
Cytotoxic Marine Alkaloid 3,10-Dibromofascaplysin Induces Apoptosis and Synergizes with Cytarabine Resulting in Leukemia Cell Death

 , , , ,
, , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
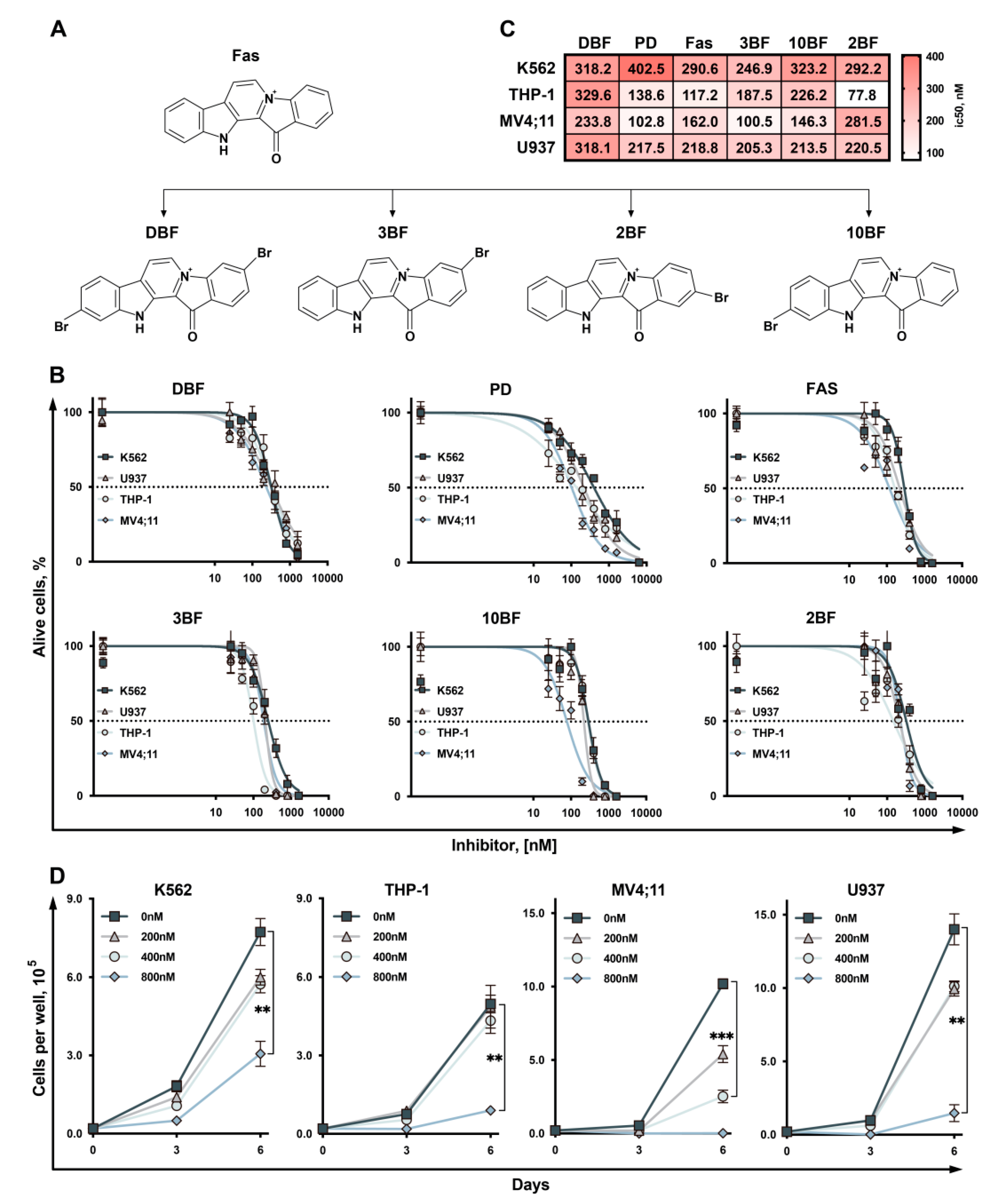
2.1. 3,10-Dibromofascaplysin (DBF) Suppress Cell Growth of Myeloid Leukemia Cells
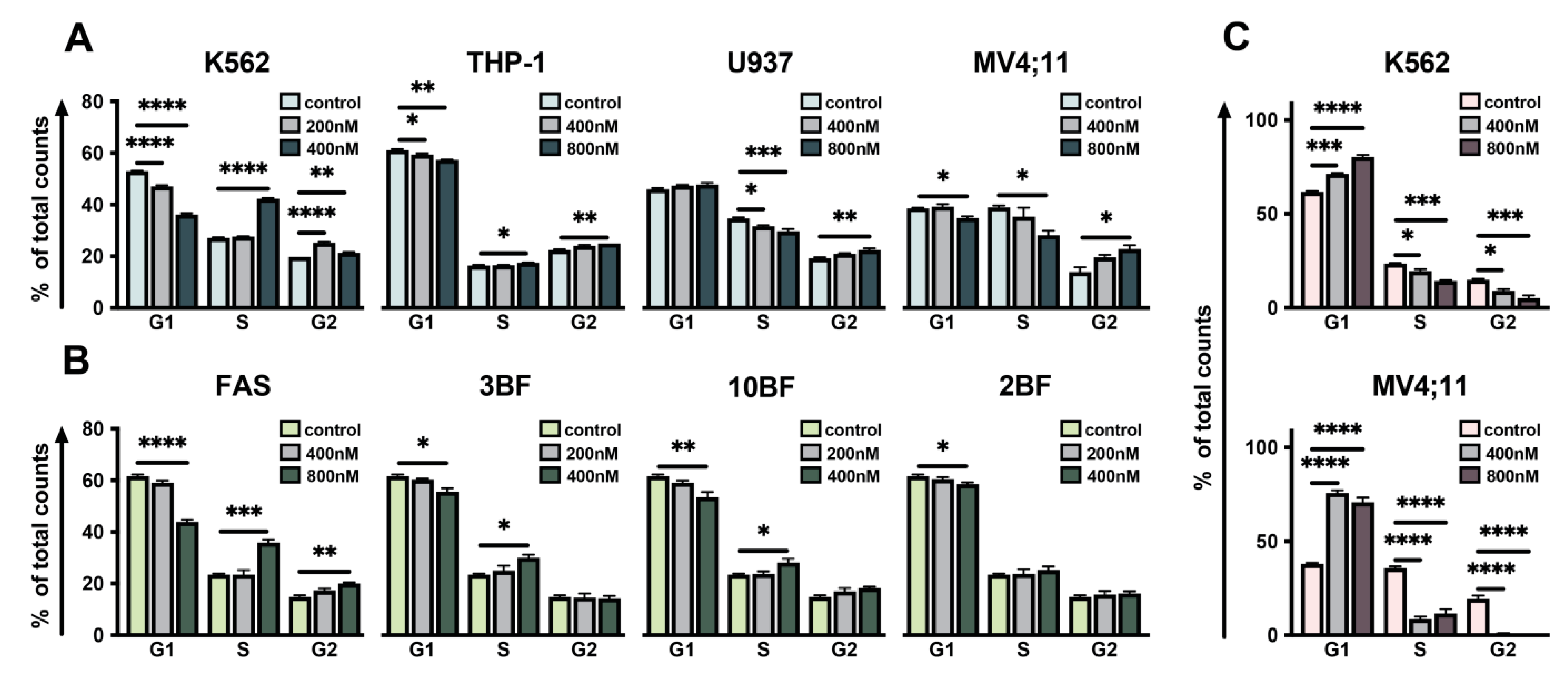
2.2. Effect of DBF on Cell Cycle of Leukemia Cells
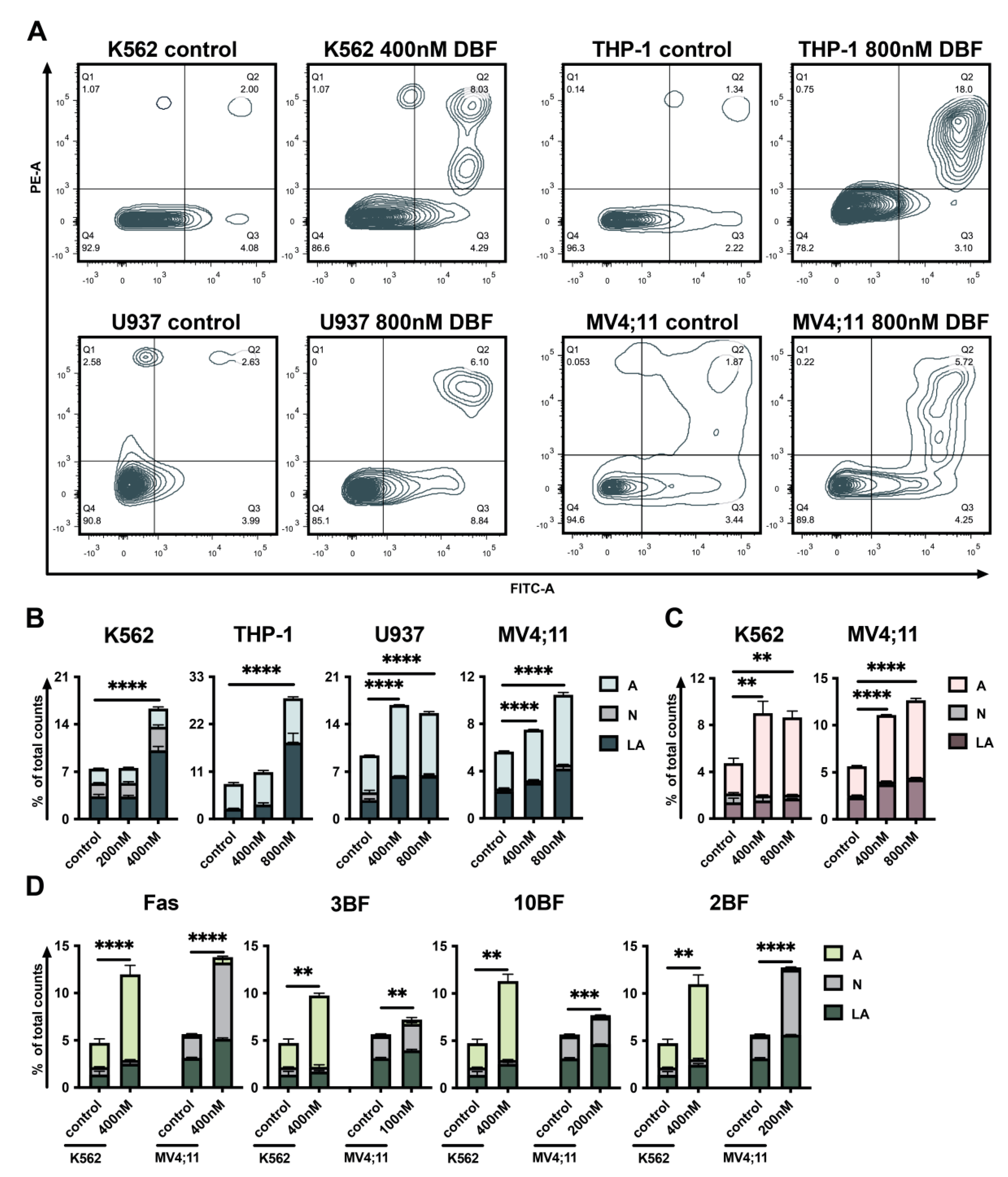
2.3. DBF Induces Apoptosis of Acute Myeloid Leukemia Cells
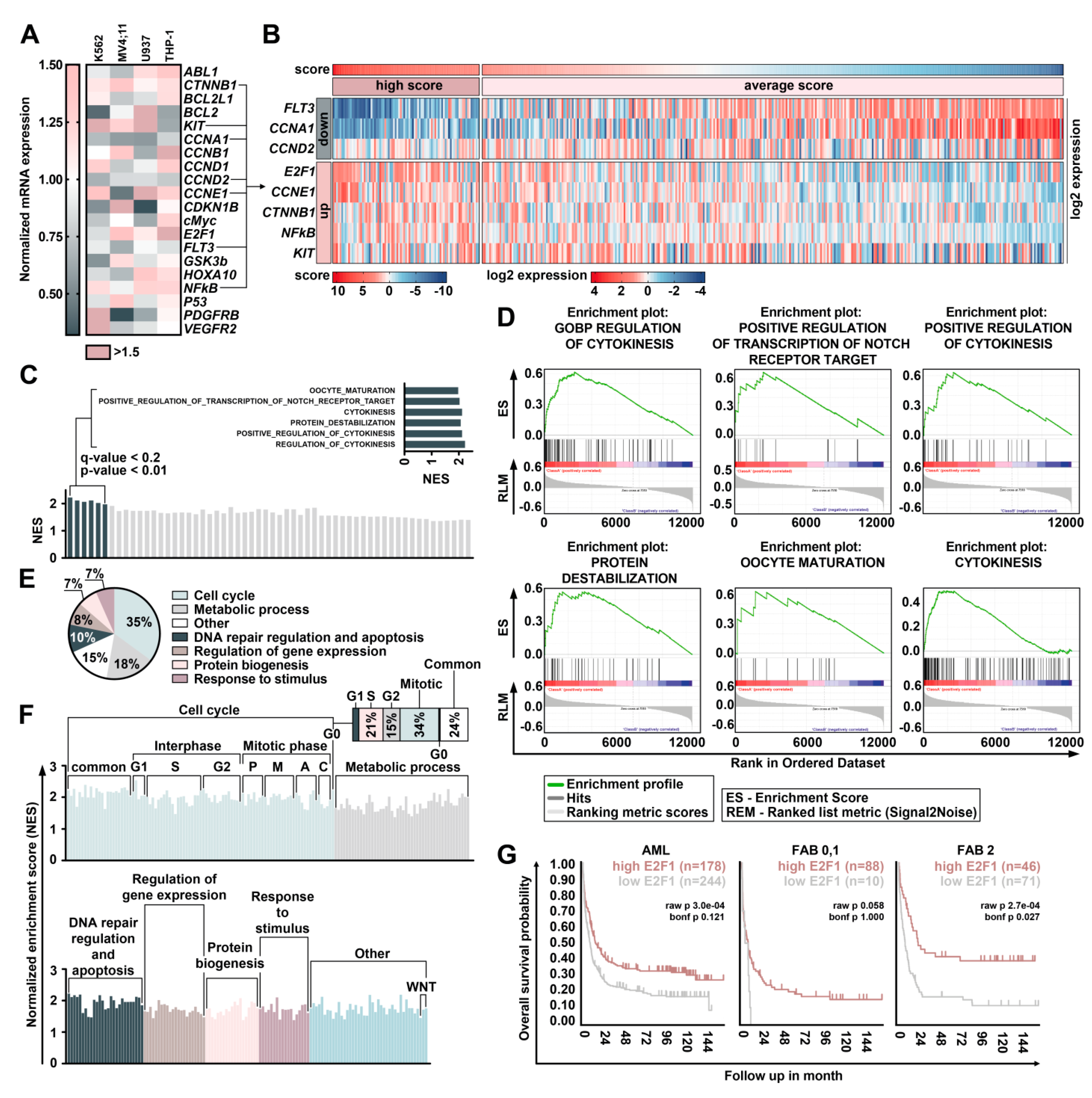
2.4. Treatment with DBF Induce Alterations in Expression of Genes Responsible for Survival of Leukemia Cells
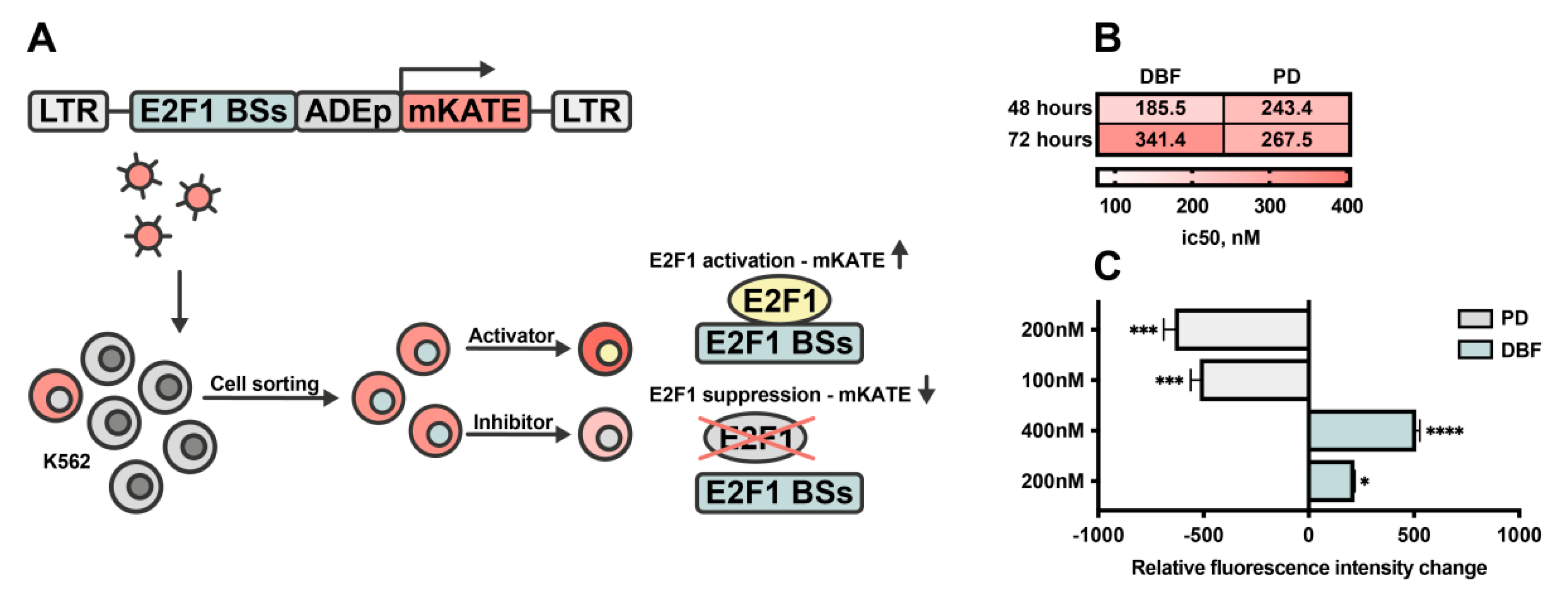
2.5. DBF Induce Activation of E2F1 Transcription Factor in Myeloid Leukemia Cells
2.6. DBF Synergistically Acts with AraC to Induce Suppression of Acute Myeloid Leukemia Growth
3. Discussion
4. Materials and Methods
4.1. Reagents and Chemicals
4.2. Cell Lines and Culture Conditions
4.3. Cell Viability Assay
4.4. RNA Extraction and Real-Time PCR
4.5. Analysis of Cell Cycle Progression
4.6. Analysis of Apoptosis
4.7. AML Samples Scoring and Gene Set Enrichment Analysis (GSEA)
4.8. E2F1 Reporter Cell Line Generation and Flow Cytometry
4.9. Drug Combination Analysis
4.10. Data and Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Gruszka, A.M.; Valli, D.; Alcalay, M. Understanding the molecular basis of acute myeloid leukemias: Where are we now? Int. J. Hematol. Oncol. 2017, 6, 43–53. [Google Scholar] [CrossRef]
- Pelcovits, A.; Niroula, R. Acute Myeloid Leukemia: A Review. R. I. Med. J. 2020, 103, 38–40. [Google Scholar]
- Petrov, I.; Suntsova, M.; Mutorova, O.; Sorokin, M.; Garazha, A.; Ilnitskaya, E.; Spirin, P.; Larin, S.; Kovalchuk, O.; Prassolov, V.; et al. Molecular pathway activation features of pediatric acute myeloid leukemia (AML) and acute lymphoblast leukemia (ALL) cells. Aging 2016, 8, 2936–2947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamaguchi, H. Gene mutations in acute myeloid leukemia. Rinsho Ketsueki 2016, 57, 2535–2542. [Google Scholar] [PubMed]
- Davis, J.R.; Benjamin, D.J.; Jonas, B.A. New and emerging therapies for acute myeloid leukaemia. J. Investig. Med. 2018, 66, 1088–1095. [Google Scholar] [CrossRef]
- Dinardo, C.D.; Wei, A.H. How I treat acute myeloid leukemia in the era of new drugs. Blood 2020, 135, 85–96. [Google Scholar] [CrossRef]
- Saxena, K.; Konopleva, M. New Treatment Options for Older Patients with Acute Myeloid Leukemia. Curr. Treat. Opt. Oncol. 2021, 22, 39. [Google Scholar] [CrossRef]
- Russo, D.; Garcia-Gutierrez, J.V.; Soverini, S.; Baccarani, M. Chronic Myeloid Leukemia Prognosis and Therapy: Criticisms and Perspectives. J. Clin. Med. 2020, 9, 1709. [Google Scholar] [CrossRef]
- Antar, A.I.; Otrock, Z.K.; Jabbour, E.; Mohty, M.; Bazarbachi, A. FLT3 inhibitors in acute myeloid leukemia: Ten frequently asked questions. Leukemia 2020, 34, 682–696. [Google Scholar] [CrossRef]
- Soncini, D.; Orecchioni, S.; Ruberti, S.; Minetto, P.; Martinuzzi, C.; Agnelli, L.; Todoerti, K.; Cagnetta, A.; Miglino, M.; Clavio, M.; et al. The new small tyrosine kinase inhibitor ARQ531 targets acute myeloid leukemia cells by disrupting multiple tumor-addicted programs. Haematologica 2019, 105, 2420–2431. [Google Scholar] [CrossRef] [Green Version]
- An, X.; Tiwari, A.K.; Sun, Y.; Ding, P.-R.; Ashby, C.R.; Chen, Z.-S. BCR-ABL tyrosine kinase inhibitors in the treatment of Philadelphia chromosome positive chronic myeloid leukemia: A review. Leuk. Res. 2010, 34, 1255–1268. [Google Scholar] [CrossRef]
- Lennartsson, J.; Rönnstrand, L. Stem Cell Factor Receptor/c-Kit: From Basic Science to Clinical Implications. Physiol. Rev. 2012, 92, 1619–1649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giles, F.J.; Cortes, J.E.; Kantarjian, H.M. Targeting the kinase activity of the BCR-ABL fusion protein in patients with chronic myeloid leukemia. Curr. Mol. Med. 2005, 5, 615–623. [Google Scholar] [CrossRef]
- Spirin, P.; Lebedev, T.; Orlova, N.; Morozov, A.; Poymenova, N.; Dmitriev, S.; Buzdin, A.; Stocking, C.; Kovalchuk, O.; Prassolov, V. Synergistic suppression of t(8;21)-positive leukemia cell growth by combining oridonin and MAPK1/ERK2 inhibitors. Oncotarget 2017, 8, 56991–57002. [Google Scholar] [CrossRef] [PubMed]
- Weisberg, E.; Meng, C.; Case, A.; Tiv, H.L.; Gokhale, P.C.; Buhrlage, S.J.; Yang, J.; Liu, X.; Wang, J.; Gray, N.; et al. Effects of the multi-kinase inhibitor midostaurin in combination with chemotherapy in models of acute myeloid leukaemia. J. Cell. Mol. Med. 2020, 24, 2968–2980. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Germann, U.A.; Furey, B.F.; Markland, W.; Hoover, R.R.; Aronov, A.M.; Roix, J.J.; Hale, M.; Boucher, D.M.; Sorrell, D.A.; Martinez-Botella, G.; et al. Targeting the MAPK signaling pathway in cancer: Promising preclinical activity with the novel selective ERK1/2 inhibitor BVD-523 (ulixertinib). Mol. Cancer Ther. 2017, 16, 2351–2363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Machado, C.B.; Da Silva, E.L.; Filho, M.O.D.M.; De Moraes, M.E.A.; Moreira-Nunes, C.A. PARP Inhibitors as Therapeutic Options for Tyrosine Kinase-dependent Leukemia: A Review. Anticancer Res. 2020, 40, 3055–3063. [Google Scholar] [CrossRef]
- Dellomo, A.J.; Baer, M.R.; Rassool, F.V. Partnering with PARP inhibitors in acute myeloid leukemia with FLT3-ITD. Cancer Lett. 2019, 454, 171–178. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Boyson, C.A.; Di Liberto, M.; Huang, X.; Hannah, J.; Dorn, D.C.; Moore, M.A.S.; Chen-Kiang, S.; Zhou, P. CDK4/6 Inhibitor PD 0332991 Sensitizes Acute Myeloid Leukemia to Cytarabine-Mediated Cytotoxicity. Cancer Res. 2015, 75, 1838–1845. [Google Scholar] [CrossRef] [Green Version]
- Seca, H.; Lima, R.T.; Lopes-Rodrigues, V.; Guimaraes, J.E.; Gabriela, G.; Vasconcelos, M.H. Targeting miR-21 Induces Autophagy and Chemosensitivity of Leukemia Cells. Curr. Drug Targets 2013, 14, 1135–1143. [Google Scholar] [CrossRef]
- Dykstra, K.M.; Fay, H.R.S.; Massey, A.C.; Yang, N.; Johnson, M.; Portwood, S.; Guzman, M.L.; Wang, E.S. Inhibiting autophagy targets human leukemic stem cells and hypoxic AML blasts by disrupting mitochondrial homeostasis. Blood Adv. 2021, 5, 2087–2100. [Google Scholar] [CrossRef] [PubMed]
- Turner, N.C.; Ro, J.; André, F.; Loi, S.; Verma, S.; Iwata, H.; Harbeck, N.; Loibl, S.; Bartlett, C.H.; Zhang, K.; et al. Palbociclib in Hormone-Receptor–Positive Advanced Breast Cancer. N. Engl. J. Med. 2015, 373, 209–219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, E.S.; Scott, L.J. Palbociclib: A Review in HR-Positive, HER2-Negative, Advanced or Metastatic Breast Cancer. Target. Oncol. 2017, 12, 373–383. [Google Scholar] [CrossRef] [PubMed]
- Mycock, K.; Zhan, L.; Taylor-Stokes, G.; Milligan, G.; Mitra, D. Real-World Palbociclib Use in HR+/HER2− Advanced Breast Cancer in Canada: The IRIS Study. Curr. Oncol. 2021, 28, 678–688. [Google Scholar] [CrossRef]
- Bharate, S.B.; Manda, S.; Mupparapu, N.; Battini, N.; Vishwakarma, R.A. Chemistry and Biology of Fascaplysin, a Potent Marine-Derived CDK-4 Inhibitor. Mini-Rev. Med. Chem. 2012, 12, 650–664. [Google Scholar] [CrossRef]
- Hamilton, G. Cytotoxic Effects of Fascaplysin against Small Cell Lung Cancer Cell Lines. Mar. Drugs 2014, 12, 1377–1389. [Google Scholar] [CrossRef] [Green Version]
- Segraves, N.L.; Robinson, S.J.; Garcia, D.; Said, S.A.; Fu, X.; Schmitz, F.J.; Pietraszkiewicz, H.; Valeriote, F.A.; Crews, P. Comparison of Fascaplysin and Related Alkaloids: A Study of Structures, Cytotoxicities, and Sources. J. Nat. Prod. 2004, 67, 783–792. [Google Scholar] [CrossRef]
- Soni, R.; Muller, L.; Furet, P.; Schoepfer, J.; Stephan, C.; Zumstein-Mecker, S.; Fretz, H.; Chaudhuri, B. Inhibition of Cyclin-Dependent Kinase 4 (Cdk4) by Fascaplysin, a Marine Natural Product. Biochem. Biophys. Res. Commun. 2000, 275, 877–884. [Google Scholar] [CrossRef]
- Oh, T.-I.; Lee, Y.-M.; Nam, T.-J.; Ko, Y.-S.; Mah, S.; Kim, J.; Kim, Y.; Reddy, R.H.; Kim, Y.J.; Hong, S.; et al. Fascaplysin Exerts Anti-Cancer Effects through the Downregulation of Survivin and HIF-1α and Inhibition of VEGFR2 and TRKA. Int. J. Mol. Sci. 2017, 18, 2074. [Google Scholar] [CrossRef]
- Sharma, S.; Guru, S.K.; Manda, S.; Kumar, A.; Mintoo, M.J.; Prasad, V.D.; Sharma, P.R.; Mondhe, D.M.; Bharate, S.B.; Bhushan, S. A marine sponge alkaloid derivative 4-chloro fascaplysin inhibits tumor growth and VEGF mediated angiogenesis by disrupting PI3K/Akt/mTOR signaling cascade. Chem. Interact. 2017, 275, 47–60. [Google Scholar] [CrossRef]
- Kuzmich, A.S.; Fedorov, S.N.; Shastina, V.V.; Shubina, L.K.; Radchenko, O.S.; Balaneva, N.N.; Zhidkov, M.E.; Park, J.-I.; Kwak, J.Y.; Stonik, V.A. The anticancer activity of 3- and 10-bromofascaplysins is mediated by caspase-8, -9, -3-dependent apoptosis. Bioorg. Med. Chem. 2010, 18, 3834–3840. [Google Scholar] [CrossRef]
- Zhidkov, M.E.; Smirnova, P.A.; Tryapkin, O.A.; Kantemirov, A.V.; Khudyakova, Y.V.; Malyarenko, O.S.; Ermakova, S.P.; Grigorchuk, V.P.; Kaune, M.; Von Amsberg, G.; et al. Total Syntheses and Preliminary Biological Evaluation of Brominated Fascaplysin and Reticulatine Alkaloids and Their Analogues. Mar. Drugs 2019, 17, 496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dyshlovoy, S.A.; Kaune, M.; Hauschild, J.; Kriegs, M.; Hoffer, K.; Busenbender, T.; Smirnova, P.A.; Zhidkov, M.E.; Poverennaya, E.V.; Oh-Hohenhorst, S.J.; et al. Efficacy and Mechanism of Action of Marine Alkaloid 3,10-Dibromofascaplysin in Drug-Resistant Prostate Cancer Cells. Mar. Drugs 2020, 18, 609. [Google Scholar] [CrossRef] [PubMed]
- Ding, L.; Cao, J.; Lin, W.; Chen, H.; Xiong, X.; Ao, H.; Yu, M.; Lin, J.; Cui, Q. The Roles of Cyclin-Dependent Kinases in Cell-Cycle Progression and Therapeutic Strategies in Human Breast Cancer. Int. J. Mol. Sci. 2020, 21, 1960. [Google Scholar] [CrossRef] [Green Version]
- Barbosa, K.; Li, S.; Adams, P.D.; Deshpande, A.J. The role of TP53 in acute myeloid leukemia: Challenges and opportunities. Genes Chromosomes Cancer 2019, 58, 875–888. [Google Scholar] [CrossRef] [Green Version]
- Hemann, M.T.; Lowe, S.W. The p53-Bcl-2 connection. Cell Death Differ. 2006, 13, 1256–1259. [Google Scholar] [CrossRef]
- Zebisch, A.; Lal, R.; Müller, M.; Lind, K.; Kashofer, K.; Girschikofsky, M.; Fuchs, D.; Wölfler, A.; Geigl, J.B.; Sill, H. Acute myeloid leukemia with TP53 germ line mutations. Blood 2016, 128, 2270–2272. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.-D.; Zhang, T.-J.; Xu, Z.-J.; Gu, Y.; Ma, J.-C.; Li, X.-X.; Guo, H.; Wen, X.-M.; Zhang, W.; Yang, L.; et al. BCL2 overexpression: Clinical implication and biological insights in acute myeloid leukemia. Diagn. Pathol. 2019, 14, 68. [Google Scholar] [CrossRef]
- Poeta, G.; Bruno, A.; Del Principe, M.; Venditti, A.; Maurillo, L.; Buccisano, F.; Stasi, R.; Neri, B.; Luciano, F.; Siniscalchi, A.; et al. Deregulation of the Mitochondrial Apoptotic Machinery and Development of Molecular Targeted Drugs in Acute Myeloid Leukemia. Curr. Cancer Drug Targets 2008, 8, 207–222. [Google Scholar] [CrossRef]
- Li, Z.; Herold, T.; He, C.; Valk, P.J.; Chen, P.; Jurinovic, V.; Mansmann, U.; Radmacher, M.D.; Maharry, K.S.; Sun, M.; et al. Identification of a 24-Gene Prognostic Signature That Improves the European LeukemiaNet Risk Classification of Acute Myeloid Leukemia: An International Collaborative Study. J. Clin. Oncol. 2013, 31, 1172–1181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mundle, S.D.; Saberwal, G. Evolving intricacies and implications of E2F1 regulation. FASEB J. 2003, 17, 569–574. [Google Scholar] [CrossRef] [PubMed]
- Krek, W.; Xu, G.; Livingston, D.M. Cyclin A-kinase regulation of E2F-1 DNA binding function underlies suppression of an S phase checkpoint. Cell 1995, 83, 1149–1158. [Google Scholar] [CrossRef] [Green Version]
- Huang, Y.; Ishiko, T.; Nakada, S.; Utsugisawa, T.; Kato, T.; Yuan, Z.M. Role for E2F in DNA damage-induced entry of cells into S phase. Cancer Res. 1997, 57, 3640–3643. [Google Scholar]
- Lin, W.-C.; Lin, F.-T.; Nevins, J.R. Selective induction of E2F1 in response to DNA damage, mediated by ATM-dependent phosphorylation. Genome Res. 2001, 15, 1833–1844. [Google Scholar]
- Maser, R.S.; Mirzoeva, O.K.; Wells, J.; Olivares, H.; Williams, B.R.; Zinkel, R.A.; Farnham, P.; Petrini, J.H.J. Mre11 Complex and DNA Replication: Linkage to E2F and Sites of DNA Synthesis. Mol. Cell. Biol. 2001, 21, 6006–6016. [Google Scholar] [CrossRef] [Green Version]
- Saldivar, J.C.; Hamperl, S.; Bocek, M.J.; Chung, M.; Bass, T.E.; Cisneros-Soberanis, F.; Samejima, K.; Xie, L.; Paulson, J.R.; Earnshaw, W.C.; et al. An intrinsic S/G2 checkpoint enforced by ATR. Science 2018, 361, 806–810. [Google Scholar] [CrossRef] [Green Version]
- Cam, H.; Dynlacht, B.D. Emerging roles for E2F: Beyond the G1/S transition and DNA replication. Cancer Cell 2003, 3, 311–316. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Guru, S.K.; Pathania, A.; Manda, S.; Kumar, A.; Bharate, S.B.; Vishwakarma, R.A.; Malik, F.; Bhushan, S. Fascaplysin Induces Caspase Mediated Crosstalk Between Apoptosis and Autophagy Through the Inhibition of PI3K/AKT/mTOR Signaling Cascade in Human Leukemia HL-60 Cells. J. Cell. Biochem. 2015, 116, 985–997. [Google Scholar] [CrossRef]
- Meng, N.; Mu, X.; Lv, X.; Wang, L.; Li, N.; Gong, Y. Autophagy represses fascaplysin-induced apoptosis and angiogenesis inhibition via ROS and p8 in vascular endothelia cells. Biomed. Pharmacother. 2019, 114, 108866. [Google Scholar] [CrossRef]
- Uras, I.; Walter, G.J.; Scheicher, R.; Bellutti, F.; Prchal-Murphy, M.; Tigan, A.S.; Valent, P.; Heidel, F.H.; Kubicek, S.; Scholl, C.; et al. Palbociclib treatment of FLT3-ITD+ AML cells uncovers a kinase-dependent transcriptional regulation of FLT3 and PIM1 by CDK6. Blood 2016, 127, 2890–2902. [Google Scholar] [CrossRef] [Green Version]
- Sorf, A.; Sucha, S.; Morell, A.; Novotna, E.; Staud, F.; Zavrelova, A.; Visek, B.; Wsol, V.; Ceckova, M. Targeting Pharmacokinetic Drug Resistance in Acute Myeloid Leukemia Cells with CDK4/6 Inhibitors. Cancers 2020, 12, 1596. [Google Scholar] [CrossRef]
- VanArsdale, T.; Boshoff, C.; Arndt, K.T.; Abraham, R.T. Molecular Pathways: Targeting the Cyclin D-CDK4/6 Axis for Cancer Treatment. Clin. Cancer Res. 2015, 21, 2905–2910. [Google Scholar] [CrossRef] [Green Version]
- Teeyapant, R.; Woerdenbag, H.J.; Kreis, P.; Hacker, J.; Wray, V.; Witte, L.; Proksch, P. Antibiotic and Cytotoxic Activity of Brominated Compounds from the Marine Sponge Verongia aerophoba. Z. Naturforsch. C J. Biosci. 1993, 48, 939–945. [Google Scholar] [CrossRef]
- Cortes, J.; Giles, F.; O’Brien, S.; Thomas, D.; Albitar, M.; Rios, M.B.; Talpaz, M.; Garcia-Manero, G.; Faderl, S.; Letvak, L.; et al. Results of imatinib mesylate therapy in patients with refractory or recurrent acute myeloid leukemia, high-risk myelodysplastic syndrome, and myeloproliferative disorders. Cancer 2003, 97, 2760–2766. [Google Scholar] [CrossRef]
- Malagola, M.; Martinelli, G.; Rondoni, M.; Paolini, S.; Gaitani, S.; Arpinati, M.; Piccaluga, P.P.; Amabile, A.M.; Basi, C.; Ottaviani, E.; et al. Imatinib mesylate in the treatment of c-kit-positive acute myeloid leukemia: Is this the real target? Blood 2005, 105, 904–905. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allen-Petersen, B.L.; Sears, R.C. Mission Possible: Advances in MYC Therapeutic Targeting in Cancer. BioDrugs 2019, 33, 539–553. [Google Scholar] [CrossRef] [Green Version]
- Leung, J.Y.; Ehmann, G.L.; Giangrande, P.H.; Nevins, J.R. A role for Myc in facilitating transcription activation by E2F1. Oncogene 2008, 27, 4172–4179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, X.-N.; Chen, J.-J.; Wang, L.-X.; Xiao, R.-Z.; Liu, L.-L.; Fang, Z.-G.; Liu, Q.; Long, Z.-J.; Lin, D.-J. Inhibition of c-Myc Overcomes Cytotoxic Drug Resistance in Acute Myeloid Leukemia Cells by Promoting Differentiation. PLoS ONE 2014, 9, e105381. [Google Scholar] [CrossRef]
- Ohanian, M.; Rozovski, U.; Kanagal-Shamanna, R.; Abruzzo, L.V.; Loghavi, S.; Kadia, T.; Futreal, A.; Bhalla, K.; Zuo, Z.; Huh, Y.O.; et al. MYC protein expression is an important prognostic factor in acute myeloid leukemia. Leuk. Lymphoma 2018, 60, 37–48. [Google Scholar] [CrossRef]
- Meng, P.; Ghosh, R. Transcription addiction: Can we garner the Yin and Yang functions of E2F1 for cancer therapy? Cell Death Dis. 2014, 5, e1360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hinds, P.W. Cdk2 dethroned as master of S phase entry. Cancer Cell 2003, 3, 305–307. [Google Scholar] [CrossRef] [Green Version]
- Xu, M.; Sheppard, K.A.; Peng, C.Y.; Yee, A.S.; Piwnica-Worms, H. Cyclin A/CDK2 binds directly to E2F-1 and inhibits the DNA-binding activity of E2F-1/DP-1 by phosphorylation. Mol. Cell. Biol. 1994, 14, 8420–8431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, Y.-P.; Liu, M.-C.; Cai, Q.; Jia, F.-C.; Wu, A.-X. A Cascade Coupling Strategy for One-Pot Total Synthesis of β-Carboline and Isoquinoline-Containing Natural Products and Derivatives. Chemistry 2013, 19, 10132–10137. [Google Scholar] [CrossRef]
- Gu, Z.; Eils, R.; Schlesner, M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics 2016, 32, 2847–2849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nissim, L.; Wu, M.-R.; Pery, E.; Binder-Nissim, A.; Suzuki, H.; Stupp, D.; Wehrspaun, C.; Tabach, Y.; Sharp, P.A.; Lu, T.K. Synthetic RNA-Based Immunomodulatory Gene Circuits for Cancer Immunotherapy. Cell 2017, 171, 1138–1150.e15. [Google Scholar] [CrossRef] [Green Version]
- Weber, K.; Bartsch, U.; Stocking, C.; Fehse, B. A multicolor panel of novel lentiviral “gene ontology” (LeGO) vectors for functional gene analysis. Mol. Ther. 2008, 16, 698–706. [Google Scholar] [CrossRef] [PubMed]
- Yadav, B.; Wennerberg, K.; Aittokallio, T.; Tang, J. Searching for Drug Synergy in Complex Dose–Response Landscapes Using an Interaction Potency Model. Comput. Struct. Biotechnol. J. 2015, 13, 504–513. [Google Scholar] [CrossRef] [Green Version]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Spirin, P.; Shyrokova, E.; Lebedev, T.; Vagapova, E.; Smirnova, P.; Kantemirov, A.; Dyshlovoy, S.A.; Amsberg, G.v.; Zhidkov, M.; Prassolov, V. Cytotoxic Marine Alkaloid 3,10-Dibromofascaplysin Induces Apoptosis and Synergizes with Cytarabine Resulting in Leukemia Cell Death. Mar. Drugs 2021, 19, 489. https://doi.org/10.3390/md19090489
Spirin P, Shyrokova E, Lebedev T, Vagapova E, Smirnova P, Kantemirov A, Dyshlovoy SA, Amsberg Gv, Zhidkov M, Prassolov V. Cytotoxic Marine Alkaloid 3,10-Dibromofascaplysin Induces Apoptosis and Synergizes with Cytarabine Resulting in Leukemia Cell Death. Marine Drugs. 2021; 19(9):489. https://doi.org/10.3390/md19090489
Chicago/Turabian StyleSpirin, Pavel, Elena Shyrokova, Timofey Lebedev, Elmira Vagapova, Polina Smirnova, Alexey Kantemirov, Sergey A. Dyshlovoy, Gunhild von Amsberg, Maxim Zhidkov, and Vladimir Prassolov. 2021. "Cytotoxic Marine Alkaloid 3,10-Dibromofascaplysin Induces Apoptosis and Synergizes with Cytarabine Resulting in Leukemia Cell Death" Marine Drugs 19, no. 9: 489. https://doi.org/10.3390/md19090489