
Endothelin-1 as a Mediator of Heme Oxygenase-1-Induced Stemness in Colorectal Cancer: Influence of p53

 , , , ,
, , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture and Reagents
2.2. Patients
2.3. Transient Transfection of HO-1
2.4. Western Blotting
2.5. ET-1 ELISA
2.6. RNA Isolation and cDNA Synthesis
2.7. Real Time PCR
2.8. DNA Extraction and p53 Mutations Analysis
2.9. ALDEFLUOR Assay
2.10. Isolation and Characterization of CSCs
2.11. Sphere Forming Assay
2.12. MTT Assay
2.13. Statistical Analysis
3. Results
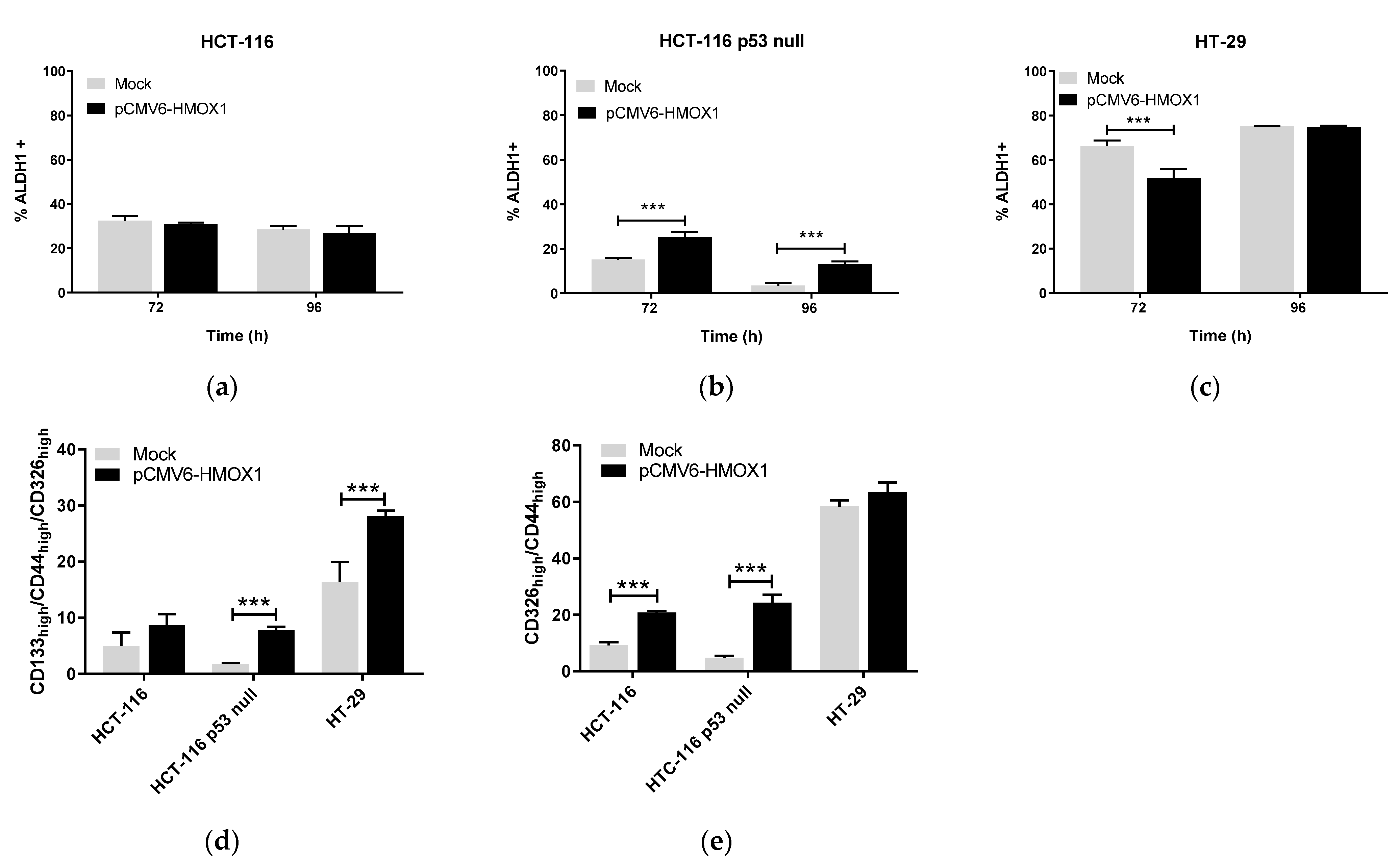
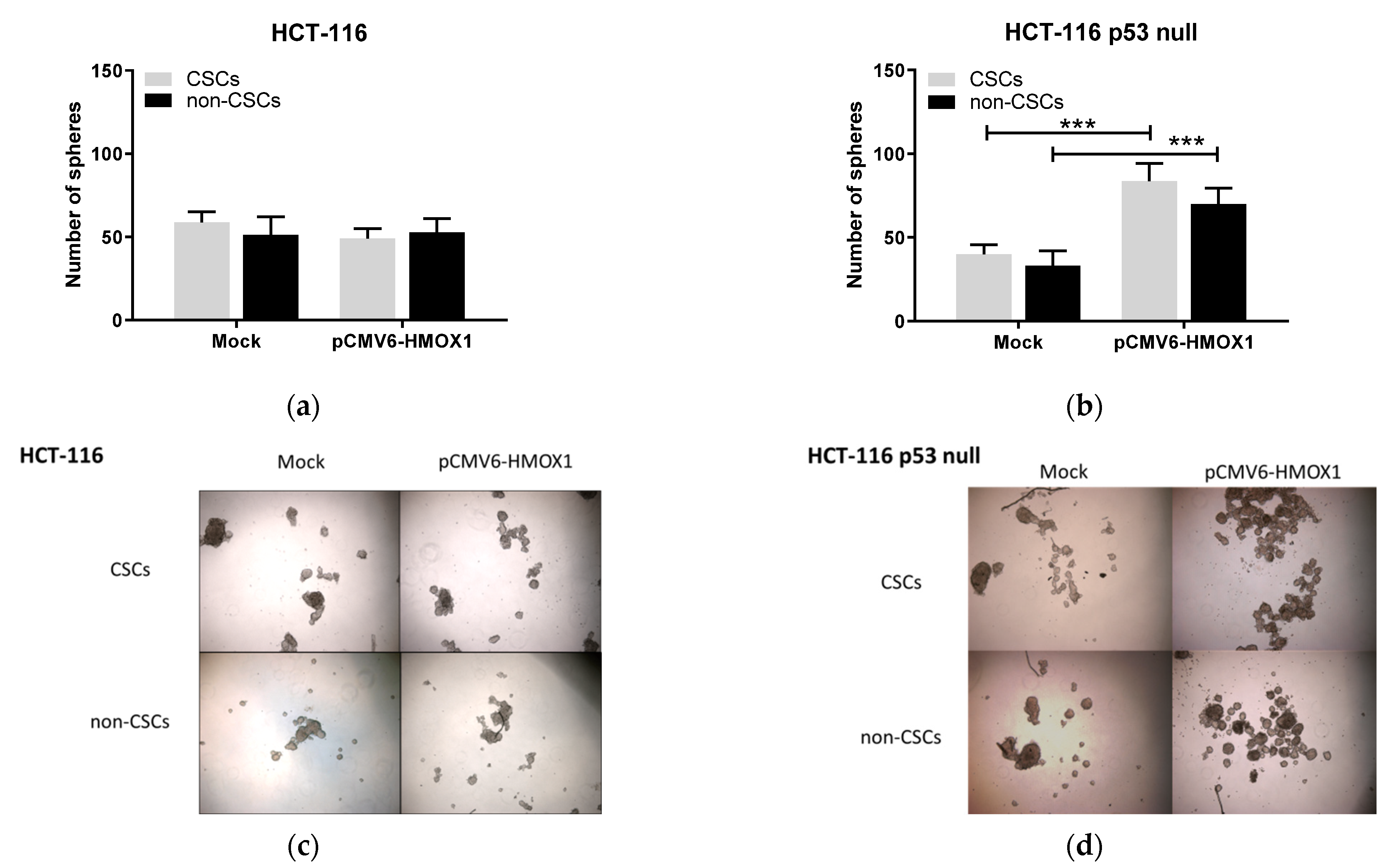
3.1. HO-1 Overexpression Induces Stemness in CRC In Vitro Regardless of p53 Status
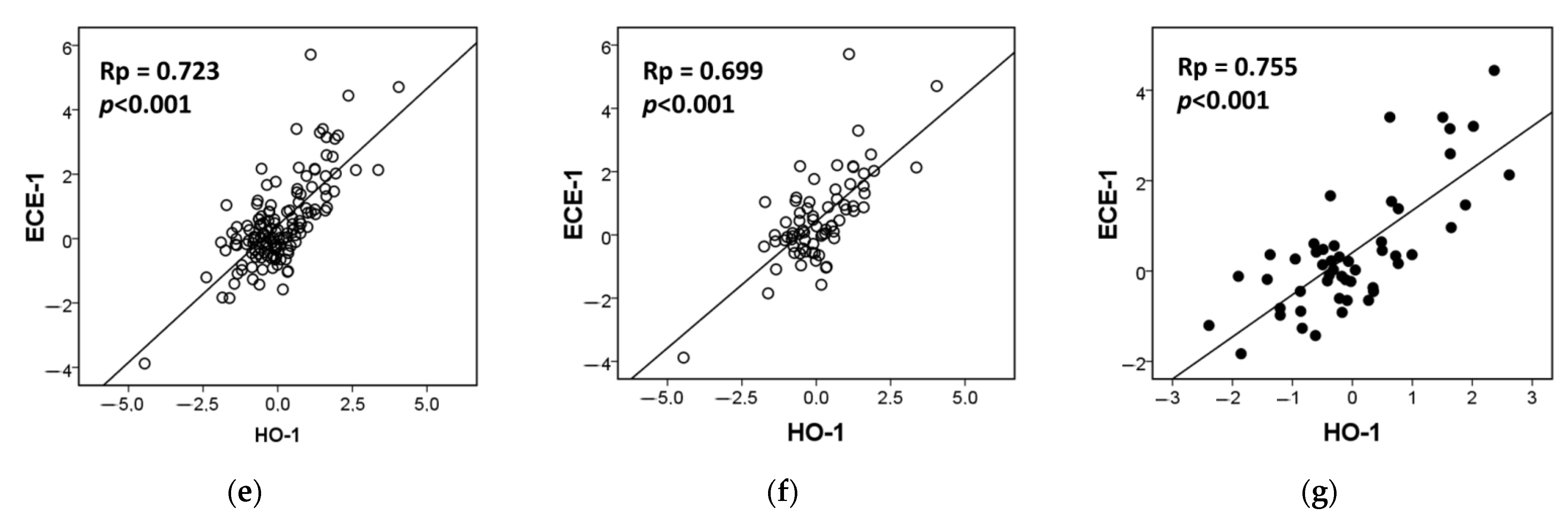
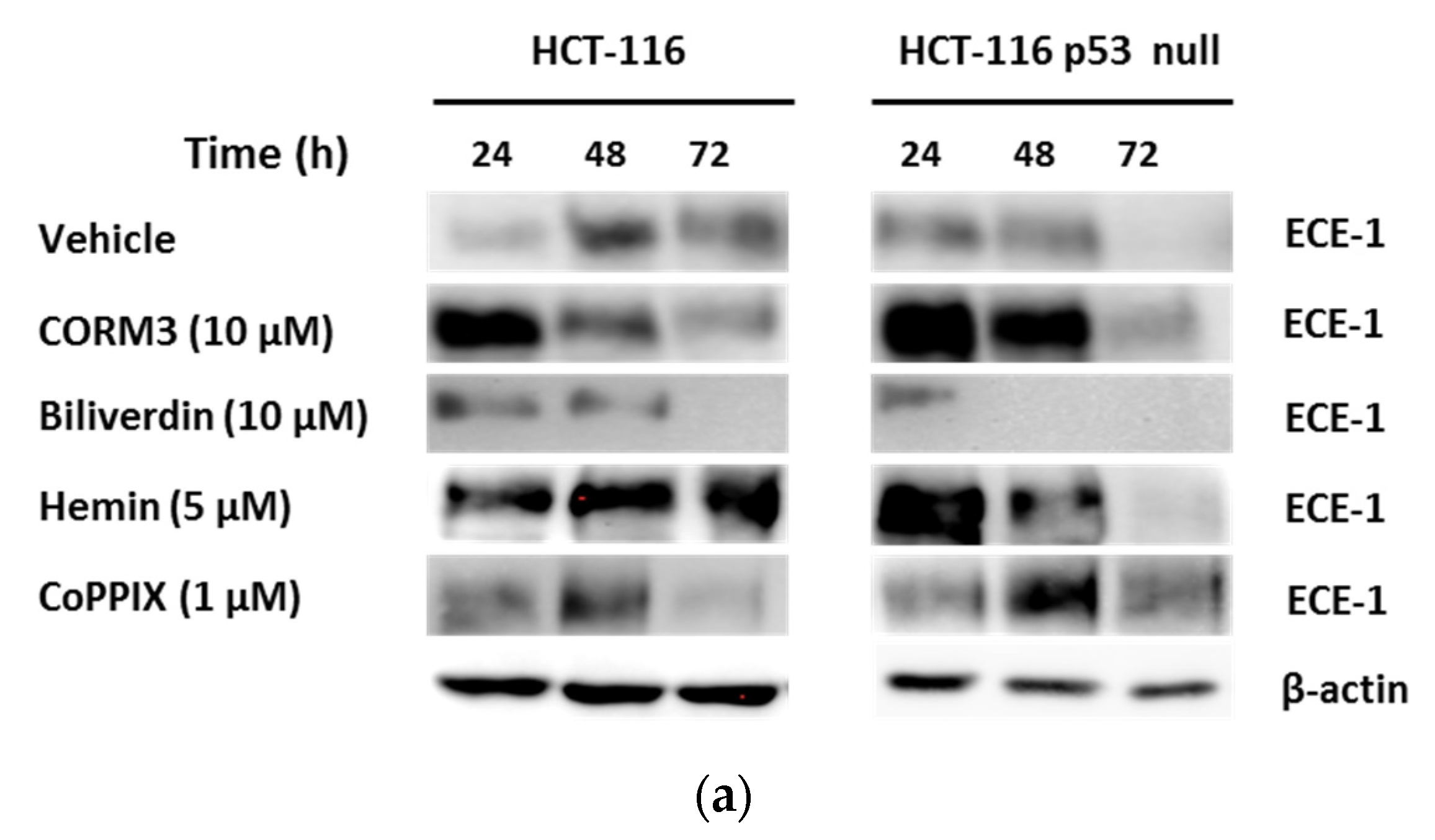
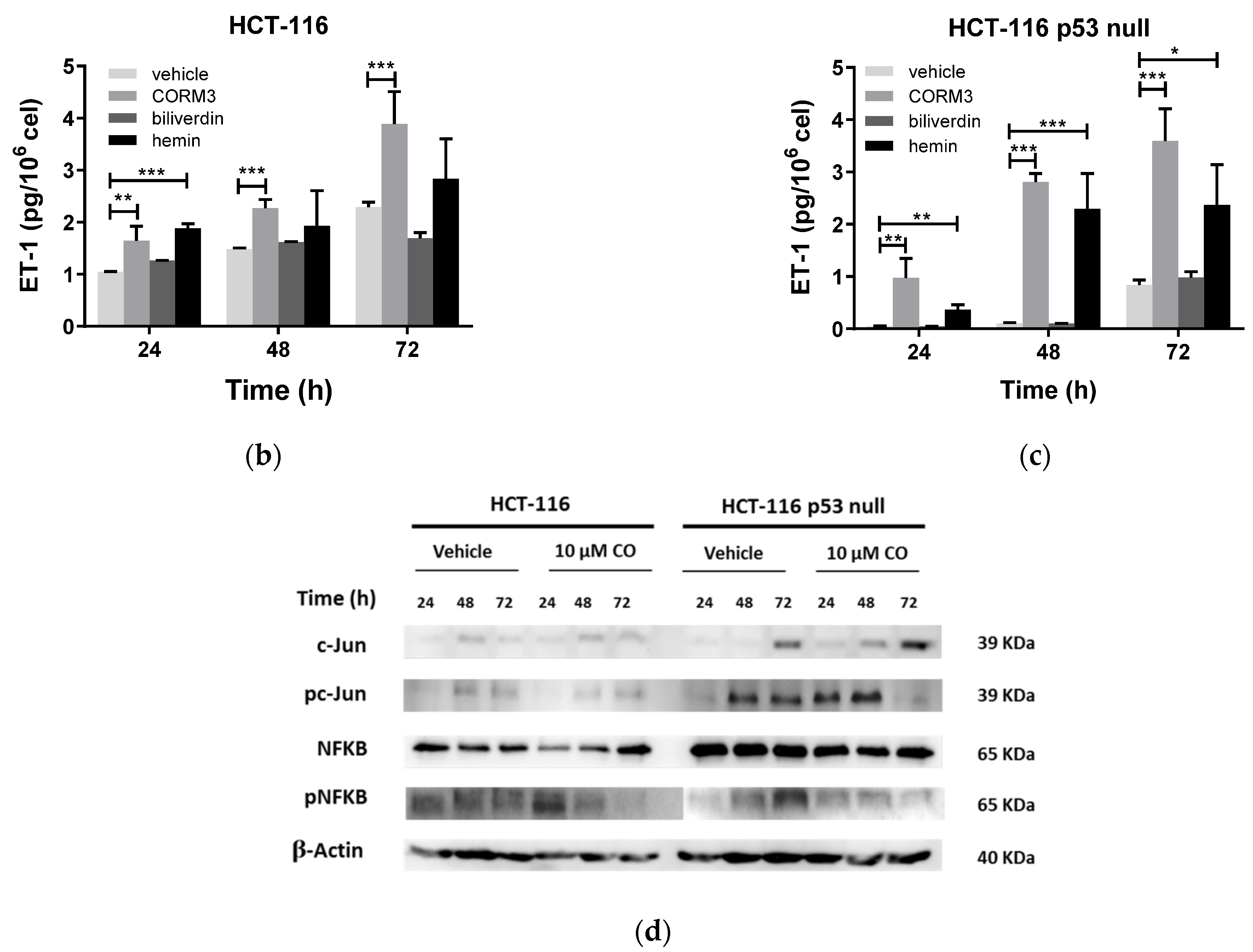
3.2. HO-1 Overexpression Induces ECE-1 Expression and ET-1 Production by CRC Cells
3.3. HO-1 Overexpression Induces Stemness in CRC Cell Lines through ECE-1/ET-1 Only in p53 Wild-Type Cells
3.4. HO-1 Overexpression Induces Resistance to 5-FU Treatment in CRC In Vitro
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [Green Version]
- Van der Jeught, K.; Xu, H.C.; Li, Y.J.; Lu, X.B.; Ji, G. Drug resistance and new therapies in colorectal cancer. World J. Gastroenterol. 2018, 24, 3834–3848. [Google Scholar] [CrossRef]
- Zeuner, A.; Todaro, M.; Stassi, G.; De Maria, R. Colorectal cancer stem cells: From the crypt to the clinic. Cell Stem Cell 2014, 15, 692–705. [Google Scholar] [CrossRef] [Green Version]
- Jozkowicz, A.; Was, H.; Dulak, J. Heme oxygenase-1 in tumors: Is it a false friend? Antioxid. Redox Signal. 2007, 9, 2099–2117. [Google Scholar] [CrossRef] [Green Version]
- Loboda, A.; Damulewicz, M.; Pyza, E.; Jozkowicz, A.; Dulak, J. Role of Nrf2/HO-1 system in development, oxidative stress response and diseases: An evolutionarily conserved mechanism. Cell. Mol. Life Sci. 2016, 73, 3221–3247. [Google Scholar] [CrossRef] [Green Version]
- Vijayan, V.; Wagener, F.; Immenschuh, S. The macrophage heme-heme oxygenase-1 system and its role in inflammation. Biochem. Pharmacol. 2018, 153, 159–167. [Google Scholar] [CrossRef]
- Rochette, L.; Zeller, M.; Cottin, Y.; Vergely, C. Redox Functions of Heme Oxygenase-1 and Biliverdin Reductase in Diabetes. Trends Endocrinol. Metab. 2018, 29, 74–85. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.T.; Lin, Z.M.; He, S.J.; Zuo, J.P. Heme oxygenase-1 as a potential therapeutic target in rheumatic diseases. Life Sci. 2018. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.M.; Lin, C.C.; Hsieh, H.L. High-Glucose-Derived Oxidative Stress-Dependent Heme Oxygenase-1 Expression from Astrocytes Contributes to the Neuronal Apoptosis. Mol. Neurobiol. 2017, 54, 470–483. [Google Scholar] [CrossRef]
- Nitti, M.; Piras, S.; Marinari, U.M.; Moretta, L.; Pronzato, M.A.; Furfaro, A.L. HO-1 Induction in Cancer Progression: A Matter of Cell Adaptation. Antioxidants. 2017, 6, 29. [Google Scholar] [CrossRef] [PubMed]
- Chiang, S.K.; Chen, S.E.; Chang, L.C. A Dual Role of Heme Oxygenase-1 in Cancer Cells. Int. J. Mol. Sci. 2018, 20, 39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biswas, C.; Shah, N.; Muthu, M.; La, P.; Fernando, A.P.; Sengupta, S.; Yang, G.; Dennery, P.A. Nuclear heme oxygenase-1 (HO-1) modulates subcellular distribution and activation of Nrf2, impacting metabolic and anti-oxidant defenses. J. Biol. Chem. 2014, 289, 26882–26894. [Google Scholar] [CrossRef] [Green Version]
- Lin, P.L.; Chang, J.T.; Wu, D.W.; Huang, C.C.; Lee, H. Cytoplasmic localization of Nrf2 promotes colorectal cancer with more aggressive tumors via upregulation of PSMD4. Free Radic. Biol. Med. 2016, 95, 121–132. [Google Scholar] [CrossRef] [PubMed]
- Chang, L.C.; Chiang, S.K.; Chen, S.E.; Yu, Y.L.; Chou, R.H.; Chang, W.C. Heme oxygenase-1 mediates BAY 11-7085 induced ferroptosis. Cancer Lett. 2018, 416, 124–137. [Google Scholar] [CrossRef]
- Becker, J.C.; Fukui, H.; Imai, Y.; Sekikawa, A.; Kimura, T.; Yamagishi, H.; Yoshitake, N.; Pohle, T.; Domschke, W.; Fujimori, T. Colonic expression of heme oxygenase-1 is associated with a better long-term survival in patients with colorectal cancer. Scand. J. Gastroenterol. 2007, 42, 852–858. [Google Scholar] [CrossRef]
- Andrés, N.C.; Fermento, M.E.; Gandini, N.A.; Romero, A.L.; Ferro, A.; Donna, L.G.; Curino, A.C.; Facchinetti, M.M. Heme oxygenase-1 has antitumoral effects in colorectal cancer: Involvement of p53. Exp. Mol. Pathol. 2014, 97, 321–331. [Google Scholar] [CrossRef]
- Lien, G.S.; Wu, M.S.; Bien, M.Y.; Chen, C.H.; Lin, C.H.; Chen, B.C. Epidermal growth factor stimulates nuclear factor-kappaB activation and heme oxygenase-1 expression via c-Src, NADPH oxidase, PI3K, and Akt in human colon cancer cells. PLoS ONE 2014, 9, e104891. [Google Scholar] [CrossRef] [Green Version]
- Seo, G.S.; Jiang, W.Y.; Chi, J.H.; Jin, H.; Park, W.C.; Sohn, D.H.; Park, P.H.; Lee, S.H. Heme oxygenase-1 promotes tumor progression and metastasis of colorectal carcinoma cells by inhibiting antitumor immunity. Oncotarget 2015, 6, 19792–19806. [Google Scholar] [CrossRef] [Green Version]
- Hellmuth, M.; Wetzler, C.; Nold, M.; Chang, J.H.; Frank, S.; Pfeilschifter, J.; Muhl, H. Expression of interleukin-8, heme oxygenase-1 and vascular endothelial growth factor in DLD-1 colon carcinoma cells exposed to pyrrolidine dithiocarbamate. Carcinogenesis 2002, 23, 1273–1279. [Google Scholar] [CrossRef] [Green Version]
- Kim, T.H.; Hur, E.G.; Kang, S.J.; Kim, J.A.; Thapa, D.; Lee, Y.M.; Ku, S.K.; Jung, Y.; Kwak, M.K. NRF2 blockade suppresses colon tumor angiogenesis by inhibiting hypoxia-induced activation of HIF-1alpha. Cancer Res. 2011, 71, 2260–2275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waghela, B.N.; Vaidya, F.U.; Pathak, C. Upregulation of NOX-2 and Nrf-2 Promotes 5-Fluorouracil Resistance of Human Colon Carcinoma (HCT-116) Cells. Biochemistry 2021, 86, 262–274. [Google Scholar] [PubMed]
- Yin, H.; Fang, J.; Liao, L.; Maeda, H.; Su, Q. Upregulation of heme oxygenase-1 in colorectal cancer patients with increased circulation carbon monoxide levels, potentially affects chemotherapeutic sensitivity. BMC Cancer 2014, 14, 1471–2407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shibue, T.; Weinberg, R.A. EMT, CSCs, and drug resistance: The mechanistic link and clinical implications. Nat. Rev. Clin. Oncol. 2017, 14, 611–629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghosh, D.; Ulasov, I.V.; Chen, L.; Harkins, L.E.; Wallenborg, K.; Hothi, P.; Rostad, S.; Hood, L.; Cobbs, C.S. TGFbeta-Responsive HMOX1 Expression Is Associated with Stemness and Invasion in Glioblastoma Multiforme. Stem Cells 2016, 34, 2276–2289. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.H.; Yoon, H.J.; Cha, Y.N.; Surh, Y.J. Role of heme oxygenase-1 and its reaction product, carbon monoxide, in manifestation of breast cancer stem cell-like properties: Notch-1 as a putative target. Free Radic. Res. 2018, 52, 1336–1347. [Google Scholar] [CrossRef]
- Jang, J.E.; Eom, J.I.; Jeung, H.K.; Chung, H.; Kim, Y.R.; Kim, J.S.; Cheong, J.W.; Min, Y.H. PERK/NRF2 and autophagy form a resistance mechanism against G9a inhibition in leukemia stem cells. J. Exp. Clinical Cancer Res. 2020, 39, 66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horinouchi, T.; Terada, K.; Higashi, T.; Miwa, S. Endothelin receptor signaling: New insight into its regulatory mechanisms. J. Pharmacol. Sci. 2013, 123, 85–101. [Google Scholar] [CrossRef] [Green Version]
- Rosano, L.; Bagnato, A. Endothelin therapeutics in cancer: Where are we? Am. J. Physiol.-Regul. Integr. Comp. Physiol. 2016, 310, R469–R475. [Google Scholar] [CrossRef] [Green Version]
- Cianfrocca, R.; Rosano, L.; Tocci, P.; Sestito, R.; Caprara, V.; Di Castro, V.; De Maria, R.; Bagnato, A. Blocking endothelin-1-receptor/beta-catenin circuit sensitizes to chemotherapy in colorectal cancer. Cell Death Differ. 2017, 24, 1811–1820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morita, T.; Kourembanas, S. Endothelial cell expression of vasoconstrictors and growth factors is regulated by smooth muscle cell-derived carbon monoxide. J. Clin. Investig. 1995, 96, 2676–2682. [Google Scholar] [CrossRef]
- Zhang, F.; Kaide, J.I.; Yang, L.; Jiang, H.; Quan, S.; Kemp, R.; Gong, W.; Balazy, M.; Abraham, N.G.; Nasjletti, A. CO modulates pulmonary vascular response to acute hypoxia: Relation to endothelin. Am. J. Physiol.-Heart Circ. Physiol. 2004, 286, H137–H144. [Google Scholar] [CrossRef]
- Liou, S.F.; Hsu, J.H.; Chen, Y.T.; Chen, I.J.; Yeh, J.L. KMUP-1 Attenuates Endothelin-1-Induced Cardiomyocyte Hypertrophy through Activation of Heme Oxygenase-1 and Suppression of the Akt/GSK-3beta, Calcineurin/NFATc4 and RhoA/ROCK Pathways. Molecules 2015, 20, 10435–10449. [Google Scholar] [CrossRef] [Green Version]
- Lin, H.C.; Su, S.L.; Lu, C.Y.; Lin, A.H.; Lin, W.C.; Liu, C.S.; Yang, Y.C.; Wang, H.M.; Lii, C.K.; Chen, H.W. Andrographolide inhibits hypoxia-induced HIF-1alpha-driven endothelin 1 secretion by activating Nrf2/HO-1 and promoting the expression of prolyl hydroxylases 2/3 in human endothelial cells. Environ. Toxicol. 2017, 32, 918–930. [Google Scholar] [CrossRef]
- Chou, A.K.; Chen, T.I.; Winardi, W.; Dai, M.H.; Chen, S.C.; Howng, S.L.; Yen, C.P.; Lin, T.K.; Jeng, A.Y.; Kwan, A.L. Functional neuroprotective effect of CGS 26303, a dual ECE inhibitor, on ischemic-reperfusion spinal cord injury in rats. Exp. Biol. Med. 2007, 232, 214–218. [Google Scholar]
- Casado, J.; Iñigo-Chaves, A.; Jiménez-Ruiz, S.M.; Ríos-Arrabal, S.; Carazo-Gallego, Á.; González-Puga, C.; Núñez, M.I.; Ruíz-Extremera, Á.; Salmerón, J.; León, J. AA-NAT, MT1 and MT2 Correlates with Cancer Stem-Like Cell Markers in Colorectal Cancer: Study of the Influence of Stage and p53 Status of Tumors. Int. J. Mol. Sci. 2017, 18, 1251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Witte, K.E.; Hertel, O.; Windmöller, B.A.; Helweg, L.P.; Höving, A.L.; Knabbe, C.; Busche, T.; Greiner, J.F.W.; Kalinowski, J.; Noll, T.; et al. Nanopore Sequencing Reveals Global Transcriptome Signatures of Mitochondrial and Ribosomal Gene Expressions in Various Human Cancer Stem-like Cell Populations. Cancers 2021, 13, 1136. [Google Scholar] [CrossRef]
- Morata-Tarifa, C.; Jiménez, G.; García, M.A.; Entrena, J.M.; Griñán-Lisón, C.; Aguilera, M.; Picon-Ruiz, M.; Marchal, J.A. Low adherent cancer cell subpopulations are enriched in tumorigenic and metastatic epithelial-to-mesenchymal transition-induced cancer stem-like cells. Sci. Rep. 2016, 6, 18772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hemmati, M.; Yousefi, B.; Bahar, A.; Eslami, M. Importance of Heme Oxygenase-1 in Gastrointestinal Cancers: Functions, Inductions, Regulations, and Signaling. J. Gastrointest. Cancer 2021, 52, 454–461. [Google Scholar] [CrossRef] [PubMed]
- Germanova, D.; Keirsse, J.; Köhler, A.; Hastir, J.F.; Demetter, P.; Delbauve, S.; Elkrim, Y.; Verset, L.; Larbanoix, L.; Preyat, N.; et al. Myeloid tumor necrosis factor and heme oxygenase-1 regulate the progression of colorectal liver metastases during hepatic ischemia-reperfusion. Int. J. Cancer 2021, 148, 1276–1288. [Google Scholar] [CrossRef]
- Sebastián, V.P.; Salazar, G.A.; Coronado-Arrázola, I.; Schultz, B.M.; Vallejos, O.P.; Berkowitz, L.; Álvarez-Lobos, M.M.; Riedel, C.A.; Kalergis, A.M.; Bueno, S.M. Heme Oxygenase-1 as a Modulator of Intestinal Inflammation Development and Progression. Front. Immunol. 2018, 9, 1956. [Google Scholar] [CrossRef] [PubMed]
- Puentes-Pardo, J.D.; Moreno-SanJuan, S.; Carazo, Á.; León, J. Heme Oxygenase-1 in Gastrointestinal Tract Health and Disease. Antioxidants 2020, 9, 214. [Google Scholar] [CrossRef]
- Wegiel, B.; Nemeth, Z.; Correa-Costa, M.; Bulmer, A.C.; Otterbein, L.E. Heme oxygenase-1: A metabolic nike. Antioxid. Redox Signal. 2014, 20, 1709–1722. [Google Scholar] [CrossRef] [Green Version]
- Wegiel, B.; Gallo, D.; Csizmadia, E.; Harris, C.; Belcher, J.; Vercellotti, G.M.; Penacho, N.; Seth, P.; Sukhatme, V.; Ahmed, A.; et al. Carbon monoxide expedites metabolic exhaustion to inhibit tumor growth. Cancer Res. 2013, 73, 7009–7021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Almeida, A.S.; Sonnewald, U.; Alves, P.M.; Vieira, H.L. Carbon monoxide improves neuronal differentiation and yield by increasing the functioning and number of mitochondria. J. Neurochem. 2016, 138, 423–435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Rogoff, H.A.; Keates, S.; Gao, Y.; Murikipudi, S.; Mikule, K.; Leggett, D.; Li, W.; Pardee, A.B.; Li, C.J. Suppression of cancer relapse and metastasis by inhibiting cancer stemness. Proc. Natl. Acad. Sci. USA 2015, 112, 1839–1844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryoo, I.G.; Kim, G.; Choi, B.H.; Lee, S.H.; Kwak, M.K. Involvement of NRF2 Signaling in Doxorubicin Resistance of Cancer Stem Cell-Enriched Colonospheres. Biomol. Ther. 2016, 24, 482–488. [Google Scholar] [CrossRef] [Green Version]
- Shiokawa, D.; Sakai, H.; Ohata, H.; Miyazaki, T.; Kanda, Y.; Sekine, S.; Narushima, D.; Hosokawa, M.; Kato, M.; Suzuki, Y.; et al. Slow-Cycling Cancer Stem Cells Regulate Progression and Chemoresistance in Colon Cancer. Cancer Res. 2020, 80, 4451–4464. [Google Scholar] [CrossRef] [PubMed]
- Sharif, T.; Dai, C.; Martell, E.; Ghassemi-Rad, M.S.; Hanes, M.R.; Murphy, P.J.; Kennedy, B.E.; Venugopal, C.; Subapanditha, M.; Giacomantonio, C.A.; et al. TAp73 Modifies Metabolism and Positively Regulates Growth of Cancer Stem-Like Cells in a Redox-Sensitive Manner. Clin. Cancer Res. 2019, 25, 2001–2017. [Google Scholar] [CrossRef]
- Leis, O.; Eguiara, A.; Lopez-Arribillaga, E.; Alberdi, M.J.; Hernandez-Garcia, S.; Elorriaga, K.; Pandiella, A.; Rezola, R.; Martin, A.G. Sox2 expression in breast tumours and activation in breast cancer stem cells. Oncogene 2012, 31, 1354–1365. [Google Scholar] [CrossRef] [Green Version]
- Abdalla, M.Y.; Ahmad, I.M.; Rachagani, S.; Banerjee, K.; Thompson, C.M.; Maurer, H.C.; Olive, K.P.; Bailey, K.L.; Britigan, B.E.; Kumar, S. Enhancing responsiveness of pancreatic cancer cells to gemcitabine treatment under hypoxia by heme oxygenase-1 inhibition. Transl. Res. 2019, 207, 56–69. [Google Scholar] [CrossRef]
- Zhan, L.; Zhang, H.; Zhang, Q.; Woods, C.G.; Chen, Y.; Xue, P.; Dong, J.; Tokar, E.J.; Xu, Y.; Hou, Y.; et al. Regulatory role of KEAP1 and NRF2 in PPARγ expression and chemoresistance in human non-small-cell lung carcinoma cells. Free Radic. Biol. Med. 2012, 53, 758–768. [Google Scholar] [CrossRef] [Green Version]
- Nam, S.Y.; Sabapathy, K. p53 promotes cellular survival in a context-dependent manner by directly inducing the expression of haeme-oxygenase-1. Oncogene 2011, 30, 4476–4486. [Google Scholar] [CrossRef]
- Lee, S.Y.; Jo, H.J.; Kim, K.M.; Song, J.D.; Chung, H.T.; Park, Y.C. Concurrent expression of heme oxygenase-1 and p53 in human retinal pigment epithelial cell line. Biochem. Biophys. Res. Commun. 2008, 365, 870–874. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.H.; Song, N.Y.; Kim, E.H.; Na, H.K.; Joe, Y.; Chung, H.T.; Surh, Y.J. 15-Deoxy-Δ12,14-prostaglandin J2 induces p53 expression through Nrf2-mediated upregulation of heme oxygenase-1 in human breast cancer cells. Free Radical Res. 2014, 48, 1018–1027. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Miguel, P.; Medrano-Andrés, D.; Griera-Merino, M.; Ortiz, A.; Rodríguez-Puyol, M.; Rodríguez-Puyol, D.; López-Ongil, S. Tweak up-regulates endothelin-1 system in mouse and human endothelial cells. Cardiovasc. Res. 2017, 113, 207–221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serizawa, F.; Patterson, E.; Potter, R.F.; Fraser, D.D.; Cepinskas, G. Pretreatment of human cerebrovascular endothelial cells with CO-releasing molecule-3 interferes with JNK/AP-1 signaling and suppresses LPS-induced proadhesive phenotype. Microcirculation 2015, 22, 28–36. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.C.; Yang, C.C.; Hsiao, L.D.; Chen, S.Y.; Yang, C.M. Heme Oxygenase-1 Induction by Carbon Monoxide Releasing Molecule-3 Suppresses Interleukin-1β-Mediated Neuroinflammation. Front. Mol. Neurosci. 2017, 10, 387. [Google Scholar] [CrossRef] [PubMed]
- Brouard, S.; Berberat, P.O.; Tobiasch, E.; Seldon, M.P.; Bach, F.H.; Soares, M.P. Heme Oxygenase-1-derived Carbon Monoxide Requires the Activation of Transcription Factor NF-κB to Protect Endothelial Cells from Tumor Necrosis Factor-α-mediated Apoptosis. J. Biol. Chem. 2002, 277, 17950–17961. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chhikara, M.; Wang, S.; Kern, S.J.; Ferreyra, G.A.; Barb, J.J.; Munson, P.J.; Danner, R.L. Carbon monoxide blocks lipopolysaccharide-induced gene expression by interfering with proximal TLR4 to NF-kappaB signal transduction in human monocytes. PLoS ONE 2009, 4, e8139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mizuguchi, S.; Capretta, A.; Suehiro, S.; Nishiyama, N.; Luke, P.; Potter, R.F.; Fraser, D.D.; Cepinskas, G. Carbon monoxide-releasing molecule CORM-3 suppresses vascular endothelial cell SOD-1/SOD-2 activity while up-regulating the cell surface levels of SOD-3 in a heparin-dependent manner. Free Radic. Biol. Med. 2010, 49, 1534–1541. [Google Scholar] [CrossRef]
- Myant, K.B.; Cammareri, P.; McGhee, E.J.; Ridgway, R.A.; Huels, D.J.; Cordero, J.B.; Schwitalla, S.; Kalna, G.; Ogg, E.L.; Athineos, D.; et al. ROS production and NF-κB activation triggered by RAC1 facilitate WNT-driven intestinal stem cell proliferation and colorectal cancer initiation. Cell Stem Cell 2013, 12, 761–773. [Google Scholar] [CrossRef] [Green Version]
- Vítek, L.; Schwertner, H.A. The heme catabolic pathway and its protective effects on oxidative stress-mediated diseases. Adv. Clin. Chem. 2007, 43, 1–57. [Google Scholar] [PubMed]
- Zheng, J.; Nagda, D.A.; Lajud, S.A.; Kumar, S.; Mouchli, A.; Bezpalko, O.; O’Malley, B.W., Jr.; Li, D. Biliverdin’s regulation of reactive oxygen species signalling leads to potent inhibition of proliferative and angiogenic pathways in head and neck cancer. Br. J. Cancer 2014, 110, 2116–2122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trachootham, D.; Alexandre, J.; Huang, P. Targeting cancer cells by ROS-mediated mechanisms: A radical therapeutic approach? Nat. Rev. Drug Discov. 2009, 8, 579–591. [Google Scholar] [CrossRef]
- Kajarabille, N.; Latunde-Dada, G.O. Programmed Cell-Death by Ferroptosis: Antioxidants as Mitigators. Int. J. Mol. Sci. 2019, 20, 4968. [Google Scholar] [CrossRef] [Green Version]
- Puglisi, M.A.; Barba, M.; Corbi, M.; Errico, M.F.; Giorda, E.; Saulnier, N.; Boninsegna, A.; Piscaglia, A.C.; Carsetti, R.; Cittadini, A.; et al. Identification of Endothelin-1 and NR4A2 as CD133-regulated genes in colon cancer cells. J. Pathol. 2011, 225, 305–314. [Google Scholar] [CrossRef]
- Pérez-Moreno, P.; Indo, S.; Niechi, I.; Huerta, H.; Cabello, P.; Jara, L.; Aguayo, F.; Varas-Godoy, M.; Burzio, V.A.; Tapia, J.C. Endothelin-converting enzyme-1c promotes stem cell traits and aggressiveness in colorectal cancer cells. Mol. Oncol. 2020, 14, 347–362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peduto Eberl, L.; Bovey, R.; Juillerat-Jeanneret, L. Endothelin-receptor antagonists are proapoptotic and antiproliferative in human colon cancer cells. Br. J. Cancer 2003, 88, 788–795. [Google Scholar] [CrossRef] [Green Version]
- Eberl, L.P.; Valdenaire, O.; Saintgiorgio, V.; Jeannin, J.F.; Juillerat-Jeanneret, L. Endothelin receptor blockade potentiates FasL-induced apoptosis in rat colon carcinoma cells. Int. J. Cancer 2000, 86, 182–187. [Google Scholar] [CrossRef]
- Eberl, L.P.; Egidy, G.; Pinet, F.; Juillerat-Jeanneret, L. Endothelin receptor blockade potentiates FasL-induced apoptosis in colon carcinoma cells via the protein kinase C-pathway. J. Cardiovasc. Pharmacol. 2000, 36, S354–S356. [Google Scholar] [CrossRef] [PubMed]
- Enevoldsen, F.C.; Sahana, J.; Wehland, M.; Grimm, D.; Infanger, M.; Krüger, M. Endothelin Receptor Antagonists: Status Quo and Future Perspectives for Targeted Therapy. J. Clin. Med. 2020, 9, 824. [Google Scholar] [CrossRef] [PubMed] [Green Version]










Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ríos-Arrabal, S.; Puentes-Pardo, J.D.; Moreno-SanJuan, S.; Szuba, Á.; Casado, J.; García-Costela, M.; Escudero-Feliu, J.; Verbeni, M.; Cano, C.; González-Puga, C.; et al. Endothelin-1 as a Mediator of Heme Oxygenase-1-Induced Stemness in Colorectal Cancer: Influence of p53. J. Pers. Med. 2021, 11, 509. https://doi.org/10.3390/jpm11060509
Ríos-Arrabal S, Puentes-Pardo JD, Moreno-SanJuan S, Szuba Á, Casado J, García-Costela M, Escudero-Feliu J, Verbeni M, Cano C, González-Puga C, et al. Endothelin-1 as a Mediator of Heme Oxygenase-1-Induced Stemness in Colorectal Cancer: Influence of p53. Journal of Personalized Medicine. 2021; 11(6):509. https://doi.org/10.3390/jpm11060509
Chicago/Turabian StyleRíos-Arrabal, Sandra, Jose D. Puentes-Pardo, Sara Moreno-SanJuan, Ágata Szuba, Jorge Casado, María García-Costela, Julia Escudero-Feliu, Michela Verbeni, Carlos Cano, Cristina González-Puga, and et al. 2021. "Endothelin-1 as a Mediator of Heme Oxygenase-1-Induced Stemness in Colorectal Cancer: Influence of p53" Journal of Personalized Medicine 11, no. 6: 509. https://doi.org/10.3390/jpm11060509