1. Introduction
Mitochondria provide aerobic eukaryotes with cellular energy by generating adenosine tri- phosphate (ATP) through the process of oxidative phosphorylation (OXPHOS). As a first step, electrons from nutrients (such as carbohydrates and fatty acids) are transferred by four complexes (CI-CIV) anchored to the mitochondrial inner membrane. This results in a transmembrane electrochemical proton gradient, which drives ATP synthesis from adenosine di-phosphate (ADP) and inorganic phosphate (Pi) by the ATP synthase (CV).
Defects of the OXPHOS system have been implicated in a broad spectrum of human diseases. Typical clinical traits include encephalopathies, cardiomyopathies, myopathies, visual/hearing, liver and renal dysfunctions [
1,
2,
3]. Many of these diseases are caused by mitochondrial DNA (mtDNA) mutations. This DNA encodes 13 protein subunits of the OXPHOS system and a number of RNAs necessary for the synthesis of these proteins inside the organelle [
4]. Pathogenic mtDNA mutations often co-exist with non-mutated mtDNA (heteroplasmy) and are highly recessive (only rare cases of dominancy were reported [
5]), which makes it difficult to know how mitochondrial function is affected in patient’s cells and tissues. Furthermore, because of the high mutability of the mitochondrial genome due to its exposure to damaging oxygen species (ROS) and the poor activity of mitochondrial DNA repair systems, it can be difficult to establish the pathogenicity of a mtDNA variant [
4]. Additionally, the effects of deleterious mtDNA mutations may be aggravated by nucleotide changes in nuclear and mitochondrial DNA that are not pathogenic per se (the so-called modifier genes) [
6,
7].
Being amenable to mitochondrial genetic transformation [
8], and owing to its good fermenting capability that enables survival after the loss of oxidative phosphorylation and inability to stably maintain heteroplasmy [
9],
Saccharomyces cerevisiae has been used as a model to investigate mtDNA mutations identified in patients. We have exploited these attributes to study
MT-ATP6 gene mutations found in patients. This gene encodes the subunit
a of ATP synthase, which is involved in moving protons through the membrane domain (F
O) of ATP synthase coupled to ATP synthesis [
10,
11,
12,
13,
14,
15]. These yeast-based studies helped to better define the functional consequences of subunit
a mutations and provided support for the pathogenicity of rare alleles identified in only a limited number of cases like m.8851T>C [
12] and m.8969G>A [
16]. Importantly, the extent to which yeast ATP synthase was affected correlated with disease severity, which reflects the strong evolutionary conservation of the regions of subunit
a where these mutations localize [
17,
18,
19,
20,
21,
22,
23,
24,
25,
26].
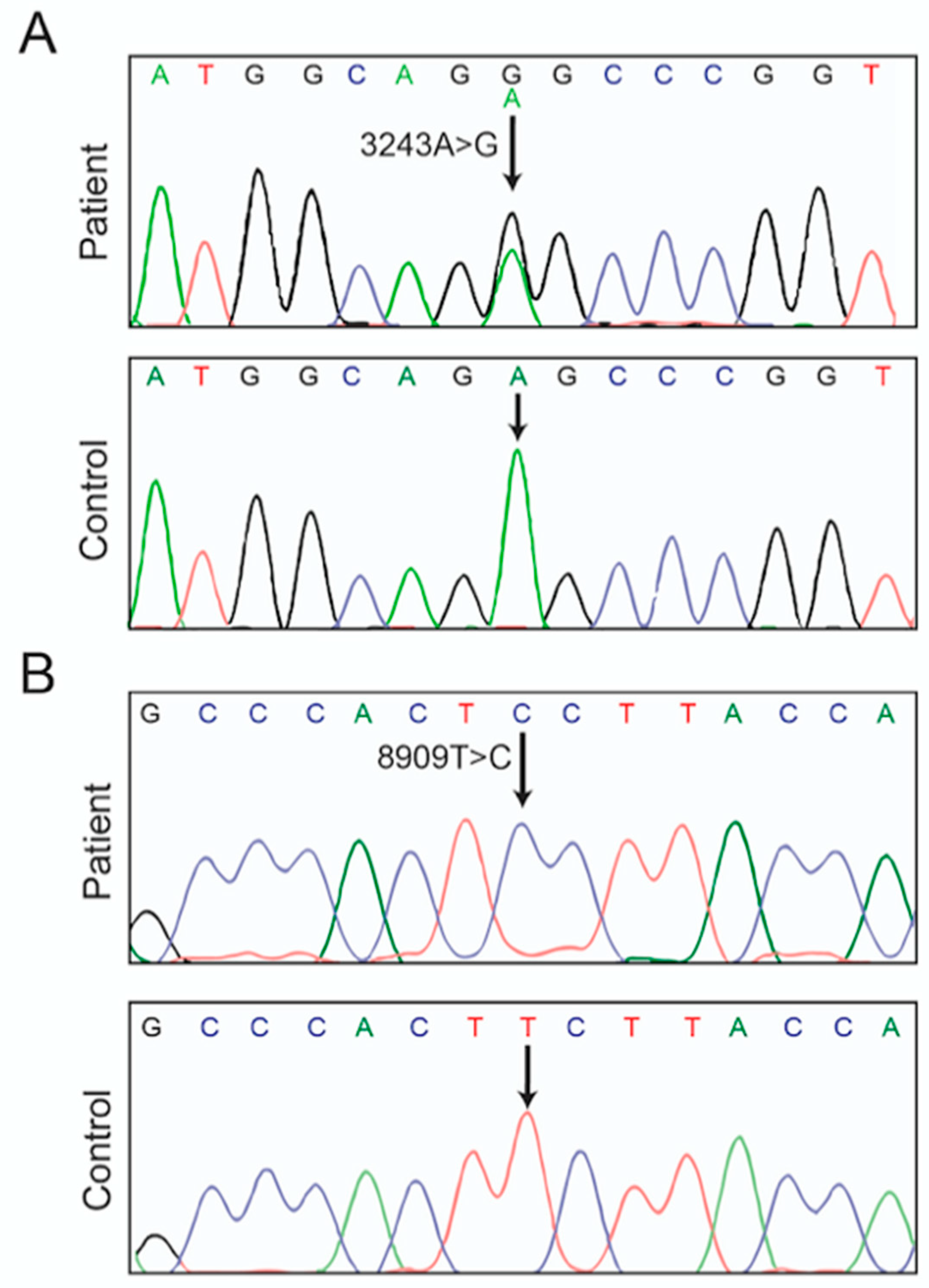
Herein we report the identification of a novel MT-ATP6 variant, m.8909T>C. It converts into serine a highly conserved phenylalanine residue of subunit a (aF128S in humans; aF145S in yeast). We detected it by entirely sequencing the mtDNA of a patient with an extreme clinical presentation including brain, kidney and muscular dysfunctions leading to premature death at the age of 14. This patient also carried the well-known pathogenic m.3243A>G mutation in mt-tRNALeu(1) (MT-TL1). In a yeast model of the m.8909T>C variant, ATP synthase assembly/stability was significantly compromised to an extent comparable to that seen previously in yeast models of other ATP6 mutations with a well-established pathogenicity. These findings indicate that alone the m.8909T>C variant has the potential to compromise human health.
2. Materials and Methods
2.1. Kidney Analyses
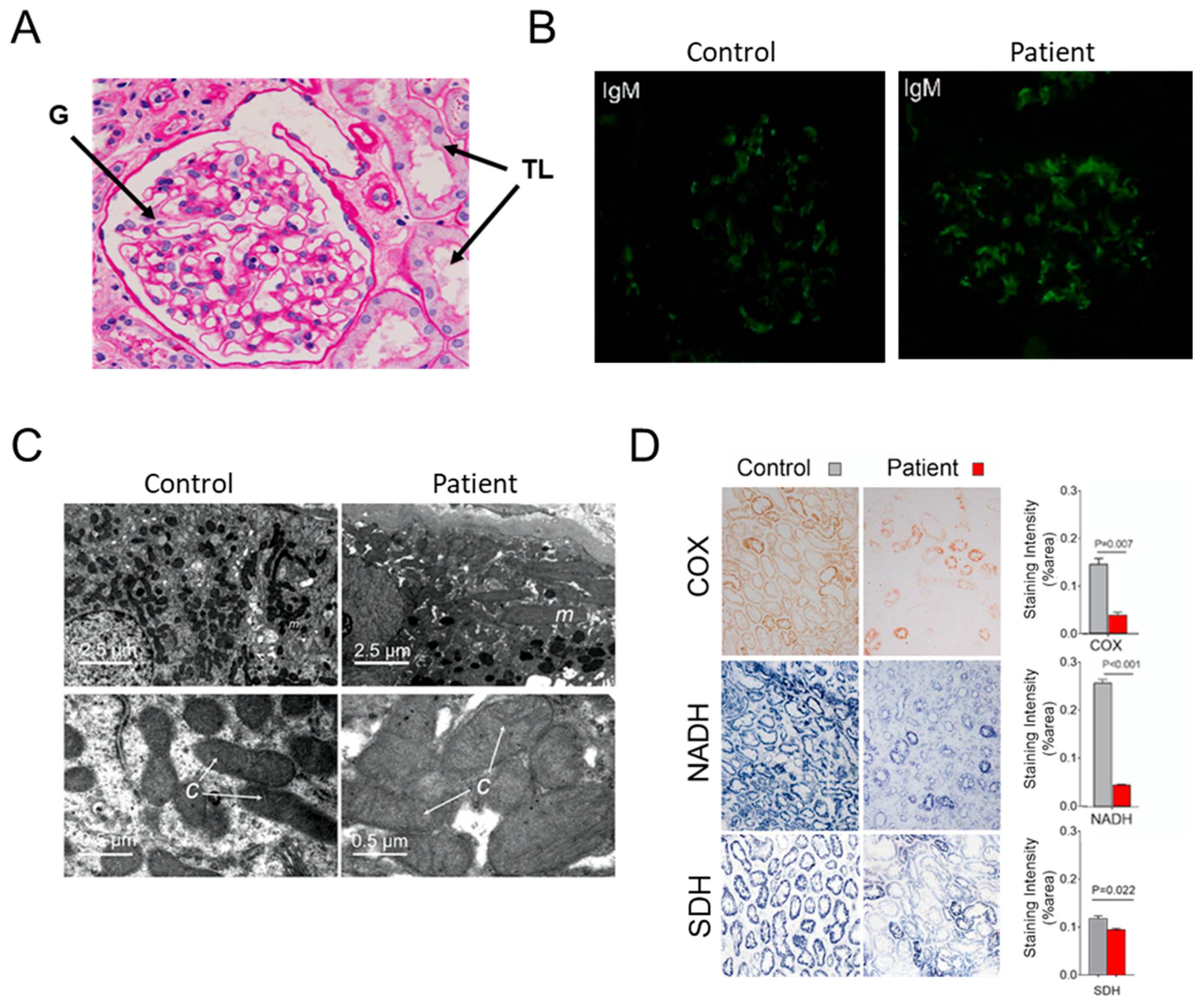
Kidney biopsies were performed under ultrasound guidance by an experienced investigator. Paraffin-embedded sections were routinely stained with periodic acid Schiff and assessed by light microscopy. Fluorescence staining for IgG, IgA, immunoglobulin M (IgM), C3c and C1q was performed on freshly frozen renal tissues and ultra-thin sections stained with uranyl acetate and lead nitrate were examined by electron microscopy as described in [
16]. Kidney biopsies were frozen in isopentane chilled with liquid nitrogen. Six-micrometer thick cryostat frozen sections were performed for enzyme histochemical staining for Complex IV (COX), Complex II (SDH) and nicotinamide adenine dinucleotide (NADH) dehydrogenase activities using specific substrates of these enzymes including Cytochrome
c (C7752, Sigma-Aldrich), β-nicotinamide adenine dinucleotide (N7410, Sigma-Aldrich, St. Louis, MO, USA) and succinic acid (S3674, Sigma-Aldrich), respectively as described [
27,
28,
29]. Optical density quantification of these activities was assessed from 20 consecutive microscopic fields in each renal section and the adjacent background regions.
2.2. Patient Consent, Ethical Committees, and Adhesion to Biosecurity and Institutional Safety Procedures
The female Chinese patient herein described was hospitalized with recurrent kidney disease and multiple systemic dysfunctions at Jinling Hospital in Nanjing (China). One hundred healthy control adults were randomly recruited from a panel of unaffected, genetically unrelated Han Chinese individuals from the same geographic region. All methods were conducted in accordance with the Ethics Committee of the Jinling Hospital and in the respect of biosafety and public health. Written informed consents were obtained from the patient and her parents as well as the 100 healthy controls. The mother didn’t consent for sequencing her mtDNA and medical examination.
2.3. mtDNA Characterization
Whole DNA was prepared from total blood, kidney and urine sediment samples with the DNA extraction kit from Qiagen (Hilden, Germany), and mtDNA was amplified as described in [
16]. The revised Cambridge reference sequence (rCRS) of
H. sapiens mitochondrial DNA (GenBank NC_012920.1) was used to identify variants of this DNA. Variant prioritization and the presence of deletion/depletion was performed as described [
16]. The abundance of mutations was assessed using a Pyromark Q24 platform (Qiagen) as described in [
16]. After purification using streptavidin Sepharose HP (GE Healthcare), and denaturation with NaOH, the PCR products were annealed and sequenced with the primers listed in
Table 1.
2.4. Media for Growing Yeast
The following media were used for growing yeast. YPAD: 1% (w/v) yeast extract, 2% (w/v) bacto peptone, 40 mg/L adenine and 2% (v/v) glucose; YPAGly: 1% (w/v) yeast extract, 2% (w/v) bacto peptone, 40 mg/L adenine and 2% (v/v) glycerol; YPGALA: 1% (w/v) yeast extract, 2% (w/v) bacto peptone, 40 mg/L adenine and 2% (v/v) galactose. Solid media contained 2% (w/v) agar.
2.5. Construction of S. cerevisiae Strain RKY108 (aF145S)
We used the QuikChange XL Site-directed Mutagenesis Kit of Stratagene to introduce an equivalent of the m.8909T>C mutation (
aF145S) in the yeast
ATP6 gene cloned in pUC19 [
30], utilizing the oligonucleotide 5′-GGTTTATATAAACATGGTTGAGTATTCTTCTCATTA
TCAGTACCTGCTGGTACACCATTACC-3′. The
atp6-F
145S gene was cloned into the plasmid pJM2 [
8]. The resulting plasmid (pRK66) was introduced into mitochondria of the strain DFS160 that totally lacks mitochondrial DNA (ρ
0) as described [
8] (see
Table 2 for complete genotypes and sources of yeast strains). The resulting mitochondrial transformants (RKY109) were crossed to the
atp6::
ARG8m deletion strain MR10 [
30] to yield strain RKY108. This strain has the MR10 nucleus and the
atp6-F
145S gene in a complete (ρ
+) mitochondrial genome. The presence of the
atp6 mutation in these clones was confirmed by DNA sequencing. No other change was detected in the
ATP6 gene. The corresponding wild type strain MR6 (WT) is a derivative of strain W303-1B in which the nuclear
ARG8 gene has been replaced with
HIS3 (
arg8::HIS3) and with the entirely sequenced mitochondrial genome of strain BY4741 [
30].
2.6. Yeast-Based Drug Assay
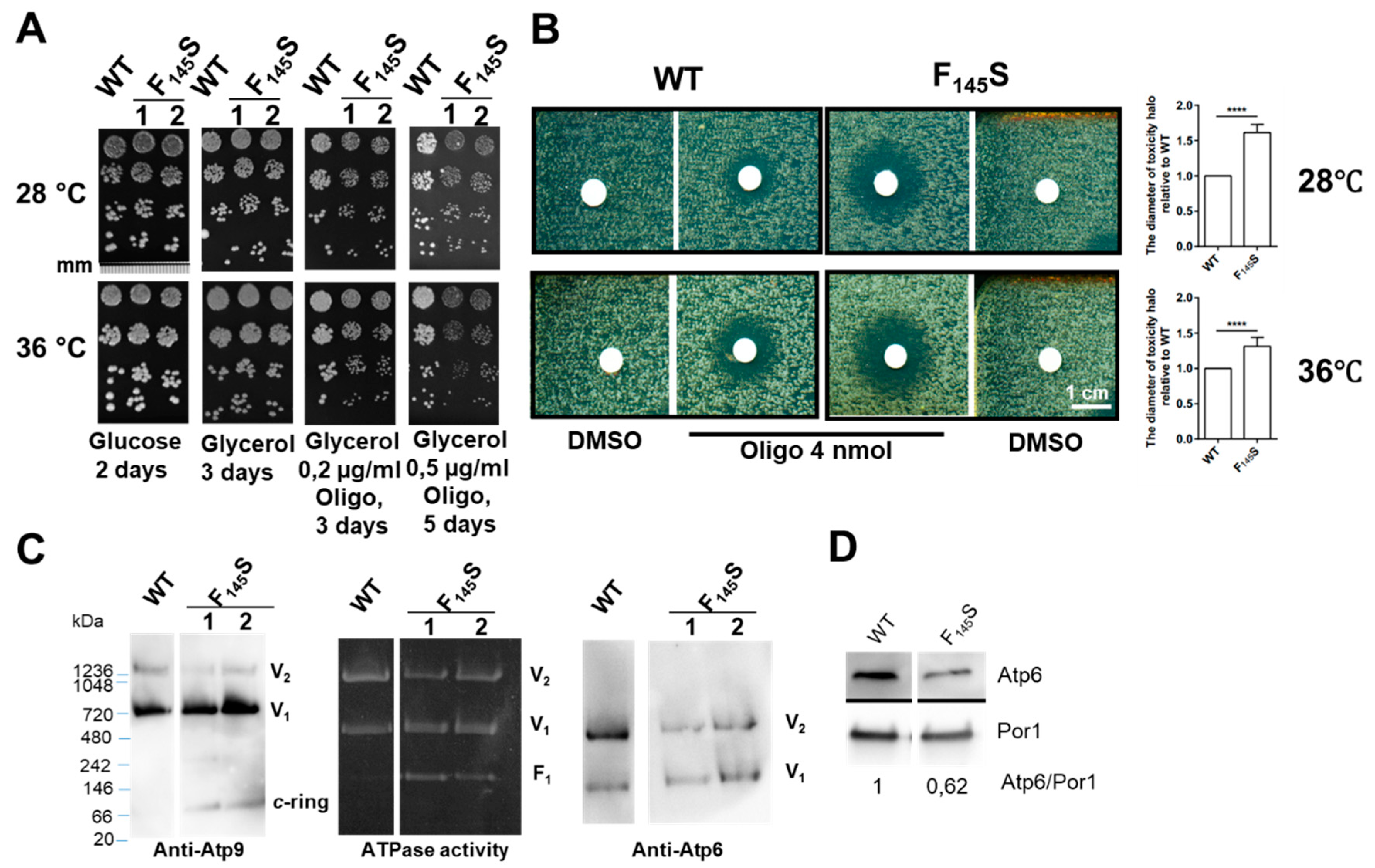
0.05 OD650nm of exponentially growing cells were homogeneously spread with sterile glass beads on a square Petri dish (12 cm × 12 cm) containing solid YPAGly medium. Sterile filters were deposited on the plate and spotted with oligomycin (purchased from Sigma, St. Louis, MO, USA) dissolved in DMSO.
2.7. Biochemical Investigation of Mitochondria
Oxygen consumption measurements in mitochondria isolated from yeast cells grown in complete galactose medium were performed according to [
32], using a Clark electrode (Heito, Paris, France). Freshly prepared mitochondria were added to 1 mL of respiration buffer (10 mM Tris-maleate pH 6.8, 0.65 M sorbitol, 0.3 mM EGTA, and 3 mM potassium phosphate) at 0.15 mg/mL in the reaction chamber maintained at 28 °C. The oxygen consumption was measured after successive additions of 4 mM NADH, 150 µM ADP, 4 µM carbonyl cyanide m-chlorophenylhydrazone (CCCP). The rate of ATP synthesis was measured in the same conditions but with 750 µM ADP. Aliquots were withdrawn every 15 s from the reaction mix and supplemented with 3.5% (
w/
v) perchloric acid and 12.5 mM EDTA. After neutralization of the samples to pH 6.5 with KOH 0.3 M/MOPS, ATP was quantified by luciferin/luciferase assay (ATPLite kit from Perkin Elmer, Waltham, MA, USA) on a LKB bioluminometer. Oligomycin (3 μg/mL) was used to determine the amount of ATP produced by ATP synthase. ATPase activity was measured in non-osmotically protected mitochondria at pH 8.4 as described [
33]. Blue native polyacrylamide gel electrophoresis (BN-PAGE) was performed as described [
34]. For this, 200 µg of mitochondrial proteins were suspended in 50 µL of extraction buffer (30 mM HEPES, 150 mM potassium acetate, 12% glycerol, 2 mM 6-aminocaproic acid, 1 mM EGTA, 2% digitonin (Sigma), one protease inhibitor cocktail tablet (Roche) (pH 7.4) and incubated for 30 min on ice. After centrifugation (14,000 rpm, 4 °C, 30 min) the supernatant containing the solubilized complexes were supplemented with 2.25 µL of loading dye (5% Serva Blue G-250, 750 mM 6-aminocaproic acid) and run into NativePAGE 3–12% Bis-Tris Gel (Invitrogen, Carlsbad, CA, USA). After transfer onto PVDF membrane the yeast ATP synthase complexes were detected with polyclonal antibodies against α-F
1 (Atp1), subunit
c (Atp9) and subunit
a (Atp6) used after 1:10,000, 1:5000, and 1:1000 dilutions respectively. The Atp1 antibodies were kindly provided by J. Velours. Anti-Atp9 antibodies were prepared by Eurogentec (Seraing, Belgium) with the synthetic peptide corresponding to the loop connecting the two transmembrane helices of Atp9 as an immunogen. The procedure used to in-gel visualize ATP synthase by its ATPase activity is described in [
31].
2.8. Amino-Acid Alignments and Subunit a Topology
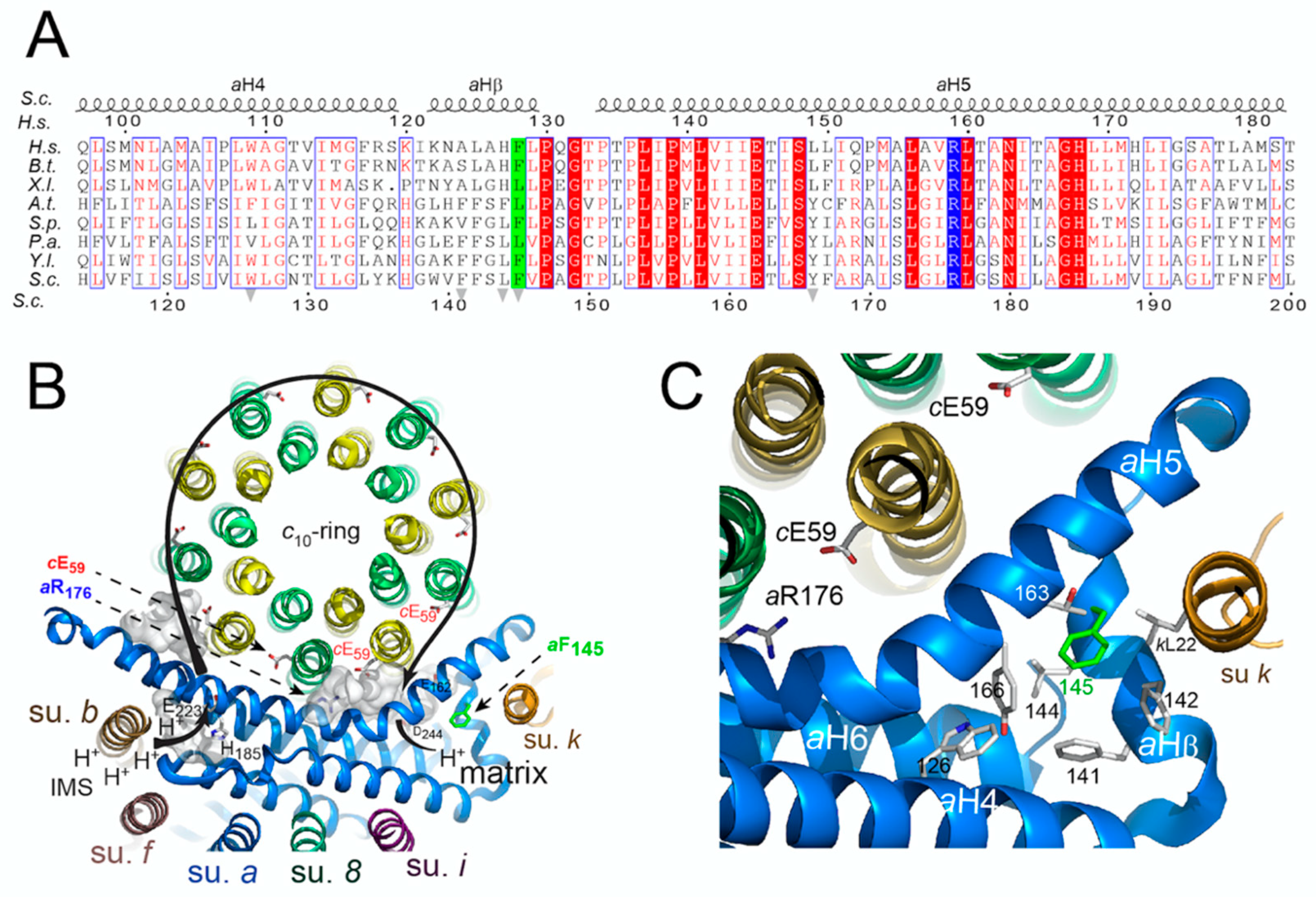
Clustal Omega [
35] was used to compare amino acid sequences of subunits
a of various species. The topology of the
aF145S mutation was investigated according to the yeast ATP synthase structure described in [
36] and drawn using PyMOL [
37].
2.9. Statistical Analyses
Chi-square test and SPSS (16.0), Chicago, IL, USA. method were used for evaluating the statistical significance of the data. All of the tests were two tailed, and p values < 0.05 were considered significant.
4. Discussion
With the advent of next generation sequencing methods, numerous novel variants of the mitochondrial DNA (mtDNA) are continuously identified in patients suffering from mitochondrial disorders. They are frequently found in only a limited number of cases, sometimes in a single individual (as with the case here reported), which makes it difficult to conclude about their pathogenicity. In a study aiming to probe the possible implication of mtDNA alterations in renal disease in the region of Nanjing, we examined more than 5000 patients with a biopsy-proven kidney dysfunction. Patients retained for entire mtDNA sequencing additionally showed at least two symptoms commonly observed in mitochondrial diseases like diabetes mellitus, deafness, neuromuscular and cardiac manifestations. Another criterion was the presence of abnormal mitochondrial ultrastructure and decreased cytochrome
c oxidase (COX) and NADH (Complex I) dehydrogenase activities in kidney biopsies. In the patient with all these features here reported, we identified a novel variant in the mitochondrial
ATP6 gene, m8909T>C. It was homoplasmic in blood, urine sediments, and kidney (
Figure 2B). It was detected in combination with the well-known pathogenic m.3243A>G mutation in a leucine tRNA gene (
MT-TL1) [
38], at a lesser abundancy (50–90%). There is no doubt that defects in mitochondrial translation induced by the m.3243A>G mutation largely contributed to the disease process and the severe decreases in CI and CIV activities, with only a minimal loss of SDH activity as was observed in other patients carrying this mutation [
58].
The m.8909T>C variant leads to replacement of a well conserved phenylalanine residue with serine in ATP synthase subunit
a (
aF128S) (
Figure 4A), which prompted us to investigate the consequences of an equivalent of this mutation (
aF145S) in yeast. Several lines of evidence demonstrate a partial impairment of ATP synthase function in the mutant compared to wild type yeast: (i) respiratory growth showed a higher sensitivity to suboptimal concentrations of oligomycin, a chemical that inhibits ATP synthase; (ii) as was observed in many yeast ATP synthase defective mutants, the rate of mitochondrial oxygen consumption was diminished; (iii) ATP was produced less rapidly; and (iv) the presence in BN gels of partial ATP synthase assemblies (free F
1 and
c-ring particles) attested for a compromised ability of the mutated subunit
a to be stably incorporated into ATP synthase (
Figure 3). According to recently published atomic structures of yeast ATP synthase [
36,
54], these effects are presumably the consequence of a disorganization of a cluster of hydrophobic residues supposedly important to help subunit
a to adopt a stable functional conformation around the
c-ring. Similar ATP synthase defects were previously observed in yeast models of
MT-ATP6 mutations with a well-established pathogenicity, like m.9176T>C [
13] and m.8993T>C [
11]. It is thus a reasonable hypothesis that the m.8909T>C variant could by itself have detrimental consequences on human health.
In line with this hypothesis, the patient here described showed a very severe clinical presentation in comparison to 35 previously reported patients suffering from kidney problems and presumed to carry only the m.3243A>G mutation (in most of them the mtDNA was not entirely sequenced). The median onset age of disease was 26 years (
Table S1). More than 50% of these patients were first diagnosed with a renal disease due to persistent proteinuria. Glomerular lesions were observed in 21 patients, FSGS in 15 patients, and 3 cases showed TIN (
Table S2) [
59,
60,
61,
62,
63,
64,
65,
66,
67,
68,
69,
70,
71,
72,
73,
74,
75,
76,
77,
78,
79,
80,
81]. At the time of diagnosis, two relatively aged patients (41 and 47 years old) developed ESRD. A total of 21 patients were recorded with a median follow-up period of 72 months (range 24–240 months). Kidney function remained stable in two patients after 24 and 72 months, respectively, and one patient died of heart failure 60 months after diagnosis. Eighteen patients evolved toward renal failure or ESRD (
Table S1) within 10-years of follow-up after diagnosis (
Figure S1). Our patient, carrying both m.8909T>C and m.3243A>G, developed a kidney disease at the age of 14 and showed an extremely rapid progression to ESRD and death after only 8 months after diagnosis of a nephropathic syndrome.
As the m.8909T>C mutation was homoplasmic in the analyzed cells and tissues, it is likely that it is maternally inherited. Unfortunately, the patient’s mother did not consent to be sequenced and followed medically. She was apparently healthy at the time her daughter was admitted at the hospital. We had no more contact with her since the death of her child. From her apparent good health, at least at the age she brought her daughter at the hospital, we may conclude that the m.8909T>C mutation has no dramatic consequences on mitochondrial function, which is consistent with the relatively mild effects of an equivalent of this mutation on yeast ATP synthase. However, when combined to a more severe allele like m.3243G>A, it may have the potential to accelerate a disease process. Previous studies already concluded that well-known pathogenic
ATP6 variants can act in synergy with other genetic determinants in patients with a very severe clinical presentation. For instance, while alone it usually provokes mild clinical phenotypes [
82], the m.9176T>C mutation was identified in a case of fulminant and fatal Leigh syndrome [
23]. As there was no major difference in the amount of mutated mtDNA in relative’s probands, it was concluded that additional mitochondrial or nuclear genetic determinants were responsible for the different phenotypic expression. A second study similarly reported a rapid clinical evolution leading to sudden infant death syndrome (SIDS) in families segregating the m.8993T>G mutation [
83] or the m.10044A>G mutation of the mt-tRNA
Gly gene [
84], which suggested that mtDNA abnormalities should be considered as contributing to SIDS. Because of a lack of data from the mother of the patient here described, it would be premature to claim that m.8909T>C is a pathogenic mutation. However, considering the very severe clinical presentation of this patient, and the detrimental consequences of m.8909T>C on yeast ATP synthase, it is a reasonable proposal that this mutation has the potential to impact human health, in particular when combined to other genetic abnormalities that compromise mitochondrial function.

 ,
, 




{kind=link}
{kind=link}
{kind=link}
{kind=link}