
Validation of Chemokine Biomarkers in Duchenne Muscular Dystrophy

Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Participants and Sample Collection
2.2. Meso Scale Discovery ELISA Assay of Chemokines
2.3. Protein Extraction from Skeletal Muscle Tissue
2.4. Data Processing and Statistical Analysis
3. Results
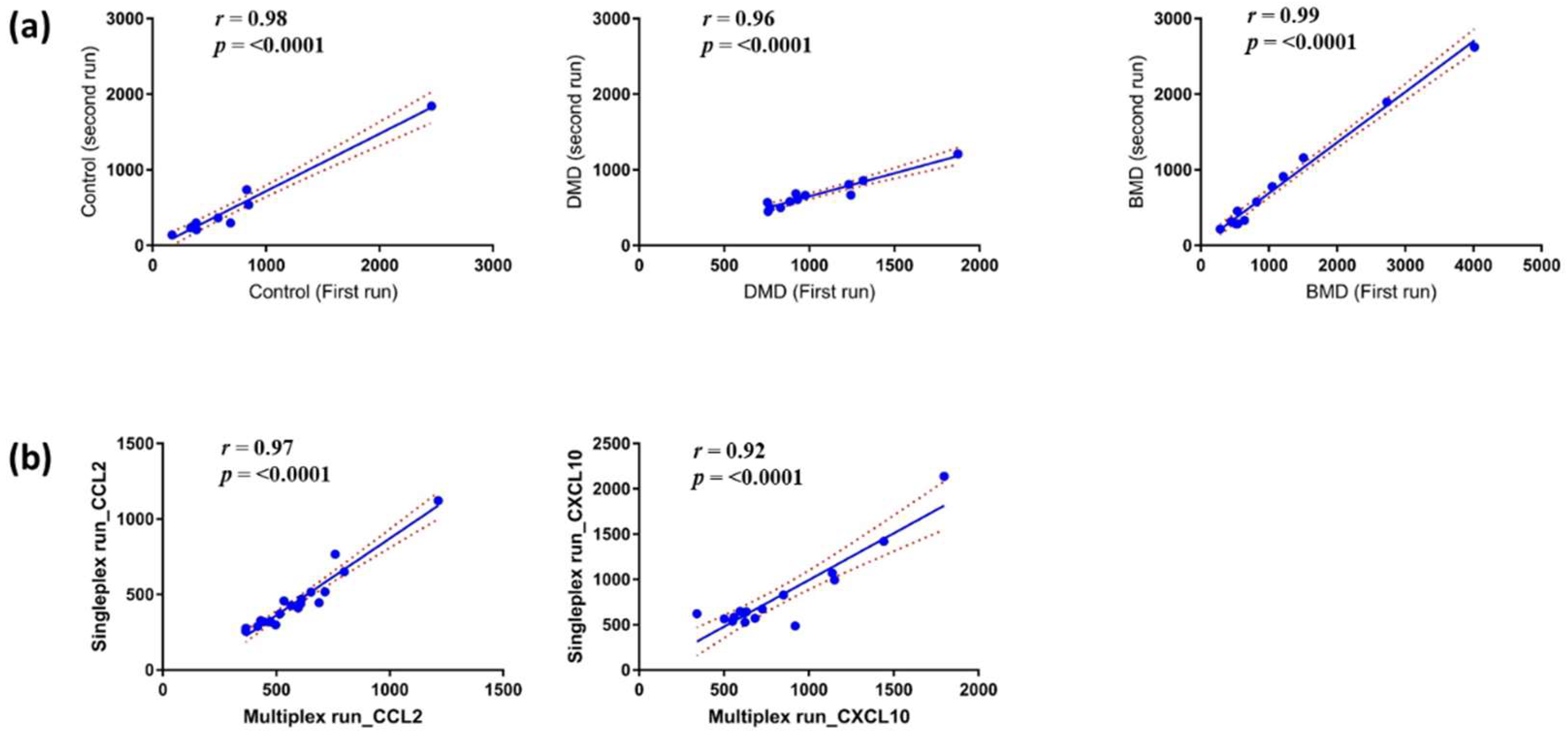
3.1. Reproducibility and Stability of the MSD ELISA Assays for Chemokines in Serum Samples
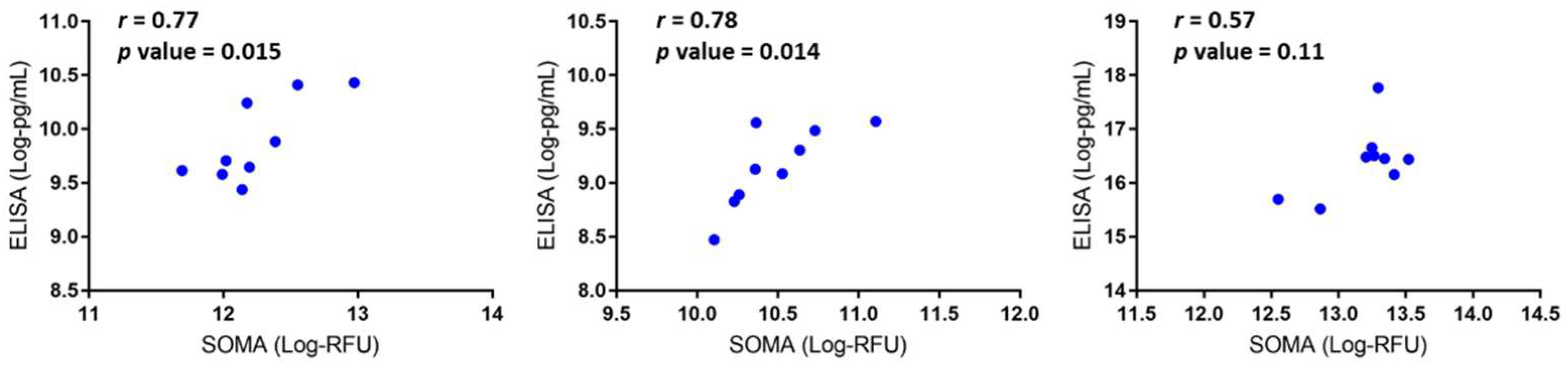
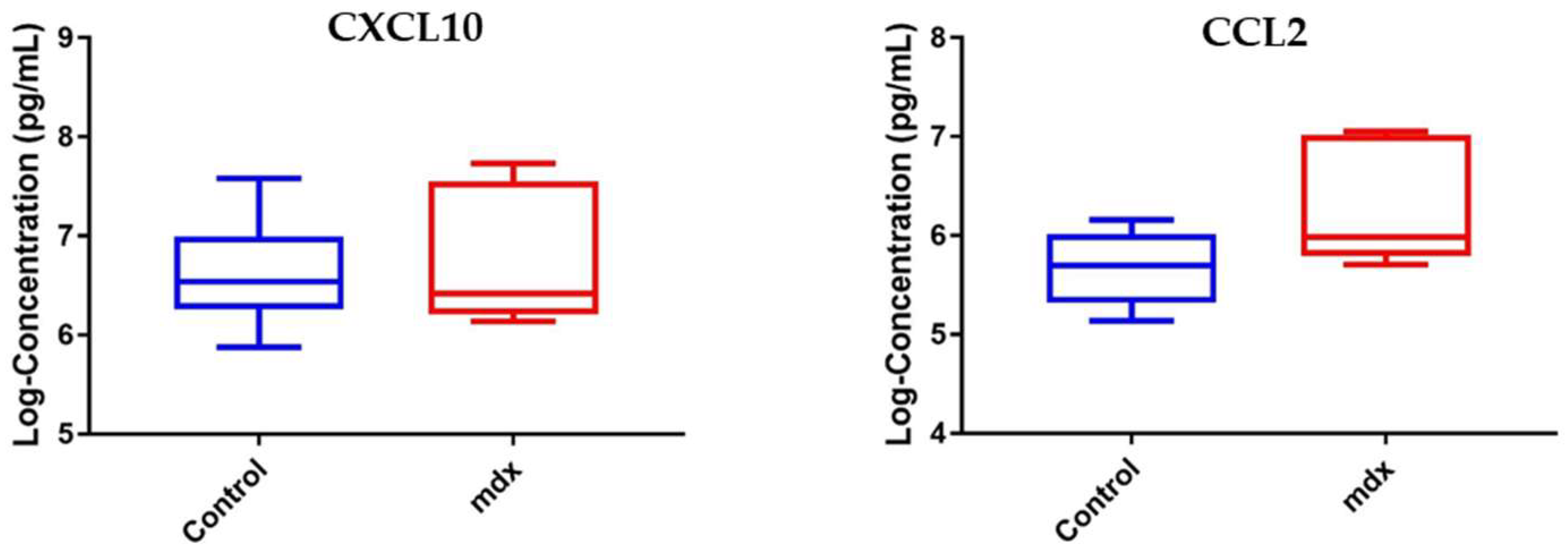
3.2. Data Validation Using Serum Samples from DMD Patients and Cross-Species Comparison
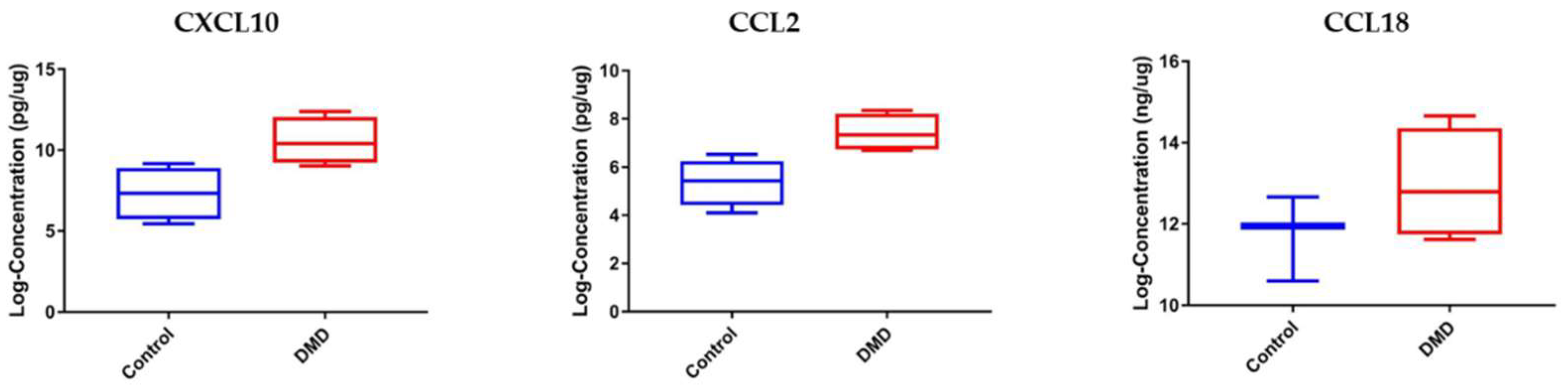
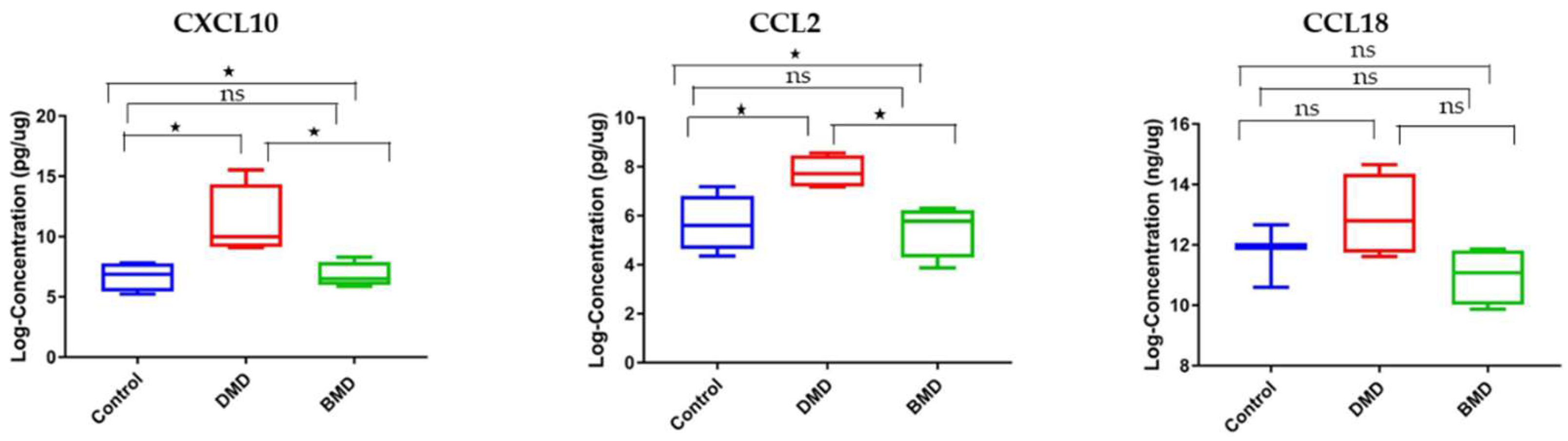
3.3. CCL2 and CXCL10 Are Elevated in Skeletal Muscle Samples of DMD Patients Compared to Healthy Controls
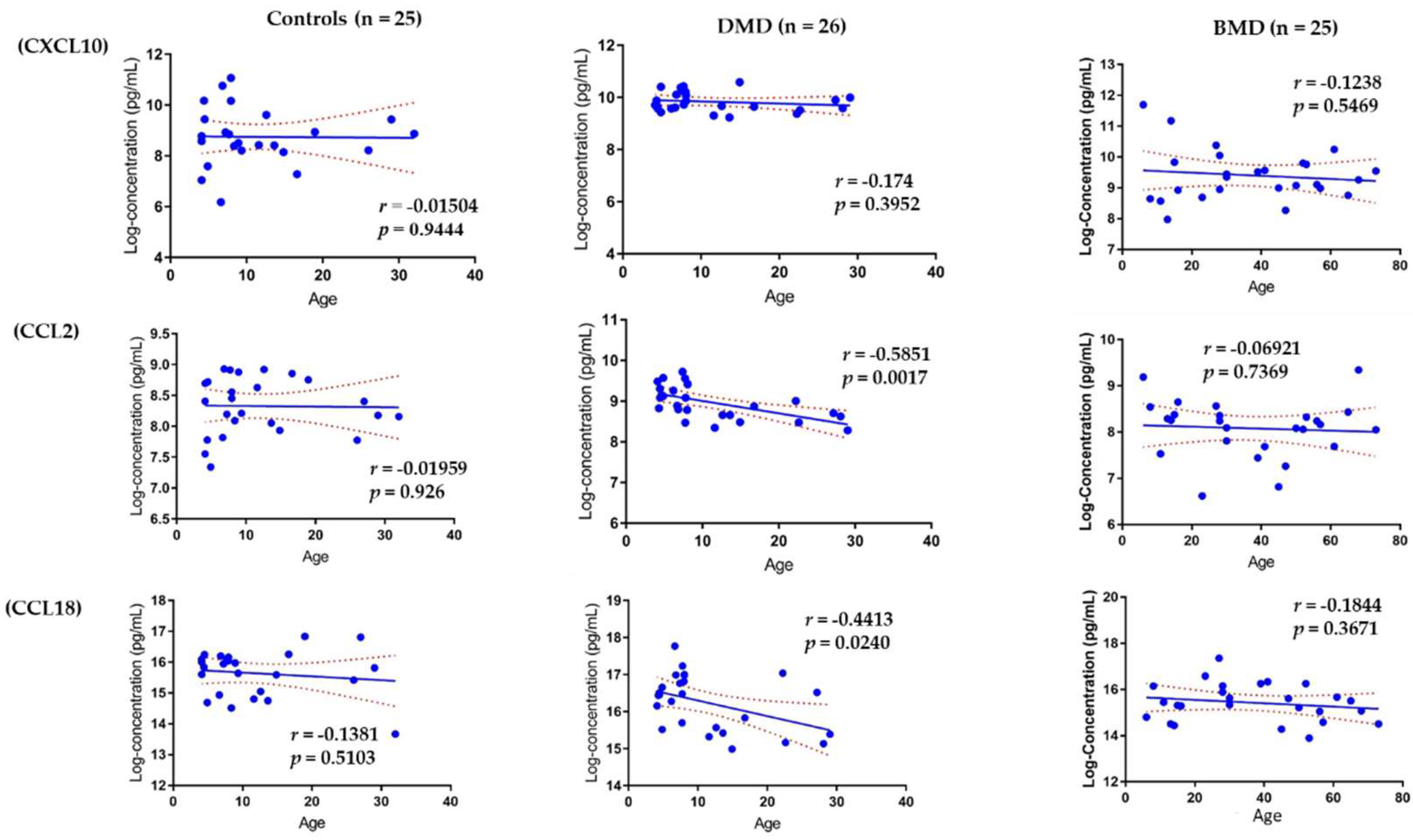
3.4. Association of Circulating Chemokines with Age
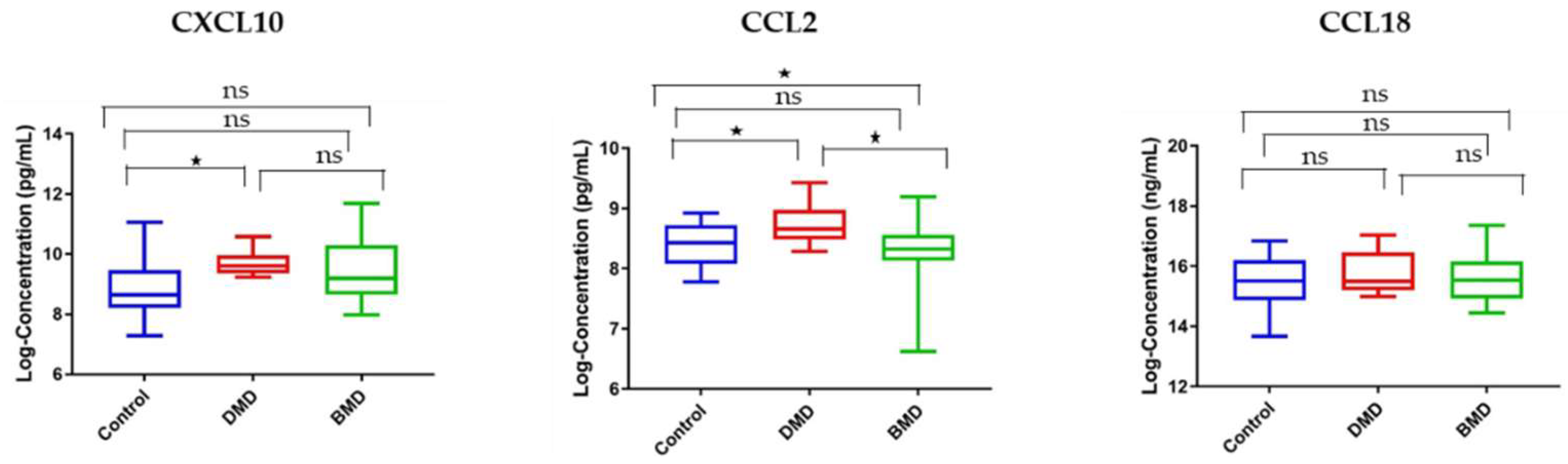
3.5. Circulating and Skeletal Muscle Chemokines Are Elevated in DMD Patients Compared to Age-Matched BMD Patients
3.6. Response of Circulating Chemokines to Glucocorticoid Treatment
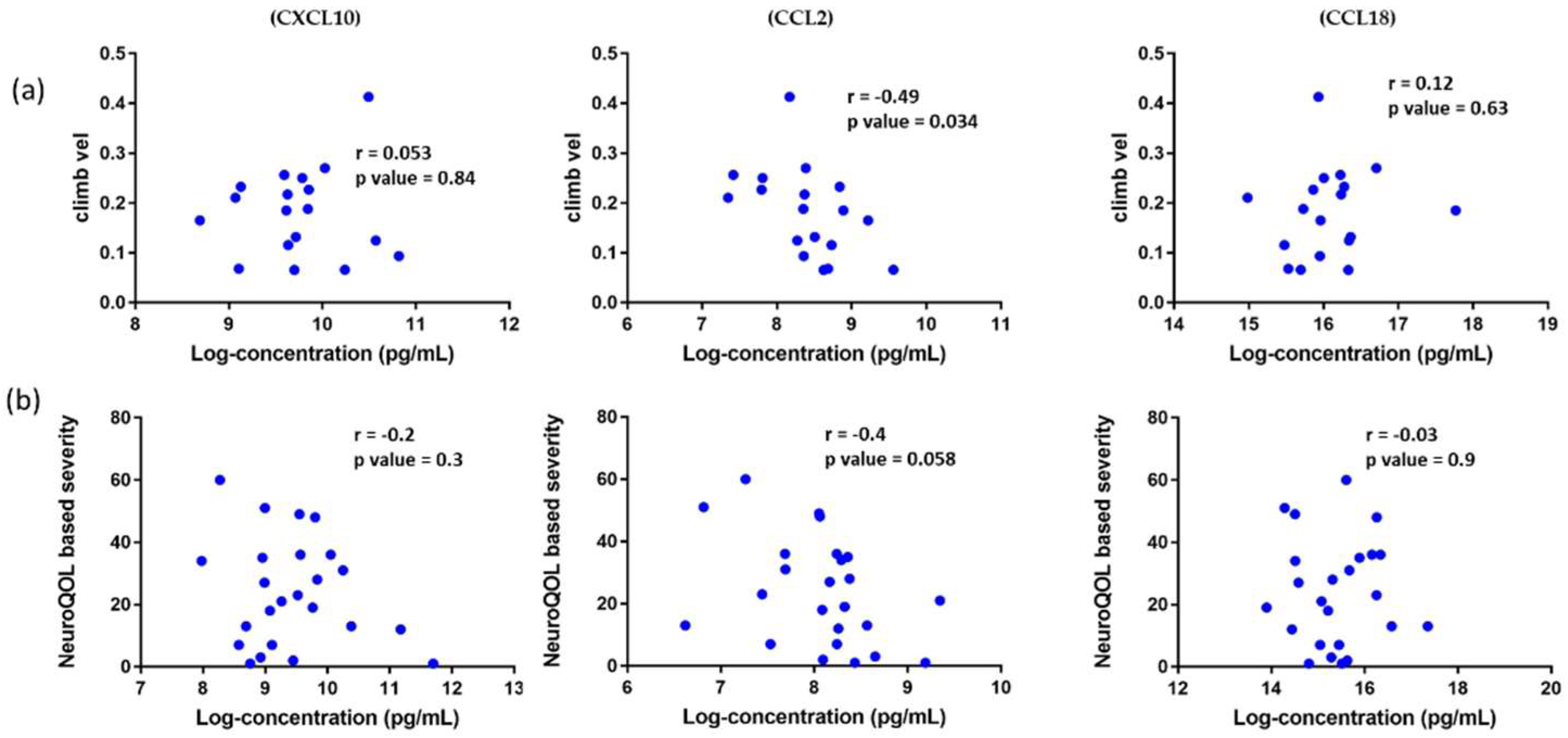
3.7. Association of Circulating Chemokines with Clinical Outcome Measures in DMD Patients and Reported Patient Outcomes in BMD Patients
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Allen, D.; Whitehead, N.P.; Froehner, S.C. Absence of Dystrophin Disrupts Skeletal Muscle Signaling: Roles of Ca2+, Reactive Oxygen Species, and Nitric Oxide in the Development of Muscular Dystrophy. Physiol. Rev. 2016, 96, 253–305. [Google Scholar] [CrossRef] [Green Version]
- Guiraud, S.; Davies, K.E. Regenerative biomarkers for Duchenne muscular dystrophy. Neural Regen. Res. 2019, 14, 1317–1320. [Google Scholar] [CrossRef] [PubMed]
- Hathout, Y.; Liang, C.; Ogundele, M.; Xu, G.; Tawalbeh, S.M.; Dang, U.J.; Hoffman, E.; Gordish-Dressman, H.; Conklin, L.S.; Anker, J.N.V.D.; et al. Disease-specific and glucocorticoid-responsive serum biomarkers for Duchenne Muscular Dystrophy. Sci. Rep. 2019, 9, 12167. [Google Scholar] [CrossRef]
- Alayi, T.D.; Tawalbeh, S.M.; Ogundele, M.; Smith, H.R.; Samsel, A.M.; Barbieri, M.L.; Hathout, Y. Tandem Mass Tag-Based Serum Proteome Profiling for Biomarker Discovery in Young Duchenne Muscular Dystrophy Boys. ACS Omega 2020, 5, 26504–26517. [Google Scholar] [CrossRef] [PubMed]
- Luster, A.D. Chemokines—Chemotactic Cytokines That Mediate Inflammation. N. Engl. J. Med. 1998, 338, 436–445. [Google Scholar] [CrossRef]
- Zlotnik, A.; Yoshie, O. Chemokines. Immunity 2000, 12, 121–127. [Google Scholar] [CrossRef] [Green Version]
- De Paepe, B.; De Bleecker, J.L. Cytokines and Chemokines as Regulators of Skeletal Muscle Inflammation: Presenting the Case of Duchenne Muscular Dystrophy. Mediat. Inflamm. 2013, 2013, 540370. [Google Scholar] [CrossRef] [Green Version]
- Deshmane, S.L.; Kremlev, S.; Amini, S.; Sawaya, B.E. Monocyte Chemoattractant Protein-1 (MCP-1): An Overview. J. Interferon Cytokine Res. 2009, 29, 313–326. [Google Scholar] [CrossRef]
- Tidball, J.G.; Welc, S.S.; Wehling-Henricks, M. Immunobiology of Inherited Muscular Dystrophies. Compr. Physiol. 2018, 8, 1313–1356. [Google Scholar] [CrossRef]
- Xuan, W.; Qu, Q.; Zheng, B.; Xiong, S.; Fan, G.-H. The chemotaxis of M1 and M2 macrophages is regulated by different chemokines. J. Leukoc. Biol. 2014, 97, 61–69. [Google Scholar] [CrossRef]
- Davies, K.E. Challenges in Duchenne muscular dystrophy. Neuromuscul. Disord. 1997, 7, 482–486. [Google Scholar] [CrossRef]
- Wilson, K.; Faelan, C.; Patterson-Kane, J.; Rudmann, D.G.; Moore, S.; Frank, D.; Charleston, J.; Tinsley, J.; Young, G.D.; Milici, A.J. Duchenne and Becker Muscular Dystrophies: A Review of Animal Models, Clinical End Points, and Biomarker Quantification. Toxicol. Pathol. 2017, 45, 961–976. [Google Scholar] [CrossRef] [Green Version]
- Morales, J.A.; Mahajan, K. Dystrophinopathies. Available online: https://www.ncbi.nlm.nih.gov/pubmed/30480978 (accessed on 21 July 2021).
- McDonald, C.M.; Henricson, E.K.; Ms, R.T.A.; Han, J.J.; Escolar, D.M.; Dpt, J.M.F.; Duong, T.; Ms, A.A.; Clemens, P.R.; Hoffman, E.; et al. The cooperative international neuromuscular research group duchenne natural history study-a longitudinal investigation in the era of glucocorticoid therapy: Design of protocol and the methods used. Muscle Nerve 2013, 48, 32–54. [Google Scholar] [CrossRef] [PubMed]
- Ning, Q.; Rangwala, S.; Shelburne, C.; Kobayashi, L.; Liu, P.; Lu, C.; Stewart, D.; Tsionsky, M.; Plisova, T.; Oberoi, P.; et al. Development and Characterization of U-PLEX® Cytokine Assays; The American Association of Immunologists: Rockville, MD, USA, 2015. [Google Scholar]
- Porter, J.D.; Guo, W.; Merriam, A.P.; Khanna, S.; Cheng, G.; Zhou, X.; Andrade, F.H.; Richmonds, C.; Kaminski, H.J. Persistent over-expression of specific CC class chemokines correlates with macrophage and T-cell recruitment in mdx skeletal muscle. Neuromuscul. Disord. 2003, 13, 223–235. [Google Scholar] [CrossRef]
- Spitali, P.; Hettne, K.; Tsonaka, R.; Charrout, M.; Bergen, J.V.D.; Koeks, Z.; Kan, H.; Hooijmans, M.T.; Roos, A.; Straub, V.; et al. Tracking disease progression non-invasively in Duchenne and Becker muscular dystrophies. J. Cachex-Sarcopenia Muscle 2018, 9, 715–726. [Google Scholar] [CrossRef]
- Hathout, Y.; Brody, E.; Clemens, P.R.; Cripe, L.; DeLisle, R.K.; Furlong, P.; Gordish-Dressman, H.; Hache, L.; Henricson, E.; Hoffman, E.; et al. Large-scale serum protein biomarker discovery in Duchenne muscular dystrophy. Proc. Natl. Acad. Sci. USA 2015, 112, 7153–7158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grounds, M.D.; Terrill, J.R.; Al-Mshhdani, B.A.; Duong, M.N.; Radley-Crabb, H.G.; Arthur, P. Biomarkers for Duchenne muscular dystrophy: Myonecrosis, inflammation and oxidative stress. Dis. Model. Mech. 2020, 13, dmm043638. [Google Scholar] [CrossRef]
- Ayoglu, B.; Chaouch, A.; Lochmüller, H.; Politano, L.; Bertini, E.; Spitali, P.; Hiller, M.; Niks, E.; Gualandi, F.; Pontén, F.; et al. Affinity proteomics within rare diseases: A BIO—NMD study for blood biomarkers of muscular dystrophies. EMBO Mol. Med. 2014, 6, 918–936. [Google Scholar] [CrossRef]
- Szigyarto, C.A.-K. Duchenne Muscular Dystrophy: Recent advances in protein biomarkers and the clinical application. Expert Rev. Proteom. 2020, 17, 365–375. [Google Scholar] [CrossRef]
- Wynn, T.A. Cellular and molecular mechanisms of fibrosis. J. Pathol. 2007, 214, 199–210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mojumdar, K.; Liang, F.; Giordano, C.; Lemaire, C.; Danialou, G.; Okazaki, T.; Bourdon, J.; Rafei, M.; Galipeau, J.; Divangahi, M.; et al. Inflammatory monocytes promote progression of Duchenne muscular dystrophy and can be therapeutically targeted via CCR 2. EMBO Mol. Med. 2014, 6, 1476–1492. [Google Scholar] [CrossRef] [PubMed]
- De Paepe, B.; Creus, K.K.; Martin, J.-J.; De Bleecker, J.L. Upregulation of chemokines and their receptors in duchenne muscular dystrophy: Potential for attenuation of myofiber necrosis. Muscle Nerve 2012, 46, 914–916. [Google Scholar] [CrossRef] [PubMed]
- Kadian, N.; Raju, K.S.R.; Rashid, M.; Malik, M.Y.; Taneja, I.; Wahajuddin, M. Comparative assessment of bioanalytical method validation guidelines for pharmaceutical industry. J. Pharm. Biomed. Anal. 2016, 126, 83–97. [Google Scholar] [CrossRef]
- Kranig, S.A.; Tschada, R.; Braun, M.; Patry, C.; Pöschl, J.; Frommhold, D.; Hudalla, H. Dystrophin deficiency promotes leukocyte recruitment in mdx mice. Pediatr. Res. 2019, 86, 188–194. [Google Scholar] [CrossRef] [PubMed]
- Signorelli, M.; Ayoglu, B.; Johansson, C.; Lochmüller, H.; Straub, V.; Muntoni, F.; Niks, E.; Tsonaka, R.; Persson, A.; Aartsma-Rus, A.; et al. Longitudinal serum biomarker screening identifies malate dehydrogenase 2 as candidate prognostic biomarker for Duchenne muscular dystrophy. J. Cachex Sarcopenia Muscle 2019, 11, 505–517. [Google Scholar] [CrossRef]
- Lu, H.; Huang, D.; Ransohoff, R.M.; Zhou, L. Acute skeletal muscle injury: CCL2 expression by both monocytes and injured muscle is required for repair. FASEB J. 2011, 25, 3344–3355. [Google Scholar] [CrossRef] [Green Version]
- Pescatori, M.; Broccolini, A.; Minetti, C.; Bertini, E.; Bruno, C.; D’Amico, A.; Bernardini, C.; Mirabella, M.; Silvestri, G.; Giglio, V.; et al. Gene expression profiling in the early phases of DMD: A constant molecular signature characterizes DMD muscle from early postnatal life throughout disease progression. FASEB J. 2007, 21, 1210–1226. [Google Scholar] [CrossRef]
- Starosta, A.; Konieczny, P. Therapeutic aspects of cell signaling and communication in Duchenne muscular dystrophy. Cell. Mol. Life Sci. 2021, 78, 4867–4891. [Google Scholar] [CrossRef]
- Tawalbeh, S.M.; Marin, W.; Morgan, G.A.; Dang, U.J.; Hathout, Y.; Pachman, L.M. Serum protein biomarkers for juvenile dermatomyositis: A pilot study. BMC Rheumatol. 2020, 4, 52. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein Name | Cnt (pg/mL) | DMD (pg/mL) | p-Value | DMD/Cnt |
|---|---|---|---|---|
| CXCL10 | 580 ± 103 | 942 ± 51 | <0.0001 | 1.62 |
| CCL2 | 333 ± 21 | 518 ± 27 | <0.0001 | 1.55 |
| CCL18 | 57,047 ± 5279 | 87,746 ± 8936 | 0.0004 | 1.54 |
| Protein Name | DMD Pre-Treat (pg/mL) | DMD Post-Treat (pg/mL) | p Value |
|---|---|---|---|
| CXCL10 | 702 ± 118 | 533 ± 82 | 0.097 |
| CCL2 | 449 ± 75 | 426 ± 64 | 0.8765 |
| CCL18 | 64,271 ± 5523 | 64,856 ± 4690 | 0.8793 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ogundele, M.; Zhang, J.S.; Goswami, M.V.; Barbieri, M.L.; Dang, U.J.; Novak, J.S.; Hoffman, E.P.; Nagaraju, K.; CINRG-DNHS Investigators; Hathout, Y. Validation of Chemokine Biomarkers in Duchenne Muscular Dystrophy. Life 2021, 11, 827. https://doi.org/10.3390/life11080827
Ogundele M, Zhang JS, Goswami MV, Barbieri ML, Dang UJ, Novak JS, Hoffman EP, Nagaraju K, CINRG-DNHS Investigators, Hathout Y. Validation of Chemokine Biomarkers in Duchenne Muscular Dystrophy. Life. 2021; 11(8):827. https://doi.org/10.3390/life11080827
Chicago/Turabian StyleOgundele, Michael, Jesslyn S. Zhang, Mansi V. Goswami, Marissa L. Barbieri, Utkarsh J. Dang, James S. Novak, Eric P. Hoffman, Kanneboyina Nagaraju, CINRG-DNHS Investigators, and Yetrib Hathout. 2021. "Validation of Chemokine Biomarkers in Duchenne Muscular Dystrophy" Life 11, no. 8: 827. https://doi.org/10.3390/life11080827