Extremely Potent Block of Bacterial Voltage-Gated Sodium Channels by µ-Conotoxin PIIIA

 , , ,
, , ,
Abstract
:1. Introduction
2. Results
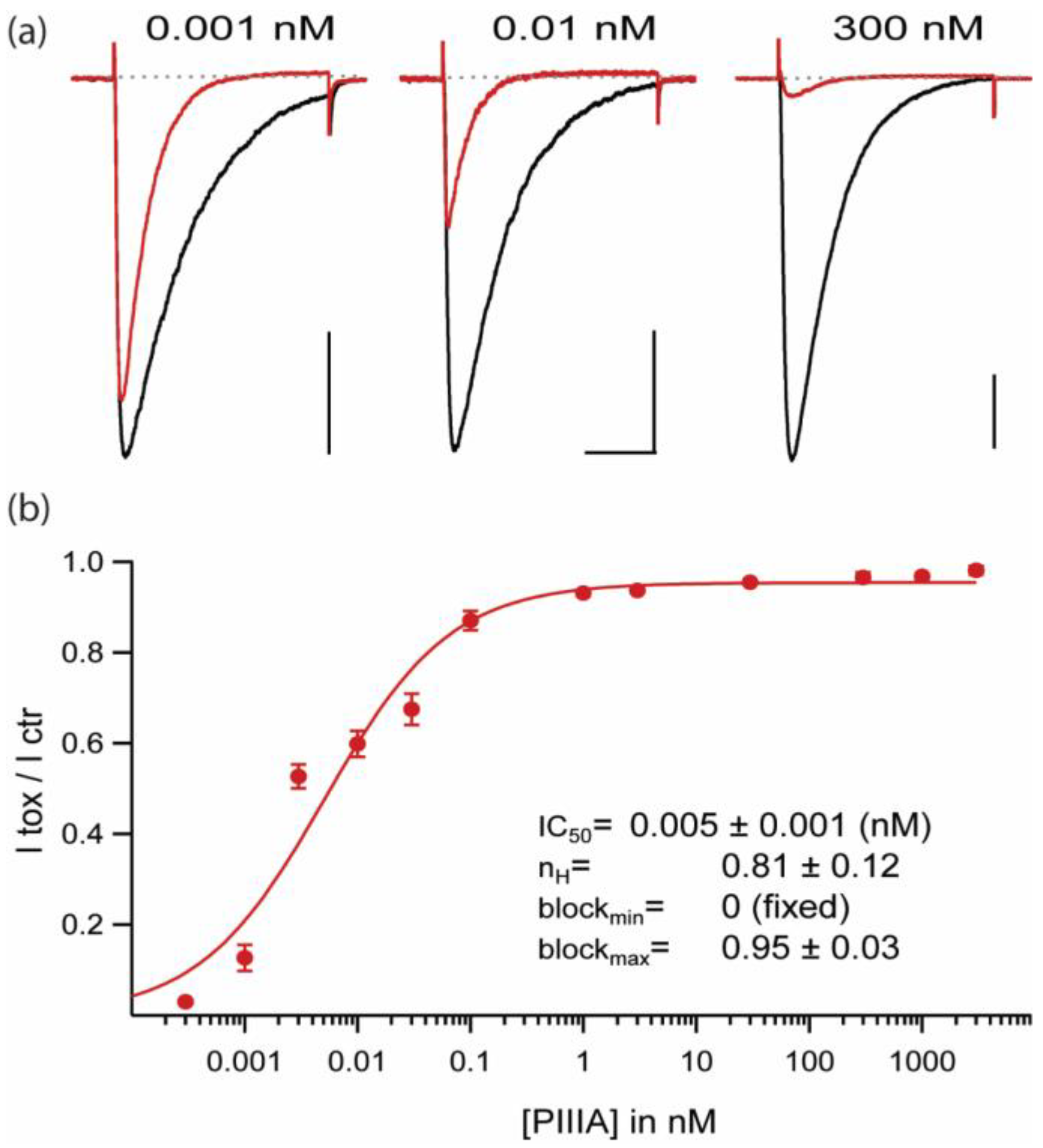
2.1. Extremely High Affinity Block of NaChBac by µ-Conotoxin PIIIA
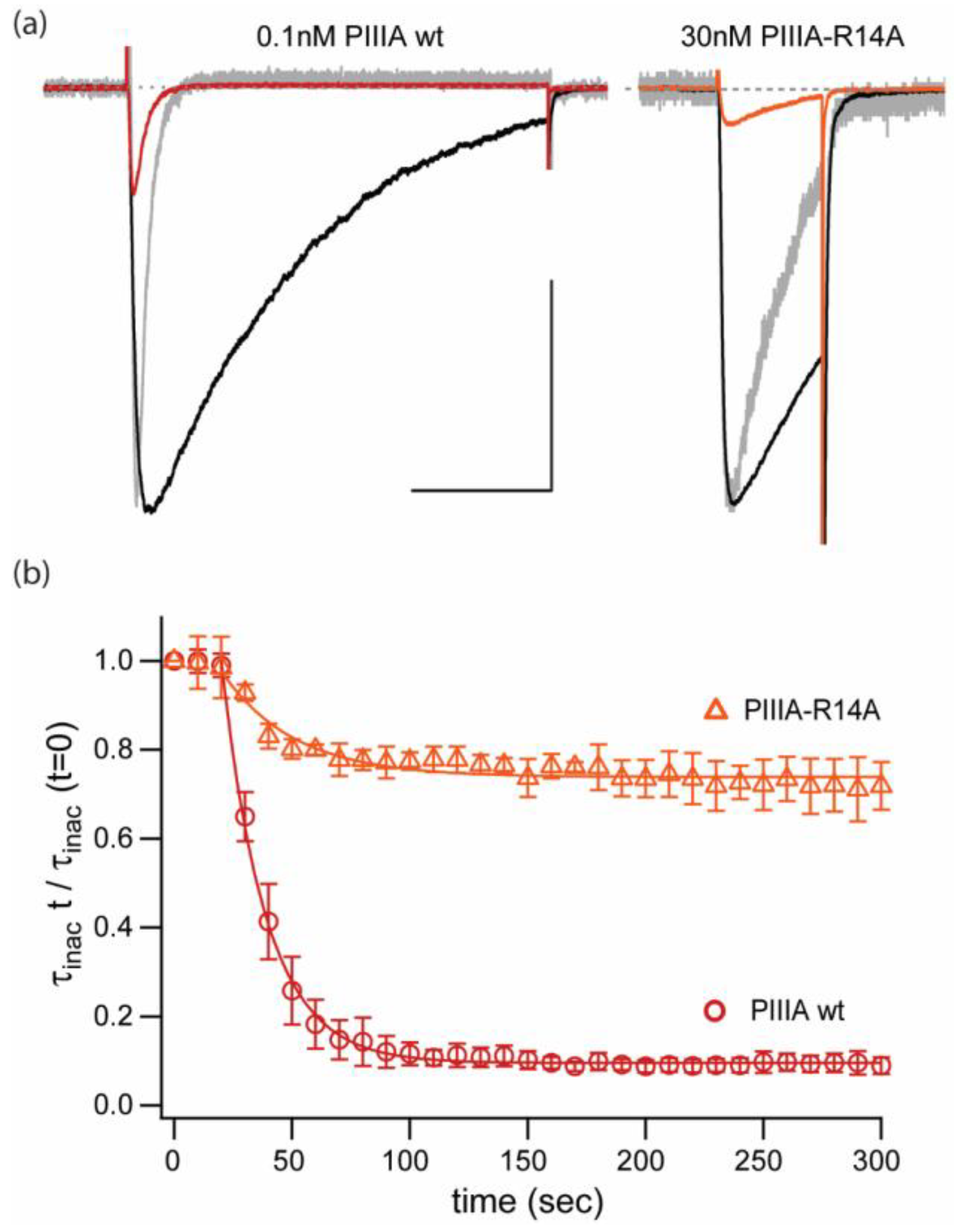
2.2. Replacement of a Key “Blocking” Residue (PIIIA-R14A) Abolishes the Speeding of Inactivation and Reduces Apparent Affinity by ~100-Fold, but Does Not Prevent Reduction of Current
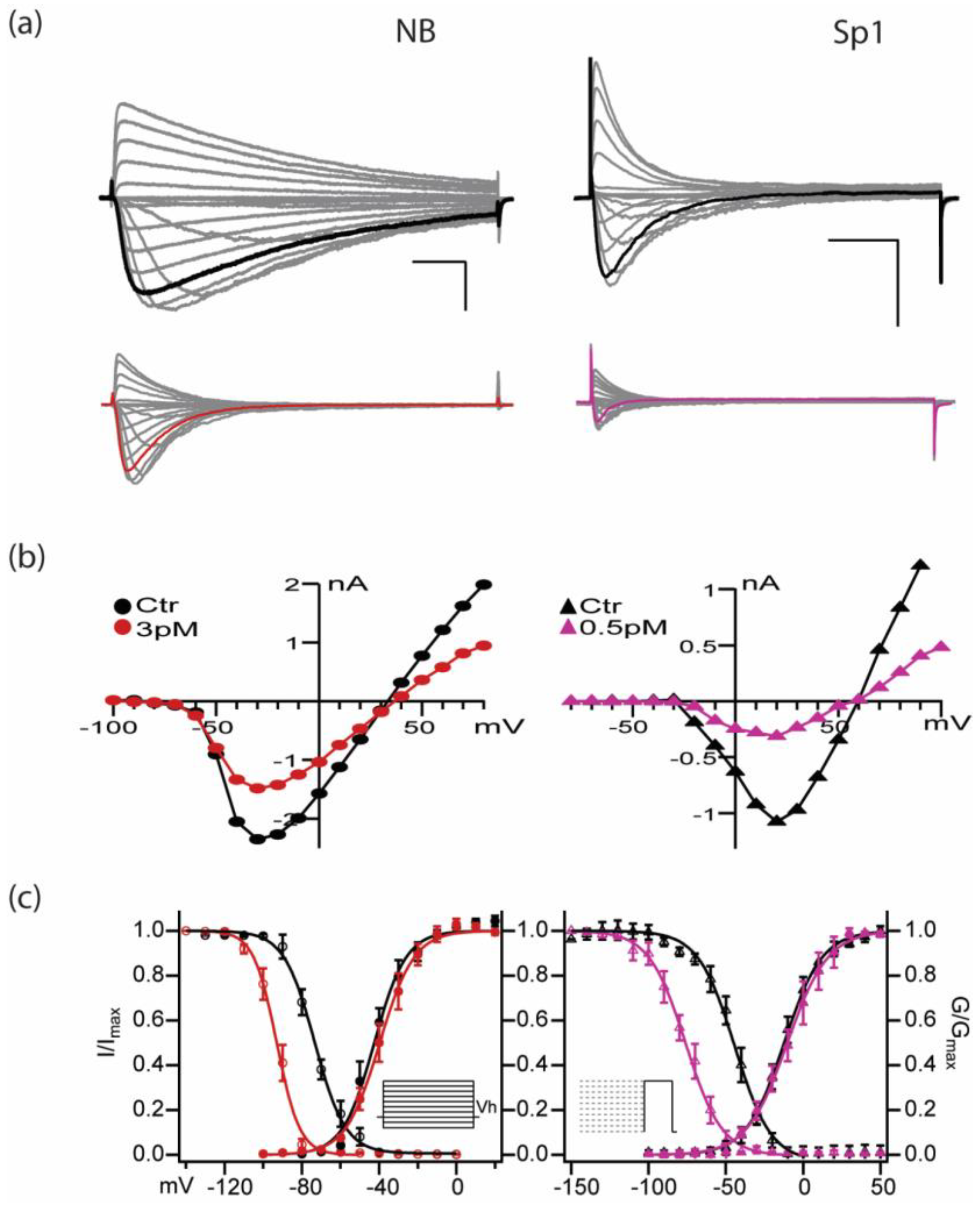
2.3. PIIIA Inhibits Navbacs from B. Halodurans and S.pomeroy: Inactivation Is Shifted toward More Negative Voltages, without a Measurable Shift in Activation
2.4. Activity- or State- Dependence of µ-Conotoxin PIIIA Inhibition of NaChBac and NavSp1, two NavBac Channels with Substantially Different Kinetics
2.5. Holding Potential and Ionic Conditions Affect PIIIA Inhibition of NavBacs. Possible Effects of PIIIA Interactions with Ions in The Pore?
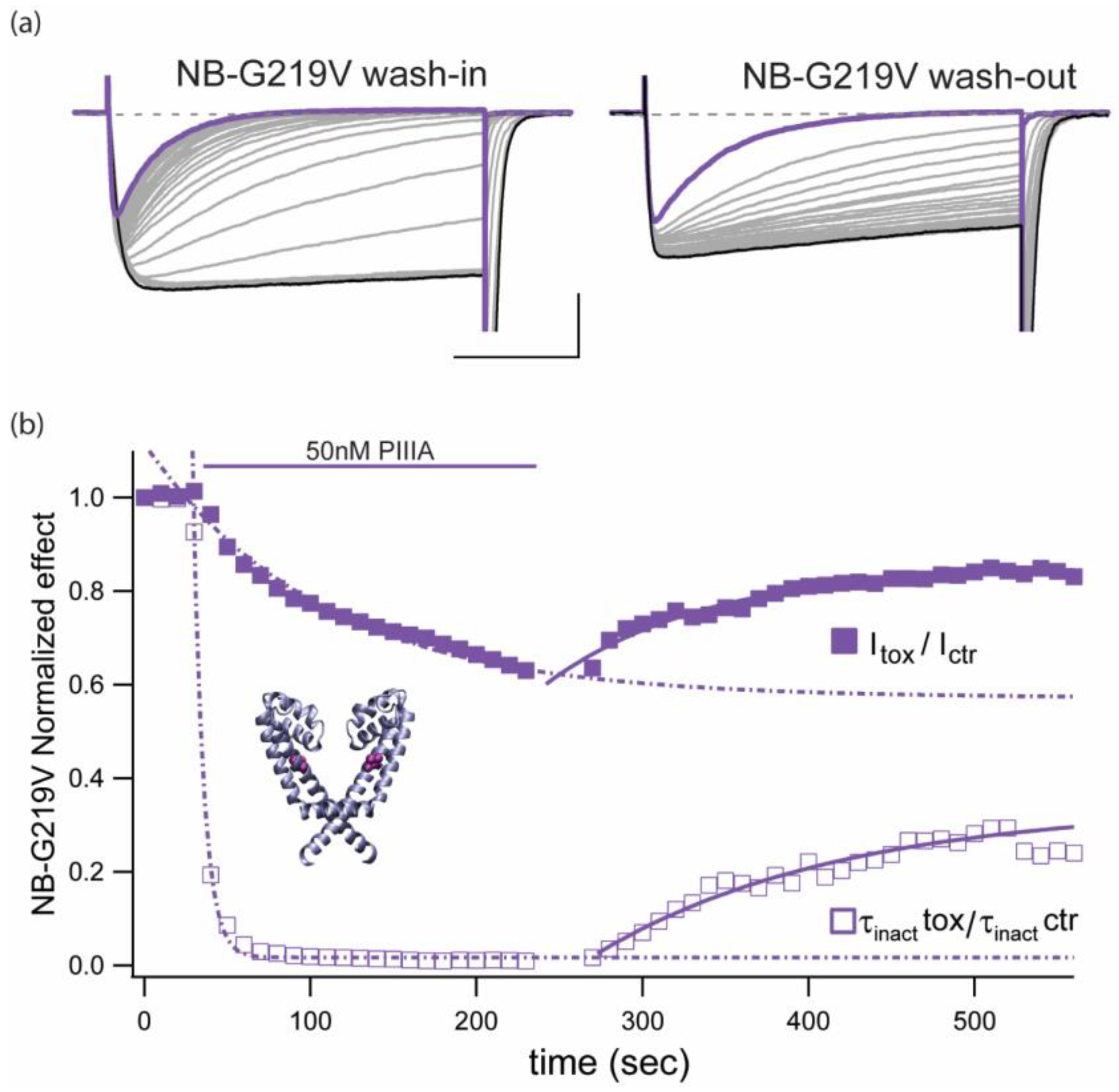
2.6. The Slowly Inactivating Mutant NaChBac-G219V is Less Vulnerable to PIIIA Peak Current Block than the Wild Type NaChBac Channel
 ), as well as inactivation-like, single-pulse decay (
), as well as inactivation-like, single-pulse decay (  ), which became complete within a 300 ms pulse, after ~200 s pulsing at 0.1 Hz (Figure 6b, τinact PIIIA: 55.9 ± 9.8 ms, n = 4).
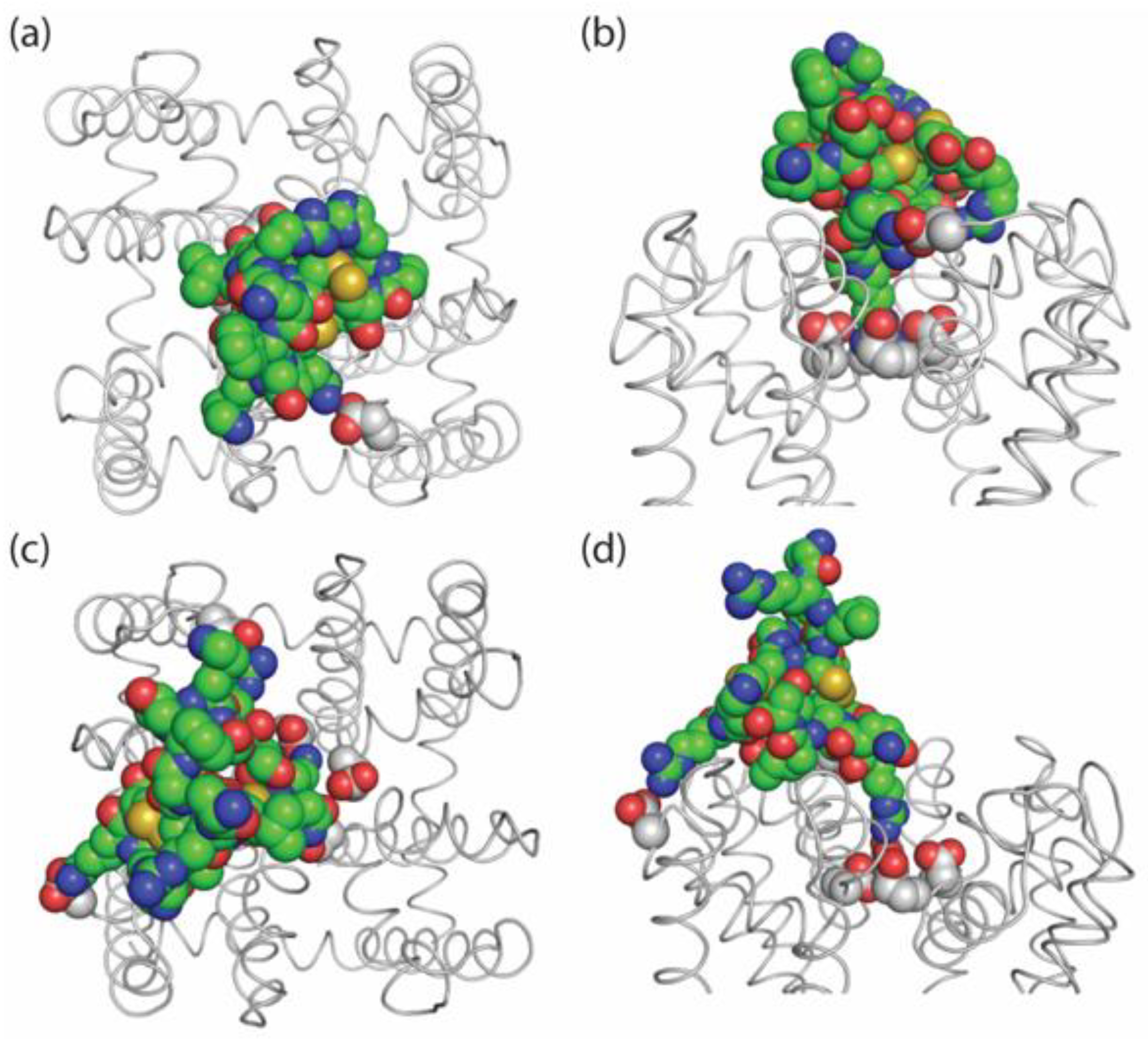
), which became complete within a 300 ms pulse, after ~200 s pulsing at 0.1 Hz (Figure 6b, τinact PIIIA: 55.9 ± 9.8 ms, n = 4). 2.7. Possible Binding Orientations of PIIIA in NaChBac
3. Discussion
3.1. Does Extremely High Affinity of PIIIA Result from Complementary Charge Arrays on PIIIA and NaChBac?
3.2. Gating Modulation (e.g., Enhanced Inactivation) vs. Physical Pore Block
3.3. Broadening the Scope of µCTX Pharmacology: Homotetramers vs. 4-Domain Channels
3.4. µCTX Pharmacology of Invertebrate Homotetrameric-NavBacs
4. Materials and Methods
4.1. Constructs and Mutagenesis
4.2. µ-Conotoxin Synthesis
4.3. Electrophysiology
4.4. Computational Modeling
4.5. Data Analysis
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A
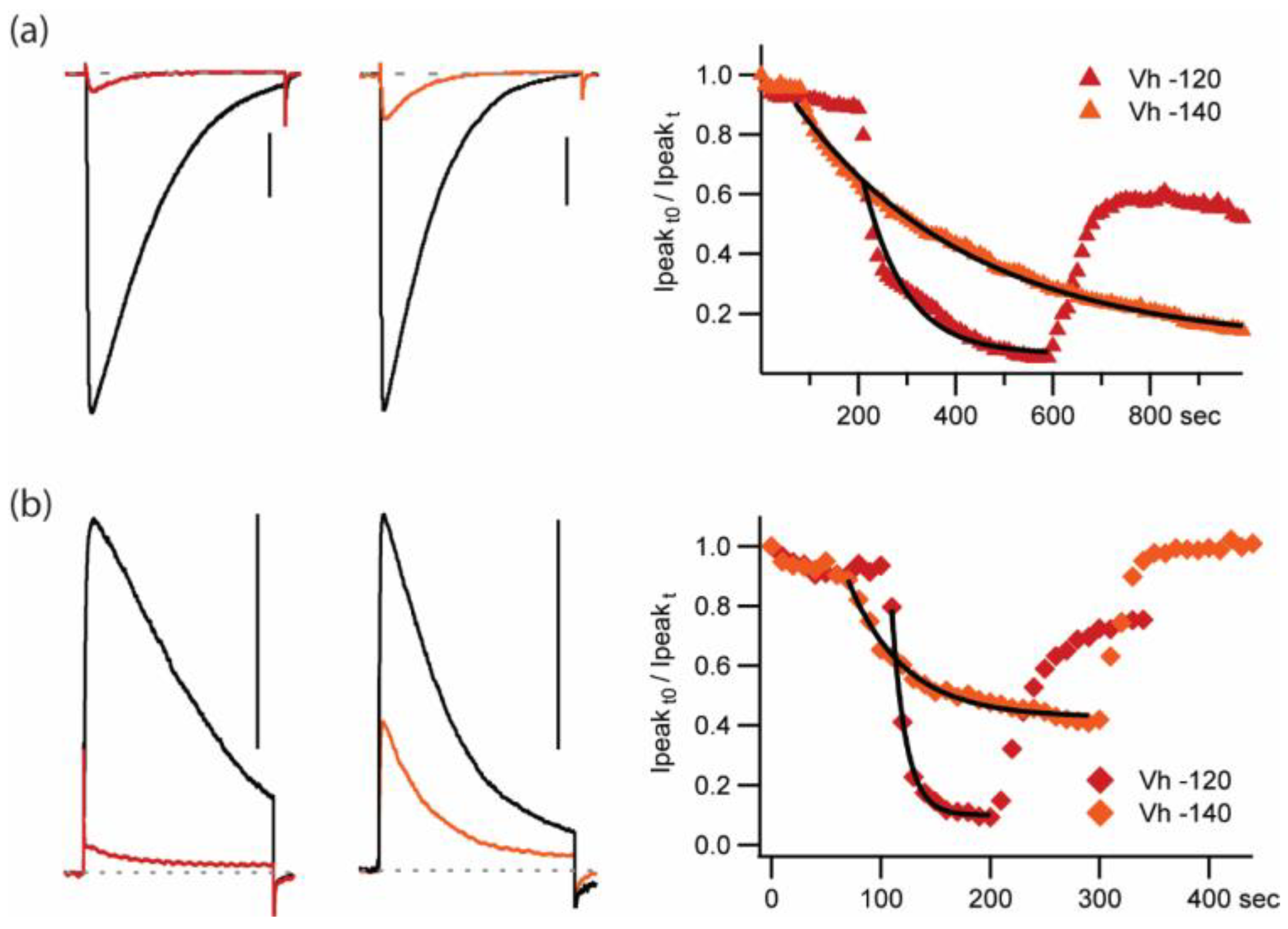
 −100 mV,
−100 mV,  −120 mV and
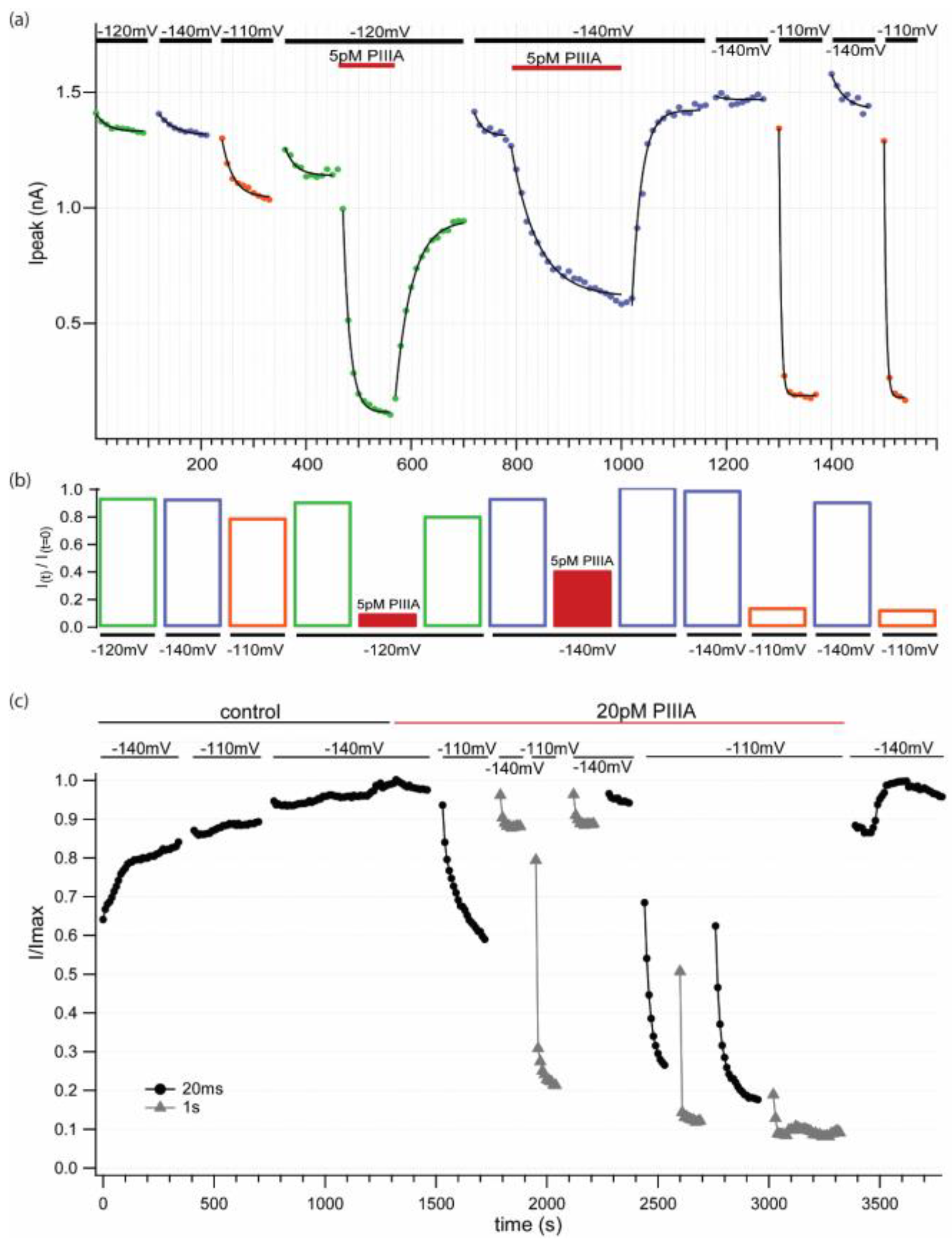
−120 mV and  −140 mV). Wash-in and washout (5 pM PIIIA). Low [PIIIA] (<0.055 pM) remaining after washing most of the toxin out of the chamber produces dramatic, rapid, reversible block and unblock in response to changes in holding potential from -140 to -110 mV. Data suggest different affinities for different pre-activated states. (b) Relative current plot for the conditions shown in (a). (c) Normalized diary plot for a second experiment where the cell was stimulated at 0.1 Hz with different −10 mV pulse durations (
−140 mV). Wash-in and washout (5 pM PIIIA). Low [PIIIA] (<0.055 pM) remaining after washing most of the toxin out of the chamber produces dramatic, rapid, reversible block and unblock in response to changes in holding potential from -140 to -110 mV. Data suggest different affinities for different pre-activated states. (b) Relative current plot for the conditions shown in (a). (c) Normalized diary plot for a second experiment where the cell was stimulated at 0.1 Hz with different −10 mV pulse durations (  20 ms,
20 ms,  1 sec) at the noted holding potentials in control and when exposed to 20 pM PIIIA.
−100 mV, −120 mV and −140 mV). Wash-in and washout (5 pM PIIIA). Low [PIIIA] (<0.055 pM) remaining after washing most of the toxin out of the chamber produces dramatic, rapid, reversible block and unblock in response to changes in holding potential from -140 to -110 mV. Data suggest different affinities for different pre-activated states. (b) Relative current plot for the conditions shown in (a). (c) Normalized diary plot for a second experiment where the cell was stimulated at 0.1 Hz with different −10 mV pulse durations ( 20 ms, 1 sec) at the noted holding potentials in control and when exposed to 20 pM PIIIA.
1 sec) at the noted holding potentials in control and when exposed to 20 pM PIIIA.
−100 mV, −120 mV and −140 mV). Wash-in and washout (5 pM PIIIA). Low [PIIIA] (<0.055 pM) remaining after washing most of the toxin out of the chamber produces dramatic, rapid, reversible block and unblock in response to changes in holding potential from -140 to -110 mV. Data suggest different affinities for different pre-activated states. (b) Relative current plot for the conditions shown in (a). (c) Normalized diary plot for a second experiment where the cell was stimulated at 0.1 Hz with different −10 mV pulse durations ( 20 ms, 1 sec) at the noted holding potentials in control and when exposed to 20 pM PIIIA.

References
- Cruz, L.J.; Gray, W.R.; Olivera, B.M.; Zeikus, R.D.; Kerr, L.; Yoshikami, D.; Moczydlowski, E. Conus geographus toxins that discriminate between neuronal and muscle sodium channels. J. Biol. Chem. 1985, 260, 9280–9288. [Google Scholar]
- Olivera, B.M.; Lecture, E.E.J. Conus venom peptides, receptor and ion channel targets, and drug design: 50 million years of neuropharmacology. Mol. Biol. Cell 1997, 8, 2101–2109. [Google Scholar] [CrossRef]
- Terlau, H.; Olivera, B.M. Conus venoms: A rich source of novel ion channel-targeted peptides. Physiol. Rev. 2004, 84, 41–68. [Google Scholar] [CrossRef]
- French, R.J.; Terlau, H. Sodium channel toxins--receptor targeting and therapeutic potential. Curr. Med. Chem. 2004, 11, 3053–3064. [Google Scholar] [CrossRef]
- French, R.J.; Yoshikami, D.; Sheets, M.F.; Olivera, B.M. The tetrodotoxin receptor of voltage-gated sodium channels--perspectives from interactions with micro-conotoxins. Mar. Drugs 2010, 8, 2153–2161. [Google Scholar] [CrossRef]
- Zhang, M.M.; Green, B.R.; Catlin, P.; Fiedler, B.; Azam, L.; Chadwick, A.; Terlau, H.; McArthur, J.R.; French, R.J.; Gulyas, J.; et al. Structure/function characterization of micro-conotoxin KIIIA, an analgesic, nearly irreversible blocker of mammalian neuronal sodium channels. J. Biol. Chem. 2007, 282, 30699–30706. [Google Scholar] [CrossRef]
- Ramirez, D.; Gonzalez, W.; Fissore, R.A.; Carvacho, I. Conotoxins as Tools to Understand the Physiological Function of Voltage-Gated Calcium (CaV) Channels. Mar. Drugs 2017, 15, 313. [Google Scholar] [CrossRef]
- Tosti, E.; Boni, R.; Gallo, A. micro-Conotoxins Modulating Sodium Currents in Pain Perception and Transmission: A Therapeutic Potential. Mar Drugs 2017, 15, 295. [Google Scholar] [CrossRef]
- Ahern, C.A.; Payandeh, J.; Bosmans, F.; Chanda, B. The hitchhiker’s guide to the voltage-gated sodium channel galaxy. J. Gen. Physiol. 2016, 147, 1–24. [Google Scholar] [CrossRef]
- Ren, D.; Navarro, B.; Xu, H.; Yue, L.; Shi, Q.; Clapham, D.E. A prokaryotic voltage-gated sodium channel. Science 2001, 294, 2372–2375. [Google Scholar] [CrossRef]
- Koishi, R.; Xu, H.; Ren, D.; Navarro, B.; Spiller, B.W.; Shi, Q.; Clapham, D.E. A superfamily of voltage-gated sodium channels in bacteria. J. Biol. Chem. 2004, 279, 9532–9538. [Google Scholar] [CrossRef]
- Scheuer, T. Bacterial sodium channels: Models for eukaryotic sodium and calcium channels. Handb. Exp. Pharmacol. 2014, 221, 269–291. [Google Scholar]
- Finol-Urdaneta, R.K.; Wang, Y.; Al-Sabi, A.; Zhao, C.; Noskov, S.Y.; French, R.J. Sodium channel selectivity and conduction: Prokaryotes have devised their own molecular strategy. J. Gen. Physiol. 2014, 143, 157–171. [Google Scholar] [CrossRef] [Green Version]
- Chen, R.; Chung, S.H. Binding modes of mu-conotoxin to the bacterial sodium channel (NaVAb). Biophys. J. 2012, 102, 483–488. [Google Scholar] [CrossRef]
- Chen, R.; Robinson, A.; Chung, S.H. Mechanism of mu-conotoxin PIIIA binding to the voltage-gated Na+ channel NaV1.4. PLoS ONE 2014, 9, e93267. [Google Scholar]
- Hui, K.; Lipkind, G.; Fozzard, H.A.; French, R.J. Electrostatic and steric contributions to block of the skeletal muscle sodium channel by mu-conotoxin. J. Gen. Physiol. 2002, 119, 45–54. [Google Scholar] [CrossRef]
- Hui, K.; McIntyre, D.; French, R.J. Conotoxins as sensors of local pH and electrostatic potential in the outer vestibule of the sodium channel. J. Gen. Physiol. 2003, 122, 63–79. [Google Scholar] [CrossRef]
- Korkosh, V.S.; Zhorov, B.S.; Tikhonov, D.B. Folding similarity of the outer pore region in prokaryotic and eukaryotic sodium channels revealed by docking of conotoxins GIIIA, PIIIA, and KIIIA in a NavAb-based model of Nav1.4. J. Gen. Physiol. 2014, 144, 231–244. [Google Scholar] [CrossRef] [Green Version]
- McArthur, J.R.; Singh, G.; O’Mara, M.L.; McMaster, D.; Ostroumov, V.; Tieleman, D.P.; French, R.J. Orientation of mu-conotoxin PIIIA in a sodium channel vestibule, based on voltage dependence of its binding. Mol. Pharmacol. 2011, 80, 219–227. [Google Scholar] [CrossRef]
- McArthur, J.R.; Ostroumov, V.; Al-Sabi, A.; McMaster, D.; French, R.J. Multiple, distributed interactions of mu-conotoxin PIIIA associated with broad targeting among voltage-gated sodium channels. Biochemistry 2011, 50, 116–124. [Google Scholar] [CrossRef]
- Zhao, Y.; Yarov-Yarovoy, V.; Scheuer, T.; Catterall, W.A. A gating hinge in Na+ channels; a molecular switch for electrical signaling. Neuron 2004, 41, 859–865. [Google Scholar] [CrossRef]
- Irie, K.; Kitagawa, K.; Nagura, H.; Imai, T.; Shimomura, T.; Fujiyoshi, Y. Comparative study of the gating motif and C-type inactivation in prokaryotic voltage-gated sodium channels. J. Biol. Chem. 2010, 285, 3685–3694. [Google Scholar] [CrossRef] [PubMed]
- Barber, A.F.; Carnevale, V.; Raju, S.G.; Amaral, C.; Treptow, W.; Klein, M.L. Hinge-bending motions in the pore domain of a bacterial voltage-gated sodium channel. Biochim. Biophys. Acta 2012, 1818, 2120–2125. [Google Scholar] [CrossRef] [Green Version]
- Sula, A.; Booker, J.; Ng, L.C.; Naylor, C.E.; DeCaen, P.G.; Wallace, B.A. The complete structure of an activated open sodium channel. Nat. Commun. 2017, 8, 14205. [Google Scholar] [CrossRef] [Green Version]
- Chen, F.; Huang, W.; Jiang, T.; Yu, R. Determination of the mu-Conotoxin PIIIA Specificity Against Voltage-Gated Sodium Channels from Binding Energy Calculations. Mar. Drugs 2018, 16, 153. [Google Scholar] [CrossRef] [PubMed]
- Mahdavi, S.; Kuyucak, S. Systematic study of binding of mu-conotoxins to the sodium channel NaV1.4. Toxins 2014, 6, 3454–3470. [Google Scholar] [CrossRef]
- Finol-Urdaneta, R.K.; McArthur, J.R.; Goldschen-Ohm, M.P.; Gaudet, R.; Tikhonov, D.B.; Zhorov, B.S.; French, R.J. Batrachotoxin acts as a stent to hold open homotetrameric prokaryotic voltage-gated sodium channels. J. Gen. Physiol. 2019, 151, 186–199. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, S.; Vyas, R.; Chalamalasetti, S.V.; Sahu, I.D.; Clatot, J.; Wan, X.; Lorigan, G.A.; Deschenes, I.; Chakrapani, S. The voltage-gated sodium channel pore exhibits conformational flexibility during slow inactivation. J. Gen. Physiol. 2018, 150, 1333–1347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Ostmeyer, J.; Cuello, L.G.; Perozo, E.; Roux, B. Rapid constriction of the selectivity filter underlies C-type inactivation in the KcsA potassium channel. J. Gen. Physiol. 2018, 150, 1408–1420. [Google Scholar] [CrossRef] [Green Version]
- Cuello, L.G.; Cortes, D.M.; Perozo, E. The gating cycle of a K(+) channel at atomic resolution. Elife 2017, 6. [Google Scholar] [CrossRef] [PubMed]
- Cuello, L.G.; Jogini, V.; Cortes, D.M.; Perozo, E. Structural mechanism of C-type inactivation in K(+) channels. Nature 2010, 466, 203–208. [Google Scholar] [CrossRef] [PubMed]
- Abderemane-Ali, F.; Findeisen, F.; Rossen, N.D.; Minor, D.L., Jr. A Selectivity Filter Gate Controls Voltage-Gated Calcium Channel Calcium-Dependent Inactivation. Neuron 2019, 101, 1134–1149. [Google Scholar] [CrossRef] [PubMed]
- Zhorov, B.S.; Tikhonov, D.B. Computational Structural Pharmacology and Toxicology of Voltage-Gated Sodium Channels. Curr. Top. Membr. 2016, 78, 117–144. [Google Scholar] [PubMed]
- Boiteux, C.; Allen, T.W. Understanding Sodium Channel Function and Modulation Using Atomistic Simulations of Bacterial Channel Structures. Curr. Top. Membr. 2016, 78, 145–182. [Google Scholar] [PubMed]
- Oakes, V.; Furini, S.; Domene, C. Voltage-Gated Sodium Channels: Mechanistic Insights From Atomistic Molecular Dynamics Simulations. Curr. Top. Membr. 2016, 78, 183–214. [Google Scholar]
- Ing, C.; Pomes, R. Simulation Studies of Ion Permeation and Selectivity in Voltage-Gated Sodium Channels. Curr. Top. Membr. 2016, 78, 215–260. [Google Scholar] [PubMed]
- Kasimova, M.A.; Granata, D.; Carnevale, V. Voltage-Gated Sodium Channels: Evolutionary History and Distinctive Sequence Features. Curr. Top. Membr. 2016, 78, 261–286. [Google Scholar]
- Winters, J.J.; Isom, L.L. Developmental and Regulatory Functions of Na(+) Channel Non-pore-forming beta Subunits. Curr. Top. Membr. 2016, 78, 315–351. [Google Scholar]
- Gawali, V.S.; Todt, H. Mechanism of Inactivation in Voltage-Gated Na(+) Channels. Curr. Top. Membr. 2016, 78, 409–450. [Google Scholar]
- Ghovanloo, M.R.; Aimar, K.; Ghadiry-Tavi, R.; Yu, A.; Ruben, P.C. Physiology and Pathophysiology of Sodium Channel Inactivation. Curr. Top. Membr. 2016, 78, 479–509. [Google Scholar]
- Barbosa, C.; Cummins, T.R. Unusual Voltage-Gated Sodium Currents as Targets for Pain. Curr. Top. Membr. 2016, 78, 599–638. [Google Scholar] [PubMed]
- DeMarco, K.R.; Clancy, C.E. Cardiac Na Channels: Structure to Function. Curr. Top. Membr. 2016, 78, 287–311. [Google Scholar] [PubMed]
- Clark, R.B.; Giles, W.R. Current-Voltage Relationship for Late Na(+) Current in Adult Rat Ventricular Myocytes. Curr. Top. Membr. 2016, 78, 451–478. [Google Scholar] [PubMed]
- Morris, C.E.; Joos, B. Nav Channels in Damaged Membranes. Curr. Top. Membr. 2016, 78, 561–597. [Google Scholar] [PubMed]
- Pan, X.; Li, Z.; Huang, X.; Huang, G.; Gao, S.; Shen, H.; Liu, L.; Lei, J.; Yan, N. Molecular basis for pore blockade of human Na(+) channel Nav1.2 by the mu-conotoxin KIIIA. Science 2019, 363, 1309–1313. [Google Scholar] [CrossRef] [PubMed]
- Pan, X.; Li, Z.; Zhou, Q.; Shen, H.; Wu, K.; Huang, X.; Chen, J.; Zhang, J.; Zhu, X.; Lei, J.; et al. Structure of the human voltage-gated sodium channel Nav1.4 in complex with beta1. Science 2018, 362. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.; Li, Z.; Jiang, Y.; Pan, X.; Wu, J.; Cristofori-Armstrong, B.; Smith, J.J.; Chin, Y.K.Y.; Lei, J.; Zhou, Q.; et al. Structural basis for the modulation of voltage-gated sodium channels by animal toxins. Science 2018, 362. [Google Scholar] [CrossRef]
- Shen, H.; Zhou, Q.; Pan, X.; Li, Z.; Wu, J.; Yan, N. Structure of a eukaryotic voltage-gated sodium channel at near-atomic resolution. Science 2017, 355. [Google Scholar] [CrossRef]
- Huang, W.; Liu, M.; Yan, S.F.; Yan, N. Structure-based assessment of disease-related mutations in human voltage-gated sodium channels. Protein Cell 2017, 8, 401–438. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Xia, M.; Li, Y.; Liu, H.; Jiang, X.; Ren, W.; Wu, J.; DeCaen, P.; Yu, F.; Huang, S.; et al. Analysis of the selectivity filter of the voltage-gated sodium channel Na(v)Rh. Cell Res. 2013, 23, 409–422. [Google Scholar] [CrossRef]
- Ekberg, J.; Craik, D.J.; Adams, D.J. Conotoxin modulation of voltage-gated sodium channels. Int. J. Biochem. Cell Biol. 2008, 40, 2363–2368. [Google Scholar] [CrossRef] [PubMed]
- Hille, B. Local anesthetics: Hydrophilic and hydrophobic pathways for the drug-receptor reaction. J. Gen. Physiol. 1977, 69, 497–515. [Google Scholar] [CrossRef] [PubMed]
- Hondeghem, L.M.; Katzung, B.G. Time- and voltage-dependent interactions of antiarrhythmic drugs with cardiac sodium channels. Biochim. Biophys. Acta 1977, 472, 373–398. [Google Scholar] [CrossRef]
- Yang, E.; Granata, D.; Eckenhoff, R.G.; Carnevale, V.; Covarrubias, M. Propofol inhibits prokaryotic voltage-gated Na(+) channels by promoting activation-coupled inactivation. J. Gen. Physiol. 2018, 150, 1299–1316. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Yang, E.; Wells, M.M.; Bondarenko, V.; Woll, K.; Carnevale, V.; Granata, D.; Klein, M.L.; Eckenhoff, R.G.; Dailey, W.P.; et al. Propofol inhibits the voltage-gated sodium channel NaChBac at multiple sites. J. Gen. Physiol. 2018, 150, 1317–1331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moczydlowski, E.; Hall, S.; Garber, S.S.; Strichartz, G.S.; Miller, C. Voltage-dependent blockade of muscle Na+ channels by guanidinium toxins. J. Gen. Physiol. 1984, 84, 687–704. [Google Scholar] [CrossRef] [PubMed]
- Moczydlowski, E.; Garber, S.S.; Miller, C. Batrachotoxin-activated Na+ channels in planar lipid bilayers. Competition of tetrodotoxin block by Na+. J. Gen. Physiol. 1984, 84, 665–686. [Google Scholar] [CrossRef]
- Leipold, E.; Ullrich, F.; Thiele, M.; Tietze, A.A.; Terlau, H.; Imhof, D.; Heinemann, S.H. Subtype-specific block of voltage-gated K(+) channels by mu-conopeptides. Biochem. Biophys. Res. Commun. 2017, 482, 1135–1140. [Google Scholar] [CrossRef]
- Kaufmann, D.; Tietze, A.A.; Tietze, D. In Silico Analysis of the Subtype Selective Blockage of KCNA Ion Channels through the micro-Conotoxins PIIIA, SIIIA, and GIIIA. Mar. Drugs 2019, 17, 180. [Google Scholar] [CrossRef]
- Paul George, A.A.; Heimer, P.; Leipold, E.; Schmitz, T.; Kaufmann, D.; Tietze, D.; Heinemann, S.H.; Imhof, D. Effect of Conformational Diversity on the Bioactivity of micro-Conotoxin PIIIA Disulfide Isomers. Mar. Drugs 2019, 17, 390. [Google Scholar] [CrossRef]
- DeCaen, P.G.; Takahashi, Y.; Krulwich, T.A.; Ito, M.; Clapham, D.E. Ionic selectivity and thermal adaptations within the voltage-gated sodium channel family of alkaliphilic Bacillus. Elife 2014, 3. [Google Scholar] [CrossRef] [PubMed]
- Prindle, A.; Liu, J.; Asally, M.; Ly, S.; Garcia-Ojalvo, J.; Suel, G.M. Ion channels enable electrical communication in bacterial communities. Nature 2015, 527, 59–63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taylor, A.R. A fast Na+/Ca2+-based action potential in a marine diatom. PLoS ONE 2009, 4, e4966. [Google Scholar] [CrossRef] [PubMed]
- Oami, K.; Naitoh, Y.; Sibaoka, T. Modification of Voltage-Sensitive Inactivation of Na+ Current by External Ca2+ in the Marine Dinoflagellate Noctiluca-Miliaris. J. Comp. Physiol. A 1995, 176, 635–640. [Google Scholar] [CrossRef]
- Helliwell, K.E.; Chrachri, A.; Koester, J.A.; Wharam, S.; Verret, F.; Taylor, A.R.; Wheeler, G.L.; Brownlee, C. Alternative Mechanisms for Fast Na(+)/Ca(2+) Signaling in Eukaryotes via a Novel Class of Single-Domain Voltage-Gated Channels. Curr. Biol. 2019, 29, 1503–1511. [Google Scholar] [CrossRef] [PubMed]
- Tikhonov, D.B.; Zhorov, B.S. Predicting Structural Details of the Sodium Channel Pore Basing on Animal Toxin Studies. Front. Pharmacol. 2018, 9, 880. [Google Scholar] [CrossRef] [PubMed]
- Margolskee, R.F.; McHendry-Rinde, B.; Horn, R. Panning transfected cells for electrophysiological studies. Biotechniques 1993, 15, 906–911. [Google Scholar] [PubMed]


 ) and PIIIA-R14A (
) and PIIIA-R14A (  ). Data represents mean ± sem from 3 different cells. The R14A substitution appears to affect differentially inhibition/block and speeding of the inactivation rate seen for the wt PIIIA conotoxin.
) and PIIIA-R14A ( ). Data represents mean ± sem from 3 different cells. The R14A substitution appears to affect differentially inhibition/block and speeding of the inactivation rate seen for the wt PIIIA conotoxin.
). Data represents mean ± sem from 3 different cells. The R14A substitution appears to affect differentially inhibition/block and speeding of the inactivation rate seen for the wt PIIIA conotoxin.
) and PIIIA-R14A ( ). Data represents mean ± sem from 3 different cells. The R14A substitution appears to affect differentially inhibition/block and speeding of the inactivation rate seen for the wt PIIIA conotoxin. NB,
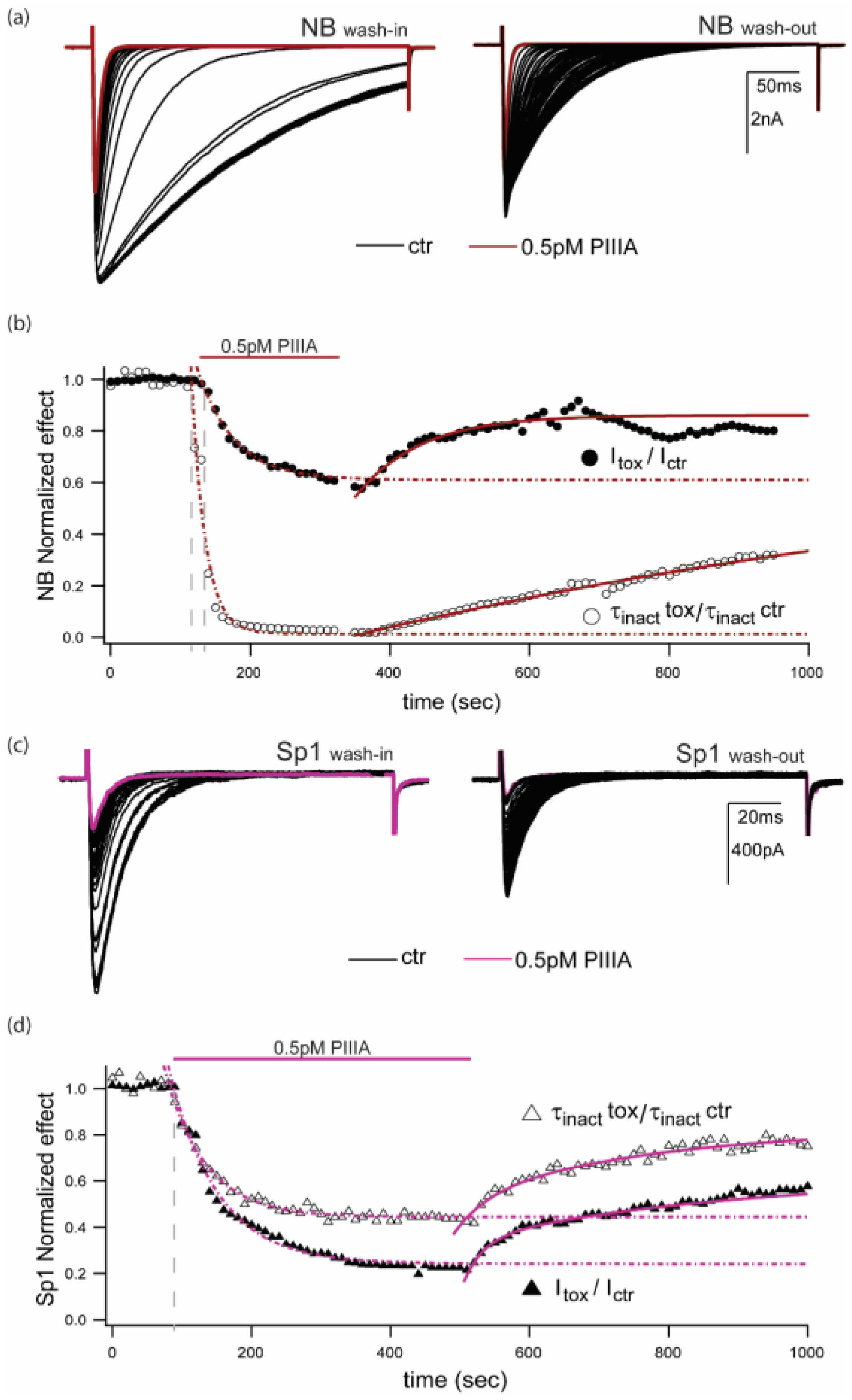
NB,  Sp1) and in the presence of PIIIA for NB (
Sp1) and in the presence of PIIIA for NB (  , left) and Sp1 (
, left) and Sp1 (  , right). (c) Left: NB and Right: Sp1 activation and inactivation plots for control (NB , Sp1 ), and in presence of PIIIA (NB , Sp1
, right). (c) Left: NB and Right: Sp1 activation and inactivation plots for control (NB , Sp1 ), and in presence of PIIIA (NB , Sp1  ). Data are presented as mean ± sem from 4 replicates per condition, parameters are compiled in Table 1. Protocols are summarized in the insets.
NB, Sp1) and in the presence of PIIIA for NB ( , left) and Sp1 ( , right). (c) Left: NB and Right: Sp1 activation and inactivation plots for control (NB , Sp1 ), and in presence of PIIIA (NB , Sp1 ). Data are presented as mean ± sem from 4 replicates per condition, parameters are compiled in Table 1. Protocols are summarized in the insets.
). Data are presented as mean ± sem from 4 replicates per condition, parameters are compiled in Table 1. Protocols are summarized in the insets.
NB, Sp1) and in the presence of PIIIA for NB ( , left) and Sp1 ( , right). (c) Left: NB and Right: Sp1 activation and inactivation plots for control (NB , Sp1 ), and in presence of PIIIA (NB , Sp1 ). Data are presented as mean ± sem from 4 replicates per condition, parameters are compiled in Table 1. Protocols are summarized in the insets.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| NaChBac | Sp1 | |||
|---|---|---|---|---|
| Control | PIIIA | Control | PIIIA | |
| mean ± sem | mean ± sem | mean = ± sem | mean ± sem | |
| Activation | ||||
| Gmax | 38.8±3.6 | 22.9 ± 4.4 * | 29.7 ± 4.8 | 12.9 ± 2.20 * |
| Vhalf, mV | −39.9±2.2 | −39.3 ± 3.4 | 5.6 ± 1.8 | 5.3 ± 1.44 |
| Slope factor, mV/e-fold | 6.7±0.6 | 8.5 ± 0.7 | 11.0 ± 1.8 | 13.3 ± 1.75 |
| SSI | ||||
| Vhalf, mV | −73.4 ± 4.5 | −94 ± 6.7 * | −44.3±2.9 | −76.1 ± 5.7 ** |
| Slope factor, mV/e-fold | −7.9 ± 0.9 | −5.9 ± 1.4 | −11.8±1.1 | −10.8 ± 1.2 |
| n | 4 | 4 | 4 | 4 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Finol-Urdaneta, R.K.; McArthur, J.R.; Korkosh, V.S.; Huang, S.; McMaster, D.; Glavica, R.; Tikhonov, D.B.; Zhorov, B.S.; French, R.J. Extremely Potent Block of Bacterial Voltage-Gated Sodium Channels by µ-Conotoxin PIIIA. Mar. Drugs 2019, 17, 510. https://doi.org/10.3390/md17090510
Finol-Urdaneta RK, McArthur JR, Korkosh VS, Huang S, McMaster D, Glavica R, Tikhonov DB, Zhorov BS, French RJ. Extremely Potent Block of Bacterial Voltage-Gated Sodium Channels by µ-Conotoxin PIIIA. Marine Drugs. 2019; 17(9):510. https://doi.org/10.3390/md17090510
Chicago/Turabian StyleFinol-Urdaneta, Rocio K., Jeffrey R. McArthur, Vyacheslav S. Korkosh, Sun Huang, Denis McMaster, Robert Glavica, Denis B. Tikhonov, Boris S. Zhorov, and Robert J. French. 2019. "Extremely Potent Block of Bacterial Voltage-Gated Sodium Channels by µ-Conotoxin PIIIA" Marine Drugs 17, no. 9: 510. https://doi.org/10.3390/md17090510