
Pharmacogenomics of Monoclonal Antibodies for the Treatment of Rheumatoid Arthritis


1
Integrated Research Institute for Drug Development, College of Pharmacy, Dongguk University-Seoul, Goyang 10326, Korea
2
College of Pharmacy, Dongguk University-Seoul, Goyang 10326, Korea
*
Author to whom correspondence should be addressed.
†
These authors contributed equally to this work.
J. Pers. Med. 2022, 12(8), 1265; https://doi.org/10.3390/jpm12081265
Submission received: 6 July 2022
/
Revised: 27 July 2022
/
Accepted: 28 July 2022
/
Published: 31 July 2022
(This article belongs to the Special Issue Personalized Medicine for Rheumatic Disorders)
Abstract
:Precision medicine refers to a highly individualized and personalized approach to patient care. Pharmacogenomics is the study of how an individual’s genomic profile affects their drug response, enabling stable and effective drug selection, minimizing side effects, and maximizing therapeutic efficacy. Rheumatoid arthritis (RA) is an autoimmune disease that causes chronic inflammation in the joints. It mainly starts in peripheral joints, such as the hands and feet, and progresses to large joints, which causes joint deformation and bone damage due to inflammation of the synovial membrane. Here, we review various pharmacogenetic studies investigating the association between clinical response to monoclonal antibody therapy and their target genetic polymorphisms. Numerous papers have reported that some single nucleotide polymorphisms (SNPs) are related to the therapeutic response of several monoclonal antibody drugs including adalimumab, infliximab, rituximab, and tocilizumab, which target tumor necrosis factor (TNF), CD20 of B-cells, and interleukin (IL)-6. Additionally, there are some pharmacogenomic studies reporting on the association between the clinical response of monoclonal antibodies having various mechanisms, such as IL-1, IL-17, IL-23, granulocyte-macrophage colony-stimulating factor (GM-CSF) and the receptor activator of nuclear factor-kappa B (RANK) inhibition. Biological therapies are currently prescribed on a “trial and error” basis for RA patients. If appropriate drug treatment is not started early, joints may deform, and long-term treatment outcomes may worsen. Pharmacogenomic approaches that predict therapeutic responses for RA patients have the potential to significantly improve patient quality of life and reduce treatment costs.
1. Introduction
Precision medicine is defined as the diagnosis and treatment tailored to the patient based on their genotype, biomarkers, phenotype, or psychosocial characteristics to minimize unnecessary adverse events and improve clinical outcomes [1,2]. Medicines manufactured with the “one-size-fits-all” approach are effective in some patients and have no or minor side effects, while others are ineffective and have strong side effects [3]. To overcome the limitations of this one-size-fits-all framework in which all individuals presenting with some constellation of symptoms receive similar treatment, the diagnosis and management of various diseases are undergoing a paradigm shift to a personalized approach that prevents or treats diseases by considering each patient’s characteristics [4]. The paradigm shift towards precision medicine enables accurate disease prediction and prevention, reduces individual side effects and inefficient prescriptions, and provides safer diagnoses and treatments [5].
Over the past few decades, human genetics research has been fueled by cutting-edge sequencing technologies that lead to a deeper understanding of the relationship between genetic variation and health [6]. Pharmacogenomics is an emerging application to adjust drug selection and dosage considering a patient’s genetic characteristics [7]. It is one aspect of clinical genomics that will have the earliest and broadest clinical implementation with the potential to impact the treatment of all patients [8]. Although several pharmacogenetic guidelines have been reported by international scientific consortia in recent years, the application of pharmacogenomics in clinical care is still limited. Various major barriers have been identified from basic pharmacogenomics research to implementation, and many coordinated international efforts are underway to overcome them [9]. Studies of previously neglected rare genetic variants and validation of their function and clinical impact through preclinical models are essential to advance pharmacogenetic knowledge.
Another technological advancement related to precision medicine is the development of “biopharmaceuticals”. The biopharmaceutical market is advancing faster than all pharmaceutical markets, with innovations such as immunotherapy, antibody-drug conjugates, and gene therapy [10]. Biopharmaceuticals rarely cause side effects and show high specificity and activity compared to conventional drugs [11]. Their properties could provide targeted therapies rather than symptomatic treatments, accelerating treatment for conditions that cannot be treated with conventional synthetic drugs. [12]. Among biopharmaceuticals, monoclonal antibodies are the most profitable and are used to cure a variety of diseases, including autoimmune diseases, angiogenesis-related diseases, cardiovascular diseases, inflammatory diseases, and cancer [13]. The top 10 best-selling biopharmaceuticals in 2017 contained eight Abs. Among this list, the mAb adalimumab (ADA), a tumor necrosis factor-alpha (TNF-α) inhibitor used to treat rheumatoid arthritis and related disorders, was the most profitable product each year, generating global sales of approximately USD 62.6 billion between 2014 and 2017 [14]. The number of newly registered monoclonal antibodies is predicted to steadily increase and dominate the biopharmaceutical market [10].
Rheumatoid arthritis (RA) is an autoimmune disease that causes chronic inflammation of the joints throughout the body. Mainly, the destruction of articular cartilage and bone damage proceed due to persistent synovitis and infiltration of immune cells in the peripheral joints of the hands and feet [15]. As the disease progresses, chronic pain caused by functional impairment and joint deformation causes physical disability, reduced quality of life, and cardiovascular and other comorbidities [16]. Nonsteroidal anti-inflammatory drugs (NSAIDs) used to treat RA do not interfere with joint damage and therefore do not cure the disease. Glucocorticoids provide rapid symptom and disease correction but are associated with serious long-term side effects [17]. Disease-modifying antirheumatic drugs (DMARDs) used to coordinate disease progression through anti-inflammatory and immunomodulatory actions are key to RA treatment [18]. Commonly used primary conventional synthetic DMARDs to treat RA include methotrexate, hydroxychloroquine, and sulfasalazine. In case of ineffectiveness, leflunomide or tacrolimus (calcineurin inhibitor) are used. In the past, immunosuppressive agents such as azathioprine or cyclosporine, and parenteral gold, penicillamine, and bucillamine were often used for general autoimmune diseases, but they are now rarely prescribed due to the development of more effective and safer drugs [19]. When faced with the limitation that a sufficient therapeutic effect cannot be obtained with the above therapeutic agents, biological DMARDs (TNF-α inhibitors, B-cell modulation, and interleukin (IL)-6R blockade) or targeted DMARDs (Janus kinase inhibitors) may be used alone or together with existing conventional synthetic DMARDs [20]. Most biological DMARDs exhibit enhanced efficacy when combined with other conventional synthetic DMARDs [21]. These drugs are designed to target inflammatory molecules, cells, and pathways that cause tissue damage in patients with RA [22]. In this study, we summarized the pharmacogenetic studies of mAb drugs among biological DMARDs of RA.
2. Adalimumab
The discovery of the role of certain cytokines, especially TNF-α, in the pathogenesis of RA has dramatically changed disease treatment [23]. TNF-α is one of the major mediators abundantly expressed in the synovial fluid and synovium of patients with RA [24]. They modulate immune responses that have a powerful impact on cellular and humoral immunity [25]. For example, TNF-α can induce both cartilage degradation and bone resorption, directly affecting osteocyte receptor activation of nuclear factor κB ligand (RANK-L) expression, and increasing osteoclast generation [26,27]. In addition, TNF-α contributes to the pathogenesis of RA by inducing the production of other inflammatory cytokines, such as interleukin (IL)-1β and IL-6, which promote inflammation in the synovial membrane and attract and accumulate leukocytes [28,29]. In the past several decades, these TNF-α blockers have shown excellent efficacy in improving the inflammation and joint destruction caused by RA, and angiogenesis inhibition has been observed in various clinical trials [30,31,32,33,34].
ADA was approved by the Food and Drug Administration (FDA) in 2002 as an anti-TNF-α drug made by producing a novel antibody protein by cloning “phage display” [35,36]. It is a fully recombinant human mAb that is structurally and functionally indistinguishable from naturally occurring human IgG1 [37]. ADA specifically binds to TNF-α and blocks interactions with p55 and p75 cell surface TNF receptors [38]. Many clinical trials indicate ADA’s efficacy and safety for RA. As a blockbuster drug, the largest number of biosimilars are already on the market or in development. Genetic polymorphisms associated with the ADA response are discussed in Table 1.
2.1. FCGR2A
Fc receptor IgG immunoglobulins (FCGRs) can bind to extracellular IgG and cause cell activation or inhibition [48]. Consequently, genetic mutations affecting the activity of FCGRs are likely to affect the therapeutic efficacy of immunoglobulin-based therapies such as anti-TNF drugs [49]. FCGR2A, which encodes an Fc receptor expressed in various immune cells but mainly in macrophages and dendritic cells, is correlated with anti-TNF treatment in RA treatment [50,51]. ADA, most commonly used to treat RA, has an IgG1 Fc portion capable of binding to FCGRs. Changes in Fc binding affinity may affect the response to these biological therapies [52].
Avila-Pedretti et al. [39] studied whether genetic mutations at the Fc receptor FCGR2A were associated with the response to the anti-TNF agent ADA. A total of 95 RA patients treated with ADA were included and genotyped for the FCGR2A polymorphism rs1081274. Response to ADA treatment was measured according to the European League Against Rheumatism response (EULAR) criteria. They measured disease activity scores (DAS) using 28 joint counts after 12 weeks of ADA treatment. There was a statistically significant association with the genotype frequency of the FCGR2A polymorphism rs1801274 according to EULAR extreme clinical response to ADA treatment (odds ratio (OR) = 2.54; confidence interval (CI)95% = 1.9–5.4; p = 0.022). There have been several reports that RA patients positive for anti-cyclic citrullinated peptide (CCP) have a differential and stronger genetic background than anti-CCP-negative patients [53,54]. The association between the FCGR2A polymorphism rs1801274 and the response to ADA in RA patients in the anti-CCP positive group was investigated, and a significant relationship was confirmed (OR = 2.56; CI95% = 1.18–5.54; p = 0.047).
2.2. DHX32
Additionally, Avila-Pedretti et al. [39] identified a microarray-based study in RA analyzing the transcriptome of synovial macrophages (GSE49604, n = 8 samples). They found DEAH (Asp-Glu-Ala-His) box polypeptide 32 gene (DHX32) was positively correlated with FCGR2A (average r2 = 0.93, p < 0.001). The DHX32 gene encodes a putative RNA helicase and is involved in lymphocyte differentiation and activation [55,56]. In particular, RNA helicase is important for innate immune inactivation of viral RNA, which may contribute to the development of autoimmune diseases such as RA [57,58]. They selected a single nucleotide polymorphism (SNP) for DHX32 and determined its association with clinical response to ADA treatment in the RA patient cohort. There was a significant association between the DHX32 SNP rs12356233 and the clinical response to ADA (OR = 2.7; CI95% = 1.3–5.61; p = 0.0064). Moreover, analysis of the anti-CCP positive group of RA patients still found a significant association with ADA (OR = 2.65.; CI95% = 1.25–5.6; p = 0.0095).
2.3. RGS12
The regulator of G protein signaling 12 gene (RGS12), which is specifically expressed in human osteoclasts, is essential for NF-κB inflammatory signaling and thus plays an important role in the progression of RA [59]. Deletion of RGS12 attenuates inflammatory pain, which may be due to dysregulation of the COX2/PGE2 signaling pathway [60]. In another microarray-based study (GSE1050, n = 8 samples) in RA analyzing the transcriptome of synovial macrophages, Avila-Pedretti et al. [39] found that the RGS12 gene was negatively correlated with FCGR2A gene expression (average r2 = −0.96, p < 0.001). The RGS12 SNP rs4690093 confirmed a nominally significant association between the clinical response to ADA (OR = 0.4; CI95% = 0.17–0.98; p = 0.04). This effect was similar when analyzing the anti-CCP positive group of RA patients (OR = 0.4; CI95% = 0.16–0.99; p = 0.049).
2.4. IL-6
Dávila-Fajardo et al. [40] conducted a study validating the reported association of the IL-6 −174G/C polymorphism rs1800795 with the anti-TNF response in an independent cohort of 199 RA patients. Patients were classified as good or moderate responders and non-responders to the EULAR criteria at 6, 12, 18, and 24 months after treatment with TNF-α inhibitors, including ADA. When comparing the allele frequencies of responders and non-responders, there were slightly more patients with the −174G/C IL-6 polymorphism in the responder group compared with the non-responder group, but this was not statistically significant (p = 0.456). It was significantly associated with good or moderate EULAR response at 12, 18, and 24 months (OR = 2.93; CI95% = 1.29–6.70; p = 0.011, OR = 5.17; CI95% = 1.80–14.85; p = 2.27 × 10−3, and OR = 14.86; CI95% = 2.91–75.91; p = 1.18 × 10−3, respectively). Their results confirm the role of the −174G/C IL-6 polymorphism as a genetic predictive marker of responsiveness to anti-TNF therapy, including ADA.
2.5. PTPN22
Potter et al. [41] conducted a study to determine whether PTPN22 genetic susceptibility mutations predicted response to ADA treatment in 68 RA patients. The difference in ADA treatment response between autoantibody-positive and -negative patients was observed, but there was no statistically significant difference using logistic regression analysis with the EULAR response criteria. No association between drug response and shared epitope or PTPN22 R620W (C1858T) polymorphism was demonstrated in ADA (p > 0.05). In summary, although genetic factors are likely to contribute to treatment response, the well-established RA susceptibility loci, shared epitope, or PTPN22 are not included.
2.6. TNF
Cuchacovich et al. [42] conducted the first study to investigate the effect of the −308 TNF-α polymorphism on the clinical response to ADA therapy in patients with RA. They genotyped 81 RA patients for the −308 TNF-α polymorphism by polymerase chain reaction-restriction fragment length polymorphism analysis. They then subdivided patients into two groups (G/A and G/G genotype), and clinical responses were compared using DAS28 at 8, 16, and 24 weeks. As a result, there were significantly more DAS28 responders in the G/G genotype group (88%) than in the G/A genotype group (68%) at week 24. Additionally, the average DAS28 improvement of the G/G genotype group was higher than that of the G/A genotype group at week 24 (2.5 and 1.8, respectively).
Seitz et al. [43] explored whether the −308 TNF-α promoter polymorphism affects the therapeutic response to ADA-containing anti-TNF-α therapy in 54 RA patients. The average improvement in DAS28 score after 24 weeks of anti-TNF-α therapy was 0.83 ± 0.15 in the A/A genotype group, 1.50 ± 0.16 in the A/G genotype group, and 2.72 ± 0.70 in the G/G genotype group (p < 0.0001). These results confirmed that RA patients with the TNF-α −308 G/G genotype responded better to anti-TNF-α treatment than RA patients with the A/A or A/G genotype.
O’Rielly et al. [44] performed a meta-analysis of TNF-α −308 G/A polymorphism rs1800629, predicting poor response to TNF-α inhibitors, including ADA, in RA patients. The results were extracted based on DAS28 or achieving at least ACR 20 response. The frequency of the A allele status was 119/531 (22%) in responders and 60/161 (37%) in non-responders of nine studies [42,43,61,62,63,64,65,66,67]. Regardless of the prescribed TNF-α inhibitors, the odds for the A allele state were significantly reduced in responders versus non-responders (OR = 0.43; CI95% = 0.28–0.68; p = 0.000245). These results indicate that retention of −308 G/A polymorphism is predictive of a decreased response to TNF-α inhibitors, including ADA.
However, not all patients respond well to TNF-α inhibitors, so Zeng et al. [45] additionally meta-analyzed 15 studies [42,43,46,61,62,63,64,65,66,67,68,69,70,71,72] with a total of 2,127 patients to evaluate the TNF-α promoter −308 G/A polymorphism. Results showed that RA patients with the G allele responded better to treatment (OR = 1.87; CI95% = 1.26–2.79; p = 0.002). A separate meta-analysis of both studies showed that individuals with the A allele were associated with a weaker response to anti-TNF-α treatment with ADA than those with the G allele. In conclusion, individualized treatment can be suggested based on the TNF-α −308 G/A polymorphic genotype of RA patients.
Miceli-Richard et al. [46] conducted a study to determine whether TNF-α gene polymorphisms (−238A/G, −308A/G, and −857C/T) are genetic predictors of the clinical response to ADA in RA patients. A total of 380 patients were treated with ADA + methotrexate (n = 182), ADA + other DMARD (n = 96), or ADA alone (n = 102), and the results were recorded as DAS28, ACR response, and the Health Assessment Questionnaire-Disability Index at 12 weeks of treatment. Of these, 152 RA patients had an ACR50 response at 12 weeks, but the three tested TNF-α polymorphisms were not significantly related to the ACR50 response. The haplotype reconstruction of the TNF-α locus revealed the GGC haplotype (−238G/−308G/−857C) was present in more than 50% of patients and was significantly associated with a lower ACR50 response at 12 weeks only in the group treated with methotrexate and ADA (p = 0.0041). These findings indicate that a single TNF-α locus haplotype (−238G/−308G/−857C) present on both chromosomes is associated with a lower response to treatment with methotrexate and ADA in patients with RA.
2.7. TNFR2
Ongaro et al. [47] assessed whether the polymorphisms 676T>G in the TNFR2 gene could affect the clinical response in 105 RA patients who received anti-TNFα therapy with ADA for one year according to the ACR criteria [73]. The percent improvement (20, 50, or 70%) of all efficacy variables included in the ACR score set represented patients with low, medium, and high response grades, respectively. They analyzed the adjusted ORs obtained by subdividing the number of patients by genotype by comparing ACR70 and ACR (50+20). As a result, after three and six months of ADA treatment, the risk of belonging to the ACR group for TG genotype patients was significantly increased by about three times compared to wild-type (TT) genotype patients (OR = 2.90; CI95% = 0.95–8.89, and OR = 2.94; CI95% = 1.15–7.56, respectively). Moreover, there was a significant increase in adjusted ORs after 3 and 12 months of ADA treatment when they compared ACR70 and ACR20 responders (OR = 3.78; CI95% = 1.07–13.31, and OR = 4.30; CI95% = 1.16–15.99, respectively). The OR values obtained for the ACR70 versus ACR (50+20) or ACR20 comparisons for the GG genotype were not significant. Because the total number of patients with the GG genotype was low (n = 8), individuals carrying the 676G allele were counted together (TG+GG). This group of patients was similar to patients with the TG genotype in both ACR70 versus ACR (50+20) or ACR20, with significant adjusted ORs after 12 months of treatment (OR = 3.50; CI95% = 0.99–12.35). Therefore, the presence of one G allele tends to be a less responsive phenotype during anti-TNFα therapy involving ADA. In conclusion, the TNFR2 676 T/G genotype is associated with a low response to anti-TNFα therapy containing ADA, so it can be a useful genetic marker for predicting various response grades to anti-TNFα therapy.
3. Infliximab
Infliximab (IFX) is the first chimeric mAb (mouse/human) designed to block and neutralize TNF-α, a major inflammatory cytokine [74]. Since its introduction in 1998, it has revolutionized the induction and maintenance of treating RA and inflammatory bowel disease, namely Crohn’s disease and ulcerative colitis [75,76]. IFX reduces serum levels of inflammatory mediators and vascular endothelial growth factors as well as TNFα inhibition. It also reduces the expression of chemokines in synovial tissues and decreases lymphocyte migration to the joints in RA patients [77]. Genetic polymorphisms associated with IFX response are summarized in Table 2.
3.1. FCGR2A and FCGR3A
Avila-Pedretti et al. [39] also studied the association between the FCGR2A polymorphism rs1081274 and clinical response to IFX in a total of 126 RA patients treated with IFX. A comparison of the frequency of the FCGR2A polymorphism rs1801274 between a global cohort of IFX-treated responders and non-responders showed no statistically significant association between clinical response to FCGR2A polymorphism rs1801274 in patients treated with IFX (OR = 0.76; CI95% = 0.44–1.32; p = 0.11). In contrast, the FCGR2A polymorphism rs1801274 was significantly associated with IFX response in the anti-CCP positive RA patients group (OR = 0.62; CI95% = 0.32–1.22; p = 0.35).
Cañete et al. [78] evaluated the relationship between the functional SNP of the FCGR2A gene and response to IFX treatment in 91 RA patients. The FCGR2A-RR genotype is a risk factor for susceptibility to autoimmune diseases, as immune complexes are less efficiently cleared from the circulation in RA patients, leading to tissue damage [82]. RA patients with the low-affinity FCGR2A-RR genotype had a significantly better ACR20 response (RR: 60% and HH-RH: 33.3%; p = 0.035), while EULAR good and moderate responses only showed a significant trend after 30 weeks of IFX treatment (RR: 38.1% and HH-RH: 25.0%). They also showed an association between the low-affinity FCGR2A-RR genotype and decreased DAS28 with three parameters, including C-reactive protein (3v-CRP), using a linear model multivariate analysis. These results suggest that IFX can be eliminated less efficiently in RA patients with low-affinity variants than in RA patients with high-affinity variants (HH or RH). They also investigated the effect on the FCGR3A polymorphism and the clinical response to IFX in patients with RA. After six weeks of follow-up, the low affinity FCGR3A allele had a significantly higher ACR50 response (FF: 24.1% and VV-VF: 2.2%; p = 0.003) and EULAR good response rate (FF: 44.8% and VV-VF: 22.9%; p = 0.040). Changes in DAS28 3v-CRP during follow-up were similar to those found in ACR and EULAR responses. In conclusion, the response to IFX treatment in RA patients is affected by the FCGR3A genotype.
3.2. RGS12
Avila-Pedretti G et al. [39] identified RGS12 as having a strong correlation with FCGR2A expression. Like the association between FCGR2A and clinical response of IFX, they found a nominally significant association between RGS12 SNP rs2857859 and response to IFX in anti-CCP positive RA patients (OR = 0.4; CI95% = 0.17–0.99; uncorrected p = 0.042). This association was not significant after several test corrections.
3.3. PTPN22
Potter C et al. [41] evaluated the role of the PTPN22 R620W (C1858T) polymorphism as a predictor of ADA as well as IFX treatment outcomes in 296 patients with RA. Compared to rheumatoid factor (RF)-negative patients, RF-positive patients showed significantly less improvement in DAS28 values after anti-TNF therapy, including IFX as well as ADA and etanercept (OR =−0.48; CI95% = −0.87–0.08; p = 0.018). Moreover, patients positive for anti-CCP antibody showed less improved DAS28 values compared to anti-CCP negative patients (OR = −0.39; CI95% = −0.71–0.07; p = 0.017). Additionally, the effects of RF and anti-CCP antibodies were evaluated using multivariate linear regression combining both antibodies with previously known predictors, such as baseline HAQ and concurrent DMARD therapy and gender. As a result, RF and anti-CCP positivity did not better predict the response to anti-TNF therapy, and there was no association between these two factors and drug response (RF: R2 = 0.17; anti-CCP: R2 = 0.17; RF + anti-CCP: R2 = 0.17). Finally, they performed a linear regression including the interaction between drug type and autoantibody status to confirm that the predictive effects of RF and anti-CCP antibodies on IFX response were the same. The results showed that the effects of RF and anti-CCP antibodies were demonstrated in RA patients treated with IFX, but the effects were not statistically significant between the two drug types. As mentioned previously, an association between the tested anti-TNF therapies, including ADA, IFX, etanercept, and the PTPN22 R620W (C1858T) polymorphism, has not been demonstrated (OR = −0.11; CI95% = −0.36–0.15; p = 0.41). Therefore, the presence of these antibodies accounts for only a small part of the change in treatment response. PTPN22 does not affect IFX efficacy.
3.4. MHC
Martinez et al. [79]. studied the association of major histocompatibility complex (MHC) polymorphisms with clinical response to IFX. They genotyped HLA-DRB1, HLA-DQA1, HLA-DQB1, MHC class I chain-related gene A (MICA) transmembrane polymorphism alleles, and TNFa-e, D6S273, HLA–B–associated transcript 2 (BAT2), and D6S2223 microsatellites in 78 RA patients who received IFX treatment. A control sample from 323 healthy individuals was also included to detect the linkage disequilibrium between maker pairs. The results showed no single allele associated with IFX response, including the TNFa/b microsatellite allele linked to the TNF promoter polymorphism. The frequency of the TNFa11;b4 mini-haplotype was increased (41% versus 16% in non-responders, p = 0.01) and that of the D6S273_3 allele was decreased in the responders (32%) versus non-responders (56%, p = 0.04). The D6S273_4/BAT2_2 pair was observed more frequently in the responders (46% versus 11% in non-responders, p = 0.001). This allele pair was only associated with the responder group when compared to the control group (46% in responders versus 17% in controls, p = 0.00002). They did not identify statistically significant differences in the frequency of MICA and D6S2223 polymorphisms and HLA-DRB1, HLA-DQA1, and HLA-DQB1 alleles in responders and non-responders.
3.5. TNF
Mugnier et al. [61] evaluated whether the TNF-α promoter −308G/A polymorphism affects the response to IFX treatment using DAS28 in 59 RA patients. After 22 weeks of IFX treatment, in 42% of RA patients in the A/A or A/G groups and 81% of RA patients in the G/G group, DAS28 showed significant improvements to 1.24 ± 1.74 and 2.29 ± 1.33, respectively (p = 0.029).
Cuchacovich et al. [63] investigated the effect of a TNF-α promoter polymorphism (G/A and G/G) circulating TNF-α levels on the IFX clinical response in 132 patients with RA. They found that serum TNF-α levels increased in patients who progressively showed significant improvement in all parameters with IFX treatment. This increase in TNF-α was a result of quantification of both free and circulating TNF-α and immune complexes of TNF-α bound to the anti-TNF-α monoclonal antibody [83]. When the two groups were analyzed separately, they found a statistically significant correlation between the ACR50 improvement criteria and the increase in TNF-α levels over time only in RA patients from the G/A group (p < 0.03).
Fonseca et al. [64] conducted a study of 22 RA patients to investigate the effect of the polymorphism at position −308 of the TNF-α gene on IFX treatment. Of all patients, 68% (n = 15) had the −308 GG genotype and 32% (n = 7) had the −308 AG genotype. After treatment with IFX for approximately 25 months, the DAS28 score of −308 GG genotype patients decreased, while the DAS28 score of −308 AG genotype patients slightly increased (p < 0.01). The Health Assessment Questionnaire was more evolved in the GG genotype group compared to that in the −308 AG group (p = 0.064).
Marotte et al. [67] studied the association between the TNF-α −308 polymorphism and clinical response to IFX treatment in 198 patients with RA. They found that the TNF-α −308 polymorphism was not associated with the ACR response to IFX. The circulating TNF-α bioactivity level was higher in the A/A or A/G genotype group compared to that of the G/G genotype group. However, the difference in TNF-α protein level according to the genotype was not confirmed.
Chatzikyriakidou et al. [70] conducted a study on the correlation between the TNFR1 gene polymorphism 36A/G, the TNF-α gene polymorphism −857C/T, −308G/A, −238G/A, and 489G/A, and the therapeutic effect in 27 RA patients treated with IFX. However, independent polymorphisms were not predictive of patient response to anti-TNF-α therapy.
Maxwell et al. [71] investigated the association between the response to infliximab by eight SNPs and DAS28 in the TNF gene region. The TNF −308 gene polymorphism was not related to the clinical IFX response, but it was negatively related to the TNF −238 G/A polymorphism (p = 0.028).
Fabris et al. [80] conducted a study to investigate whether the −238 G/G or +489 A/A TNF-α genotype in 66 patients with severe RA who received IFX differed from patients with mild-moderate RA. Patients with severe RA had a −238 G/G genotype in 100% of cases, whereas 92.8% of patients with mild-to-moderate RA and 92.5% of healthy individuals had a −238 G/G genotype (OR = 11.7; CI95% = 0.6–216; p = 0.03). The +489 A/A genotype was less frequent in patients than in the control group (OR = 4.2; CI95% = 0.97–18.4; p = 0.045). The −238 A/G genotype did not occur in patients with severe RA, and the mild-to-moderate RA genotype had the same frequency as the control group. Thus, −238 G/G homozygosity is associated with severe RA and the +489 A/A genotype may protect against poor RA outcomes.
3.6. TNFR1 and TNFR2
As mentioned earlier, Chatzikyriakidou et al. [70] reported that five independent polymorphisms were not predictive of patient response to IFX treatment. However, when they performed complex genotyping of both TNFR2 and TNF-α gene polymorphisms, they found a statistically significant difference between good and poor IFX responders in the genotype association distribution of 676T/G and −857C/T (p = 0.008). Good responders more frequently carried the TNFR2 allele 676T in homozygosity, with homozygosity of the TNF-α allele −857C/T compared with poor responders. The combination of 676T/G (TNFR2) and −857C/T (TNF-α) can be used to predict the efficacy of infliximab treatment.
Swierkot et al. [81] evaluated if five SNPs within the TNF-α and TNF receptor encoding genes (TNFA: G−308A, G−238A, C−857T; TNFR1A G36A; and TNFR1B T676G) affect the efficacy of TNF-α inhibitor therapy, including IFX, in 280 patients with RA. At 24 weeks of treatment, 45% of all patients achieved low disease activity or remission. After six months, lower disease activity or remission was observed more frequently in patients homozygous for the TNFR1A 36 allele than in patients homozygous for GG (52% versus 34%, p = 0.04) Additionally, at 24 weeks of treatment, the subgroup of RA patients homozygous for the TNFA-856T variant had a significantly lower DAS28 score compared to RA patients carrying the C allele (p = 0.045). No other polymorphisms were associated with EULAR responses at 12 and 24 weeks of treatment. In conclusion, homozygosity for the TNFR1A 36A allele and of TNFA −857T variant was associated with better response to anti-TNF therapy, including IFX.
4. Rituximab
B-cell targeting was first proposed as an RA treatment method in the 1990s based on the hypothesis that autoantibodies, such as rheumatoid factor, promote the survival of B-cells and thereby propagate chronic inflammation [84]. In addition, they can act as antigen-presenting cells through interaction with T-cells, which can stimulate inflammation [85]. Synovial B-cells are mainly part of the B-cell–T-cell aggregates that are closely related to the expression of factors such as A proliferation-inducing ligand (APPRIL), B-cell-activating factor (BAFF), and chemokines [86]. These molecules are key components of humoral adaptive immunity and are potential therapeutic targets for RA [87].
Rituximab (RTX) is a chimeric mAb with a specific affinity for CD20, which is expressed on most malignant B-cells [88]. It is approved to treat blood B-cell malignancies and non-hematologic B-cell-mediated diseases such as RA [89,90]. RTX binds to CD20 via a crystallizable fragment (Fc) and is reorganized in lipid rafts [91]. Thereafter, antibody-dependent cell-mediated cytotoxicity occurs due to the interaction of the Fcγ receptor expressed on the surface of effector cells (macrophages, granulocytes, and natural killer cells) with the Fc portion of RTX [92]. RTX in vivo mainly acts through immune-mediated mechanisms, including complement-dependent cytotoxicity involving NK cells and phagocytosis by macrophages and neutrophils [93,94]. The mechanisms of action of rituximab are only partly understood [95]. Table 3 described the genetic polymorphisms associated with RTX response.
4.1. FCGR2A and FCGR3
Jiménez Morales et al. [96] aimed to evaluate the effects of FCGR2A rs1801274 and FCGR3A rs396991 gene polymorphisms on response to RTX, EULAR response, remission, low disease activity (LDA), and DAS28 improvement in 55 patients diagnosed with RA and treated with RTX for 6, 12, and 18 months. Results showed that patients receiving RTX and carrying the T allele for FCGR2A rs1801274 gene polymorphism had a higher EULAR response at 6 months (OR = 4.86; CI95% = 1.11–21.12; p = 0.035), 12 months (OR = 4.66; CI95% = 0.90–24.12; p = 0.066) and 18 months (OR = 2.48; CI95% = 0.35–17.31; p = 0.357), a higher remission at 6 months (OR = 10.625; CI95% = 1.07–105.47; p = 0.044), and a higher improvement in DAS28 at 12 months (B = 0.782; CI95% = −0.15–1.71; p = 0.098) and 18 months (B = 1.414; CI95% = 0.19–2.63; p = 0.025). In addition, patients carrying the FCGR3A rs396991-G allele and receiving RTX had improved LDA (OR = 4.904; CI95% = 0.84–28.48; p = 0.077) and DAS28 (B = −1.083; CI95% = −1.98–−0.18; p = 0.021) at 18 months. These results suggest that the FCGR2A rs1801274 and FCGR3A rs396991 gene polymorphisms are good predictors of response to RTX treatment.
Quartuccio et al. [97] studied whether the FCGR3A −158 V/F polymorphism could affect the response to RTX in 212 RA patients. The FCGR3A genotype was not associated with a good/moderate EULAR response after four months of RTX treatment (p = 0.09). However, a significant difference between the VV and VF or FF genotypes was associated with a good/moderate EULAR response after six months of RTX treatment (p = 0.015 and p = 0.018, respectively), but not between the VF and FF genotypes (p = 0.96). RA patients with the VV genotype were associated with a good/moderate EULAR response after six months of RTX treatment by univariate logistic regression analysis (OR = 4.4; CI95% = 1.4–13.5; p = 0.01).
Ruyssen-Witrand et al. [98] evaluated the association between single nucleotide polymorphisms in the FCGR3A gene and response to RTX treatment in 111 patients with RA who did not respond to or tolerate anti-TNF therapy. Of all RA patients, 81% (n = 90) had a response, 27% (n = 30) of which had a good response. Patients with RA carrying the FCGR3A −158V allele showed a statistically significant association with a higher response rate (OR = 4.6; CI95% = 1.5–13.6; p = 0.006). In multivariate analysis, V allele retention was independently associated with response to RTX (OR = 3.8; CI95% = 1.2–11.7; p = 0.023).
Similarly, Pál et al. [99] assessed the relationship between the FCGR3A polymorphism and the treatment outcome of RTX therapy in 52 RA patients. The distribution of FCGR3A genotypes was: VV: 15% (n = 8); VF: 65% (n = 34), FF: 19% (n = 10). A DAS28 reduction was shown in RA patients with three FCGR3A genotypes (VV: 1.98 ± 0.54, p = 0.008; VF: 2.07 ± 0.23, p < 0.001; FF: 1.59 ± 0.52, p = 0.014). Significant differences in DAS28 reduction on RTX treatment were identified between the VF heterozygote and the FF homozygote (p = 0.032) and between the heterozygote and VV+FF homozygotes (p = 0.017). Moreover, VV and VF patients achieved significant LDA compared to FF patients (VV: 62.5%; VF: 64.7%; FF: 30%).
However, conflicting results have been reported. Sarsour et al. [100] also conducted a study to determine if the FCGR3A polymorphism was associated with RTX efficacy in patients with RA. Longitudinal patient outcomes were assessed using the Clinical Disease Activity Index (CDAI) in 158 RTX-treated and 390 RA-treated TNF-α antagonists as controls. Similar changes in CDAI were observed for the three FCGR3A genotypes in the RTX-treated (VV: 4.56; VF: 7.44; FF: 4.75; p > 0.05) and TNF-α antagonist-treated patients (VV: 5.12; VF: 6.77; FF: 4.36; p > 0.05). The FCGR3A genotype was not significantly associated with treatment response in RTX-treated patients compared to TNF-α antagonist-treated patients (p = 0.86).
4.2. BAFF
Ruyssen-Witrand et al. [101] also determined whether BAFF polymorphisms were correlated with response to RTX treatment in 115 patients with RA. After 24 weeks of RTX treatment, the BAFF −871 C/C genotype was associated with a higher EULAR response rate than the T/T genotype (OR = 6.9; CI95% = 1.6–29.6; p = 0.03). In the multivariate analysis, the C allele was independently related to the response to RTX (OR = 4.1; CI95% = 1.3–12.7; p = 0.017).
4.3. IL-6
Fabris et al. [102] evaluated the effect of IL-6 −174 G/C polymorphism on response to RTX. Treatment response was assessed using both EULAR and ACR criteria after six months of RTX treatment in 142 RA patients. According to the EULAR criteria, patients with the IL-6 −174 C/C genotype showed less of a response to RTX than those with the GC/CC genotype (OR = 3.196; CI95% = 1.204–8.485; p = 0.0234), and similar results were found when evaluating the response based on the ACR criteria.
4.4. TGFβ1
Daïen et al. [103] conducted a study on the association between the TGFβ1 SNPs and responsiveness to RTX in 63 RA patients. Of these, 44 patients were defined as responders and 19 as non-responders. Both TGFβ1-10 (rs1800470) and TGFβ1-25 (rs1800471) were associated with clinical responses (OR = 1.6; CI95% = 1.2–2.3; p = 0.002 and OR = 1.6; CI95% = 1.3–1.9; p = 0.025, respectively). Additionally, the combination of the two SNPs elicited a much better RTX response (OR = 2.6; CI95% = 1.4–4.6; p = 0.008). In addition, they researched the association between the RTX clinical response and genes coding for cytokines involved in synovitis (IL-10: rs1800896; LTA: rs909253 and rs1041981; TNF-α: rs1800629, rs80267959, and rs1799724; TNFR2: rs1061622) and genes related to RA susceptibility (TRAF1: rs1081848; STAT4: rs7574865; TNFAIP3: rs6920220, and PTPN22L: rs2476601), but no associations were noted.
5. Tocilizumab
IL-6 plays a pivotal role in the aforementioned interaction between B-cells and T-cells. It promotes differentiation of Ig-producing plasma cells, leading to hypergammaglobulinemia, associated with chronic inflammation, and also contributes to the sustained maintenance of B-cell subpopulation plasmablasts, known as precursors of plasma cells [104]. These surviving plasmablasts are a source of autoantibodies, such as anti-citrullinated protein/peptide antibodies, and may accompany related autoimmune and chronic inflammatory diseases [105].
Tocilizumab (TCZ) is a humanized monoclonal antibody that acts as an IL-6R antagonist [106]. Potential immunological effects of TCZ include induction and expansion of B-regulatory cells, decreased expression of pro-inflammatory cytokines and chemokine genes, and increased expression of healing genes in synovial fluid [107]. It is approved for use in juvenile idiopathic polyarthritis and giant cell arteritis [108]. TCZ may be used as monotherapy in the treatment of adult patients with moderate-to-severe active RA who have had an inadequate response to one or more DMARDs or TNF-α inhibitors [109]. We show the genes related to the response of RA to tocilizumab in Table 4.
5.1. FCGR2A and FCGR3A
Jiménez Morales et al. [96] evaluated the effects of FCGR2A rs1801274 and FCGR3A rs396991 gene polymorphisms on response to TCZ. A retrospective prospective cohort study was conducted on 87 patients with RA who received TCZ treatment for 6, 12, and 18 months. No association was found between the FCGR2A rs1801274 gene polymorphism and response to TCZ. In contrast, patients carrying the FCGR3A rs39699-TT genotype had a higher EULAR response (OR = 5.075; CI95% = 1.20–21.33; p = 0.027) at 12 months. Therefore, patients carrying the genotype TT for rs1801274 FCGR3A would be better candidates for TCZ treatment.
5.2. HLA-DRB1
The human leukocyte antigen (HLA)-DRB1 is the most strongly known genetic risk factor for the complex genetic etiology of RA, a group of alleles referred to as the shared epitope [112]. Wang et al. [110] performed the first genome-wide association study to identify genetic factors related to TCZ response of 1,683 patients with RA in six clinical studies. They identified putative associations with eight loci (rs11052877, rs4910008, rs9594987, rs10108210, rs703297, rs703505, rs1560011, and rs7055107) previously involved as risk alleles for RA or not linked to responses to other therapies. None of these polymorphisms are clearly associated with the IL-6 pathway, and there is no association between HLA-DRB1 (shared epitope) with TCZ response.
5.3. IL-6R
Maldonado-Montoro et al. [111] examined the clinical parameters and rs12083537, rs2228145, rs4329505, rs11265618 gene polymorphisms of IL6R on TCZ EULAR response, LDA, and DAS28 improvement. They investigated a historic cohort of 77 patients with RA who had been treated with TCZ for 12 months. Of all gene polymorphisms, none were associated with EULAR response, remission, and DAS28 improvement. The AA genotype for rs12083537 (OR = 1.4; CI95% = 1.13–2.01; p = 0.021) and the CC genotype for rs11265618 (OR = 1.3; CI95% = 1.13–1.77; p = 0.031) are predictors of good response LDA. As a result of multivariate analysis, the AA genotype for rs12083537 (OR = 13.0; CI95% = 2.31–72.91; p = 0.004) and the CC genotype for rs11265618 (OR = 12.15; CI95% = 2.18–67.81; p = 0.004) were indicated. In conclusion, the IL6R polymorphisms AA genotype for rs12083537 and CC genotype for rs11265618 were significantly associated with higher LDA rates.
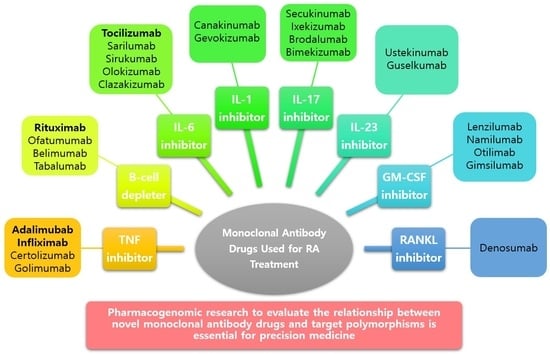
6. Newer Monoclonal Antibody Drugs and Genetic Polymorphisms of Their Targets
To provide precision medicine to individuals with a better understanding of the pathophysiology of RA, novel monoclonal antibody drugs that can treat RA through various mechanisms are being developed [113]. The association between various SNPs and response to biologic therapy in RA has been reported in several pharmacological studies [114]. Studies on the association of genetic polymorphisms with new targets and susceptibility to RA are being actively reported (Table 5).
IL-1, the first interleukin identified, is a major proinflammatory cytokine and a known pathogenic factor of auto-inflammation, auto-immunity, or infection. It is mainly secreted by macrophages, monocytes, and dendritic cells [156,157]. Canakinumab is a fully human anti-IL-1β monoclonal antibody drug that selectively neutralizes the bioactivity of IL-1β [158]. Another IL-11β monoclonal antibody, gevokizumab, was also studied in a phase lla study [159].
IL-17 is a pro-inflammatory cytokine mainly secreted by T helper 17 (Th17) cells and other T-cells [160]. This family includes six members from IL-17A to IL-17F [161]. IL-17 induces activation of the nuclear factor-κB, mitogen-activated protein kinases pathways, and the phosphoinositide-3 kinase pathway, leading to many inflammatory genes, mainly neutrophil-specific chemokines [162]. An excess of IL-17, which plays an important role in host defense, is observed in many chronic inflammatory and autoimmune diseases, including RA. It also contributes to tissue destruction [163]. Secukinumab and ixekizumab are humanized monoclonal antibody drugs against IL-17A, and brodalumab is a human immunoglobulin G2 monoclonal antibody drug against IL-17R. Several studies have reported that they are effective in treating RA when administered to patients who have no experience in biologic therapy or are resistant to anti-TNF therapy [164,165,166,167]. Recently, it has been reported that bimekizumab, developed for the dual blockade of IL-17A and IL-17F, effectively treats RA patients with an inadequate response to certolizumab pegol [168].
IL-23, a member of the IL-12 cytokine family, is a heterodimeric cytokine composed of an IL-23 p19 subunit and an IL-12/30 p40 subunit. It is mainly secreted by activated macrophages or dendritic cells [169]. They are known to induce differentiation of Th0 cells into Th17 cells and stimulate the production of pro-inflammatory cytokines such as TNF-α, IL-1β, IL-21, and IL-17 [170]. Ustekinumab is a human IgG1 monoclonal antibody drug that binds to the p40 subunit shared with IL-12 and IL-23 and blocks IL-12 and IL-23 signaling pathways through the inhibition of IL-12Rβ1 binding [171]. Guselkumab is the first IL-23 inhibitor approved for the treatment of severe plaque psoriasis by selectively targeting IL-23 [172]. However, the administration of both drugs to RA patients who did not respond to methotrexate treatment did not result in significant clinical improvement compared to the control group [173].
Granulocyte-macrophage colony-stimulating factor (GM-CSF) belongs to the colony-stimulating factor family of hematopoietic growth factors and is mainly produced by T-cells and stromal cells [174]. They are essential for regulating the function of mature bone marrow cells, such as macrophages, and induce the expression of HLA class Il antigens in synovial cells of RA patients [175]. Drugs targeting cytokines such as lenzilumab, namilumab, otilimab, and gimsilumab are being developed to block the GM-CSF pathway [176]. In particular, mavrilimumab, a human IgG4 monoclonal antibody against GM-CSF receptor α, showed an excellent clinical response and safety profile in RA patients [177].
The receptor activator of nuclear factor kappa B (RANKL) ligand belongs to the TNF superfamily and is an activated T-cell generator that regulates dendritic cell survival. Subsequent studies have reported that it is essential for osteoclast development [178,179]. Denosumab is a fully human anti-RANKL monoclonal antibody that competitively suppresses RANKL-RANK binding to inhibit osteoclast formation [180].
7. Conclusions and Future Challenges
Although the exact etiology of RA is still unknown, many researchers emphasize that it is caused by a combination of genetic and environmental factors. Several novel monoclonal antibodies that specify a target through various mechanisms have been developed and are showing excellent effects in the treatment of RA. In addition, recent review papers on sex and gender differences in RA treatment [181] and on different types of oral microbes involved in inducing RA progression [182] have suggested the possibility of more precise customization of treatment for individual patients. However, pharmacogenomic studies on the association between the new monoclonal antibody drugs and various genetic polymorphisms are insufficient. Moreover, factors involved in immunity and inflammation are very diverse, and there is currently no clear direction for personalized medicine [183]. Therefore, further research is essential, and this will increase the safety and efficacy of the newly developed monoclonal antibody drugs, enabling more complete precision medicine. This review reveals the complex genetic basis for the response to monoclonal antibody drugs among biological DMARDs used to treat RA, and it may contribute to important advances in understanding the molecular mechanisms involved in these therapies.
Author Contributions
Conceptualization, S.H.L., K.K. and C.-I.C.; investigation, S.H.L. and K.K.; resources, S.H.L. and K.K.; data Curation, S.H.L.; writing—original draft preparation, S.H.L. and K.K.; writing—review and editing, S.H.L. and C.-I.C.; supervision, C.-I.C.; project administration, C.-I.C.; funding acquisition, C.-I.C. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIT) (No. 2019R1C1C1006213).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Jameson, J.L.; Longo, D.L. Precision Medicine—Personalized, Problematic, and Promising. N. Engl. J. Med. 2015, 372, 2229–2234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- König, I.R.; Fuchs, O.; Hansen, G.; von Mutius, E.; Kopp, M.V. What Is Precision Medicine? Eur. Respir. J. 2017, 50, 1700391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kohler, S. Precision Medicine—Moving Away from One-Size-Fits-All. Quest 2018, 14, 12–15. [Google Scholar] [CrossRef]
- McDonald, E.S.; Clark, A.S.; Tchou, J.; Zhang, P.; Freedman, G.M. Clinical Diagnosis and Management of Breast Cancer. J. Nucl. Med. 2016, 57, 9S–16S. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Sanzo, M.; Cipolloni, L.; Borro, M.; La Russa, R.; Santurro, A.; Scopetti, M.; Simmaco, M.; Frati, P. Clinical Applications of Personalized Medicine: A New Paradigm and Challenge. Curr. Pharm. Biotechnol. 2017, 18, 194–203. [Google Scholar] [CrossRef]
- van der Wouden, C.H.; Böhringer, S.; Cecchin, E.; Cheung, K.-C.; Dávila-Fajardo, C.L.; Deneer, V.H.M.; Dolžan, V.; Ingelman-Sundberg, M.; Jönsson, S.; Karlsson, M.O.; et al. Generating Evidence for Precision Medicine: Considerations Made by the Ubiquitous Pharmacogenomics Consortium When Designing and Operationalizing the PREPARE Study. Pharm. Genom. 2020, 30, 131–144. [Google Scholar] [CrossRef]
- Cecchin, E.; Stocco, G. Pharmacogenomics and Personalized Medicine. Genes 2020, 11, 679. [Google Scholar] [CrossRef] [PubMed]
- Weinshilboum, R.M.; Wang, L. Pharmacogenomics: Precision Medicine and Drug Response. Mayo Clin. Proc. 2017, 92, 1711–1722. [Google Scholar] [CrossRef] [PubMed]
- Chenoweth, M.J.; Giacomini, K.M.; Pirmohamed, M.; Hill, S.L.; van Schaik, R.H.N.; Schwab, M.; Shuldiner, A.R.; Relling, M.V.; Tyndale, R.F. Global Pharmacogenomics Within Precision Medicine: Challenges and Opportunities. Clin. Pharmacol. Ther. 2020, 107, 57–61. [Google Scholar] [CrossRef] [PubMed]
- Kesik-Brodacka, M. Progress in Biopharmaceutical Development. Biotechnol. Appl. Biochem. 2018, 65, 306–322. [Google Scholar] [CrossRef] [Green Version]
- Mitragotri, S.; Burke, P.A.; Langer, R. Overcoming the Challenges in Administering Biopharmaceuticals: Formulation and Delivery Strategies. Nat. Rev. Drug Discov. 2014, 13, 655–672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Minghetti, P.; Rocco, P.; Cilurzo, F.; Vecchio, L.D.; Locatelli, F. The Regulatory Framework of Biosimilars in the European Union. Drug Discov. Today 2012, 17, 63–70. [Google Scholar] [CrossRef] [PubMed]
- Sivaccumar, J.; Sandomenico, A.; Vitagliano, L.; Ruvo, M. Monoclonal Antibodies: A Prospective and Retrospective View. Curr. Med. Chem. 2021, 28, 435–471. [Google Scholar] [CrossRef]
- Walsh, G. Biopharmaceutical Benchmarks 2018. Nat. Biotechnol. 2018, 36, 1136–1145. [Google Scholar] [CrossRef]
- Smolen, J.S.; Aletaha, D. Rheumatoid Arthritis Therapy Reappraisal: Strategies, Opportunities and Challenges. Nat. Rev. Rheumatol. 2015, 11, 276–289. [Google Scholar] [CrossRef] [PubMed]
- Smolen, J.S.; Aletaha, D.; McInnes, I.B. Rheumatoid Arthritis. Lancet 2016, 388, 2023–2038. [Google Scholar] [CrossRef]
- Kirwan, J.R. The Effect of Glucocorticoids on Joint Destruction in Rheumatoid Arthritis. The Arthritis and Rheumatism Council Low-Dose Glucocorticoid Study Group. N. Engl. J. Med. 1995, 333, 142–146. [Google Scholar] [CrossRef] [PubMed]
- Donahue, K.E.; Gartlehner, G.; Jonas, D.E.; Lux, L.J.; Thieda, P.; Jonas, B.L.; Hansen, R.A.; Morgan, L.C.; Lohr, K.N. Systematic Review: Comparative Effectiveness and Harms of Disease-Modifying Medications for Rheumatoid Arthritis. Ann. Intern. Med. 2008, 148, 124–134. [Google Scholar] [CrossRef]
- Cho, S.-K.; Sung, Y.-K. Treatment strategy for patients with rheumatoid arthritis. J. Korean Med. Assoc. 2020, 63, 422–430. [Google Scholar] [CrossRef]
- Burmester, G.R.; Pope, J.E. Novel Treatment Strategies in Rheumatoid Arthritis. Lancet 2017, 389, 2338–2348. [Google Scholar] [CrossRef]
- Schoels, M.; Aletaha, D.; Smolen, J.S.; Wong, J.B. Comparative Effectiveness and Safety of Biological Treatment Options after Tumour Necrosis Factor α Inhibitor Failure in Rheumatoid Arthritis: Systematic Review and Indirect Pairwise Meta-Analysis. Ann. Rheum. Dis. 2012, 71, 1303–1308. [Google Scholar] [CrossRef] [PubMed]
- Abbasi, M.; Mousavi, M.J.; Jamalzehi, S.; Alimohammadi, R.; Bezvan, M.H.; Mohammadi, H.; Aslani, S. Strategies toward Rheumatoid Arthritis Therapy; the Old and the New. J. Cell. Physiol. 2019, 234, 10018–10031. [Google Scholar] [CrossRef] [PubMed]
- Radner, H.; Aletaha, D. Anti-TNF in Rheumatoid Arthritis: An Overview. Wien. Med. Wochenschr. 2015, 165, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Saxne, T.; Palladino, M.A.; Heinegård, D.; Talal, N.; Wollheim, F.A. Detection of Tumor Necrosis Factor Alpha but Not Tumor Necrosis Factor Beta in Rheumatoid Arthritis Synovial Fluid and Serum. Arthritis Rheum. 1988, 31, 1041–1045. [Google Scholar] [CrossRef] [PubMed]
- Feldmann, M.; Maini, R.N. Lasker Clinical Medical Research Award. TNF Defined as a Therapeutic Target for Rheumatoid Arthritis and Other Autoimmune Diseases. Nat. Med. 2003, 9, 1245–1250. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.-J.; Anzaghe, M.; Schülke, S. Update on the Pathomechanism, Diagnosis, and Treatment Options for Rheumatoid Arthritis. Cells 2020, 9, 880. [Google Scholar] [CrossRef] [Green Version]
- Bertolini, D.R.; Nedwin, G.E.; Bringman, T.S.; Smith, D.D.; Mundy, G.R. Stimulation of Bone Resorption and Inhibition of Bone Formation in Vitro by Human Tumour Necrosis Factors. Nature 1986, 319, 516–518. [Google Scholar] [CrossRef]
- Brennan, F.M.; McInnes, I.B. Evidence That Cytokines Play a Role in Rheumatoid Arthritis. J. Clin. Investig. 2008, 118, 3537–3545. [Google Scholar] [CrossRef] [Green Version]
- Osta, B.; Benedetti, G.; Miossec, P. Classical and Paradoxical Effects of TNF-α on Bone Homeostasis. Front. Immunol. 2014, 5, 48. [Google Scholar] [CrossRef]
- Lipsky, P.E.; van der Heijde, D.M.; St Clair, E.W.; Furst, D.E.; Breedveld, F.C.; Kalden, J.R.; Smolen, J.S.; Weisman, M.; Emery, P.; Feldmann, M.; et al. Infliximab and Methotrexate in the Treatment of Rheumatoid Arthritis. Anti-Tumor Necrosis Factor Trial in Rheumatoid Arthritis with Concomitant Therapy Study Group. N. Engl. J. Med. 2000, 343, 1594–1602. [Google Scholar] [CrossRef] [Green Version]
- Weinblatt, M.E.; Keystone, E.C.; Furst, D.E.; Moreland, L.W.; Weisman, M.H.; Birbara, C.A.; Teoh, L.A.; Fischkoff, S.A.; Chartash, E.K. Adalimumab, a Fully Human Anti-Tumor Necrosis Factor Alpha Monoclonal Antibody, for the Treatment of Rheumatoid Arthritis in Patients Taking Concomitant Methotrexate: The ARMADA Trial. Arthritis Rheum. 2003, 48, 35–45. [Google Scholar] [CrossRef] [PubMed]
- Elliott, M.J.; Maini, R.N.; Feldmann, M.; Kalden, J.R.; Antoni, C.; Smolen, J.S.; Leeb, B.; Breedveld, F.C.; Macfarlane, J.D.; Bijl, H. Randomised Double-Blind Comparison of Chimeric Monoclonal Antibody to Tumour Necrosis Factor Alpha (CA2) versus Placebo in Rheumatoid Arthritis. Lancet 1994, 344, 1105–1110. [Google Scholar] [CrossRef]
- Moreland, L.W.; Baumgartner, S.W.; Schiff, M.H.; Tindall, E.A.; Fleischmann, R.M.; Weaver, A.L.; Ettlinger, R.E.; Cohen, S.; Koopman, W.J.; Mohler, K.; et al. Treatment of Rheumatoid Arthritis with a Recombinant Human Tumor Necrosis Factor Receptor (P75)-Fc Fusion Protein. N. Engl. J. Med. 1997, 337, 141–147. [Google Scholar] [CrossRef] [PubMed]
- Szekanecz, Z.; Besenyei, T.; Paragh, G.; Koch, A.E. Angiogenesis in Rheumatoid Arthritis. Autoimmunity 2009, 42, 563–573. [Google Scholar] [CrossRef] [Green Version]
- Pelechas, E.; Voulgari, P.V.; Drosos, A.A. Preclinical Discovery and Development of Adalimumab for the Treatment of Rheumatoid Arthritis. Expert Opin. Drug Discov. 2021, 16, 227–234. [Google Scholar] [CrossRef] [PubMed]
- Smith, G.P. Filamentous Fusion Phage: Novel Expression Vectors That Display Cloned Antigens on the Virion Surface. Science 1985, 228, 1315–1317. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Chadwick, L.; Mysler, E.; Moots, R.J. Review of Biosimilar Trials and Data on Adalimumab in Rheumatoid Arthritis. Curr. Rheumatol. Rep. 2018, 20, 57. [Google Scholar] [CrossRef] [PubMed]
- Cvetković, R.S.; Scott, L.J. Adalimumab. BioDrugs 2006, 20, 293–311. [Google Scholar] [CrossRef] [PubMed]
- Avila-Pedretti, G.; Tornero, J.; Fernández-Nebro, A.; Blanco, F.; González-Alvaro, I.; Cañete, J.D.; Maymó, J.; Alperiz, M.; Fernández-Gutiérrez, B.; Olivé, A.; et al. Variation at FCGR2A and Functionally Related Genes Is Associated with the Response to Anti-TNF Therapy in Rheumatoid Arthritis. PLoS ONE 2015, 10, e0122088. [Google Scholar] [CrossRef] [Green Version]
- Dávila-Fajardo, C.L.; Márquez, A.; Pascual-Salcedo, D.; Moreno Ramos, M.J.; García-Portales, R.; Magro, C.; Alegre-Sancho, J.J.; Balsa, A.; Cabeza-Barrera, J.; Raya, E.; et al. Confirmation of -174G/C Interleukin-6 Gene Promoter Polymorphism as a Genetic Marker Predicting Antitumor Necrosis Factor Treatment Outcome. Pharm. Genom. 2014, 24, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Potter, C.; Hyrich, K.L.; Tracey, A.; Lunt, M.; Plant, D.; Symmons, D.P.M.; Thomson, W.; Worthington, J.; Emery, P.; Morgan, A.W.; et al. Association of Rheumatoid Factor and Anti-Cyclic Citrullinated Peptide Positivity, but Not Carriage of Shared Epitope or PTPN22 Susceptibility Variants, with Anti-Tumour Necrosis Factor Response in Rheumatoid Arthritis. Ann. Rheum. Dis. 2009, 68, 69–74. [Google Scholar] [CrossRef] [Green Version]
- Cuchacovich, M.; Soto, L.; Edwardes, M.; Gutierrez, M.; Llanos, C.; Pacheco, D.; Sabugo, F.; Alamo, M.; Fuentealba, C.; Villanueva, L.; et al. Tumour Necrosis Factor (TNF)Alpha -308 G/G Promoter Polymorphism and TNFalpha Levels Correlate with a Better Response to Adalimumab in Patients with Rheumatoid Arthritis. Scand. J. Rheumatol. 2006, 35, 435–440. [Google Scholar] [CrossRef]
- Seitz, M.; Wirthmüller, U.; Möller, B.; Villiger, P.M. The -308 Tumour Necrosis Factor-Alpha Gene Polymorphism Predicts Therapeutic Response to TNFalpha-Blockers in Rheumatoid Arthritis and Spondyloarthritis Patients. Rheumatology 2007, 46, 93–96. [Google Scholar] [CrossRef] [Green Version]
- O’Rielly, D.D.; Roslin, N.M.; Beyene, J.; Pope, A.; Rahman, P. TNF-Alpha-308 G/A Polymorphism and Responsiveness to TNF-Alpha Blockade Therapy in Moderate to Severe Rheumatoid Arthritis: A Systematic Review and Meta-Analysis. Pharm. J. 2009, 9, 161–167. [Google Scholar] [CrossRef]
- Zeng, Z.; Duan, Z.; Zhang, T.; Wang, S.; Li, G.; Gao, J.; Ye, D.; Xu, S.; Xu, J.; Zhang, L.; et al. Association between Tumor Necrosis Factor-α (TNF-α) Promoter -308 G/A and Response to TNF-α Blockers in Rheumatoid Arthritis: A Meta-Analysis. Mod. Rheumatol. 2013, 23, 489–495. [Google Scholar] [CrossRef]
- Miceli-Richard, C.; Comets, E.; Verstuyft, C.; Tamouza, R.; Loiseau, P.; Ravaud, P.; Kupper, H.; Becquemont, L.; Charron, D.; Mariette, X. A Single Tumour Necrosis Factor Haplotype Influences the Response to Adalimumab in Rheumatoid Arthritis. Ann. Rheum. Dis. 2008, 67, 478–484. [Google Scholar] [CrossRef]
- Ongaro, A.; De Mattei, M.; Pellati, A.; Caruso, A.; Ferretti, S.; Masieri, F.F.; Fotinidi, M.; Farina, I.; Trotta, F.; Padovan, M. Can Tumor Necrosis Factor Receptor II Gene 676T>G Polymorphism Predict the Response Grading to Anti-TNFalpha Therapy in Rheumatoid Arthritis? Rheumatol. Int. 2008, 28, 901–908. [Google Scholar] [CrossRef]
- Sibéril, S.; Dutertre, C.-A.; Boix, C.; Bonnin, E.; Ménez, R.; Stura, E.; Jorieux, S.; Fridman, W.-H.; Teillaud, J.-L. Molecular Aspects of Human FcgammaR Interactions with IgG: Functional and Therapeutic Consequences. Immunol. Lett. 2006, 106, 111–118. [Google Scholar] [CrossRef]
- Musolino, A.; Naldi, N.; Bortesi, B.; Pezzuolo, D.; Capelletti, M.; Missale, G.; Laccabue, D.; Zerbini, A.; Camisa, R.; Bisagni, G.; et al. Immunoglobulin G Fragment C Receptor Polymorphisms and Clinical Efficacy of Trastuzumab-Based Therapy in Patients with HER-2/Neu-Positive Metastatic Breast Cancer. J. Clin. Oncol. 2008, 26, 1789–1796. [Google Scholar] [CrossRef]
- Guilliams, M.; Bruhns, P.; Saeys, Y.; Hammad, H.; Lambrecht, B.N. The Function of Fcγ Receptors in Dendritic Cells and Macrophages. Nat. Rev. Immunol. 2014, 14, 94–108. [Google Scholar] [CrossRef] [PubMed]
- Montes, A.; Perez-Pampin, E.; Narváez, J.; Cañete, J.D.; Navarro-Sarabia, F.; Moreira, V.; Fernández-Nebro, A.; Del Carmen Ordóñez, M.; de la Serna, A.R.; Magallares, B.; et al. Association of FCGR2A with the Response to Infliximab Treatment of Patients with Rheumatoid Arthritis. Pharm. Genom. 2014, 24, 238–245. [Google Scholar] [CrossRef] [PubMed]
- Arora, T.; Padaki, R.; Liu, L.; Hamburger, A.E.; Ellison, A.R.; Stevens, S.R.; Louie, J.S.; Kohno, T. Differences in Binding and Effector Functions between Classes of TNF Antagonists. Cytokine 2009, 45, 124–131. [Google Scholar] [CrossRef] [PubMed]
- Kurreeman, F.; Liao, K.; Chibnik, L.; Hickey, B.; Stahl, E.; Gainer, V.; Li, G.; Bry, L.; Mahan, S.; Ardlie, K.; et al. Genetic Basis of Autoantibody Positive and Negative Rheumatoid Arthritis Risk in a Multi-Ethnic Cohort Derived from Electronic Health Records. Am. J. Hum. Genet. 2011, 88, 57–69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bossini-Castillo, L.; de Kovel, C.; Kallberg, H.; van ’t Slot, R.; Italiaander, A.; Coenen, M.; Tak, P.P.; Posthumus, M.D.; Wijmenga, C.; Huizinga, T.; et al. A Genome-Wide Association Study of Rheumatoid Arthritis without Antibodies against Citrullinated Peptides. Ann. Rheum. Dis. 2015, 74, e15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdelhaleem, M. The Novel Helicase Homologue DDX32 Is Down-Regulated in Acute Lymphoblastic Leukemia. Leuk. Res. 2002, 26, 945–954. [Google Scholar] [CrossRef]
- Abdelhaleem, M.; Sun, T.-H.; Ho, M. DHX32 Expression Suggests a Role in Lymphocyte Differentiation. Anticancer Res. 2005, 25, 2645–2648. [Google Scholar]
- Meylan, E.; Tschopp, J. Toll-like Receptors and RNA Helicases: Two Parallel Ways to Trigger Antiviral Responses. Mol. Cell 2006, 22, 561–569. [Google Scholar] [CrossRef]
- Baccala, R.; Gonzalez-Quintial, R.; Lawson, B.R.; Stern, M.E.; Kono, D.H.; Beutler, B.; Theofilopoulos, A.N. Sensors of the Innate Immune System: Their Mode of Action. Nat. Rev. Rheumatol. 2009, 5, 448–456. [Google Scholar] [CrossRef]
- Yuan, G.; Yang, S.; Ng, A.; Fu, C.; Oursler, M.J.; Xing, L.; Yang, S. RGS12 Is a Novel Critical NF-ΚB Activator in Inflammatory Arthritis. iScience 2020, 23, 101172. [Google Scholar] [CrossRef]
- Yuan, G.; Yang, S.; Gautam, M.; Luo, W.; Yang, S. Macrophage Regulator of G-Protein Signaling 12 Contributes to Inflammatory Pain Hypersensitivity. Ann. Transl. Med. 2021, 9, 448. [Google Scholar] [CrossRef]
- Mugnier, B.; Balandraud, N.; Darque, A.; Roudier, C.; Roudier, J.; Reviron, D. Polymorphism at Position -308 of the Tumor Necrosis Factor Alpha Gene Influences Outcome of Infliximab Therapy in Rheumatoid Arthritis. Arthritis Rheum. 2003, 48, 1849–1852. [Google Scholar] [CrossRef]
- Padyukov, L.; Lampa, J.; Heimbürger, M.; Ernestam, S.; Cederholm, T.; Lundkvist, I.; Andersson, P.; Hermansson, Y.; Harju, A.; Klareskog, L.; et al. Genetic Markers for the Efficacy of Tumour Necrosis Factor Blocking Therapy in Rheumatoid Arthritis. Ann. Rheum. Dis. 2003, 62, 526–529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cuchacovich, M.; Ferreira, L.; Aliste, M.; Soto, L.; Cuenca, J.; Cruzat, A.; Gatica, H.; Schiattino, I.; Pérez, C.; Aguirre, A.; et al. Tumour Necrosis Factor-Alpha (TNF-Alpha) Levels and Influence of -308 TNF-Alpha Promoter Polymorphism on the Responsiveness to Infliximab in Patients with Rheumatoid Arthritis. Scand. J. Rheumatol. 2004, 33, 228–232. [Google Scholar] [CrossRef] [PubMed]
- Fonseca, J.E.; Carvalho, T.; Cruz, M.; Nero, P.; Sobral, M.; Mourão, A.F.; Cavaleiro, J.; Ligeiro, D.; Abreu, I.; Carmo-Fonseca, M.; et al. Polymorphism at Position -308 of the Tumour Necrosis Factor Alpha Gene and Rheumatoid Arthritis Pharmacogenetics. Ann. Rheum. Dis. 2005, 64, 793–794. [Google Scholar] [CrossRef] [Green Version]
- Kang, C.P.; Lee, K.W.; Yoo, D.H.; Kang, C.; Bae, S.C. The Influence of a Polymorphism at Position -857 of the Tumour Necrosis Factor Alpha Gene on Clinical Response to Etanercept Therapy in Rheumatoid Arthritis. Rheumatology 2005, 44, 547–552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guis, S.; Balandraud, N.; Bouvenot, J.; Auger, I.; Toussirot, E.; Wendling, D.; Mattei, J.-P.; Nogueira, L.; Mugnier, B.; Legeron, P.; et al. Influence of -308 A/G Polymorphism in the Tumor Necrosis Factor Alpha Gene on Etanercept Treatment in Rheumatoid Arthritis. Arthritis Rheum. 2007, 57, 1426–1430. [Google Scholar] [CrossRef] [PubMed]
- Marotte, H.; Arnaud, B.; Diasparra, J.; Zrioual, S.; Miossec, P. Association between the Level of Circulating Bioactive Tumor Necrosis Factor Alpha and the Tumor Necrosis Factor Alpha Gene Polymorphism at -308 in Patients with Rheumatoid Arthritis Treated with a Tumor Necrosis Factor Alpha Inhibitor. Arthritis Rheum. 2008, 58, 1258–1263. [Google Scholar] [CrossRef]
- Balog, A.; Gál, J.; Gyulai, Z.; Zsilák, S.; Mándi, Y. Tumour Necrosis Factor-Alpha and Heat-Shock Protein 70-2 Gene Polymorphisms in a Family with Rheumatoid Arthritis. Acta Microbiol. Immunol. Hung. 2004, 51, 263–269. [Google Scholar] [CrossRef]
- Criswell, L.A.; Lum, R.F.; Turner, K.N.; Woehl, B.; Zhu, Y.; Wang, J.; Tiwari, H.K.; Edberg, J.C.; Kimberly, R.P.; Moreland, L.W.; et al. The Influence of Genetic Variation in the HLA-DRB1 and LTA-TNF Regions on the Response to Treatment of Early Rheumatoid Arthritis with Methotrexate or Etanercept. Arthritis Rheum. 2004, 50, 2750–2756. [Google Scholar] [CrossRef]
- Chatzikyriakidou, A.; Georgiou, I.; Voulgari, P.V.; Venetsanopoulou, A.I.; Drosos, A.A. Combined Tumour Necrosis Factor-Alpha and Tumour Necrosis Factor Receptor Genotypes Could Predict Rheumatoid Arthritis Patients’ Response to Anti-TNF-Alpha Therapy and Explain Controversies of Studies Based on a Single Polymorphism. Rheumatology 2007, 46, 1034–1035. [Google Scholar] [CrossRef] [Green Version]
- Maxwell, J.R.; Potter, C.; Hyrich, K.L.; Biologics in Rheumatoid Arthritis Genetics and Genomics Study Syndicate; Barton, A.; Worthington, J.; Isaacs, J.D.; Morgan, A.W.; Wilson, A.G. Association of the Tumour Necrosis Factor-308 Variant with Differential Response to Anti-TNF Agents in the Treatment of Rheumatoid Arthritis. Hum. Mol. Genet. 2008, 17, 3532–3538. [Google Scholar] [CrossRef] [Green Version]
- Pavy, S.; Toonen, E.J.M.; Miceli-Richard, C.; Barrera, P.; van Riel, P.L.C.M.; Criswell, L.A.; Mariette, X.; Coenen, M.J.H. Tumour Necrosis Factor Alpha -308G->A Polymorphism Is Not Associated with Response to TNFalpha Blockers in Caucasian Patients with Rheumatoid Arthritis: Systematic Review and Meta-Analysis. Ann. Rheum. Dis. 2010, 69, 1022–1028. [Google Scholar] [CrossRef]
- Felson, D.T.; Anderson, J.J.; Boers, M.; Bombardier, C.; Furst, D.; Goldsmith, C.; Katz, L.M.; Lightfoot, R.; Paulus, H.; Strand, V. American College of Rheumatology. Preliminary Definition of Improvement in Rheumatoid Arthritis. Arthritis Rheum. 1995, 38, 727–735. [Google Scholar] [CrossRef]
- Lichtenstein, L.; Ron, Y.; Kivity, S.; Ben-Horin, S.; Israeli, E.; Fraser, G.M.; Dotan, I.; Chowers, Y.; Confino-Cohen, R.; Weiss, B. Infliximab-Related Infusion Reactions: Systematic Review. J. Crohns Colitis 2015, 9, 806–815. [Google Scholar] [CrossRef] [Green Version]
- Hanauer, S.B.; Feagan, B.G.; Lichtenstein, G.R.; Mayer, L.F.; Schreiber, S.; Colombel, J.F.; Rachmilewitz, D.; Wolf, D.C.; Olson, A.; Bao, W.; et al. Maintenance Infliximab for Crohn’s Disease: The ACCENT I Randomised Trial. Lancet 2002, 359, 1541–1549. [Google Scholar] [CrossRef]
- Rutgeerts, P.; Sandborn, W.J.; Feagan, B.G.; Reinisch, W.; Olson, A.; Johanns, J.; Travers, S.; Rachmilewitz, D.; Hanauer, S.B.; Lichtenstein, G.R.; et al. Infliximab for Induction and Maintenance Therapy for Ulcerative Colitis. N. Engl. J. Med. 2005, 353, 2462–2476. [Google Scholar] [CrossRef] [Green Version]
- Markham, A.; Lamb, H.M. Infliximab. Drugs 2000, 59, 1341–1359. [Google Scholar] [CrossRef]
- Cañete, J.D.; Suárez, B.; Hernández, M.V.; Sanmartí, R.; Rego, I.; Celis, R.; Moll, C.; Pinto, J.A.; Blanco, F.J.; Lozano, F. Influence of Variants of Fc Gamma Receptors IIA and IIIA on the American College of Rheumatology and European League Against Rheumatism Responses to Anti-Tumour Necrosis Factor Alpha Therapy in Rheumatoid Arthritis. Ann. Rheum. Dis. 2009, 68, 1547–1552. [Google Scholar] [CrossRef]
- Martinez, A.; Salido, M.; Bonilla, G.; Pascual-Salcedo, D.; Fernandez-Arquero, M.; de Miguel, S.; Balsa, A.; de la Concha, E.G.; Fernandez-Gutierrez, B. Association of the Major Histocompatibility Complex with Response to Infliximab Therapy in Rheumatoid Arthritis Patients. Arthritis Rheum. 2004, 50, 1077–1082. [Google Scholar] [CrossRef]
- Fabris, M.; Di Poi, E.; D’Elia, A.; Damante, G.; Sinigaglia, L.; Ferraccioli, G. Tumor Necrosis Factor-Alpha Gene Polymorphism in Severe and Mild-Moderate Rheumatoid Arthritis. J. Rheumatol. 2002, 29, 29–33. [Google Scholar] [PubMed]
- Swierkot, J.; Bogunia-Kubik, K.; Nowak, B.; Bialowas, K.; Korman, L.; Gebura, K.; Kolossa, K.; Jeka, S.; Wiland, P. Analysis of Associations between Polymorphisms within Genes Coding for Tumour Necrosis Factor (TNF)-Alpha and TNF Receptors and Responsiveness to TNF-Alpha Blockers in Patients with Rheumatoid Arthritis. Jt. Bone Spine 2015, 82, 94–99. [Google Scholar] [CrossRef]
- van Sorge, N.M.; van der Pol, W.-L.; van de Winkel, J.G.J. FcgammaR Polymorphisms: Implications for Function, Disease Susceptibility and Immunotherapy. Tissue Antigens 2003, 61, 189–202. [Google Scholar] [CrossRef] [PubMed]
- Barrera, P.; Joosten, L.A.; den Broeder, A.A.; van de Putte, L.B.; van Riel, P.L.; van den Berg, W.B. Effects of Treatment with a Fully Human Anti-Tumour Necrosis Factor Alpha Monoclonal Antibody on the Local and Systemic Homeostasis of Interleukin 1 and TNFalpha in Patients with Rheumatoid Arthritis. Ann. Rheum. Dis. 2001, 60, 660–669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edwards, J.C.; Cambridge, G.; Abrahams, V.M. Do Self-Perpetuating B Lymphocytes Drive Human Autoimmune Disease? Immunology 1999, 97, 188–196. [Google Scholar] [CrossRef]
- Dörner, T.; Jacobi, A.M.; Lipsky, P.E. B Cells in Autoimmunity. Arthritis Res. Ther. 2009, 11, 247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McInnes, I.B.; Schett, G. The Pathogenesis of Rheumatoid Arthritis. N. Engl. J. Med. 2011, 365, 2205–2219. [Google Scholar] [CrossRef] [Green Version]
- Mota, P.; Reddy, V.; Isenberg, D. Improving B-Cell Depletion in Systemic Lupus Erythematosus and Rheumatoid Arthritis. Expert Rev. Clin. Immunol. 2017, 13, 667–676. [Google Scholar] [CrossRef]
- Salles, G.; Barrett, M.; Foà, R.; Maurer, J.; O’Brien, S.; Valente, N.; Wenger, M.; Maloney, D.G. Rituximab in B-Cell Hematologic Malignancies: A Review of 20 Years of Clinical Experience. Adv. Ther. 2017, 34, 2232–2273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- García-Muñoz, R.; Quero, C.; Pérez-Persona, E.; Domingo-García, A.; Pérez-López, C.; Villaescusa-de-la-Rosa, T.; Martínez-Castro, A.M.; Arguiñano-Pérez, J.M.; Parra-Cuadrado, J.F.; Panizo, C. Safety of Switching from Intravenous to Subcutaneous Rituximab during First-Line Treatment of Patients with Non-Hodgkin Lymphoma: The Spanish Population of the MabRella Study. Br. J. Haematol. 2020, 188, 661–673. [Google Scholar] [CrossRef] [Green Version]
- Cylwik, B.; Gruszewska, E.; Gindzienska-Sieskiewicz, E.; Kowal-Bielecka, O.; Chrostek, L. Serum Profile of Transferrin Isoforms in Rheumatoid Arthritis Treated with Biological Drugs. Clin. Biochem. 2019, 74, 31–35. [Google Scholar] [CrossRef]
- von Borstel, A.; Land, J.; Abdulahad, W.H.; Rutgers, A.; Stegeman, C.A.; Diepstra, A.; Heeringa, P.; Sanders, J.S. CD27+CD38hi B Cell Frequency During Remission Predicts Relapsing Disease in Granulomatosis With Polyangiitis Patients. Front. Immunol. 2019, 10, 2221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Golay, J.; Semenzato, G.; Rambaldi, A.; Foà, R.; Gaidano, G.; Gamba, E.; Pane, F.; Pinto, A.; Specchia, G.; Zaja, F.; et al. Lessons for the Clinic from Rituximab Pharmacokinetics and Pharmacodynamics. mAbs 2013, 5, 826–837. [Google Scholar] [CrossRef] [PubMed]
- Weiner, G.J. Rituximab: Mechanism of Action. Semin. Hematol. 2010, 47, 115–123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uchida, J.; Hamaguchi, Y.; Oliver, J.A.; Ravetch, J.V.; Poe, J.C.; Haas, K.M.; Tedder, T.F. The Innate Mononuclear Phagocyte Network Depletes B Lymphocytes through Fc Receptor-Dependent Mechanisms during Anti-CD20 Antibody Immunotherapy. J. Exp. Med. 2004, 199, 1659–1669. [Google Scholar] [CrossRef] [PubMed]
- Bergantini, L.; d’Alessandro, M.; Cameli, P.; Vietri, L.; Vagaggini, C.; Perrone, A.; Sestini, P.; Frediani, B.; Bargagli, E. Effects of Rituximab Therapy on B Cell Differentiation and Depletion. Clin. Rheumatol. 2020, 39, 1415–1421. [Google Scholar] [CrossRef] [PubMed]
- Jiménez Morales, A.; Maldonado-Montoro, M.; Martínez de la Plata, J.E.; Pérez Ramírez, C.; Daddaoua, A.; Alarcón Payer, C.; Expósito Ruiz, M.; García Collado, C. FCGR2A/FCGR3A Gene Polymorphisms and Clinical Variables as Predictors of Response to Tocilizumab and Rituximab in Patients With Rheumatoid Arthritis. J. Clin. Pharmacol. 2019, 59, 517–531. [Google Scholar] [CrossRef]
- Quartuccio, L.; Fabris, M.; Pontarini, E.; Salvin, S.; Zabotti, A.; Benucci, M.; Manfredi, M.; Biasi, D.; Ravagnani, V.; Atzeni, F.; et al. The 158VV Fcgamma Receptor 3A Genotype Is Associated with Response to Rituximab in Rheumatoid Arthritis: Results of an Italian Multicentre Study. Ann. Rheum. Dis. 2014, 73, 716–721. [Google Scholar] [CrossRef] [PubMed]
- Ruyssen-Witrand, A.; Rouanet, S.; Combe, B.; Dougados, M.; Le Loët, X.; Sibilia, J.; Tebib, J.; Mariette, X.; Constantin, A. Fcγ Receptor Type IIIA Polymorphism Influences Treatment Outcomes in Patients with Rheumatoid Arthritis Treated with Rituximab. Ann. Rheum. Dis. 2012, 71, 875–877. [Google Scholar] [CrossRef]
- Pál, I.; Szamosi, S.; Hodosi, K.; Szekanecz, Z.; Váróczy, L. Effect of Fcγ-Receptor 3a (FCGR3A) Gene Polymorphisms on Rituximab Therapy in Hungarian Patients with Rheumatoid Arthritis. RMD Open 2017, 3, e000485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarsour, K.; Greenberg, J.; Johnston, J.A.; Nelson, D.R.; O’Brien, L.A.; Oddoux, C.; Ostrer, H.; Pearlman, A.; Reed, G. The Role of the FcGRIIIa Polymorphism in Modifying the Association between Treatment and Outcome in Patients with Rheumatoid Arthritis Treated with Rituximab versus TNF-α Antagonist Therapies. Clin. Exp. Rheumatol. 2013, 31, 189–194. [Google Scholar]
- Ruyssen-Witrand, A.; Rouanet, S.; Combe, B.; Dougados, M.; Le Loët, X.; Sibilia, J.; Tebib, J.; Mariette, X.; Constantin, A. Association between -871C>T Promoter Polymorphism in the B-Cell Activating Factor Gene and the Response to Rituximab in Rheumatoid Arthritis Patients. Rheumatology 2013, 52, 636–641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fabris, M.; Quartuccio, L.; Lombardi, S.; Benucci, M.; Manfredi, M.; Saracco, M.; Atzeni, F.; Morassi, P.; Cimmino, M.A.; Pontarini, E.; et al. Study on the Possible Role of the -174G>C IL-6 Promoter Polymorphism in Predicting Response to Rituximab in Rheumatoid Arthritis. Reumatismo 2010, 62, 253–258. [Google Scholar] [CrossRef] [PubMed]
- Daïen, C.I.; Fabre, S.; Rittore, C.; Soler, S.; Daïen, V.; Tejedor, G.; Cadart, D.; Molinari, N.; Daurès, J.-P.; Jorgensen, C.; et al. TGF Beta1 Polymorphisms Are Candidate Predictors of the Clinical Response to Rituximab in Rheumatoid Arthritis. Jt. Bone Spine 2012, 79, 471–475. [Google Scholar] [CrossRef] [PubMed]
- Chihara, N.; Aranami, T.; Sato, W.; Miyazaki, Y.; Miyake, S.; Okamoto, T.; Ogawa, M.; Toda, T.; Yamamura, T. Interleukin 6 Signaling Promotes Anti-Aquaporin 4 Autoantibody Production from Plasmablasts in Neuromyelitis Optica. Proc. Natl. Acad. Sci. USA 2011, 108, 3701–3706. [Google Scholar] [CrossRef] [Green Version]
- Narazaki, M.; Tanaka, T.; Kishimoto, T. The Role and Therapeutic Targeting of IL-6 in Rheumatoid Arthritis. Expert Rev. Clin. Immunol. 2017, 13, 535–551. [Google Scholar] [CrossRef]
- Mihara, M.; Kasutani, K.; Okazaki, M.; Nakamura, A.; Kawai, S.; Sugimoto, M.; Matsumoto, Y.; Ohsugi, Y. Tocilizumab Inhibits Signal Transduction Mediated by Both MIL-6R and SIL-6R, but Not by the Receptors of Other Members of IL-6 Cytokine Family. Int. Immunopharmacol. 2005, 5, 1731–1740. [Google Scholar] [CrossRef]
- Snir, A.; Kessel, A.; Haj, T.; Rosner, I.; Slobodin, G.; Toubi, E. Anti-IL-6 Receptor Antibody (Tocilizumab): A B Cell Targeting Therapy. Clin. Exp. Rheumatol. 2011, 29, 697–700. [Google Scholar] [CrossRef] [Green Version]
- Scott, L.J. Tocilizumab: A Review in Rheumatoid Arthritis. Drugs 2017, 77, 1865–1879. [Google Scholar] [CrossRef] [Green Version]
- Dhillon, S. Intravenous Tocilizumab: A Review of Its Use in Adults with Rheumatoid Arthritis. BioDrugs Clin. Immunother. Biopharm. Gene Ther. 2014, 28, 75–106. [Google Scholar] [CrossRef]
- Wang, J.; Bansal, A.T.; Martin, M.; Germer, S.; Benayed, R.; Essioux, L.; Lee, J.S.; Begovich, A.; Hemmings, A.; Kenwright, A.; et al. Genome-Wide Association Analysis Implicates the Involvement of Eight Loci with Response to Tocilizumab for the Treatment of Rheumatoid Arthritis. Pharm. J. 2013, 13, 235–241. [Google Scholar] [CrossRef] [Green Version]
- Maldonado-Montoro, M.; Cañadas-Garre, M.; González-Utrilla, A.; Ángel Calleja-Hernández, M. Influence of IL6R Gene Polymorphisms in the Effectiveness to Treatment with Tocilizumab in Rheumatoid Arthritis. Pharm. J. 2018, 18, 167–172. [Google Scholar] [CrossRef] [PubMed]
- Gregersen, P.K.; Silver, J.; Winchester, R.J. The Shared Epitope Hypothesis. An Approach to Understanding the Molecular Genetics of Susceptibility to Rheumatoid Arthritis. Arthritis Rheum. 1987, 30, 1205–1213. [Google Scholar] [CrossRef] [PubMed]
- Guo, Q.; Wang, Y.; Xu, D.; Nossent, J.; Pavlos, N.J.; Xu, J. Rheumatoid Arthritis: Pathological Mechanisms and Modern Pharmacologic Therapies. Bone Res. 2018, 6, 15. [Google Scholar] [CrossRef]
- Pallio, G.; Mannino, F.; Irrera, N.; Eid, A.H.; Squadrito, F.; Bitto, A. Polymorphisms Involved in Response to Biological Agents Used in Rheumatoid Arthritis. Biomolecules 2020, 10, 1203. [Google Scholar] [CrossRef]
- Kaijzel, E.L.; van Dongen, H.; Bakker, A.M.; Breedveld, F.C.; Huizinga, T.W.J.; Verweij, C.L. Relationship of Polymorphisms of the Interleukin-1 Gene Cluster to Occurrence and Severity of Rheumatoid Arthritis. Tissue Antigens 2002, 59, 122–126. [Google Scholar] [CrossRef] [PubMed]
- Camargo, J.F.; Correa, P.A.; Castiblanco, J.; Anaya, J.-M. Interleukin-1beta Polymorphisms in Colombian Patients with Autoimmune Rheumatic Diseases. Genes Immun. 2004, 5, 609–614. [Google Scholar] [CrossRef] [PubMed]
- Buchs, N.; di Giovine, F.S.; Silvestri, T.; Vannier, E.; Duff, G.W.; Miossec, P. IL-1B and IL-1Ra Gene Polymorphisms and Disease Severity in Rheumatoid Arthritis: Interaction with Their Plasma Levels. Genes Immun. 2001, 2, 222–228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arman, A.; Yilmaz, B.; Coker, A.; Inanc, N.; Direskeneli, H. Interleukin-1 Receptor Antagonist (IL-1RN) and Interleukin-1B Gene Polymorphisms in Turkish Patients with Rheumatoid Arthritis. Clin. Exp. Rheumatol. 2006, 24, 643–648. [Google Scholar]
- Tolusso, B.; Pietrapertosa, D.; Morelli, A.; De Santis, M.; Gremese, E.; Farina, G.; Carniello, S.G.; Del Frate, M.; Ferraccioli, G. IL-1B and IL-1RN Gene Polymorphisms in Rheumatoid Arthritis: Relationship with Protein Plasma Levels and Response to Therapy. Pharmacogenomics 2006, 7, 683–695. [Google Scholar] [CrossRef]
- Nordang, G.B.N.; Viken, M.K.; Hollis-Moffatt, J.E.; Merriman, T.R.; Førre, Ø.T.; Helgetveit, K.; Kvien, T.K.; Lie, B.A. Association Analysis of the Interleukin 17A Gene in Caucasian Rheumatoid Arthritis Patients from Norway and New Zealand. Rheumatology 2009, 48, 367–370. [Google Scholar] [CrossRef] [Green Version]
- Paradowska-Gorycka, A.; Wojtecka-Lukasik, E.; Trefler, J.; Wojciechowska, B.; Lacki, J.K.; Maslinski, S. Association between IL-17F Gene Polymorphisms and Susceptibility to and Severity of Rheumatoid Arthritis (RA). Scand. J. Immunol. 2010, 72, 134–141. [Google Scholar] [CrossRef]
- Han, L.; Lee, H.S.; Yoon, J.H.; Choi, W.S.; Park, Y.G.; Nam, S.W.; Lee, J.Y.; Park, W.S. Association of IL-17A and IL-17F Single Nucleotide Polymorphisms with Susceptibility to Osteoarthritis in a Korean Population. Gene 2014, 533, 119–122. [Google Scholar] [CrossRef] [PubMed]
- Erkol İnal, E.; Görükmez, O.; Dündar, Ü.; Görükmez, Ö.; Yener, M.; Özemri Sağ, Ş.; Yakut, T. The Influence of Polymorphisms of Interleukin-17A and -17F Genes on Susceptibility and Activity of Rheumatoid Arthritis. Genet. Test. Mol. Biomark. 2015, 19, 461–464. [Google Scholar] [CrossRef] [PubMed]
- Shen, L.; Zhang, H.; Yan, T.; Zhou, G.; Liu, R. Association between Interleukin 17A Polymorphisms and Susceptibility to Rheumatoid Arthritis in a Chinese Population. Gene 2015, 566, 18–22. [Google Scholar] [CrossRef] [PubMed]
- Bogunia-Kubik, K.; Świerkot, J.; Malak, A.; Wysoczańska, B.; Nowak, B.; Białowąs, K.; Gębura, K.; Korman, L.; Wiland, P. IL-17A, IL-17F and IL-23R Gene Polymorphisms in Polish Patients with Rheumatoid Arthritis. Arch. Immunol. Ther. Exp. 2015, 63, 215–221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carvalho, C.N.; do Carmo, R.F.; Duarte, A.L.P.; Carvalho, A.A.T.; Leão, J.C.; Gueiros, L.A. IL-17A and IL-17F Polymorphisms in Rheumatoid Arthritis and Sjögren’s Syndrome. Clin. Oral Investig. 2016, 20, 495–502. [Google Scholar] [CrossRef] [PubMed]
- Pawlik, A.; Kotrych, D.; Malinowski, D.; Dziedziejko, V.; Czerewaty, M.; Safranow, K. IL17A and IL17F Gene Polymorphisms in Patients with Rheumatoid Arthritis. BMC Musculoskelet. Disord. 2016, 17, 208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Louahchi, S.; Allam, I.; Berkani, L.; Boucharef, A.; Abdesemed, A.; Khaldoun, N.; Nebbab, A.; Ladjouze, A.; Djidjik, R. Association Study of Single Nucleotide Polymorphisms of IL23R and IL17 in Rheumatoid Arthritis in the Algerian Population. Acta Reumatol. Port. 2016, 41, 151–157. [Google Scholar] [PubMed]
- Marwa, O.S.; Kalthoum, T.; Wajih, K.; Kamel, H. Association of IL17A and IL17F Genes with Rheumatoid Arthritis Disease and the Impact of Genetic Polymorphisms on Response to Treatment. Immunol. Lett. 2017, 183, 24–36. [Google Scholar] [CrossRef]
- Gomes da Silva, I.I.F.; Angelo, H.D.; Rushansky, E.; Mariano, M.H.; de Mascena Diniz Maia, M.; de Souza, P.R.E. Interleukin (IL)-23 Receptor, IL-17A and IL-17F Gene Polymorphisms in Brazilian Patients with Rheumatoid Arthritis. Arch. Immunol. Ther. Exp. 2017, 65, 537–543. [Google Scholar] [CrossRef]
- de la Peña, M.G.; Cruz, R.M.; Guerrero, E.G.; López, A.G.; Molina, G.P.; González, N.E.H. Polymorphism Rs2275913 of Interleukin-17A Is Related to More Intensive Therapy with Disease-Modifying Anti Rheumatic Drugs in Mexican Patients with Rheumatoid Arthritis. Acta Reumatol. Port. 2017, 42, 155–161. [Google Scholar]
- Lee, Y.H.; Bae, S.-C. Associations between Circulating IL-17 Levels and Rheumatoid Arthritis and between IL-17 Gene Polymorphisms and Disease Susceptibility: A Meta-Analysis. Postgrad. Med. J. 2017, 93, 465–471. [Google Scholar] [CrossRef] [PubMed]
- Dhaouadi, T.; Chahbi, M.; Haouami, Y.; Sfar, I.; Abdelmoula, L.; Ben Abdallah, T.; Gorgi, Y. IL-17A, IL-17RC Polymorphisms and IL17 Plasma Levels in Tunisian Patients with Rheumatoid Arthritis. PLoS ONE 2018, 13, e0194883. [Google Scholar] [CrossRef] [PubMed]
- Agonia, I.; Couras, J.; Cunha, A.; Andrade, A.J.; Macedo, J.; Sousa-Pinto, B. IL-17, IL-21 and IL-22 Polymorphisms in Rheumatoid Arthritis: A Systematic Review and Meta-Analysis. Cytokine 2020, 125, 154813. [Google Scholar] [CrossRef]
- Nisar, H.; Pasha, U.; Mirza, M.U.; Abid, R.; Hanif, K.; Kadarmideen, H.N.; Sadaf, S. Impact of IL-17F 7488T/C Functional Polymorphism on Progressive Rheumatoid Arthritis: Novel Insight from the Molecular Dynamic Simulations. Immunol. Investig. 2021, 50, 416–426. [Google Scholar] [CrossRef]
- Amin, A.; Sheikh, N.; Mukhtar, M.; Saleem, T.; Akhtar, T.; Fatima, N.; Mehmood, R. Association of Interleukin-17 Gene Polymorphisms with the Onset of Rheumatoid Arthritis. Immunobiology 2021, 226, 152045. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.; Li, Y.; Li, L.; Zhang, G.; Zhang, F.; Tang, Y.; Zhou, L.; Yang, Y.; Li, J. Association between the Interleukin (IL)-17A Rs2275913 Polymorphism and Rheumatoid Arthritis Susceptibility: A Meta-Analysis and Trial Sequential Analysis. J. Int. Med. Res. 2021, 49, 3000605211053233. [Google Scholar] [CrossRef] [PubMed]
- Shao, M.; Xu, S.; Yang, H.; Xu, W.; Deng, J.; Chen, Y.; Gao, X.; Guan, S.; Xu, S.; Shuai, Z.; et al. Association between IL-17A and IL-17F Gene Polymorphism and Susceptibility in Inflammatory Arthritis: A Meta-Analysis. Clin. Immunol. 2020, 213, 108374. [Google Scholar] [CrossRef]
- Orozco, G.; Rueda, B.; Robledo, G.; García, A.; Martín, J. Investigation of the IL23R Gene in a Spanish Rheumatoid Arthritis Cohort. Hum. Immunol. 2007, 68, 681–684. [Google Scholar] [CrossRef] [PubMed]
- Faragó, B.; Magyari, L.; Sáfrány, E.; Csöngei, V.; Járomi, L.; Horvatovich, K.; Sipeky, C.; Maász, A.; Radics, J.; Gyetvai, A.; et al. Functional Variants of Interleukin-23 Receptor Gene Confer Risk for Rheumatoid Arthritis but Not for Systemic Sclerosis. Ann. Rheum. Dis. 2008, 67, 248–250. [Google Scholar] [CrossRef] [PubMed]
- Park, J.H.; Kim, Y.J.; Park, B.L.; Bae, J.S.; Shin, H.D.; Bae, S.-C. Lack of Association between Interleukin 23 Receptor Gene Polymorphisms and Rheumatoid Arthritis Susceptibility. Rheumatol. Int. 2009, 29, 781–786. [Google Scholar] [CrossRef] [PubMed]
- Hamdy, G.; Darweesh, H.; Khattab, E.A.; Fawzy, S.; Fawzy, E.; Sheta, M. Evidence of Association of Interleukin-23 Receptor Gene Polymorphisms with Egyptian Rheumatoid Arthritis Patients. Hum. Immunol. 2015, 76, 417–420. [Google Scholar] [CrossRef] [PubMed]
- Tagiev, A.F.; Surin, V.L.; Osokina, A.V.; Luk’ianenko, A.V.; Smirnova, O.V.; Tsetaeva, N.V.; Mikhaĭlova, E.A.; Isaev, V.G.; Grineva, N.I. Polymorphism at codon 117 of the granulocyte-macrophage colony-stimulating factor (GM-CSF) gene. Genetika 1995, 31, 1370–1374. [Google Scholar] [PubMed]
- He, J.-Q.; Jian, R.; Moira, C.-Y.; Allan, B.; Helen, D.-W.; Peter, P.; Andrew, S. Polymorphisms of the GM-CSF Genes and the Development of Atopic Diseases in at-Risk Children. Chest 2003, 123, 438S. [Google Scholar] [CrossRef] [PubMed]
- Rafatpanah, H.; Bennett, E.; Pravica, V.; McCoy, M.J.; David, T.J.; Hutchinson, I.V.; Arkwright, P.D. Association between Novel GM-CSF Gene Polymorphisms and the Frequency and Severity of Atopic Dermatitis. J. Allergy Clin. Immunol. 2003, 112, 593–598. [Google Scholar] [CrossRef]
- Saeki, H.; Tsunemi, Y.; Asano, N.; Nakamura, K.; Sekiya, T.; Hirai, K.; Kakinuma, T.; Fujita, H.; Kagami, S.; Tamaki, K. Analysis of GM-CSF Gene Polymorphisms (3606T/C and 3928C/T) in Japanese Patients with Atopic Dermatitis. Clin. Exp. Dermatol. 2006, 31, 278–280. [Google Scholar] [CrossRef]
- Wilkowska, A.; Gleń, J.; Zabłotna, M.; Trzeciak, M.; Ryduchowska, M.; Sobjanek, M.; Nedoszytko, B.; Nowicki, R.; Sokołowska-Wojdyło, M. The Association of GM-CSF-677A/C Promoter Gene Polymorphism with the Occurrence and Severity of Atopic Dermatitis in a Polish Population. Int. J. Dermatol. 2014, 53, e172–e174. [Google Scholar] [CrossRef]
- Abdelaal, E.B.; Abdelsamie, H.M.; Attia, S.M.; Amr, K.S.; Eldahshan, R.M.; Elsaie, M.L. Association of a Novel Granulocyte-Macrophage Colony-Stimulating Factor (GM-CSF)-3928C/T and GM-CSF(3606T⁄C) Promoter Gene Polymorphisms with the Pathogenesis and Severity of Acne Vulgaris: A Case-Controlled Study. J. Cosmet. Dermatol. 2021, 20, 3679–3683. [Google Scholar] [CrossRef]
- Assmann, G.; Koenig, J.; Pfreundschuh, M.; Epplen, J.T.; Kekow, J.; Roemer, K.; Wieczorek, S. Genetic Variations in Genes Encoding RANK, RANKL, and OPG in Rheumatoid Arthritis: A Case-Control Study. J. Rheumatol. 2010, 37, 900–904. [Google Scholar] [CrossRef]
- Xu, S.; Ma, X.-X.; Hu, L.-W.; Peng, L.-P.; Pan, F.-M.; Xu, J.-H. Single Nucleotide Polymorphism of RANKL and OPG Genes May Play a Role in Bone and Joint Injury in Rheumatoid Arthritis. Clin. Exp. Rheumatol. 2014, 32, 697–704. [Google Scholar]
- Yang, H.; Liu, W.; Zhou, X.; Rui, H.; Zhang, H.; Liu, R. The Association between RANK, RANKL and OPG Gene Polymorphisms and the Risk of Rheumatoid Arthritis: A Case-Controlled Study and Meta-Analysis. Biosci. Rep. 2019, 39, BSR20182356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdi, S.; Bukhari, I.; Ansari, M.G.A.; BinBaz, R.A.; Mohammed, A.K.; Hussain, S.D.; Aljohani, N.; Al-Daghri, N.M. Association of Polymorphisms in RANK and RANKL Genes with Osteopenia in Arab Postmenopausal Women. Dis. Markers 2020, 2020, 1285216. [Google Scholar] [CrossRef] [PubMed]
- Wielińska, J.; Kolossa, K.; Świerkot, J.; Dratwa, M.; Iwaszko, M.; Bugaj, B.; Wysoczańska, B.; Chaszczewska-Markowska, M.; Jeka, S.; Bogunia-Kubik, K. Polymorphisms within the RANK and RANKL Encoding Genes in Patients with Rheumatoid Arthritis: Association with Disease Progression and Effectiveness of the Biological Treatment. Arch. Immunol. Ther. Exp. 2020, 68, 24. [Google Scholar] [CrossRef] [PubMed]
- Łacina, P.; Butrym, A.; Humiński, M.; Dratwa, M.; Frontkiewicz, D.; Mazur, G.; Bogunia-Kubik, K. Association of RANK and RANKL Gene Polymorphism with Survival and Calcium Levels in Multiple Myeloma. Mol. Carcinog. 2021, 60, 106–112. [Google Scholar] [CrossRef] [PubMed]
- Abdi, S.; Binbaz, R.A.; Mohammed, A.K.; Ansari, M.G.A.; Wani, K.; Amer, O.E.; Alnaami, A.M.; Aljohani, N.; Al-Daghri, N.M. Association of RANKL and OPG Gene Polymorphism in Arab Women with and without Osteoporosis. Genes 2021, 12, 200. [Google Scholar] [CrossRef] [PubMed]
- Gabay, C.; Lamacchia, C.; Palmer, G. IL-1 Pathways in Inflammation and Human Diseases. Nat. Rev. Rheumatol. 2010, 6, 232–241. [Google Scholar] [CrossRef]
- Schett, G.; Dayer, J.-M.; Manger, B. Interleukin-1 Function and Role in Rheumatic Disease. Nat. Rev. Rheumatol. 2016, 12, 14–24. [Google Scholar] [CrossRef] [PubMed]
- Alten, R.; Gomez-Reino, J.; Durez, P.; Beaulieu, A.; Sebba, A.; Krammer, G.; Preiss, R.; Arulmani, U.; Widmer, A.; Gitton, X.; et al. Efficacy and Safety of the Human Anti-IL-1beta Monoclonal Antibody Canakinumab in Rheumatoid Arthritis: Results of a 12-Week, Phase II, Dose-Finding Study. BMC Musculoskelet. Disord. 2011, 12, 153. [Google Scholar] [CrossRef] [PubMed]
- Geiler, J.; McDermott, M.F. Gevokizumab, an Anti-IL-1β MAb for the Potential Treatment of Type 1 and 2 Diabetes, Rheumatoid Arthritis and Cardiovascular Disease. Curr. Opin. Mol. Ther. 2010, 12, 755–769. [Google Scholar] [PubMed]
- Miossec, P.; Korn, T.; Kuchroo, V.K. Interleukin-17 and Type 17 Helper T Cells. N. Engl. J. Med. 2009, 361, 888–898. [Google Scholar] [CrossRef] [Green Version]
- Weaver, C.T.; Hatton, R.D.; Mangan, P.R.; Harrington, L.E. IL-17 Family Cytokines and the Expanding Diversity of Effector T Cell Lineages. Annu. Rev. Immunol. 2007, 25, 821–852. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaffen, S.L.; Jain, R.; Garg, A.V.; Cua, D.J. The IL-23-IL-17 Immune Axis: From Mechanisms to Therapeutic Testing. Nat. Rev. Immunol. 2014, 14, 585–600. [Google Scholar] [CrossRef] [PubMed]
- Noack, M.; Miossec, P. Selected Cytokine Pathways in Rheumatoid Arthritis. Semin. Immunopathol. 2017, 39, 365–383. [Google Scholar] [CrossRef] [PubMed]
- Kunwar, S.; Dahal, K.; Sharma, S. Anti-IL-17 Therapy in Treatment of Rheumatoid Arthritis: A Systematic Literature Review and Meta-Analysis of Randomized Controlled Trials. Rheumatol. Int. 2016, 36, 1065–1075. [Google Scholar] [CrossRef]
- Wu, D.; Hou, S.-Y.; Zhao, S.; Hou, L.-X.; Jiao, T.; Xu, N.-N.; Zhang, N. Meta-Analysis of IL-17 Inhibitors in Two Populations of Rheumatoid Arthritis Patients: Biologic-Naïve or Tumor Necrosis Factor Inhibitor Inadequate Responders. Clin. Rheumatol. 2019, 38, 2747–2756. [Google Scholar] [CrossRef]
- Huang, Y.; Fan, Y.; Liu, Y.; Xie, W.; Zhang, Z. Efficacy and Safety of Secukinumab in Active Rheumatoid Arthritis with an Inadequate Response to Tumor Necrosis Factor Inhibitors: A Meta-Analysis of Phase III Randomized Controlled Trials. Clin. Rheumatol. 2019, 38, 2765–2776. [Google Scholar] [CrossRef]
- Martin, D.A.; Churchill, M.; Flores-Suarez, L.; Cardiel, M.H.; Wallace, D.; Martin, R.; Phillips, K.; Kaine, J.L.; Dong, H.; Salinger, D.; et al. A Phase Ib Multiple Ascending Dose Study Evaluating Safety, Pharmacokinetics, and Early Clinical Response of Brodalumab, a Human Anti-IL-17R Antibody, in Methotrexate-Resistant Rheumatoid Arthritis. Arthritis Res. Ther. 2013, 15, R164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glatt, S.; Taylor, P.C.; McInnes, I.B.; Schett, G.; Landewé, R.; Baeten, D.; Ionescu, L.; Strimenopoulou, F.; Watling, M.I.L.; Shaw, S. Efficacy and Safety of Bimekizumab as Add-on Therapy for Rheumatoid Arthritis in Patients with Inadequate Response to Certolizumab Pegol: A Proof-of-Concept Study. Ann. Rheum. Dis. 2019, 78, 1033–1040. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kleinschek, M.A.; Muller, U.; Brodie, S.J.; Stenzel, W.; Kohler, G.; Blumenschein, W.M.; Straubinger, R.K.; McClanahan, T.; Kastelein, R.A.; Alber, G. IL-23 Enhances the Inflammatory Cell Response in Cryptococcus Neoformans Infection and Induces a Cytokine Pattern Distinct from IL-12. J. Immunol. 2006, 176, 1098–1106. [Google Scholar] [CrossRef] [Green Version]
- Bunte, K.; Beikler, T. Th17 Cells and the IL-23/IL-17 Axis in the Pathogenesis of Periodontitis and Immune-Mediated Inflammatory Diseases. Int. J. Mol. Sci. 2019, 20, 3394. [Google Scholar] [CrossRef] [Green Version]
- Luo, J.; Wu, S.-J.; Lacy, E.R.; Orlovsky, Y.; Baker, A.; Teplyakov, A.; Obmolova, G.; Heavner, G.A.; Richter, H.-T.; Benson, J. Structural Basis for the Dual Recognition of IL-12 and IL-23 by Ustekinumab. J. Mol. Biol. 2010, 402, 797–812. [Google Scholar] [CrossRef] [PubMed]
- Machado, Á.; Torres, T. Guselkumab for the Treatment of Psoriasis. BioDrugs Clin. Immunother. Biopharm. Gene Ther. 2018, 32, 119–128. [Google Scholar] [CrossRef]
- Smolen, J.S.; Agarwal, S.K.; Ilivanova, E.; Xu, X.L.; Miao, Y.; Zhuang, Y.; Nnane, I.; Radziszewski, W.; Greenspan, A.; Beutler, A.; et al. A Randomised Phase II Study Evaluating the Efficacy and Safety of Subcutaneously Administered Ustekinumab and Guselkumab in Patients with Active Rheumatoid Arthritis despite Treatment with Methotrexate. Ann. Rheum. Dis. 2017, 76, 831–839. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kondo, N.; Kuroda, T.; Kobayashi, D. Cytokine Networks in the Pathogenesis of Rheumatoid Arthritis. Int. J. Mol. Sci. 2021, 22, 10922. [Google Scholar] [CrossRef]
- Haworth, C.; Brennan, F.M.; Chantry, D.; Turner, M.; Maini, R.N.; Feldmann, M. Expression of Granulocyte-Macrophage Colony-Stimulating Factor in Rheumatoid Arthritis: Regulation by Tumor Necrosis Factor-Alpha. Eur. J. Immunol. 1991, 21, 2575–2579. [Google Scholar] [CrossRef] [PubMed]
- Crotti, C.; Biggioggero, M.; Becciolini, A.; Agape, E.; Favalli, E.G. Mavrilimumab: A Unique Insight and Update on the Current Status in the Treatment of Rheumatoid Arthritis. Expert Opin. Investig. Drugs 2019, 28, 573–581. [Google Scholar] [CrossRef] [PubMed]
- Di Franco, M.; Gerardi, M.C.; Lucchino, B.; Conti, F. Mavrilimumab: An Evidence Based Review of Its Potential in the Treatment of Rheumatoid Arthritis. Core Evid. 2014, 9, 41–48. [Google Scholar] [CrossRef] [Green Version]
- Anderson, D.M.; Maraskovsky, E.; Billingsley, W.L.; Dougall, W.C.; Tometsko, M.E.; Roux, E.R.; Teepe, M.C.; DuBose, R.F.; Cosman, D.; Galibert, L. A Homologue of the TNF Receptor and Its Ligand Enhance T-Cell Growth and Dendritic-Cell Function. Nature 1997, 390, 175–179. [Google Scholar] [CrossRef]
- Tanaka, S.; Tanaka, Y. RANKL as a Therapeutic Target of Rheumatoid Arthritis. J. Bone Miner. Metab. 2021, 39, 106–112. [Google Scholar] [CrossRef]
- Chiu, Y.G.; Ritchlin, C.T. Denosumab: Targeting the RANKL Pathway to Treat Rheumatoid Arthritis. Expert Opin. Biol. Ther. 2017, 17, 119–128. [Google Scholar] [CrossRef] [Green Version]
- Maranini, B.; Bortoluzzi, A.; Silvagni, E.; Govoni, M. Focus on Sex and Gender: What We Need to Know in the Management of Rheumatoid Arthritis. J. Pers. Med. 2022, 12, 499. [Google Scholar] [CrossRef] [PubMed]
- Bellando-Randone, S.; Russo, E.; Venerito, V.; Matucci-Cerinic, M.; Iannone, F.; Tangaro, S.; Amedei, A. Exploring the Oral Microbiome in Rheumatic Diseases, State of Art and Future Prospective in Personalized Medicine with an AI Approach. J. Pers. Med. 2021, 11, 625. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, Y.; Kanayama, N.; Nakayama, Y.; Matsushima, N. Current Status, Issues and Future Prospects of Personalized Medicine for Each Disease. J. Pers. Med. 2022, 12, 444. [Google Scholar] [CrossRef] [PubMed]

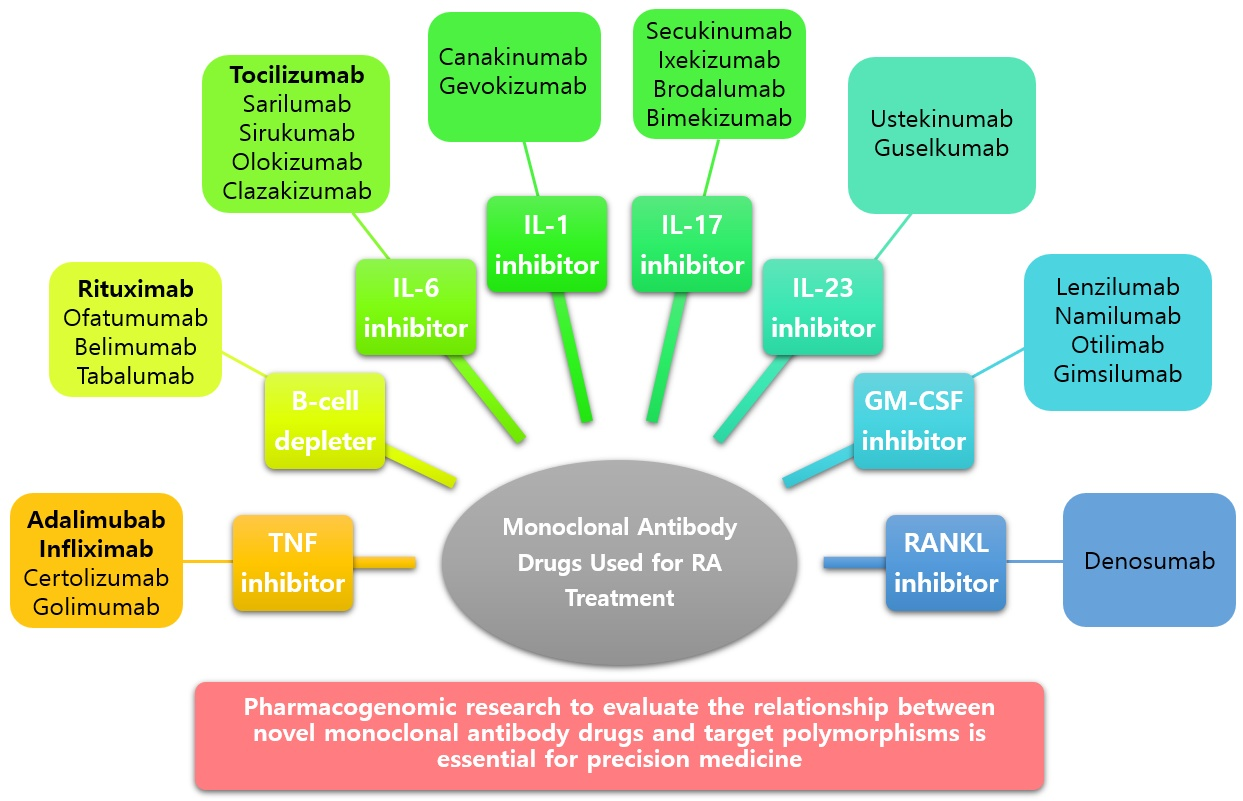{kind=link}
Table 1.
Genetic polymorphisms known to affect adalimumab response in patients with rheumatoid arthritis (RA).
Table 1.
Genetic polymorphisms known to affect adalimumab response in patients with rheumatoid arthritis (RA).
| Biological Agent | Gene | Polymorphism | Clinical Outcome(s) | Refs. |
|---|---|---|---|---|
| Adalimumab | FCGR2A | rs1801274 | Significantly associated with the clinical response | [39] |
| DHX32 | rs12356233 | |||
| RGS12 | rs4690093 | Nominally significantly associated with the response | ||
| IL-6 | rs1800795 (−174 G/C) | Significantly associated with a better response | [40] | |
| PTPN22 | 1858 C/T | No effect on efficacy adalimumab | [41] | |
| TNF | rs1800629 (−308 G/A) | TNF-308 G/G genotype associated with better clinical effect than TNF −308 A/G | [42,43,44,45] | |
| −238A/G, −308A/G and −857C/T | TNF-α locus haplotype (−238G/−308G/−857C) was associated with a lower response | [46] | ||
| TNFR2 | 676 T/G | TNFR2 676 T/T genotype associated with a better clinical response than TNFR2 676 T/G | [47] |
DHX32, DEAH (Asp-Glu-Ala-His)-box polypeptide 32; FCGR2A, Fc fragment of immunoglobulin G receptor IIa; IL-6, interleukin 6; PTPN22, protein tyrosine phosphatase non-receptor type 22; RGS12, regulator of G protein signaling 12; TNF, tumor necrosis factor; TNFR2, tumor necrosis factor receptor 2.
Table 2.
Genetic polymorphisms known to affect infliximab response in patients with RA.
| Biological Agent | Gene | Polymorphism | Clinical Outcome(s) | Refs. |
|---|---|---|---|---|
| Infliximab | FCGR2A | rs1801274 | No effect on response to infliximab | [39] |
| H131R | FCGR2A-RR was associated with a better response compared to RH or HH | [78] | ||
| FCGR3A | V158F | FCGR3A-FF was associated with a better response compared to VV or VF | ||
| RGS12 | rs2857859 | Nominally significantly associated with the response | [39] | |
| PTPN22 | 1858 C/T | No effect on response to infliximab | [41] | |
| MHC | MHC polymorphisms | TNFa11;b4 associated with responders | [79] | |
| TNF | rs1800629 (−308 G/A) | TNF-308 G/G associated with a better response than TNF −308 A/A or A/G | [61,63,64] | |
| −308 G/A | No effect on response to infliximab | [67] | ||
| −308 G/A, −238 G/A, 489 G/A, −857 C/T | [70] | |||
| rs361525 (−238 G/A) | TNF −238G/A was associated with poorer response | [71] | ||
| −238 G/G, +489 A/A | TNF −238 G/G was associated with severe RA | [80] | ||
| −308 G/A, −238 G/A | No effect on response to infliximab | [81] | ||
| TNFR1 | 36 A/A, 676 T/G | TNFR1A 36 A/A was associated with better response compared to G/G and TNFR1B 676 T/G was not associated with response to infliximab | ||
| 36 A/G | No effect on response to infliximab | [70] | ||
| TNFR2 | 676 T/G | A combination of 676 T/G (TNFR2) and −857 C/T (TNF-α) could be used for prognosis of clinical response to infliximab |
FCGR2A, Fc fragment of immunoglobulin G receptor IIa; FCGR3A, Fc fragment of immunoglobulin G receptor IIIa; MHC, major histocompatibility complex; PTPN22, protein tyrosine phosphatase non-receptor type 22; RGS12, regulator of G protein signaling 12; TNF, tumor necrosis factor; TNFR1, tumor necrosis factor receptor 1; TNFR2, tumor necrosis factor receptor 2.
Table 3.
Genetic polymorphisms known to affect rituximab response in patients with RA.
| Biological Agent | Gene | Polymorphism | Clinical Outcome(s) | Refs. |
|---|---|---|---|---|
| Rituximab | FCGR2A | rs1801274 | FCGR2A rs1801274-T/T genotype was associated with better clinical response to rituximab | [96] |
| FCGR3A | rs396991 | FCGR3A rs396991-G allele genotype was associated with better response to rituximab | ||
| −158 V/F | FCGR3A −158 V/V was associated with a better response than V/F or F/F | [97] | ||
| FCGR3A −158 V allele was independently associated with better response to rituximab | [98] | |||
| FCGR3A −158 V/V and V/F was associated with a better response than F/F | [99] | |||
| FCGR3A −158 V/F was not associated with CDAI response to rituximab | [100] | |||
| BAFF | −871 C/T | BAFF −871 C/C was associated with a better response than T/T | [101] | |
| IL-6 | rs1800795 (−174 G/C) | IL-6 −174 GG/GC genotypes was associated with better responses than with −174 C/C genotypes | [102] | |
| TGFβ1 | rs1800470, rs1800471 | TGFβ1 SNPs was associated with good response to rituximab | [103] |
BAFF, B-cell-activating factor; CDAI, Clinical Disease Activity Index; FCGR2A, Fc fragment of immunoglobulin G receptor IIa; FCGR3A, Fc fragment of immunoglobulin G receptor IIIa; IL-6, interleukin 6; SNP, single nucleotide polymorphism; TGFβ1, Transforming growth factor beta-1.
Table 4.
Genetic polymorphisms known to affect tocilizumab response in patients with RA.
| Biological Agent | Gene | Polymorphism | Clinical Outcome(s) | Refs. |
|---|---|---|---|---|
| Tocilizumab | FCGR2A | rs1801274 | Not associated with response to tocilizumab | [96] |
| FCGR3A | rs396991 | FCGR3A rs396991-T/T genotype is associated with better EULAR response to tocilizumab | ||
| HLA-DRB1 | rs11052877, rs4910008, rs9594987, rs10108210, rs703297, rs703505, rs1560011, rs7055107 | The shared epitope HLA-DRB1 had no association with tocilizumab response | [110] | |
| IL-6R | rs12083537, rs2228145, rs4329505, rs11265618 | rs12083537-A/A and rs11265618-C/C were associated with higher LDA rates | [111] |
EULAR, European League Against Rheumatism; FCGR2A, Fc fragment of immunoglobulin G receptor IIa; FCGR3A, Fc fragment of immunoglobulin G receptor IIIa; HLA-DRB1, human leukocyte antigen-beta chain 1; IL-6R, interleukin 6 receptor; LDA, low disease activity.
Table 5.
Newer monoclonal antibody drugs and genetic polymorphisms of their targets.
| Biological Agent | Polymorphism | Clinical Outcome(s) | Refs. |
|---|---|---|---|
| Canakinumab, Gevokizumab | IL-1α | rs17561 | [115] |
| IL-1β | rs16944 | [115] | |
| rs16944, rs1143634 | [116] | ||
| [117] | |||
| [118] | |||
| [119] | |||
| IL-1Ra (IL-1RN) | 2018 C/T | [117] | |
| 5111 T/C, 2017 T/C | [115] | ||
| Secukinumab, Ixekizumab, Bimekizumab | IL-17A | rs4711998, rs8193036, rs3819024, rs2275913, rs7747909 | [120] |
| rs2275913, rs3804513, rs3748067, rs1974226 | [121] | ||
| rs2275913 | [122] | ||
| [123] | |||
| rs2275913, rs3819024, rs3819025, rs4711998, rs8193036, rs8193037, rs3804513 | [124] | ||
| rs2275913, rs3804513 | [125] | ||
| rs2275913 | [126] | ||
| [127] | |||
| [128] | |||
| [129] | |||
| [130] | |||
| [131] | |||
| rs2275913, rs3819024, rs4711998, rs8193036 | [132] | ||
| rs2275913 | [133] | ||
| [134] | |||
| [135] | |||
| [136] | |||
| [137] | |||
| [138] | |||
| IL-17F | rs763780, rs2397084 | [121] | |
| rs763780 | [122] | ||
| rs763780, rs2397084 | [123] | ||
| rs763780 | [125] | ||
| [126] | |||
| rs763780, rs11465553, rs2397084 | [127] | ||
| rs763780, rs2397084 | [128] | ||
| [129] | |||
| rs763780 | [130] | ||
| rs763780, rs2397084 | [132] | ||
| [134] | |||
| rs763780 | [135] | ||
| rs763780, rs2397084 | [136] | ||
| rs763780 | [138] | ||
| Brodalumab | IL-17RC | rs708567 | [133] |
| Ustekinumab, Guselkumab | IL-23R | rs11209026, rs134315, rs10489629, rs7517847 | [125] |
| rs11209026, rs1343151, rs10489629 | [128] | ||
| rs10889677 | [130] | ||
| rs1004819, rs10489629, rs11209026, rs1343151, rs10889677, rs11209032, rs1495965 | [139] | ||
| rs10889677, rs2201841, rs1884444 | [140] | ||
| rs1004819, rs7517847, rs10489629, rs2201841, rs1343151, rs11209032, rs1495965 | [141] | ||
| rs11209026, rs2201841, rs10889677 | [142] | ||
| Lenzilumab, Namilumab, Otilimab, Gimsilumab | GM-CSF | 177 T/C | [143] |
| 545 G/A, 3606 T/C, 3928 C/T | [144] | ||
| 677 C/C | [145] | ||
| 3606 T/C, 3928 C/T | [146] | ||
| 677 A/C | [147] | ||
| 3928 C/T, 3606 T/C | [148] | ||
| Denosumab | RANKL | rs9533156, rs2277438, rs1054016 | [149] |
| rs2277438 | [150] | ||
| rs2277438, rs7984870 | [151] | ||
| rs2277438, rs9533156 | [152] | ||
| rs7325635, rs7988338 | [153] | ||
| [154] | |||
| rs2277438, rs9533156 | [155] |
IL-1α, interleukin-1 alpha; IL-1β, interleukin-1 beta; IL-1Ra (or IL-1RN), interleukin-1 receptor antagonist; IL-17A, interleukin-17A; IL-17F, interleukin-17F; IL-17RC, interleukin-17 receptor C; IL-23, interleukin-23 receptor; GM-CSF, granulocyte-macrophage colony-stimulating factor; RANKL, Receptor activator of nuclear factors κB ligand.
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Lim, S.H.; Kim, K.; Choi, C.-I. Pharmacogenomics of Monoclonal Antibodies for the Treatment of Rheumatoid Arthritis. J. Pers. Med. 2022, 12, 1265. https://doi.org/10.3390/jpm12081265
AMA Style
Lim SH, Kim K, Choi C-I. Pharmacogenomics of Monoclonal Antibodies for the Treatment of Rheumatoid Arthritis. Journal of Personalized Medicine. 2022; 12(8):1265. https://doi.org/10.3390/jpm12081265
Chicago/Turabian StyleLim, Sung Ho, Khangyoo Kim, and Chang-Ik Choi. 2022. "Pharmacogenomics of Monoclonal Antibodies for the Treatment of Rheumatoid Arthritis" Journal of Personalized Medicine 12, no. 8: 1265. https://doi.org/10.3390/jpm12081265
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.