
Fiber-Templated 3D Calcium-Phosphate Scaffolds for Biomedical Applications: The Role of the Thermal Treatment Ambient on Physico-Chemical Properties

 ,
,  , , , , and
, , , , and 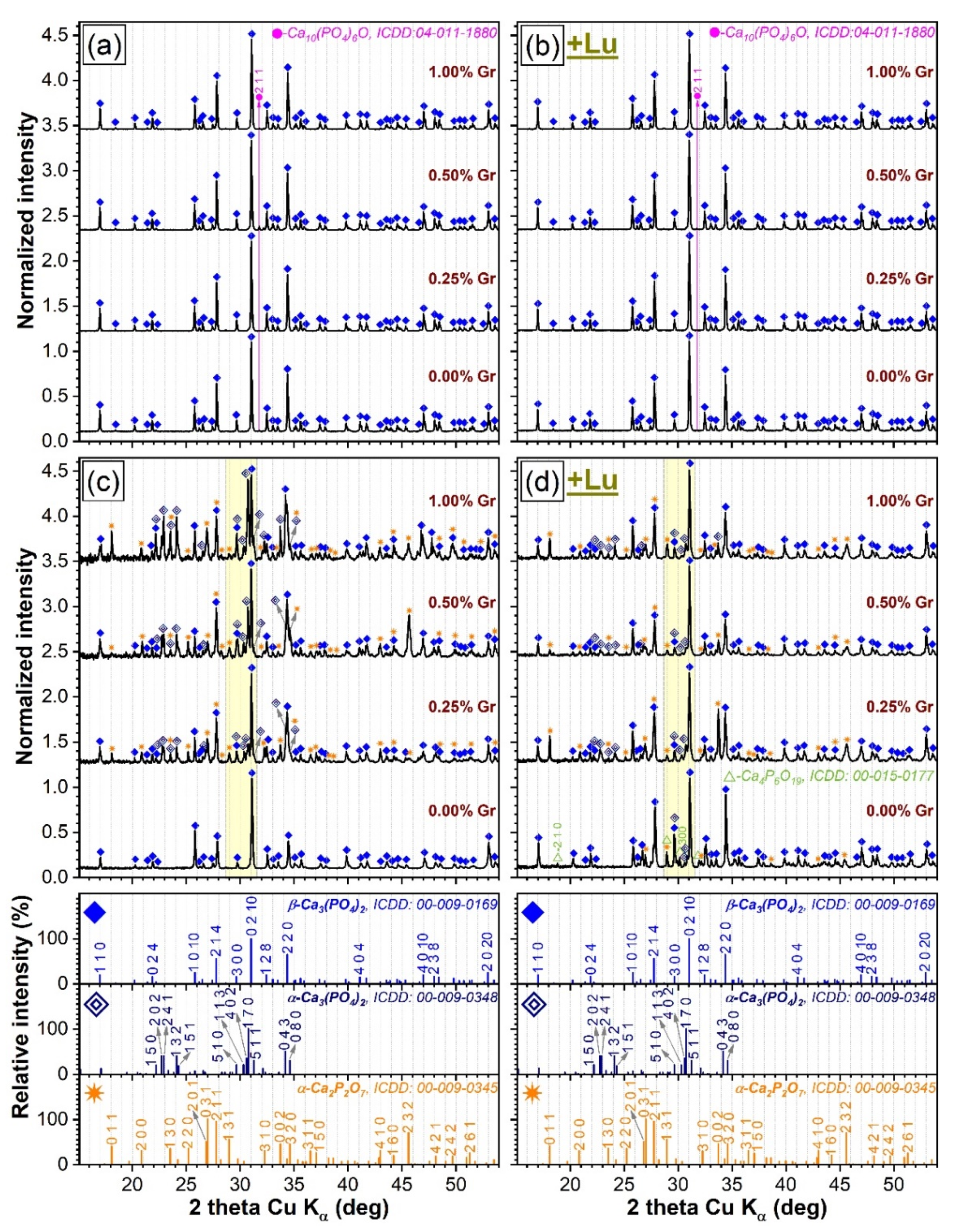{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Preparation
2.2. Physico-Chemical Characterization
2.2.1. X-ray Diffraction Investigation
2.2.2. FTIR Spectroscopy Analysis
2.2.3. Morpho-Compositional SEM/EDS Evaluation
2.2.4. Dimensional Shrinkage and Mass Loss Determination
2.2.5. Nano-Computed Tomography Reconstruction Analysis
2.2.6. Contact Angle Measurements
2.2.7. Mechanical Properties Evaluation
3. Results and Discussion
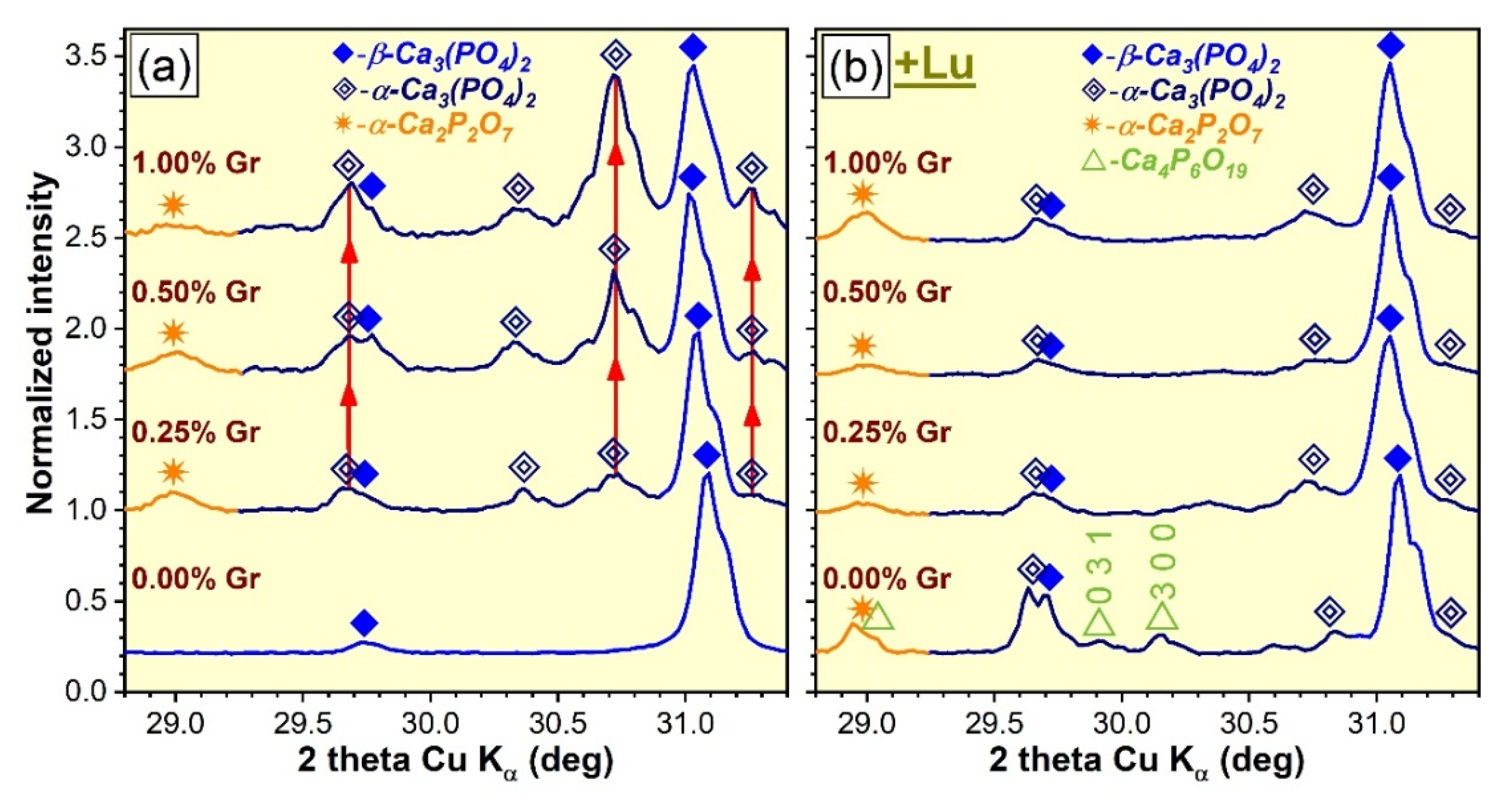
3.1. XRD Investigation
- (i).
- The DCPD phase, enclosed in HA/DCPD mixtures, was occasionally found to partially or fully transform, by dehydration and condensation, into γ-Ca2P2O7 at temperatures in the range of 500–600 °C, which further converts into the more stable β-Ca2P2O7 and α-Ca2P2O7 forms at ~700–800 °C and 1000–1200 °C, respectively, in air ambient [57,58,59] and at slightly more elevated temperatures in nitrogen [50]. Concurrently, up to 1200 °C, HA was found to gradually decompose into β-TCP or a mixture of β-TCP and α-TCP [19,60]. The high temperature de-hydroxylation of HA (with the intermediate formation of oxyapatite, regardless of the sintering ambient [51,52]) is known to be the main promoter of its conversion to β-TCP [53,61,62]. Thereby, the presence of the minor oxyapatite phase for the air sintered specimens (Figure 1a,b) could be interpreted as an indication that the de-hydroxylation process of HA already began, suggesting that its decomposition can be expected at temperatures in excess of 1200 °C. In addition to de-hydroxylation, the interaction of HA with the Ca2P2O7 counterpart when the sintering temperature is increased is considered to be a second contributor to the decomposition into β-TCP [62]. However, the origin of the TCP phases cannot be solely ascribed to HA as the hypothesis of thermal decomposition of DCPD into both Ca2P2O7 and TCP phases was also advanced [50]. The appearance of the α-TCP phase is now well-established to originate from the displacive transformation of β-TCP at temperatures of 1100–1200 °C or higher [2,28,29,53,63]. The hexaphosphate phase, although adventitiously formed in reduced amounts, is also a subsidiary effect of the DCPD sintering, yet was previously signaled in air ambient [18] and not in nitrogen.
- (ii).
- The dissimilarity in crystalline phase composition of the samples sintered in air ambient (Figure 1a,b), with respect to those processed in nitrogen (regardless of their compactness) (Figure 1c,d), is undoubtedly enabled by the sintering environment. This was to be expected since scientific literature evidence advocates that a less-reactive ambient alone, such as nitrogen, favors an accentuated de-hydroxylation of the HA and, hence, a diminished structural stability at high temperatures (i.e., 1200 °C), shifting the equilibrium to both β- and α-TCP phases [12,52].
- (iii).
- The presence of Gr can be viewed as a supplemental adjuvant (until its complete consumption) for the β-TCP → α-TCP conversion under nitrogen ambient. This is markedly evident for the CP-type specimens (Figure 2a), composed of closely packed ceramic particles admixed with Gr. This phenomenon can be ascribed to the generation of a higher thermal gradient (as Gr has a significantly higher thermal conductivity (3000 W/m·K) with respect to CaPs (1.1–1.25 W/m·K) [10,25,64]), which in turn, can foster an accelerated phase transformation [25,52].
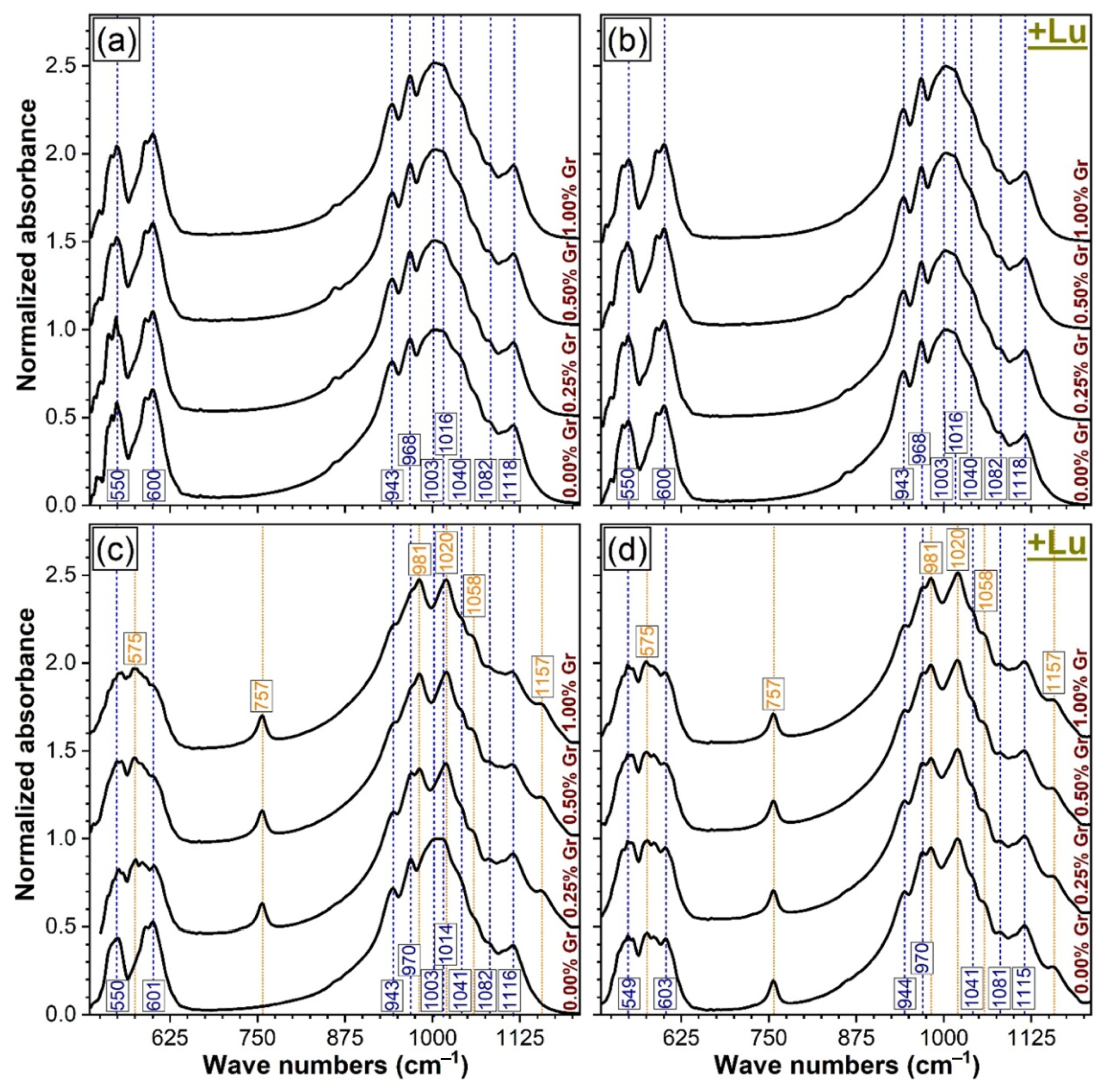
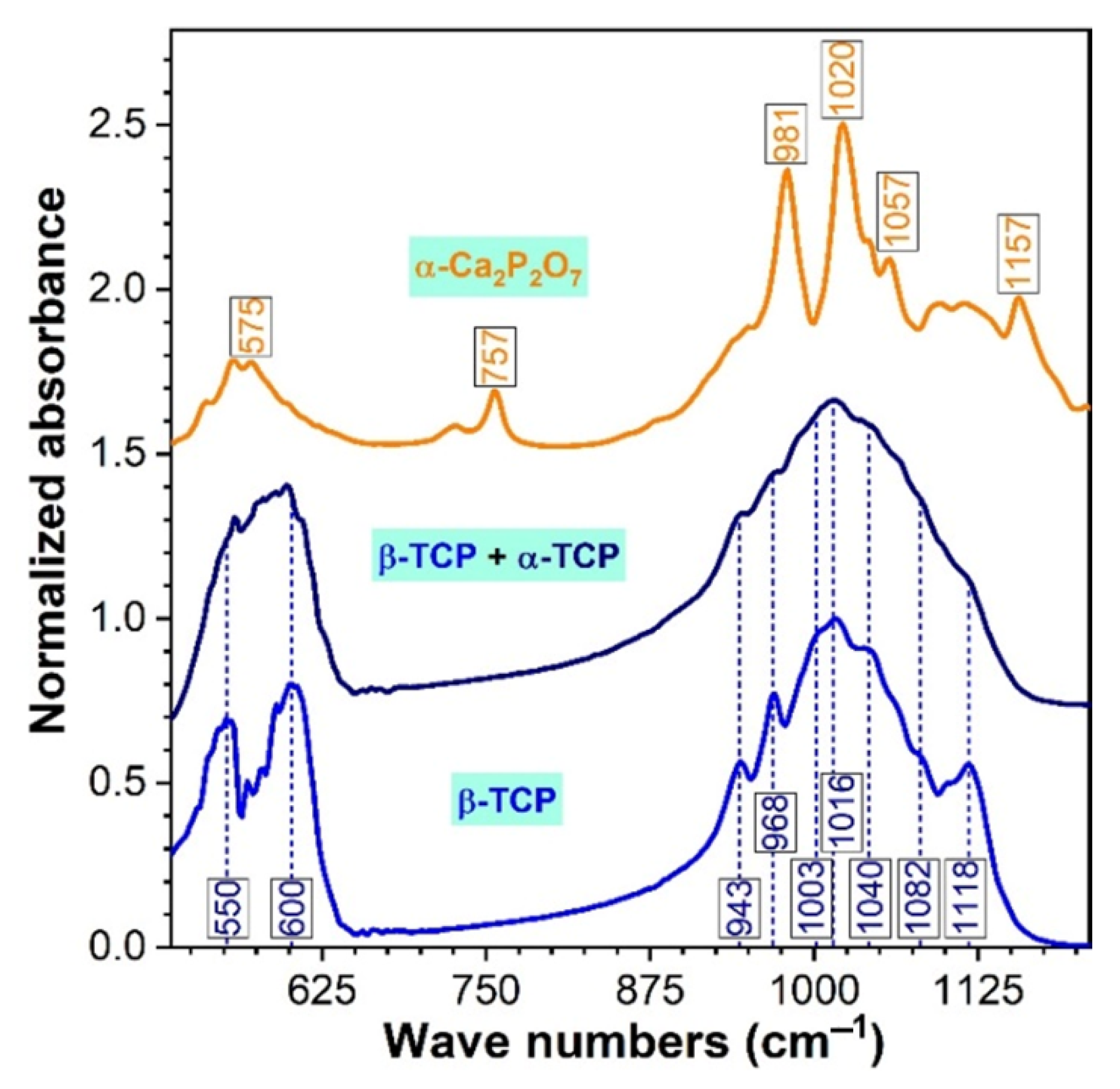
3.2. FTIR Spectroscopy Analysis
3.3. Morpho-Compositional SEM/EDS Evaluation
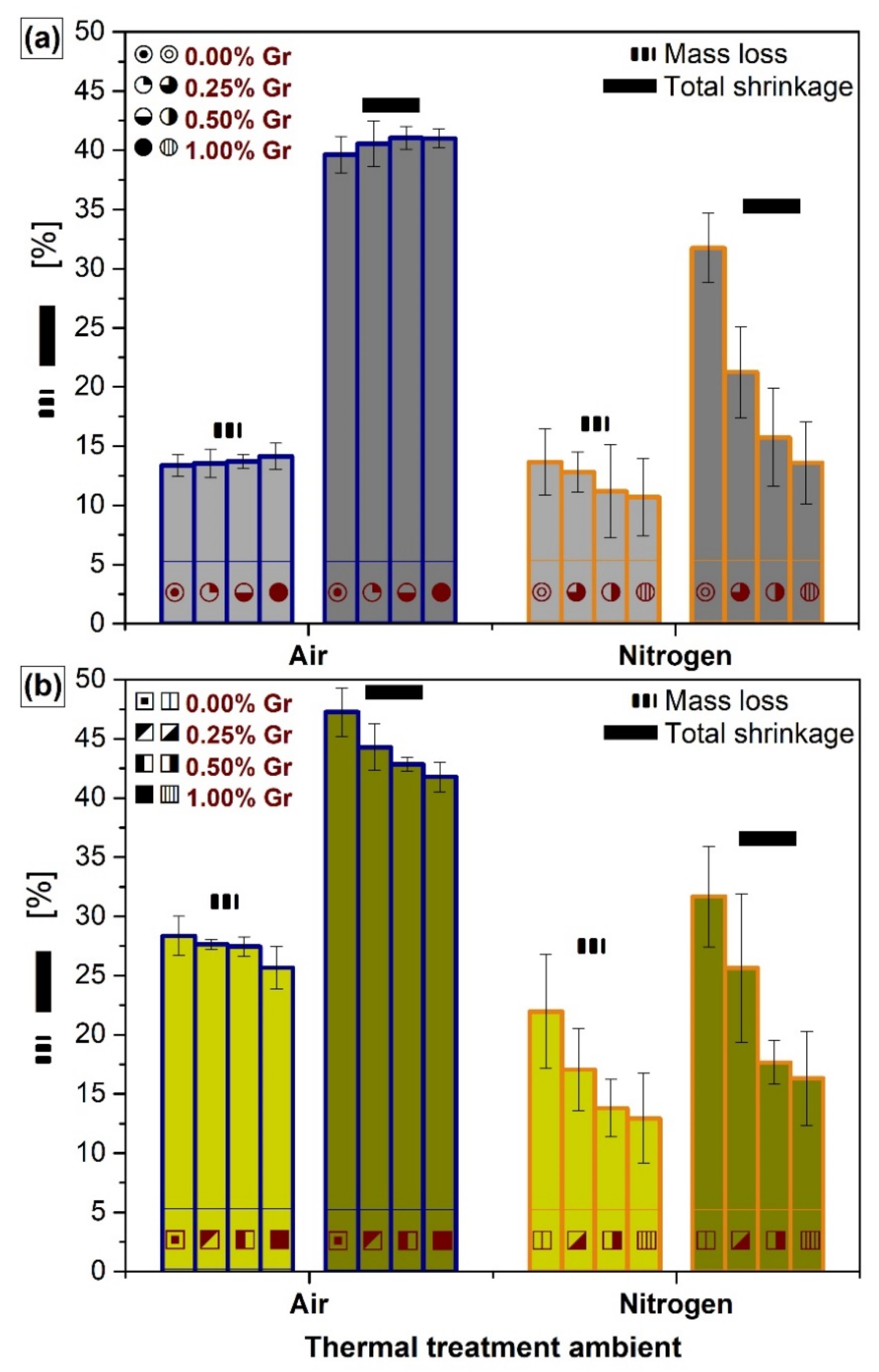
3.4. Dimensional Shrinkage and Mass Loss Determination
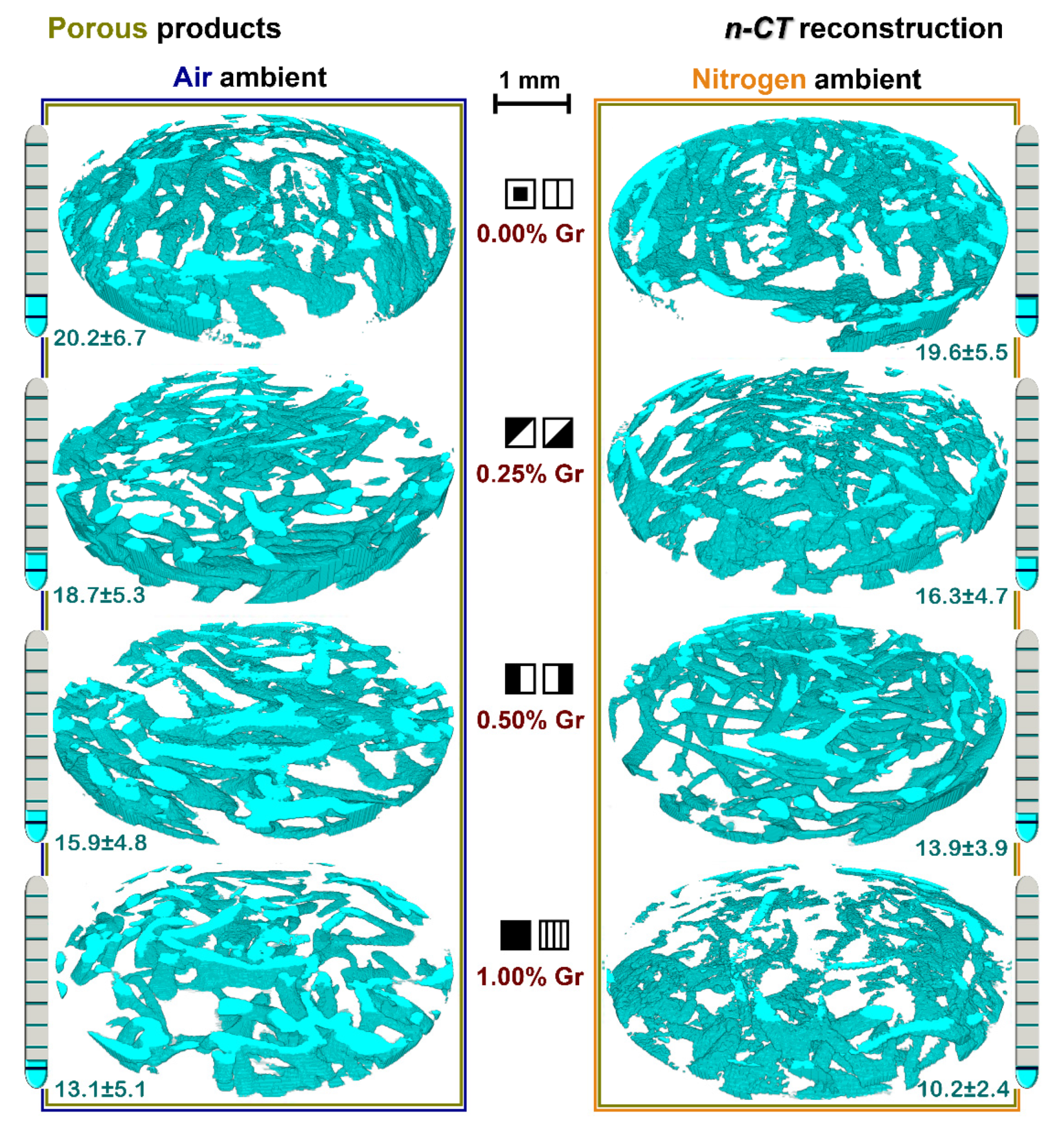
3.5. Nano-CT Reconstruction Analysis
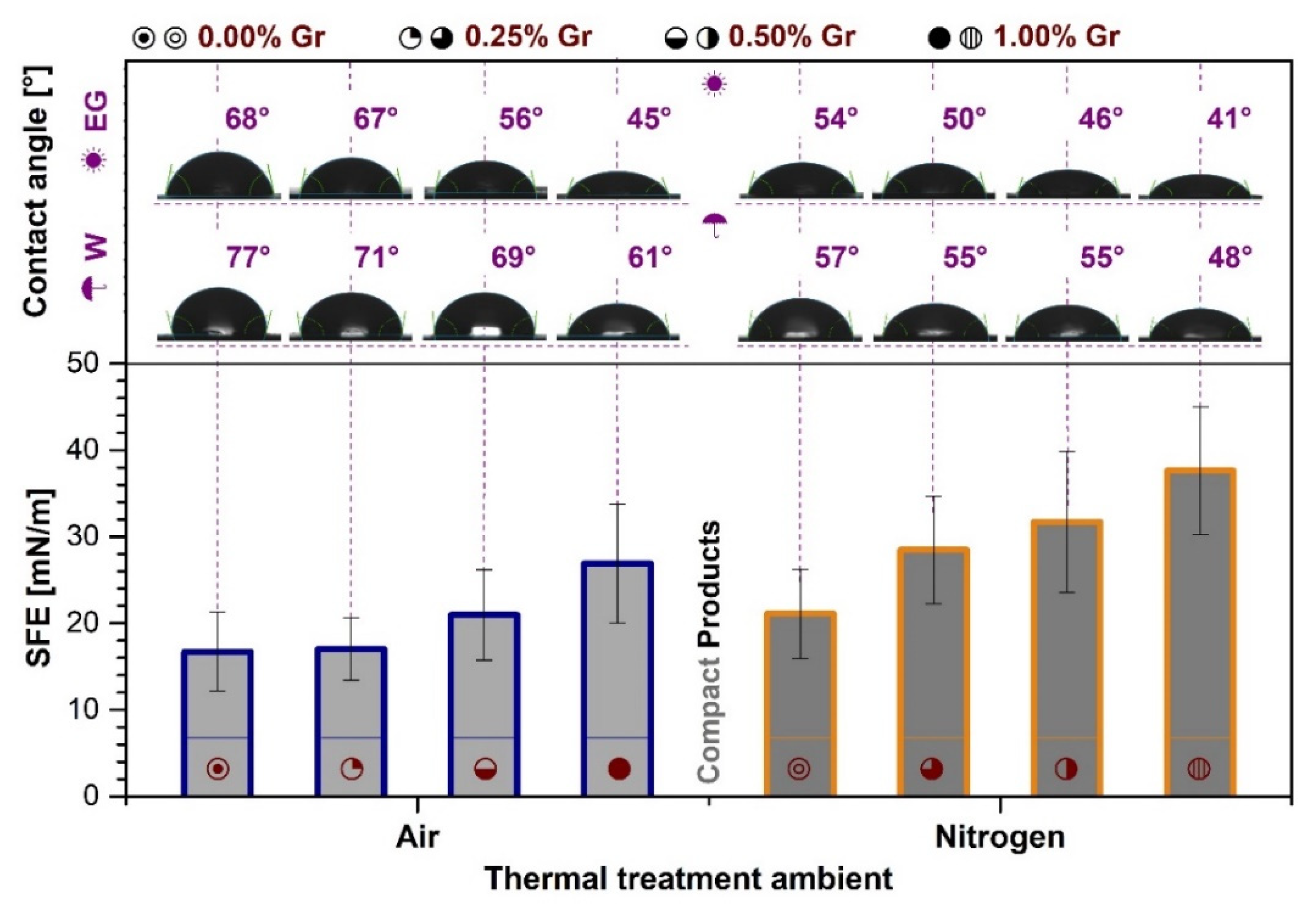
3.6. Contact Angle Measurements
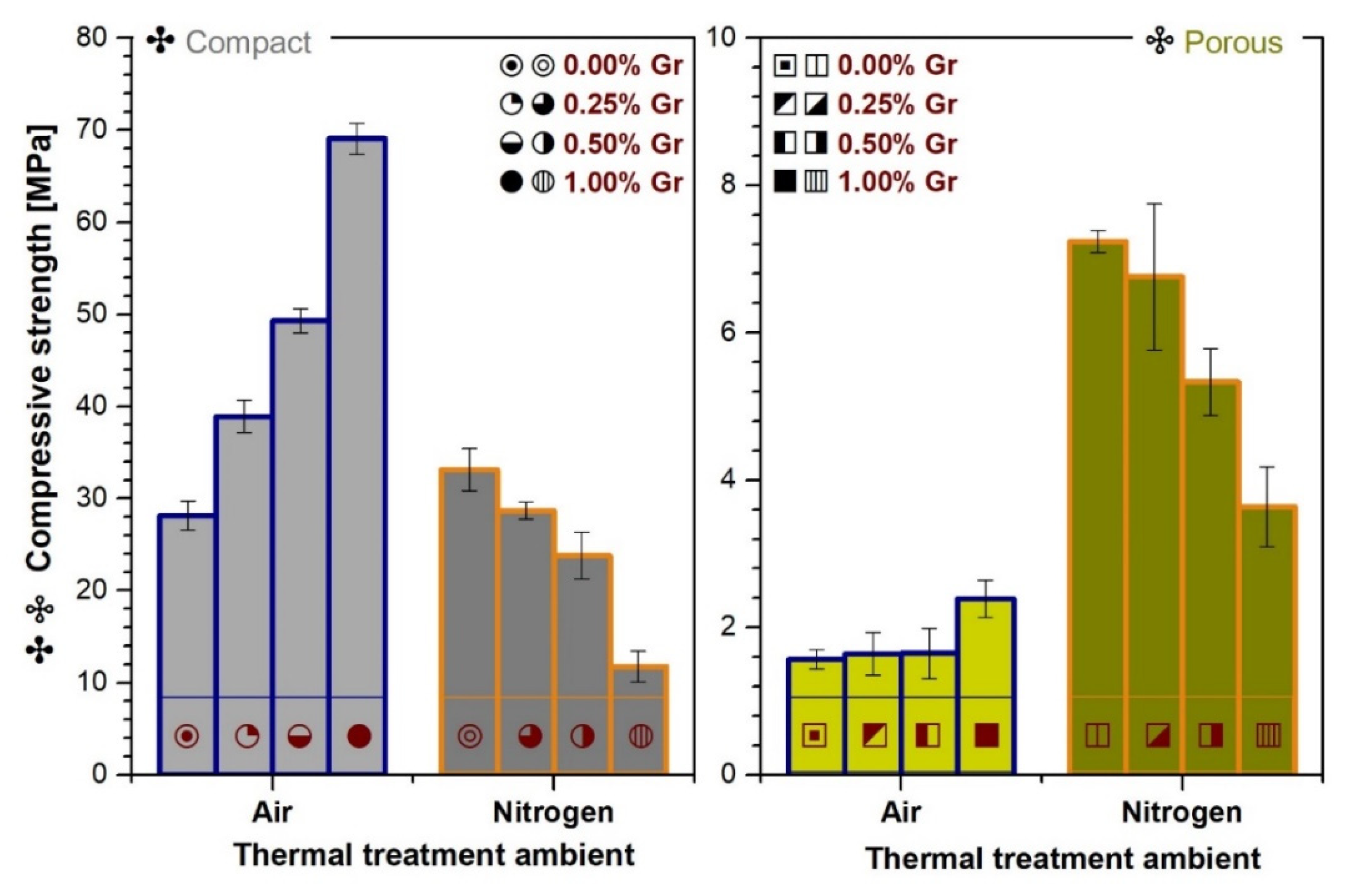
3.7. Mechanical Properties Evaluation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Eliaz, N.; Metoki, N. Calcium phosphate bioceramics: A review of their history, structure, properties, coating technologies and biomedical applications. Materials 2017, 10, 334. [Google Scholar] [CrossRef] [Green Version]
- Champion, E. Sintering of calcium phosphate bioceramics. Acta Biomater. 2013, 9, 5855–5875. [Google Scholar] [CrossRef]
- De Groot, K. Bioceramics of calcium phosphate. J. Clin. Eng. 1984, 9, 52. [Google Scholar] [CrossRef]
- Mocanu, A.-C.; Miculescu, F.; Miculescu, M.; Ciocoiu, R.C.; Pandele, A.M.; Stan, G.E.; Cîmpean, A.; Voicu, Ș.I.; Ciocan, L.-T. Comprehensive analysis of compatible natural fibre as sacrificial porogen template for tailored ceramic 3D bioproducts destined for hard tissue reconstruction. Ceram. Int. 2020, 47, 5318–5334. [Google Scholar] [CrossRef]
- Lu, J.; Yu, H.; Chen, C. Biological properties of calcium phosphate biomaterials for bone repair: A review. RSC Adv. 2018, 8, 2015–2033. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Wu, W.; Qian, C.; Xiao, W.; Zhu, H.; Guo, J.; Meng, Z.; Zhu, J.; Ge, Z.; Cui, W. Advanced biomaterials for repairing and reconstruction of mandibular defects. Mater. Sci. Eng. C 2019, 103, 109858. [Google Scholar] [CrossRef]
- Tecu, C.; Antoniac, I.; Goller, G.; Yavas, B.; Gheorghe, D.; Antoniac, A.; Ciuca, I.; Semenescu, A.; Raiciu, A.D.; Cristescu, I. The Sintering Behaviour and Mechanical Properties of Hydroxyapatite-Based Composites for Bone Tissue Regeneration. Mater. Plast. 2019, 56, 644–648. [Google Scholar] [CrossRef]
- Dee, P.; You, H.Y.; Teoh, S.-H.; Le Ferrand, H. Bioinspired approaches to toughen calcium phosphate-based ceramics for bone repair. J. Mech. Behav. Biomed. Mater. 2020, 104078. [Google Scholar] [CrossRef]
- Li, M.; Xiong, P.; Yan, F.; Li, S.; Ren, C.; Yin, Z.; Li, A.; Li, H.; Ji, X.; Zheng, Y. An overview of graphene-based hydroxyapatite composites for orthopedic applications. Bioact. Mater. 2018, 3, 1–18. [Google Scholar] [CrossRef]
- Mocanu, A.-C.; Miculescu, F.; Stan, G.E.; Ciocoiu, R.-C.; Corobea, M.C.; Miculescu, M.; Ciocan, L.T. Preliminary Studies on Graphene-Reinforced 3D Products Obtained by the One-Stage Sacrificial Template Method for Bone Reconstruction Applications. J. Funct. Biomater. 2021, 12, 13. [Google Scholar] [CrossRef]
- Miculescu, F.; Miculescu, M.; Ciocan, L.; Ernuteanu, A.; Antoniac, I.; Pencea, I.; Matei, E. Comparative Studies Regarding Heavy Elements Concentration In Human Cortical Bone. Dig. J. Nanomater. Biostruct. 2011, 6, 1117–1127. [Google Scholar]
- Feng, C.; Zhang, K.; He, R.; Ding, G.; Xia, M.; Jin, X.; Xie, C. Additive manufacturing of hydroxyapatite bioceramic scaffolds: Dispersion, digital light processing, sintering, mechanical properties, and biocompatibility. J. Adv. Ceram. 2020, 9, 360–373. [Google Scholar] [CrossRef]
- Logesh, M.; Marimuthu, A.; Ballamurugan, A. Fabrication of graphene incorporated biphasic calcium phosphate composite and evaluation of impact of graphene in the in-vitro biomineralization process. Mater. Chem. Phys. 2019, 232, 75–81. [Google Scholar] [CrossRef]
- Canillas, M.; Pena, P.; Antonio, H.; Rodríguez, M.A. Calcium phosphates for biomedical applications. Boletín Soc. Española Cerámica Vidr. 2017, 56, 91–112. [Google Scholar] [CrossRef]
- Cotrut, C.M.; Vladescu, A.; Dinu, M.; Vranceanu, D.M. Influence of deposition temperature on the properties of hydroxyapatite obtained by electrochemical assisted deposition. Ceram. Int. 2018, 44, 669–677. [Google Scholar] [CrossRef]
- Topsakal, A.; Ekren, N.; Kilic, O.; Oktar, F.N.; Mahirogullari, M.; Ozkan, O.; Sasmazel, H.T.; Turk, M.; Bogdan, I.M.; Stan, G.E. Synthesis and characterization of antibacterial drug loaded β-tricalcium phosphate powders for bone engineering applications. J. Mater. Sci. Mater. Med. 2020, 31, 1–17. [Google Scholar] [CrossRef]
- Lee, J.H.; Chang, B.-S.; Jeung, U.-O.; Park, K.-W.; Kim, M.-S.; Lee, C.-K. The first clinical trial of beta-calcium pyrophosphate as a novel bone graft extender in instrumented posterolateral lumbar fusion. Clin. Orthop. Surg. 2011, 3, 238–244. [Google Scholar] [CrossRef] [Green Version]
- Anastasiou, A.; Strafford, S.; Posada-Estefan, O.; Thomson, C.; Hussain, S.; Edwards, T.; Malinowski, M.; Hondow, N.; Metzger, N.; Brown, C. β-pyrophosphate: A potential biomaterial for dental applications. Mater. Sci. Eng. C 2017, 75, 885–894. [Google Scholar] [CrossRef]
- Krut’ko, V.; Kulak, A.; Musskaya, O.; Safronova, T.; Putlyaev, V. Calcium phosphate foam ceramic based on hydroxyapatite–brushite powder mixture. Glass Ceram. 2019, 76, 113–118. [Google Scholar] [CrossRef]
- Vladescu, A.; Badea, M.; Padmanabhan, S.C.; Paraschiv, G.; Floroian, L.; Gaman, L.; Morris, M.A.; Marty, J.-L.; Cotrut, C.M. Nanomaterials for Medical Applications and Their Antimicrobial Advantages. In Materials for Biomedical Engineering; Elsevier: Amsterdam, The Netherlands, 2019; pp. 409–431. [Google Scholar] [CrossRef]
- Rau, J.V.; Fadeeva, I.V.; Fomin, A.S.; Barbaro, K.; Galvano, E.; Ryzhov, A.P.; Murzakhanov, F.; Gafurov, M.; Orlinskii, S.; Antoniac, I. Sic Parvis Magna: Manganese-substituted tricalcium phosphate and its biophysical properties. ACS Biomater. Sci. Eng. 2019, 5, 6632–6644. [Google Scholar] [CrossRef]
- Wang, G.; Zheng, L.; Zhao, H.; Miao, J.; Sun, C.; Liu, H.; Huang, Z.; Yu, X.; Wang, J.; Tao, X. Construction of a fluorescent nanostructured chitosan-hydroxyapatite scaffold by nanocrystallon induced biomimetic mineralization and its cell biocompatibility. ACS Appl. Mater. Interfaces 2011, 3, 1692–1701. [Google Scholar] [CrossRef]
- John, Ł.; Janeta, M.; Szafert, S. Designing of macroporous magnetic bioscaffold based on functionalized methacrylate network covered by hydroxyapatites and doped with nano-MgFe2O4 for potential cancer hyperthermia therapy. Mater. Sci. Eng. C 2017, 78, 901–911. [Google Scholar] [CrossRef]
- Pepla, E.; Besharat, L.K.; Palaia, G.; Tenore, G.; Migliau, G. Nano-hydroxyapatite and its applications in preventive, restorative and regenerative dentistry: A review of literature. Ann. Stomatol. 2014, 5, 108, PMC4252862. [Google Scholar] [CrossRef]
- Baradaran, S.; Moghaddam, E.; Nasiri-Tabrizi, B.; Basirun, W.J.; Mehrali, M.; Sookhakian, M.; Hamdi, M.; Alias, Y. Characterization of nickel-doped biphasic calcium phosphate/graphene nanoplatelet composites for biomedical application. Mater. Sci. Eng. C 2015, 49, 656–668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mocanu, A.-C.; Stan, G.E.; Maidaniuc, A.; Miculescu, M.; Antoniac, I.V.; Ciocoiu, R.-C.; Voicu, Ș.I.; Mitran, V.; Cîmpean, A.; Miculescu, F. Naturally-derived biphasic calcium phosphates through increased phosphorus-based reagent amounts for biomedical applications. Materials 2019, 12, 381. [Google Scholar] [CrossRef] [Green Version]
- Jurgelane, I.; Buss, A.; Putnina, M.; Loca, D.; Locs, J. Effect of sintering temperature on sorption properties and compressive strength of calcium phosphate ceramic granules. Mater. Lett. 2021, 282, 128858. [Google Scholar] [CrossRef]
- Yetmez, M. Sintering behavior and mechanical properties of biphasic calcium phosphate ceramics. Adv. Mater. Sci. Eng. 2014, 2014, 871749. [Google Scholar] [CrossRef] [Green Version]
- Lukić, M.; Stojanović, Z.; Škapin, S.D.; Maček-Kržmanc, M.; Mitrić, M.; Marković, S.; Uskoković, D. Dense fine-grained biphasic calcium phosphate (BCP) bioceramics designed by two-step sintering. J. Eur. Ceram. Soc. 2011, 31, 19–27. [Google Scholar] [CrossRef]
- Lee, K.-S.; Han, H.-S.; Kim, Y.-C.; Lo Han, J.; Seung, R.-H.; Lee, H.-S.; Chang, J.-S.; Lee, D.-H. Evaluation of porous β-calcium pyrophosphate as bioresorbable bone graft substitute material. Mater. Res. Innov. 2015, 19, 86–90. [Google Scholar] [CrossRef]
- Abbasi, N.; Hamlet, S.; Love, R.M.; Nguyen, N.-T. Porous scaffolds for bone regeneration. J. Sci. Adv. Mater. Dev. 2020, 5, 1–9. [Google Scholar] [CrossRef]
- Babaie, E.; Bhaduri, S.B. Fabrication aspects of porous biomaterials in orthopedic applications: A review. ACS Biomater. Sci. Eng. 2017, 4, 1–39. [Google Scholar] [CrossRef]
- Maidaniuc, A.; Miculescu, F.; Ciocoiu, R.C.; Butte, T.M.; Pasuk, I.; Stan, G.E.; Voicu, S.I.; Ciocan, L.T. Effect of the processing parameters on surface, physico-chemical and mechanical features of bioceramics synthesized from abundant carp fish bones. Ceram. Int. 2020, 46, 10159–10171. [Google Scholar] [CrossRef]
- Miculescu, F.; Mocanu, A.C.; Stan, G.E.; Miculescu, M.; Maidaniuc, A.; Cîmpean, A.; Mitran, V.; Voicu, S.I.; Machedon-Pisu, T.; Ciocan, L.T. Influence of the modulated two-step synthesis of biogenic hydroxyapatite on biomimetic products’ surface. Appl. Surf. Sci. 2018, 438, 147–157. [Google Scholar] [CrossRef]
- Miculescu, F.; Mocanu, A.-C.; Dascălu, C.A.; Maidaniuc, A.; Batalu, D.; Berbecaru, A.; Voicu, S.I.; Miculescu, M.; Thakur, V.K.; Ciocan, L.T. Facile synthesis and characterization of hydroxyapatite particles for high value nanocomposites and biomaterials. Vacuum 2017, 146, 614–622. [Google Scholar] [CrossRef] [Green Version]
- Stevulova, N.; Estokova, A.; Cigasova, J.; Schwarzova, I.; Kacik, F.; Geffert, A. Thermal degradation of natural and treated hemp hurds under air and nitrogen atmosphere. J. Therm. Anal. Calorim. 2017, 128, 1649–1660. [Google Scholar] [CrossRef] [Green Version]
- Tanobe, V.O.; Sydenstricker, T.H.; Munaro, M.; Amico, S.C. A comprehensive characterization of chemically treated Brazilian sponge-gourds (Luffa cylindrica). Polym. Test. 2005, 24, 474–482. [Google Scholar] [CrossRef]
- Miculescu, F.; Stan, G.; Ciocan, L.; Miculescu, M.; Berbecaru, A.; Antoniac, I. Cortical Bone as Resource for Producing Biomimetic Materials for Clinical Use. Dig. J. Nanomater. Biostruct. 2012, 7, 1667–1677. [Google Scholar]
- Mocanu, A.-C.; Miculescu, M.; Machedon-Pisu, T.; Maidaniuc, A.; Ciocoiu, R.C.; Ioniță, M.; Pasuk, I.; Stan, G.E.; Miculescu, F. Internal and external surface features of newly developed porous ceramics with random interconnected 3D channels by a fibrous sacrificial porogen method. Appl. Surf. Sci. 2019, 489, 226–238. [Google Scholar] [CrossRef]
- Mafu, L.D.; Neomagus, H.W.; Everson, R.C.; Strydom, C.A.; Carrier, M.; Okolo, G.N.; Bunt, J.R. Chemical and structural characterization of char development during lignocellulosic biomass pyrolysis. Bioresour. Technol. 2017, 243, 941–948. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Zhang, S.; Wang, Y.; Sun, X.; Sun, K. Reduced graphene oxide/carbon nanotubes reinforced calcium phosphate cement. Ceram. Int. 2017, 43, 13083–13088. [Google Scholar] [CrossRef]
- Khosravani, M.R. Composite materials manufacturing processes. Appl. Mech. Mater. 2012, 110–116, 1361–1367. [Google Scholar] [CrossRef]
- Syama, S.; Mohanan, P.V. Safety and biocompatibility of graphene: A new generation nanomaterial for biomedical application. Int. J. Biol. Macromol. 2016, 86, 546–555. [Google Scholar] [CrossRef]
- Baudín, C.; Benet, T.; Pena, P. Effect of graphene on setting and mechanical behaviour of tricalcium phosphate bioactive cements. J. Mech. Behav. Biomed. Mater. 2019, 89, 33–47. [Google Scholar] [CrossRef]
- Basirun, W.J.; Nasiri-Tabrizi, B.; Baradaran, S. Overview of hydroxyapatite–graphene nanoplatelets composite as bone graft substitute: Mechanical behavior and in-vitro biofunctionality. Crit. Rev. Solid State Mater. 2018, 43, 177–212. [Google Scholar] [CrossRef]
- Lalwani, G.; D’Agati, M.; Khan, A.M.; Sitharaman, B. Toxicology of graphene-based nanomaterials. Adv. Drug Deliv. Rev. 2016, 105, 109–144. [Google Scholar] [CrossRef] [Green Version]
- Baradaran, S.; Moghaddam, E.; Basirun, W.J.; Mehrali, M.; Sookhakian, M.; Hamdi, M.; Moghaddam, M.N.; Alias, Y. Mechanical properties and biomedical applications of a nanotube hydroxyapatite-reduced graphene oxide composite. Carbon 2014, 69, 32–45. [Google Scholar] [CrossRef]
- Orlov, N.; Kiseleva, A.; Milkin, P.; Evdokimov, P.; Putlyaev, V. Reaction Sintering of Bioceramic Based on Substituted Calcium Phosphates CaMPO 4 (M = K, Na). Inorg. Mater. Appl. Res. 2020, 11, 394–402. [Google Scholar] [CrossRef]
- Frasnelli, M.; Sglavo, V.M. Flash sintering of tricalcium phosphate (TCP) bioceramics. J. Eur. Ceram. Soc. 2018, 38, 279–285. [Google Scholar] [CrossRef]
- Frost, R.L.; Palmer, S.J. Thermal stability of the ‘cave’mineral brushite CaHPO4·2H2O–Mechanism of formation and decomposition. Thermochim. Acta 2011, 521, 14–17. [Google Scholar] [CrossRef] [Green Version]
- Liao, C.-J.; Lin, F.-H.; Chen, K.-S.; Sun, J.-S. Thermal decomposition and reconstitution of hydroxyapatite in air atmosphere. Biomaterials 1999, 20, 1807–1813. [Google Scholar] [CrossRef]
- Lukić, M.J.; Kuzmanović, M.; Sezen, M.; Bakan, F.; Egelja, A.; Veselinović, L. Inert atmosphere processing of hydroxyapatite in the presence of lithium iron phosphate. J. Eur. Ceram. Soc. 2018, 38, 2120–2133. [Google Scholar] [CrossRef] [Green Version]
- Nilen, R.W.N.; Richter, P.W. The thermal stability of hydroxyapatite in biphasic calcium phosphate ceramics. J. Mater. Sci. Mater. Med. 2008, 19, 1693–1702. [Google Scholar] [CrossRef] [PubMed]
- Mitran, V.; Ion, R.; Miculescu, F.; Necula, M.G.; Mocanu, A.-C.; Stan, G.E.; Antoniac, I.V.; Cimpean, A. Osteoblast cell response to naturally derived calcium phosphate-based materials. Materials 2018, 11, 1097. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, Y.; Yu, C.x.; Li, Y.; Zhu, Q.; Zhou, L.; Cao, C.; Yu, T.; Du, F. Research on the changes in wettability of rice (Oryza sativa.) leaf surfaces at different development stages using the OWRK method. Pest Manag. Sci. 2014, 70, 462–469. [Google Scholar] [CrossRef] [PubMed]
- Żenkiewicz, M. Methods for the calculation of surface free energy of solids. J. Achiev. Mater. Manuf. Eng. 2007, 24, 137–145. [Google Scholar]
- Schofield, P.; Knight, K.; Van der Houwen, J.; Valsami-Jones, E. The role of hydrogen bonding in the thermal expansion and dehydration of brushite, di-calcium phosphate dihydrate. Phys. Chem. Miner. 2004, 31, 606–624. [Google Scholar] [CrossRef]
- Webb, N. The crystal structure of β-Ca2P2O. Acta Cryst. 1966, 21, 942–948. [Google Scholar] [CrossRef]
- Ming, C.; Greish, Y.; El-Ghannam, A. Crystallization behavior of silica-calcium phosphate biocomposites: XRD and FTIR studies. J. Mater. Sci. Mater. Med. 2004, 15, 1227–1235. [Google Scholar] [CrossRef]
- Minh, D.P.; Martinez, M.G.; Nzihou, A.; Sharrock, P. Thermal behavior of apatitic calcium phosphates synthesized from calcium carbonate and orthophosphoric acid or potassium dihydrogen orthophosphate. J. Therm. Anal. Calorim. 2013, 112, 1145–1155. [Google Scholar] [CrossRef] [Green Version]
- White, A.A.; Kinloch, I.A.; Windle, A.H.; Best, S.M. Optimization of the sintering atmosphere for high-density hydroxyapatite–carbon nanotube composites. J. R. Soc. Interface 2010, 7, S529–S539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tonsuaadu, K.; Gross, K.A.; Plūduma, L.; Veiderma, M. A review on the thermal stability of calcium apatites. J. Therm. Anal. Calorim. 2012, 110, 647–659. [Google Scholar] [CrossRef]
- Carrodeguas, R.G.; De Aza, S. α-Tricalcium phosphate: Synthesis, properties and biomedical applications. Acta Biomater. 2011, 7, 3536–3546. [Google Scholar] [CrossRef]
- Bento, A.; Almond, D.; Brown, S.; Turner, I. Thermal and optical characterization of the calcium phosphate biomaterial hydroxyapatite. J. Appl. Phys. 1996, 79, 6848–6852. [Google Scholar] [CrossRef]
- Zhao, Y.; Sun, K.-N.; Wang, W.-L.; Wang, Y.-X.; Sun, X.-L.; Liang, Y.-J.; Sun, X.-N.; Chui, P.-F. Microstructure and anisotropic mechanical properties of graphene nanoplatelet toughened biphasic calcium phosphate composite. Ceram. Int. 2013, 39, 7627–7634. [Google Scholar] [CrossRef]
- Nosrati, H.; Sarraf-Mamoory, R.; Behnagh, A.K.; Emameh, R.Z.; Aidun, A.; Le, D.Q.S.; Perez, M.C.; Bünger, C.E. Comparison of the effect of argon, hydrogen, and nitrogen gases on the reduced graphene oxide-hydroxyapatite nanocomposites characteristics. BMC Chem. 2020, 14, 59. [Google Scholar] [CrossRef] [PubMed]
- Ryu, H.-S.; Youn, H.-J.; Hong, K.S.; Chang, B.-S.; Lee, C.-K.; Chung, S.-S. An improvement in sintering property of β-tricalcium phosphate by addition of calcium pyrophosphate. Biomaterials 2002, 23, 909–914. [Google Scholar] [CrossRef]
- Lilley, K.; Gbureck, U.; Wright, A.; Farrar, D.; Barralet, J. Cement from nanocrystalline hydroxyapatite: Effect of calcium phosphate ratio. J. Mater. Sci. Mater. Med. 2005, 16, 1185–1190. [Google Scholar] [CrossRef]
- Galea, L.; Alexeev, D.; Bohner, M.; Doebelin, N.; Studart, A.R.; Aneziris, C.G.; Graule, T. Textured and hierarchically structured calcium phosphate ceramic blocks through hydrothermal treatment. Biomaterials 2015, 67, 93–103. [Google Scholar] [CrossRef] [PubMed]
- Miculescu, F.; Ciocan, L.; Miculescu, M.; Ernuteanu, A. Effect of heating process on micro structure level of cortical bone prepared for compositional analysis. Dig. J. Nanomater. Biostruct. 2011, 6, 225–233. [Google Scholar]
- Fernandez, M.P.; Witte, F.; Tozzi, G. Applications of X-ray computed tomography for the evaluation of biomaterial-mediated bone regeneration in critical-sized defects. J. Microsc. 2020, 277, 179–196. [Google Scholar] [CrossRef] [PubMed]
- Miculescu, F.; Jepu, I.; Porosnicu, C.; Lungu, C.; Miculescu, M.; Burhala, B. A Study On The Influence of the Primary Electron Beam on Nanodimensional Layers Analysis. Dig. J. Nanomater. Biostruct. 2011, 6, 335–345. [Google Scholar]
- Khosravani, M.R.; Reinicke, T. On the Use of X-ray Computed Tomography in Assessment of 3D-Printed Components. J. Nondestruct. Eval. 2020, 39, 1–17. [Google Scholar] [CrossRef]
- Bossa, N.; Chaurand, P.; Vicente, J.; Borschneck, D.; Levard, C.; Aguerre-Chariol, O.; Rose, J. Micro-and nano-X-ray computed-tomography: A step forward in the characterization of the pore network of a leached cement paste. Cem. Concr. Res. 2015, 67, 138–147. [Google Scholar] [CrossRef]
- Zhang, Y.-H.; Lyu, S.-S.; Liu, S.-T.; Chen, Y.-X.; Qin, W.-L.; Ye, Y.; Chen, X.-G. Thermal destruction of luffa sponge under air and nitrogen. J. Therm. Anal. Calorim. 2017, 128, 53–60. [Google Scholar] [CrossRef]
- Bobbert, F.; Zadpoor, A. Effects of bone substitute architecture and surface properties on cell response, angiogenesis, and structure of new bone. J. Mater. Chem. B 2017, 5, 6175–6192. [Google Scholar] [CrossRef] [Green Version]
- Nakamura, M.; Hori, N.; Ando, H.; Namba, S.; Toyama, T.; Nishimiya, N.; Yamashita, K. Surface free energy predominates in cell adhesion to hydroxyapatite through wettability. Mater. Sci. Eng. C. 2016, 62, 283–292. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.H.; Ha, H.J.; Ko, Y.K.; Yoon, S.J.; Rhee, J.M.; Kim, M.S.; Lee, H.B.; Khang, G. Correlation of proliferation, morphology and biological responses of fibroblasts on LDPE with different surface wettability. J. Biomater. Sci. Polym. Ed. 2007, 18, 609–622. [Google Scholar] [CrossRef] [PubMed]
- Stan, G.E.; Tite, T.; Popa, A.-C.; Chirica, I.M.; Negrila, C.C.; Besleaga, C.; Zgura, I.; Sergentu, A.C.; Popescu-Pelin, G.; Cristea, D. The Beneficial Mechanical and Biological Outcomes of Thin Copper-Gallium Doped Silica-Rich Bio-Active Glass Implant-Type Coatings. Coatings 2020, 10, 1119. [Google Scholar] [CrossRef]
- PeŠŠková, V.; Kubies, D.; Hulejova, H.; Himmlova, L. The influence of implant surface properties on cell adhesion and proliferation. J. Mater. Sci. Mater. Med. 2007, 18, 465–473. [Google Scholar] [CrossRef]
- Webb, K.; Hlady, V.; Tresco, P.A. Relative importance of surface wettability and charged functional groups on NIH 3T3 fibroblast attachment, spreading, and cytoskeletal organization. J. Biomed. Mater. Res. 1998, 41, 422–430. [Google Scholar] [CrossRef] [Green Version]
- Mitra, J.; Tripathi, G.; Sharma, A.; Basu, B. Scaffolds for bone tissue engineering: Role of surface patterning on osteoblast response. RSC Adv. 2013, 3, 11073–11094. [Google Scholar] [CrossRef]
- Descamps, M.; Boilet, L.; Moreau, G.; Tricoteaux, A.; Lu, J.; Leriche, A.; Lardot, V.; Cambier, F. Processing and properties of biphasic calcium phosphates bioceramics obtained by pressureless sintering and hot isostatic pressing. J. Eur. Ceram. Soc. 2013, 33, 1263–1270. [Google Scholar] [CrossRef]
- Lee, H.; Jang, T.-S.; Song, J.; Kim, H.-E.; Jung, H.-D. The production of porous hydroxyapatite scaffolds with graded porosity by sequential freeze-casting. Materials 2017, 10, 367. [Google Scholar] [CrossRef] [PubMed]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mocanu, A.-C.; Miculescu, F.; Stan, G.E.; Pandele, A.-M.; Pop, M.A.; Ciocoiu, R.C.; Voicu, Ș.I.; Ciocan, L.-T. Fiber-Templated 3D Calcium-Phosphate Scaffolds for Biomedical Applications: The Role of the Thermal Treatment Ambient on Physico-Chemical Properties. Materials 2021, 14, 2198. https://doi.org/10.3390/ma14092198
Mocanu A-C, Miculescu F, Stan GE, Pandele A-M, Pop MA, Ciocoiu RC, Voicu ȘI, Ciocan L-T. Fiber-Templated 3D Calcium-Phosphate Scaffolds for Biomedical Applications: The Role of the Thermal Treatment Ambient on Physico-Chemical Properties. Materials. 2021; 14(9):2198. https://doi.org/10.3390/ma14092198
Chicago/Turabian StyleMocanu, Aura-Cătălina, Florin Miculescu, George E. Stan, Andreea-Mădălina Pandele, Mihai Alin Pop, Robert Cătălin Ciocoiu, Ștefan Ioan Voicu, and Lucian-Toma Ciocan. 2021. "Fiber-Templated 3D Calcium-Phosphate Scaffolds for Biomedical Applications: The Role of the Thermal Treatment Ambient on Physico-Chemical Properties" Materials 14, no. 9: 2198. https://doi.org/10.3390/ma14092198