increases the Activity of Secretory Hydrolases and Enhances Antimicrobial Features

 , , and
, , and 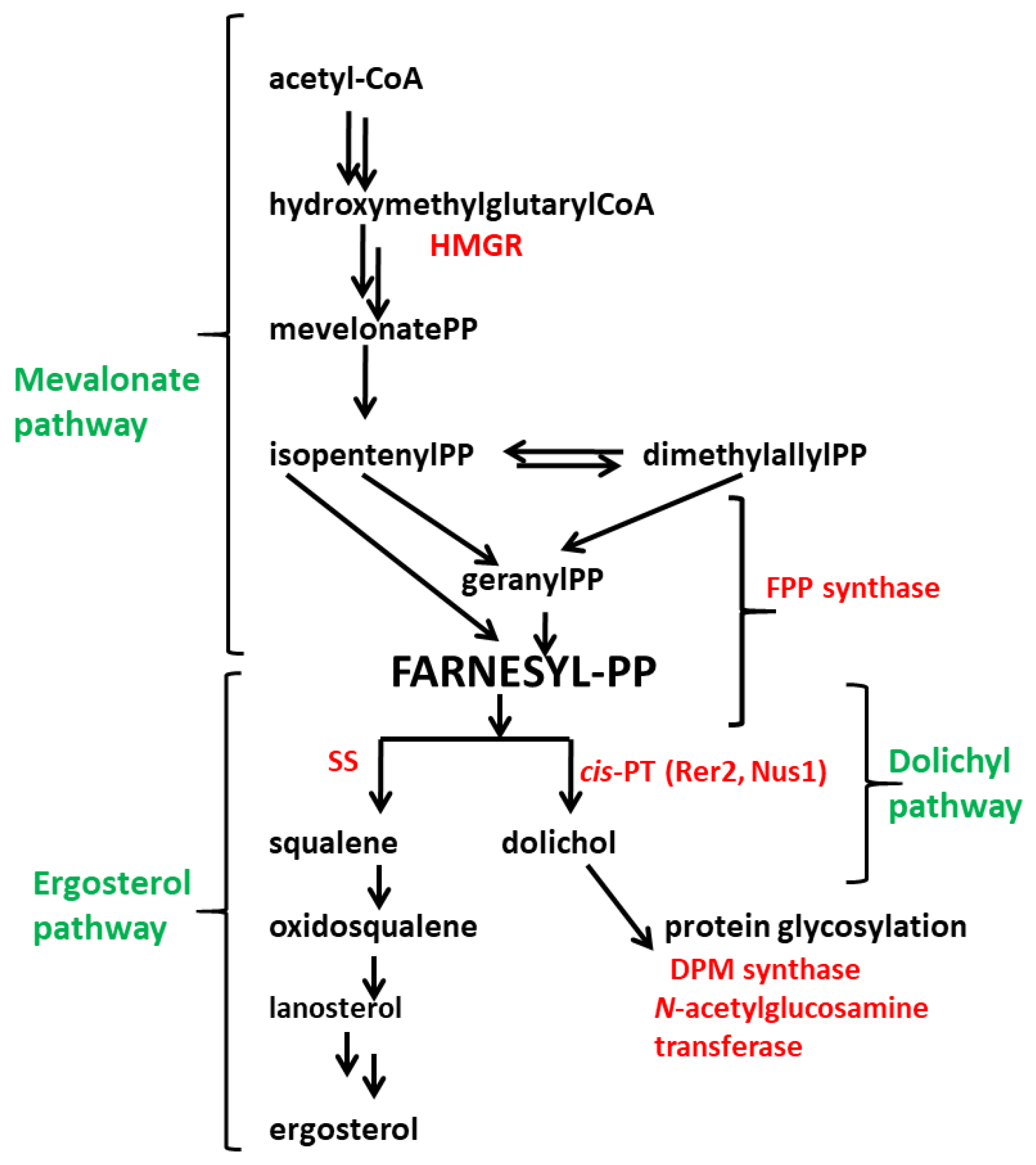{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Strains and Cultivation Media
2.2. Analysis of Fungal Growth
2.3. Expression of the S. cerevisiae RER2 Gene in T. atroviride
2.4. Nucleic Acids Isolation
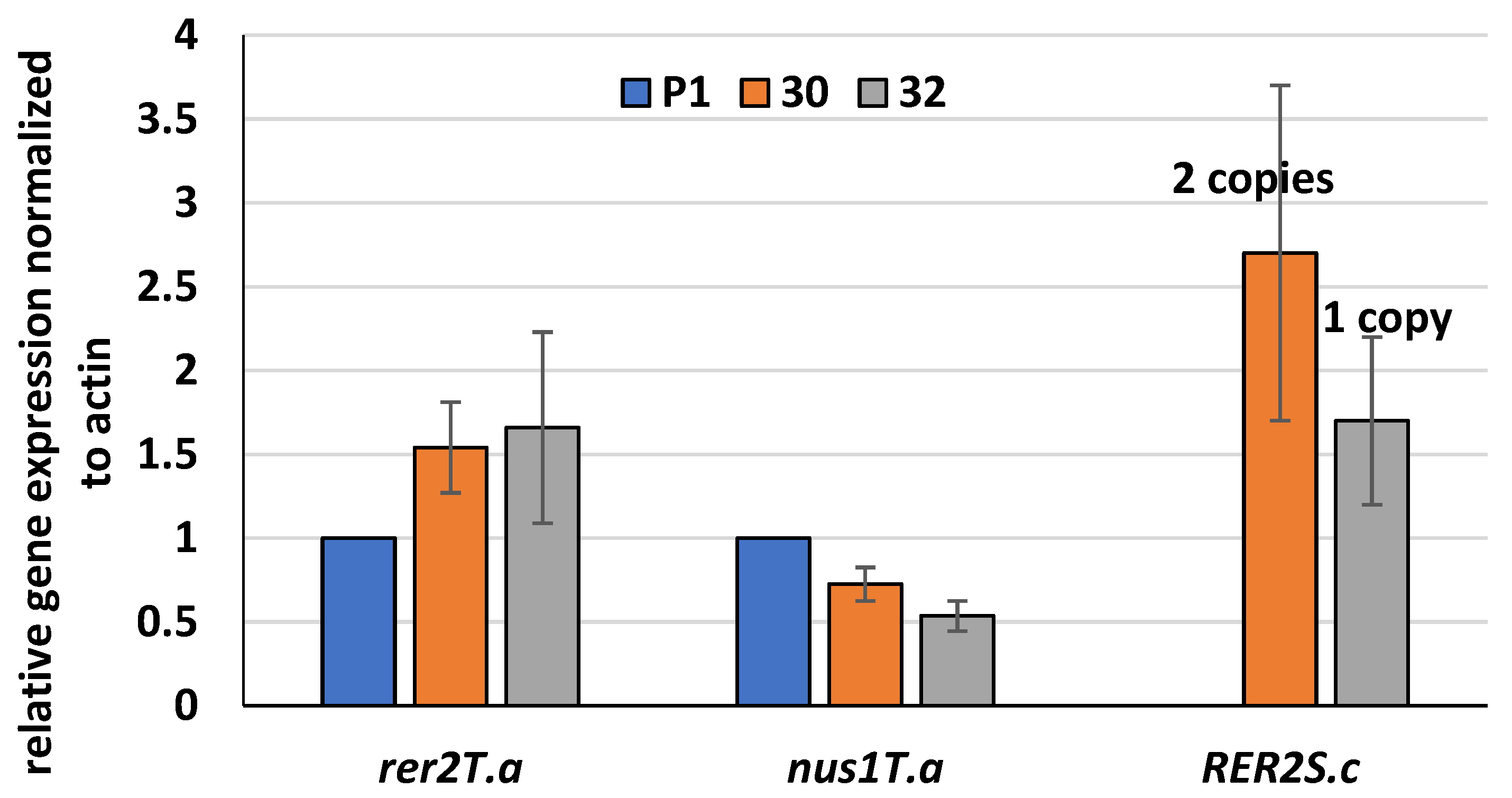
2.5. RER2 Copies Integrated into T. atroviride Genome
2.6. Quantitative Reverse Transcription PCR (RT-qPCR)
2.7. Cell Membrane Preparation
2.8. Cis-Prenyltransferase (EC 2.5.1.20) Activity
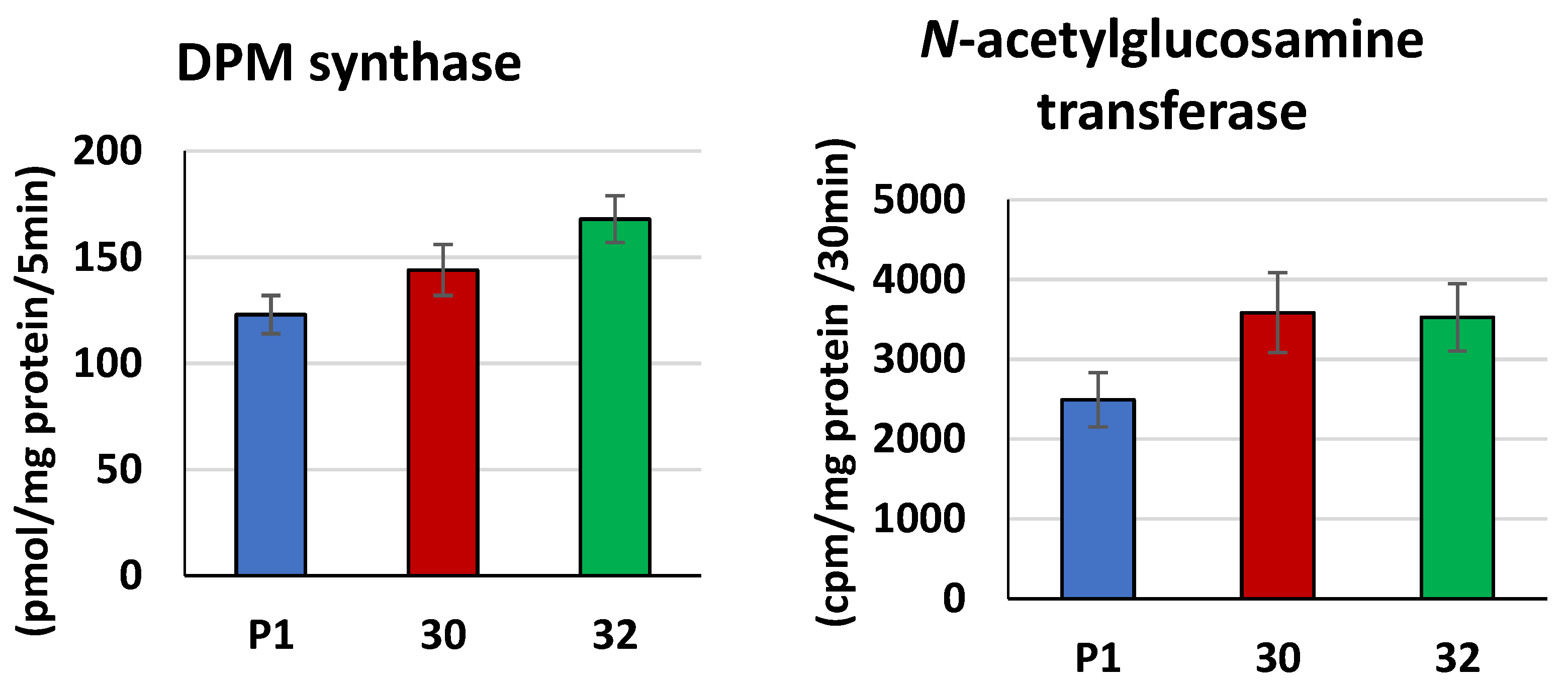
2.9. DPM Synthase (EC 2.4.1.83) Activity
2.10. N-acetylglucosamine Transferase (EC 2.7.8.15) Activity
2.11. FPP Synthase (EC2.5.1.10) Activity
2.12. Squalene Synthase (EC 2.5.1.21) Activity
2.13. Protein Concentration Assay
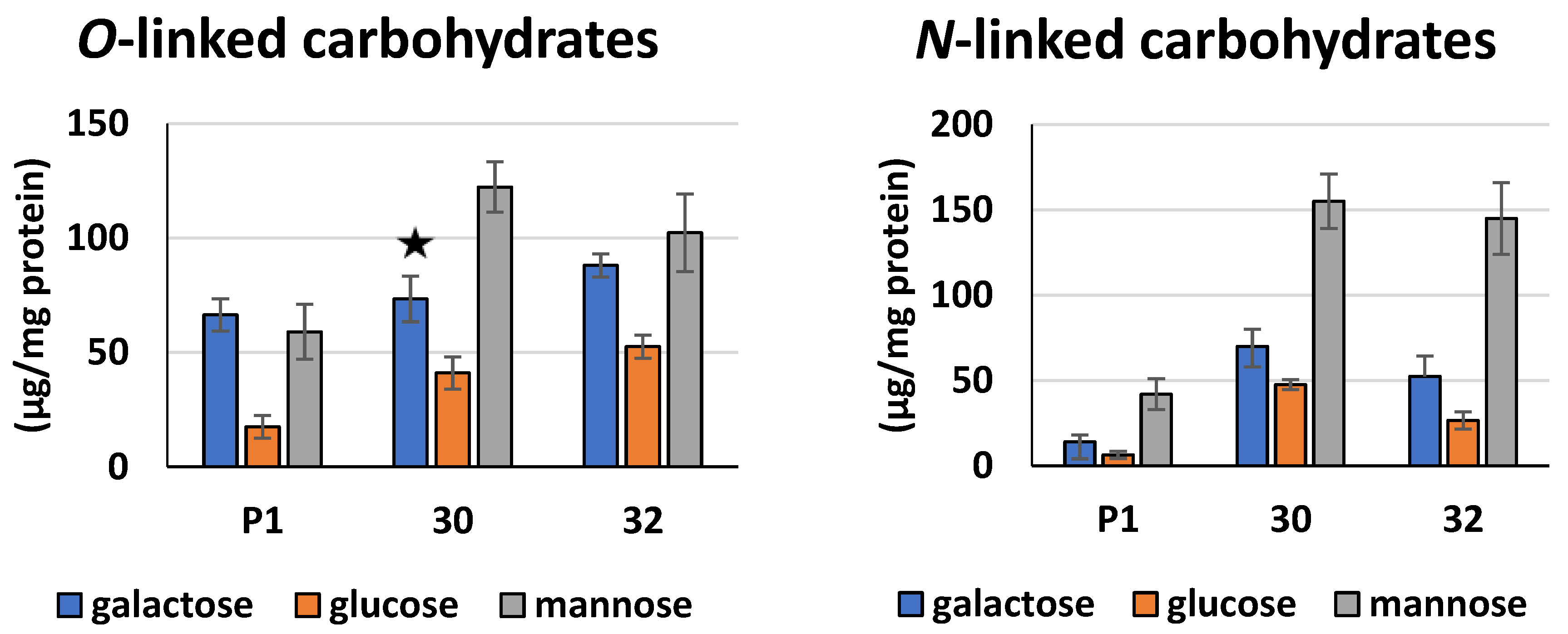
2.14. Identification and Quantification of Saccharides Bound to Secreted Proteins
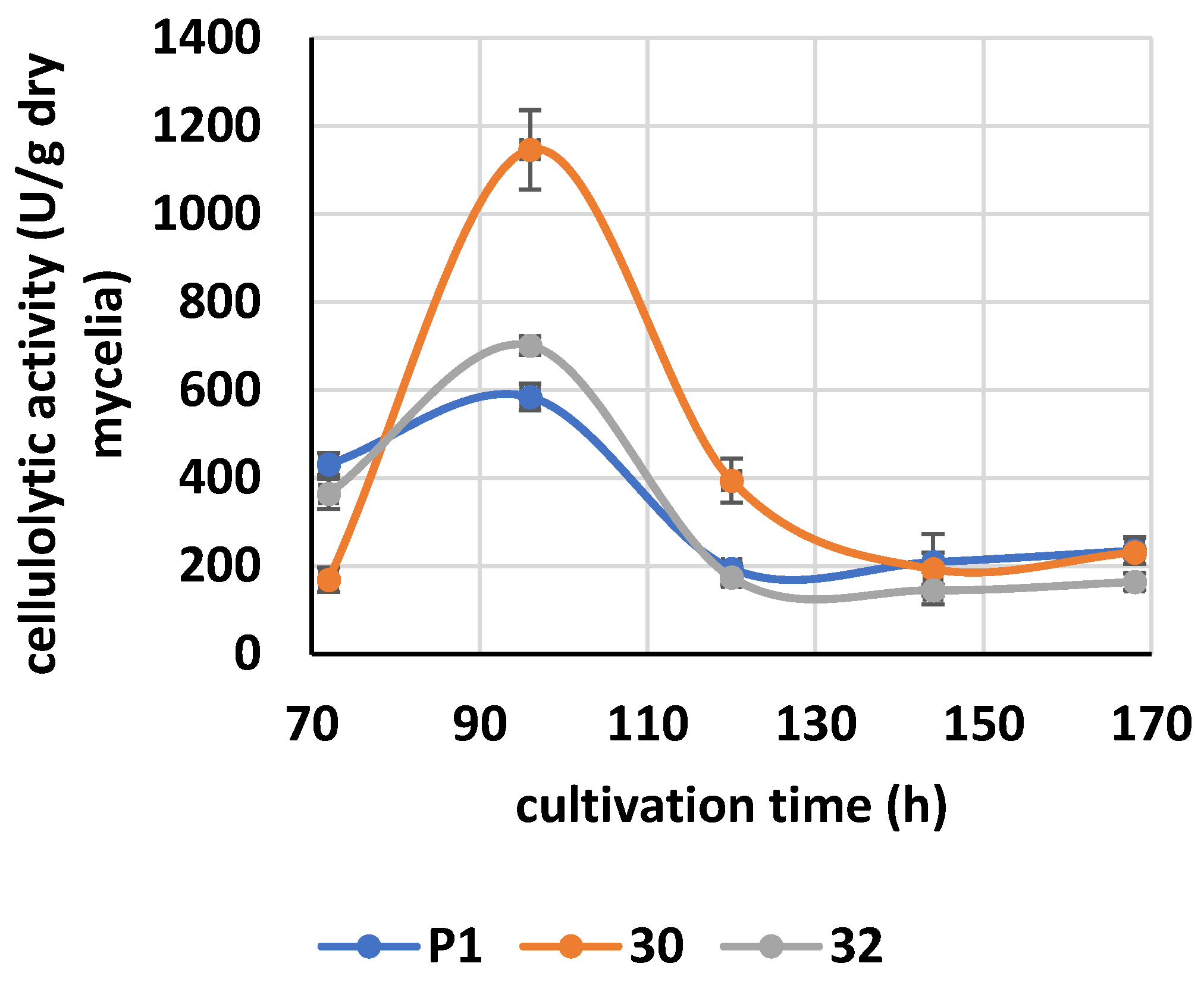
2.15. Cellulase Activity
2.16. Extraction and Purification of Polyisoprenoids
2.17. Squalene Determination
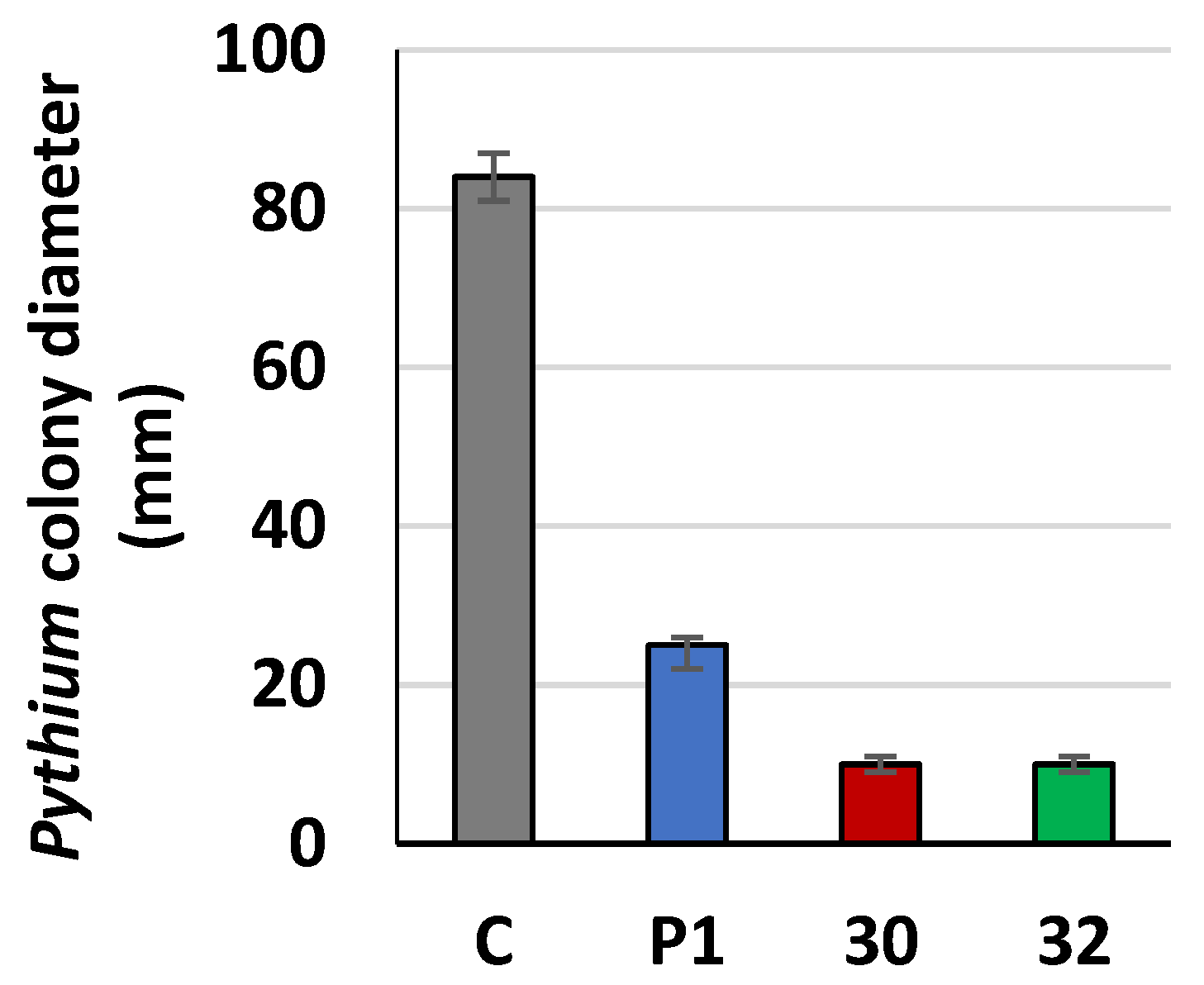
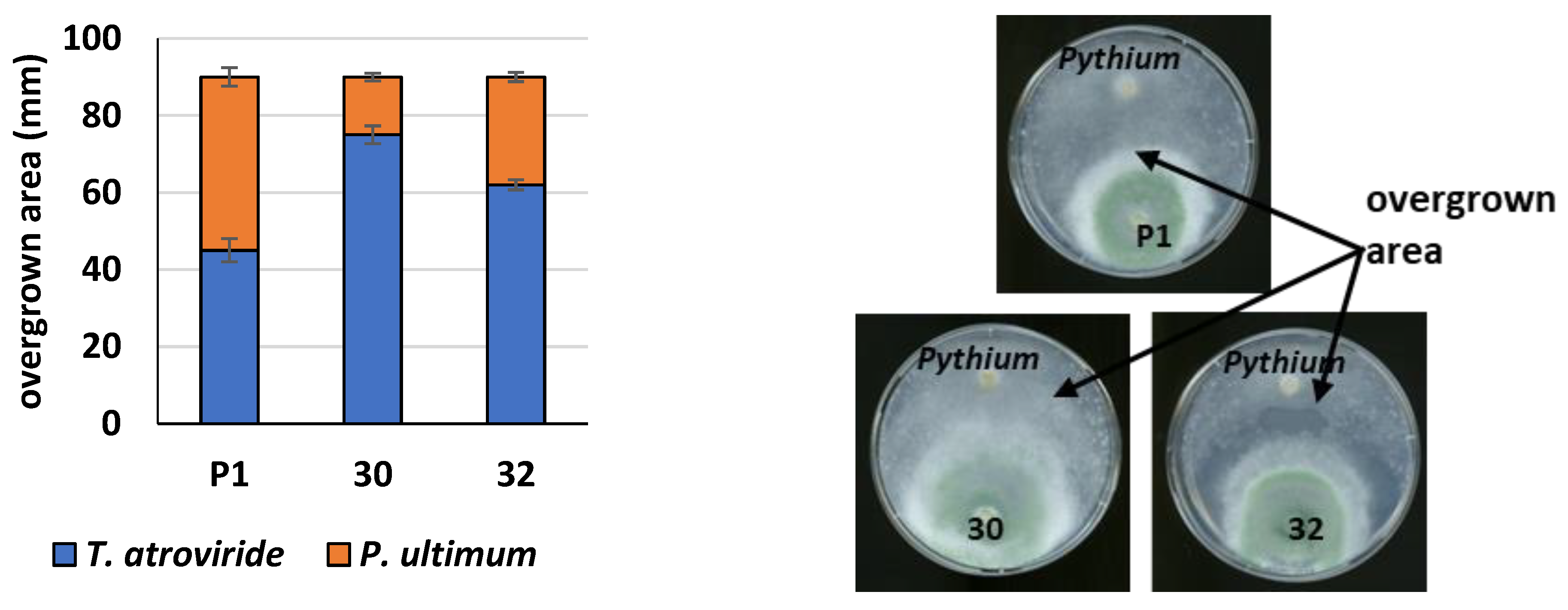
2.18. Plate Confrontation Assay
2.19. Growth Inhibition of Fungal Pathogen by Agents Secreted by Trichoderma to Cultivation Medium
2.20. Statistical Analysis
3. Results
3.1. Expression of S. cerevisiae RER2 Gene in T. atroviride
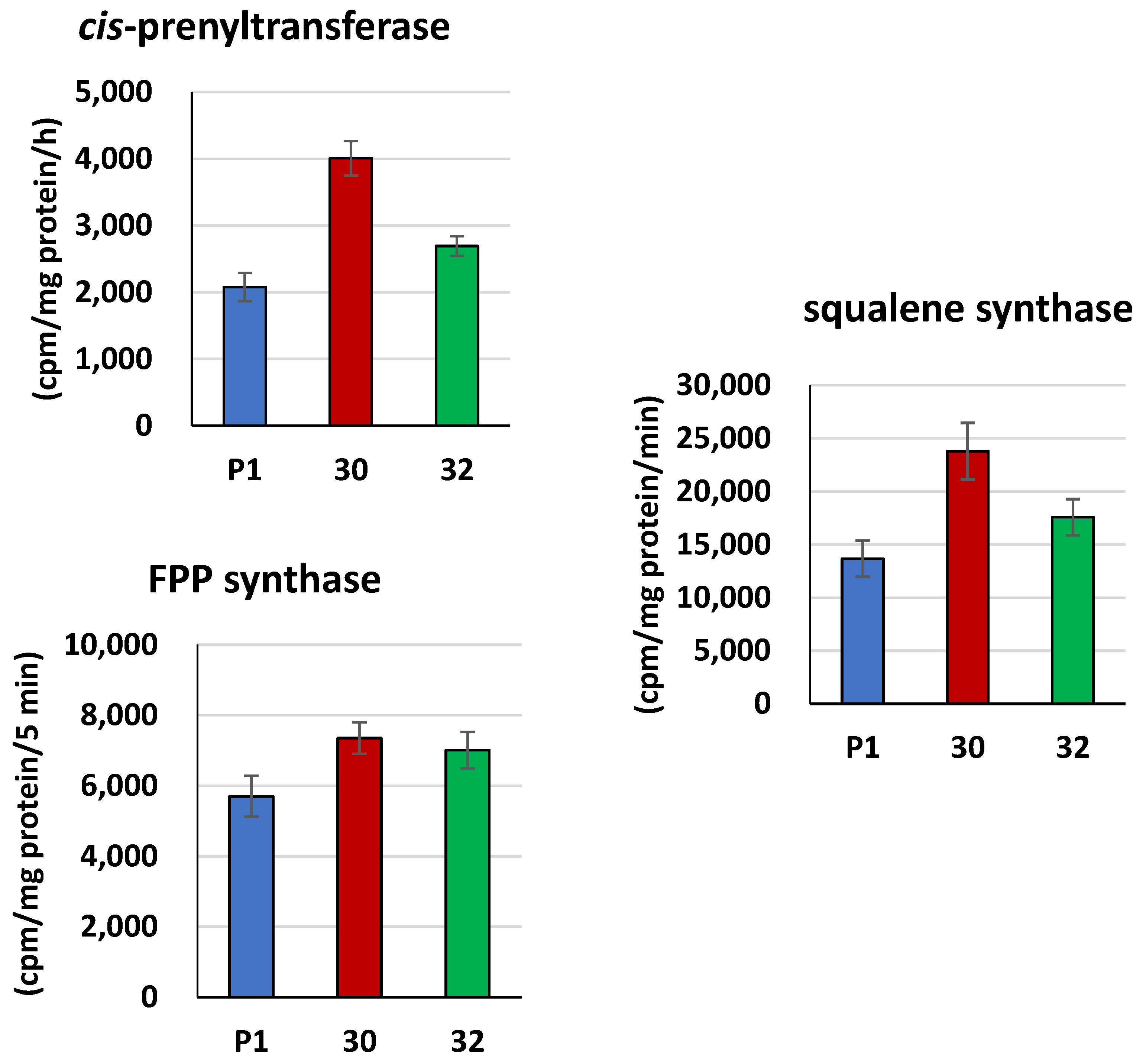
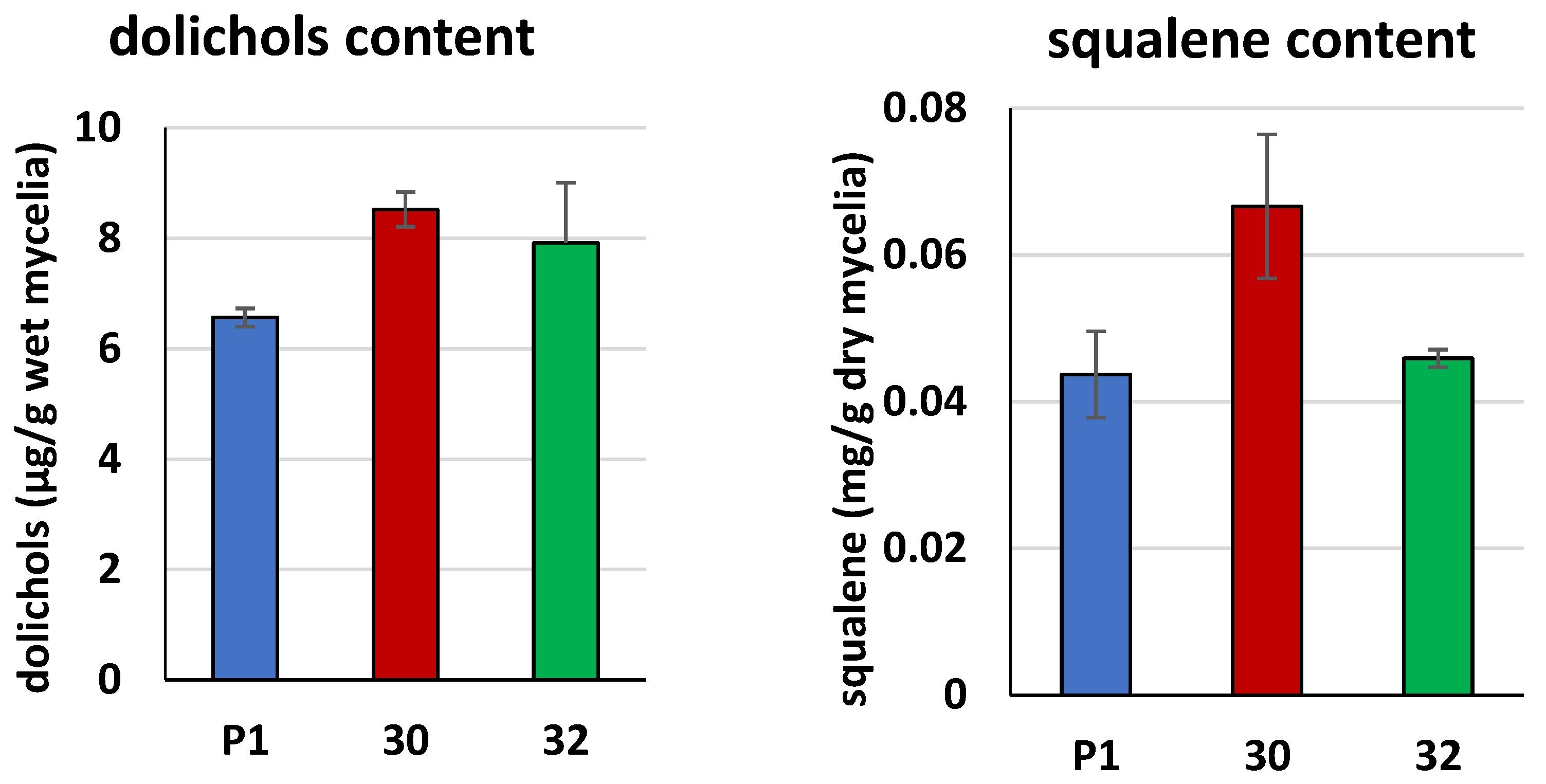
3.2. Biochemical Characterization of T. atroviride RER2 Transformants
3.3. Cellulolytic Activity of RER2-Expressing T. atroviride
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Benitez, T.; Rincon, A.M.; Limon, M.C.; Codon, A.C. Biocontrol mechanisms of Trichoderma strains. Internat. Microbiol. 2004, 7, 249–260. [Google Scholar]
- Atanasova, L.; Le Crom, S.; Gruber, S.; Coulpier, F.; Seidl-Seiboth, V.; Kubicek, C.P.; Druzhinina, I.S. Comparative transcriptomics reveals different strategies of Trichoderma mycoparasitism. BMC Genom. 2013, 14, 121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palamarczyk, G.; Maras, M.; Contreras, R.; Kruszewska, J. Protein secretion and glycosylation in Trichoderma. In Trichoderma and Glocladium; Kubicek, C.P., Harman, G.E., Eds.; Taylor and Francis Ltd.: London, UK, 1998; Volume 1, pp. 121–138. [Google Scholar]
- Hui, J.P.; White, T.C.; Thibault, P. Identification of glycan structure and glycosylation sites in cellobiohydrolase II and endoglucanases I and II from Trichoderma reesei. Glycobiology 2002, 12, 837–849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parodi, A.J. Protein glucosylation and its role in protein folding. Annu. Rev. Biochem. 2000, 69, 69–93. [Google Scholar] [CrossRef] [PubMed]
- Williamson, G.; Belshaw, N.J.; Williamson, M.P. O-glycosylation in Aspergillus glucoamylase. Conformation and role in binding. Biochem. J. 1992, 282, 423–428. [Google Scholar] [CrossRef] [Green Version]
- Guillemette, T.; van Peij, N.N.M.E.; Goosen, T.; Lanthaler, K.; Robson, G.D.; van den Hondel, C.A.M.J.J.; Stam, H.; Archer, D.B. Genomic analysis of the secretion stress response in the enzyme-producing cell factory Aspergillus niger. BMC Genom. 2007, 8, 158. [Google Scholar] [CrossRef] [Green Version]
- Pakula, T.M.; Laxell, M.; Huuskonen, A.; Usitalo, J.; Saloheimo, M.; Penttila, M. The effects of drugs inhibiting protein secretion in the filamentous fungus Trichoderma reesei. Evidence for down-regulation of genes that encode secreted proteins in the stressed cells. J. Biol. Chem. 2003, 278, 45011–45020. [Google Scholar] [CrossRef] [Green Version]
- Eneyskaya, E.V.; Kulminskaya, A.A.; Savelev, A.N.; Shabalin, K.A.; Golubev, A.M.; Neustroev, K.N. α-Mannosidase from Trichoderma reesei participates in the post-secretory deglycosylation of glycoproteins. Biochem. Biophys. Res. Comm. 1998, 245, 43–49. [Google Scholar] [CrossRef]
- Harrison, M.J.; Wathugala, I.M.; Tenkanen, M.; Packer, N.H.; Nevalainen, K.M.H. Glycosylation of acetylxylan esterase from Trichoderma reesei. Glycobiology 2002, 12, 291–298. [Google Scholar] [CrossRef] [Green Version]
- Xu, C.; Ng, D.T.W. Glycosylation-directed quality control of protein folding. Nat. Rev. Mol. Cell Biol. 2015, 16, 742–752. [Google Scholar] [CrossRef]
- Kruszewska, J.; Butterweck, A.H.; Kurzątkowski, W.; Migdalski, A.; Kubicek, C.P.; Palamarczyk, G. Overexpression of the Saccharomyces cerevisiae mannosylphosphodolichol synthase—Encoding gene in Trichoderma reesei results in an increased level of protein secretion and abnormal cell ultrastructure. Appl. Environm. Microbiol. 1999, 65, 2382–2387. [Google Scholar] [CrossRef] [PubMed]
- Perlińska-Lenart, U.; Orłowski, J.; Laudy, A.E.; Zdebska, E.; Palamarczyk, G.; Kruszewska, J.S. Glycoprotein hypersecretion alters the cell wall in Trichoderma reesei strains expressing the Saccharomyces cerevisiae dolichylphosphate mannose synthase gene. Appl. Environm. Microbiol. 2006, 72, 7778–7784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zakrzewska, A.; Palamarczyk, G.; Krotkiewski, H.; Zdebska, E.; Saloheimo, M.; Penttilä, M.; Kruszewska, J.S. Overexpression of the gene encoding GTP-mannose-1-phosphate guanyltransferase, mpg1, increases cellular GDP-mannose levels and protein mannosylation in Trichoderma reesei. Appl. Environm. Microbiol. 2003, 69, 4383–4389. [Google Scholar] [CrossRef] [Green Version]
- Daleo, G.R.; Hopp, H.E.; Romero, P.A.; Lezica, R. Biosynthesis of dolichol phosphate by subcellular fractions from liver. FEBS Lett. 1977, 81, 411–414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adair, W.L.J.; Cafmeyer, N. Characterization of the Saccharomyces cerevisiae cis-prenyltransferase required for dolichyl phosphate biosynthesis. Arch. Biochem. Biophys. 1987, 259, 589–596. [Google Scholar] [CrossRef]
- Yanish-Perron, C.; Vieira, J.; Messing, J. Improved M13 phage cloning vectors and host strains: Nucleotide sequences of the M13mp18 and pUC 19 vectors. Gene 1985, 33, 103–119. [Google Scholar] [CrossRef] [PubMed]
- Perlińska-Lenart, U.; Bańkowska, R.; Palamarczyk, G.; Kruszewska, J.S. Overexpression of the Saccharomyces cerevisiae RER2 gene in Trichoderma reesei affects dolichol dependent enzymes and protein glycosylation. Fungal Genet. Biol. 2006, 43, 422–429. [Google Scholar] [CrossRef]
- Mach, R.L.; Schindler, M.; Kubicek, C.P. Transformation of Trichoderma reesei based on hygromycin B resistance using homologous expression signals. Curr. Genet. 1994, 25, 567–570. [Google Scholar] [CrossRef]
- Chomczynski, P.; Sacchi, N. Single-step method of RNA isolation by acid guanidinum thiocyanate-phenol-chloroform extraction. Anal. Biochem. 1987, 162, 156–159. [Google Scholar] [CrossRef]
- Sambrook, J.; Fritsch, E.F.; Maniatis, T. Molecular Cloning: A Laboratory Manual, 2nd ed.; Cold Spring Harbor Laboratory: Cold Spring Harbor, NY, USA, 1989; Volume 1, pp. 7.37–7.52. [Google Scholar]
- Joshi, M.; Pittman, K.H.; Haisch, C.; Verbanac, K. Real-time PCR to determine transgene copy number and to quantitate the biolocalization of adoptively transferred cells from EGFP-transgenic mice. Biotechniques 2008, 45, 247–258. [Google Scholar] [CrossRef] [Green Version]
- Ma, L.; Chung, W.K. Quantitative analysis of copy number variants based on Real-Time LightCycler PCR. Curr. Protoc. Hum. Genet. 2015, 80, 7.21.1–7.21.8. [Google Scholar] [CrossRef] [PubMed]
- Salas-Marina, M.; Isordia-Jasso, M.I.; Islas-Osuna, M.A.; Delgado-Sanchez, P.; Jimenez-Bremont, J.F.; Rodroguez-Kessler, M.; Rosales-Saavedra, M.T.; Herrera-Estrella, A.; Casas-Flores, S. The Epl1 and Sm1 proteins from Trichoderma atroviride and Trichoderma virens differentially modulate systemic disease resistance against different life style pathogens in Solanum lycopersicum. Front. Plant Sci. 2015, 6, 77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grabińska, K.; Sosińska, G.; Orłowski, J.; Świeżewska, E.; Berges, T.; Karst, F.; Palamarczyk, G. Functional relationships between the Saccharomyces cerevisiae cis-prenyltransferases required for dolichol biosynthesis. Acta Biochim. Pol. 2005, 52, 221–232. [Google Scholar] [CrossRef] [Green Version]
- Danilov, L.L.; Chojnacki, T. A simple procedure for preparing dolichyl monophosphate by the use of POCl3. FEBS Lett. 1981, 131, 310–312. [Google Scholar] [CrossRef] [Green Version]
- Kruszewska, J.; Messner, R.; Kubicek, C.P.; Palamarczyk, G. O-glycosylation of proteins by membrane fraction of Trichoderma reesei QM9414. J. Gen. Microbiol. 1989, 135, 301–307. [Google Scholar]
- Palamarczyk, G.; Hemming, F.W. The formation of mono-N-acethylhexosamine derivatives of dolichol phosphate by pig liver microsomal fractions. Biochem. J. 1975, 148, 243–251. [Google Scholar] [CrossRef]
- Szkopińska, A.; Grabińska, K.; Delourme, D.; Karst, F.; Rytka, J.; Palamarczyk, G. Polyprenol formation in the yeast Saccharomyces cerevisiae: Effect of farnesyl diphosphate synthase overexpression. J. Lipid Res. 1997, 38, 962–968. [Google Scholar] [CrossRef]
- Karst, F.; Płochocka, D.; Meyer, S.; Szkopińska, A. Farnesyl diphosphate synthase activity affects ergosterol level and proliferation of yeast Saccharomyces cerevisiae. Cell Biol. Int. 2004, 28, 193–197. [Google Scholar] [CrossRef]
- Graczyk, S.; Perlińska-Lenart, U.; Górka-Nieć, W.; Lichota, R.; Piłsyk, S.; Zembek, P.; Lenart, J.; Bernat, P.; Gryz, E.; Augustyniak, J.; et al. Increased activity of the sterol branch of the mevalonate pathway elevates glycosylation of secretory proteins and improves antifungal properties of Trichoderma atroviride. Fungal Genet. Biol. 2020, 137, 103334. [Google Scholar] [CrossRef]
- Shechter, I.; Klinger, E.; Rucker, M.L.; Engstrom, R.G.; Spirito, J.A.; Islam, M.A.; Boettcher, B.R.; Weinstein, D.B. Solubilization, purification and characterization of a truncated form of rat hepatic squalene synthase. J. Biol. Chem. 1992, 267, 8628–8635. [Google Scholar] [CrossRef]
- Lowry, O.H.; Rosebrough, N.J.; Farr, A.L.; Randall, R.J. Protein measurement with the Folin phenol reagent. J. Biol. Chem. 1951, 193, 265–275. [Google Scholar] [CrossRef] [PubMed]
- Dubois, M.; Gilles, K.A.; Hamilton, J.K.; Robers, P.A.; Smith, F. Colorimetric method for determination of sugar and related substrates. Anal. Biochem. 1956, 28, 350–356. [Google Scholar]
- Ma, J.; Stoter, G.; Verweij, J.; Shellens, J.H. Comparison of ethanol plasma-protein precipitation with plasma ultrafiltration and trichloroacetic acid protein precipitation for the measurement of unbound platinum concentrations. Cancer Chemother. Pharmacol. 1996, 38, 391–394. [Google Scholar] [CrossRef] [PubMed]
- Duk, M.; Ugorski, M.; Lisowska, E. β-elimination of O-glycans from glycoproteins transferred to Immobilon P membranes: Method and some applications. Anal. Biochem. 1997, 253, 98–102. [Google Scholar] [CrossRef] [PubMed]
- Zdebska, E.; Kościelak, J. A single-sample method for determination of carbohydrate and protein contents in glycoprotein bands separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis. Anal. Biochem. 1999, 275, 171–179. [Google Scholar] [CrossRef]
- Bernfeld, P. Amylases, α and β. Methods Enzymol. 1955, 1, 149–158. [Google Scholar]
- Gawarecka, K.; Swiezewska, E. Analysis of plant polyisoprenoids. Methods Mol. Biol. 2014, 1153, 135–147. [Google Scholar]
- Bernat, P.; Gajewska, E.; Szewczyk, R.; Słaba, M.; Długoński, J. Tributyltin (TBT) induces oxidative stress and modifies lipid profile in the filamentous fungus Cunninghamella elegans. Environ. Sci. Pollut. Res. Int. 2014, 214, 228–4235. [Google Scholar] [CrossRef] [Green Version]
- Sato, M.; Sato, K.; Nishikawa, S.; Hirata, A.; Kato, J.; Nakano, A. The yeast RER2 gene, identified by endoplasmic reticulum protein localization mutations, encodes cis-prenyltransferase, a key enzyme in dolichol synthesis. Mol. Cell. Biol. 1999, 19, 471–483. [Google Scholar] [CrossRef] [Green Version]
- Montero, M.; Sanz, L.; Rey, M.; Llobell, A.; Monte, E. Cloning and characterization of bgn16·3, coding for a β-1,6-glucanase expressed during Trichoderma harzianum mycoparasitism. J. Appl. Microbiol. 2007, 103, 1291–1300. [Google Scholar] [CrossRef]
- Park, E.J.; Grabińska, K.A.; Guan, Z.; Stranecky, V.; Hartmannova, H.; Hodanova, K.; Beresova, V.; Sovova, J.; Jozsef, L.; Ondruskova, N.; et al. Mutation of Nogo-B receptor, a subunit of cis-prenyltransferase, causes a congenital disorder of glycosylation. Cell Metab. 2014, 20, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kubicek, C.P.; Panda, T.; Schreferl-Kunar, G.; Messner, R.; Gruber, F. O-linked—But not N-linked—Glycosylation is necessary for secretion of endoglucanase I and II by Trichoderma reesei. Can. J. Microbiol. 1987, 33, 698–703. [Google Scholar] [CrossRef]
- Harrison, M.J.; Nouwens, A.S.; Jardine, D.R.; Zachara, N.E.; Gooley, A.A.; Nevalainen, H.; Packer, N.H. Modified glycosylation of cellobiohydrolase I from a high cellulase-producing mutant strain of Trichoderma reesei. Eur. J. Biochem. 1998, 256, 119–127. [Google Scholar] [CrossRef] [PubMed]
- Latijnhouwers, M.; de Wit, P.J.G.M.; Govers, F. Oomycetes and fungi: Similar weaponry to attack plants. Trends Microbiol. 2003, 11, 462–469. [Google Scholar] [CrossRef] [PubMed]
- Zembek, P.; Perlińska-Lenart, U.; Brunner, K.; Reithner, B.; Palamarczyk, G.; Mach, R.L.; Kruszewska, J.S. Elevated activity of dolichyl phosphate mannose synthase enhances biocontrol abilities of Trichoderma atroviride. Mol. Plant-Microbe Interact. 2011, 24, 1522–1529. [Google Scholar] [CrossRef]
- Baek, J.M.; Howell, C.R.; Kenerley, C.M. The role of an extracellular chitinase from Trichoderma virens Gv29-8 in the biocontrol of Rhizoctonia solani. Curr. Genet. 1999, 35, 41–50. [Google Scholar] [CrossRef]
- Djonovic, S.; Pozo, M.J.; Kenerley, C.M. Tvbgn3, a β-1,6-glucanase from the biocontrol fungus Trichoderma virens, is involved in mycoparasitism and control of Pythum ultimum. Appl. Environm. Microbiol. 2006, 72, 7661–7670. [Google Scholar] [CrossRef]









Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Perlińska-Lenart, U.; Graczyk, S.; Piłsyk, S.; Lenart, J.; Lipko, A.; Swiezewska, E.; Bernat, P.; Kruszewska, J.S. Expression of Saccharomyces cerevisiae RER2 Gene Encoding Cis-Prenyltransferase in Trichoderma atroviride Increases the Activity of Secretory Hydrolases and Enhances Antimicrobial Features. J. Fungi 2023, 9, 38. https://doi.org/10.3390/jof9010038
Perlińska-Lenart U, Graczyk S, Piłsyk S, Lenart J, Lipko A, Swiezewska E, Bernat P, Kruszewska JS. Expression of Saccharomyces cerevisiae RER2 Gene Encoding Cis-Prenyltransferase in Trichoderma atroviride Increases the Activity of Secretory Hydrolases and Enhances Antimicrobial Features. Journal of Fungi. 2023; 9(1):38. https://doi.org/10.3390/jof9010038
Chicago/Turabian StylePerlińska-Lenart, Urszula, Sebastian Graczyk, Sebastian Piłsyk, Jacek Lenart, Agata Lipko, Ewa Swiezewska, Przemysław Bernat, and Joanna S. Kruszewska. 2023. "Expression of Saccharomyces cerevisiae RER2 Gene Encoding Cis-Prenyltransferase in Trichoderma atroviride Increases the Activity of Secretory Hydrolases and Enhances Antimicrobial Features" Journal of Fungi 9, no. 1: 38. https://doi.org/10.3390/jof9010038