
Study of Structure–Activity Relationships of the Marine Alkaloid Fascaplysin and Its Derivatives as Potent Anticancer Agents

 , , , ,
, , , ,
Abstract
:1. Introduction
2. Results and Discussion
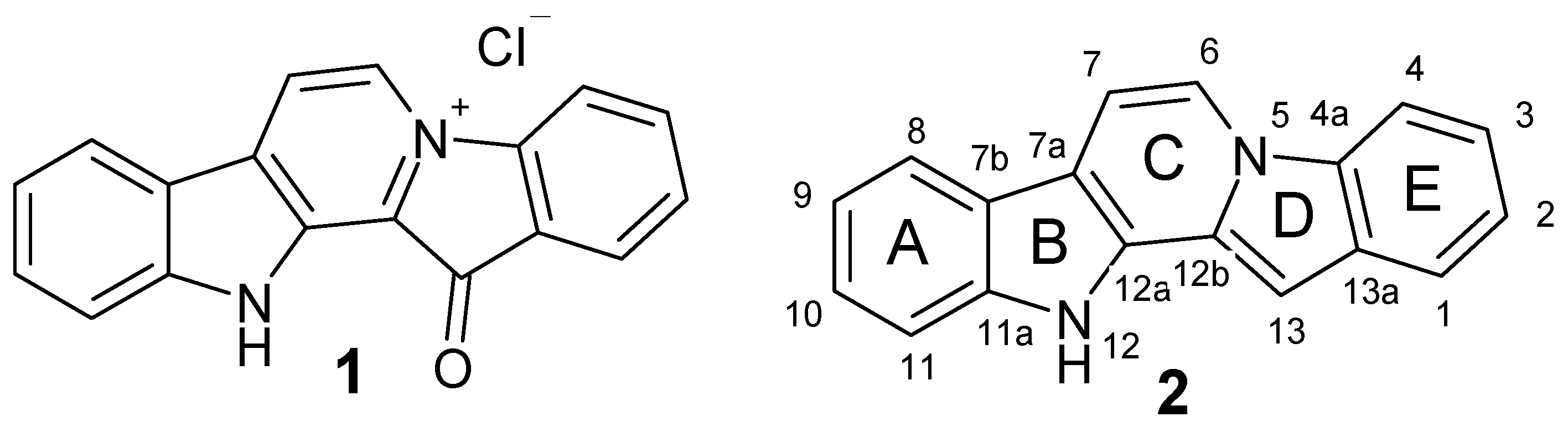
2.1. Chemistry
2.2. Biological Studies
2.2.1. Activity and Selectivity of the Synthesized Compounds in Human Prostate Cancer Cells
2.2.2. Interaction of Fascaplysin and its Derivatives with DNA
3. Materials and Methods
3.1. Chemistry
3.1.1. Preparation of Mixture of Tryptamines 26 and 27
3.1.2. Preparation of Substituted 1-Benzoyl-β-Carbolines 23a-b, 28a-b, 35a-b
3.1.3. Preparation of Substituted 1-Benzoyl-β-Carbolines 29a, 30a, 30g
3.1.4. Synthesis of Compound 30d
3.1.5. Preparation of Compounds 29b, 30b
3.1.6. Synthesis of Compound 30c
3.1.7. Preparation of Compound 30f
3.1.8. Quaternization of Substituted 1-Benzoyl-β-carbolines
3.1.9. General Procedure for the Condensation of Indigo with Methylene Active Compounds
3.1.10. Preparation of Compounds 16–18
3.2. Biological Assay
3.2.1. Reagents
3.2.2. Cell Lines and Culture Conditions
3.2.3. MTT Assay
3.2.4. In Vitro Trypan Blue-Based Viability Assay
3.2.5. Analysis of Cell Cycle Progression and DNA Fragmentation
3.2.6. Thiazole Orange Displacement (DNA Intercalation) Assay
3.2.7. Data and Statistical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Mayer, A.; Guerrero, A.; Rodríguez, A.; Taglialatela-Scafati, O.; Nakamura, F.; Fusetani, N. Marine Pharmacology in 2016–2017: Marine Compounds with Antibacterial, Antidiabetic, Antifungal, Anti-Inflammatory, Antiprotozoal, Antituberculosis and Antiviral Activities; Affecting the Immune and Nervous Systems, and Other Miscellaneous Mechanisms of Action. Mar. Drugs 2021, 19, 49. [Google Scholar] [CrossRef] [PubMed]
- Dyshlovoy, S.A.; Honecker, F. Marine Compounds and Cancer: Updates 2020. Mar. Drugs 2020, 18, 643. [Google Scholar] [CrossRef] [PubMed]
- Dyshlovoy, S.A.; Honecker, F. Marine Compounds and Cancer: The First Two Decades of XXI Century. Mar. Drugs 2019, 18, 20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roll, D.M.; Ireland, C.M.; Lu, H.S.M.; Clardy, J. Fascaplysin, an unusual antimicrobial pigment from the marine sponge Fascaplysinopsis sp. J. Org. Chem. 1988, 53, 3276–3278. [Google Scholar] [CrossRef]
- Bharate, S.B.; Manda, S.; Mupparapu, N.; Battini, N.; Vishwakarma, R.A. Chemistry and Biology of Fascaplysin, a Potent Marine-Derived CDK-4 Inhibitor. Mini-Rev. Med. Chem. 2012, 12, 650–664. [Google Scholar] [CrossRef]
- Popov, A.M.; Stonik, V.A. Physiological activity of fascaplisine—An unusual pigment from tropical sea fishes. Antibiot. Chemother. 1991, 36, 12–14. [Google Scholar]
- Hamilton, G. Cytotoxic Effects of Fascaplysin against Small Cell Lung Cancer Cell Lines. Mar. Drugs 2014, 12, 1377–1389. [Google Scholar] [CrossRef] [Green Version]
- Rath, B.; Hochmair, M.; Plangger, A.; Hamilton, G. Anticancer Activity of Fascaplysin against Lung Cancer Cell and Small Cell Lung Cancer Circulating Tumor Cell Lines. Mar. Drugs 2018, 16, 383. [Google Scholar] [CrossRef] [Green Version]
- Yan, X.; Chen, H.; Lu, X.; Wang, F.; Xu, W.; Jin, H.; Zhu, P. Fascaplysin exert anti-tumor effects through apoptotic and anti-angiogenesis pathways in sarcoma mice model. Eur. J. Pharm. Sci. 2011, 43, 251–259. [Google Scholar] [CrossRef]
- Soni, R.; Muller, L.; Furet, P.; Schoepfer, J.; Stephan, C.; Zumstein-Mecker, S.; Fretz, H.; Chaudhuri, B. Inhibition of Cyclin-Dependent Kinase 4 (Cdk4) by Fascaplysin, a Marine Natural Product. Biochem. Biophys. Res. Commun. 2000, 275, 877–884. [Google Scholar] [CrossRef]
- Hörmann, A.; Chaudhuri, B.; Fretz, H. DNA binding properties of the marine sponge pigment fascaplysin. Bioorgan. Med. Chem. 2001, 9, 917–921. [Google Scholar] [CrossRef]
- Lu, X.-L.; Zheng, Y.-L.; Chen, H.-M.; Yan, X.-J.; Wang, F.; Xu, W.-F. Anti-proliferation of human cervical cancer HeLa cell line by fascaplysin through apoptosis induction. Acta Pharm. Sin. 2009, 44, 980–986. [Google Scholar]
- Aubry, C.; Jenkins, P.R.; Mahale, S.; Chaudhuri, B.; Maréchal, J.-D.; Sutcliffe, M.J. New fascaplysin-based CDK4-specific inhibitors: Design, synthesis and biological activity. Chem. Commun. 2004, 35, 1696–1697. [Google Scholar] [CrossRef] [PubMed]
- Aubry, C.; Wilson, A.J.; Jenkins, P.R.; Mahale, S.; Chaudhuri, B.; Maréchal, J.-D.; Sutcliffe, M.J. Design, synthesis and biological activity of new CDK4-specific inhibitors, based on fascaplysin. Org. Biomol. Chem. 2006, 4, 787–801. [Google Scholar] [CrossRef] [PubMed]
- Mahale, S.; Aubry, C.; Wilson, A.J.; Jenkins, P.R.; Maréchal, J.-D.; Sutcliffe, M.J.; Chaudhuri, B. CA224, a non-planar analogue of fascaplysin, inhibits Cdk4 but not Cdk2 and arrests cells at G0/G1 inhibiting pRB phosphorylation. Bioorgan. Med. Chem. Lett. 2006, 16, 4272–4278. [Google Scholar] [CrossRef]
- García, M.D.; Wilson, A.J.; Emmerson, D.P.G.; Jenkins, P.R.; Mahale, S.; Chaudhuri, B. Synthesis, crystal structure and biological activity of β-carboline based selective CDK4-cyclin D1 inhibitors. Org. Biomol. Chem. 2006, 4, 4478–4484. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, P.R.; Wilson, J.; Emmerson, D.; Garcia, M.D.; Smith, M.R.; Gray, S.J.; Britton, R.G.; Mahale, S.; Chaudhuri, B. Design, synthesis and biological evaluation of new tryptamine and tetrahydro-β-carboline-based selective inhibitors of CDK4. Bioorgan. Med. Chem. 2008, 16, 7728–7739. [Google Scholar] [CrossRef]
- Aubry, C.; Wilson, A.J.; Emmerson, D.; Murphy, E.; Chan, Y.Y.; Dickens, M.P.; García, M.D.; Jenkins, P.R.; Mahale, S.; Chaudhuri, B. Fascaplysin-inspired diindolyls as selective inhibitors of CDK4/cyclin D1. Bioorgan. Med. Chem. 2009, 17, 6073–6084. [Google Scholar] [CrossRef]
- Mahale, S.; Bharate, S.B.; Manda, S.; Joshi, P.; Bharate, S.S.; Jenkins, P.R.; Vishwakarma, R.A.; Chaudhuri, B. Biphenyl-4-carboxylic Acid [2-(1H-Indol-3-yl)-ethyl]-methylamide (CA224), a Nonplanar Analogue of Fascaplysin, Inhibits Cdk4 and Tubulin Polymerization: Evaluation of in Vitro and in Vivo Anticancer Activity. J. Med. Chem. 2014, 57, 9658–9672. [Google Scholar] [CrossRef]
- Oh, T.-I.; Lee, Y.-M.; Nam, T.-J.; Ko, Y.-S.; Mah, S.; Kim, J.; Kim, Y.; Reddy, R.H.; Kim, Y.J.; Hong, S.; et al. Fascaplysin Exerts Anti-Cancer Effects through the Downregulation of Survivin and HIF-1α and Inhibition of VEGFR2 and TRKA. Int. J. Mol. Sci. 2017, 18, 2074. [Google Scholar] [CrossRef]
- Kumar, S.; Guru, S.K.; Pathania, A.S.; Manda, S.; Kumar, A.; Bharate, S.B.; Vishwakarma, R.A.; Malik, F.; Bhushan, S. Fascaplysin Induces Caspase Mediated Crosstalk Between Apoptosis and Autophagy Through the Inhibition of PI3K/AKT/mTOR Signaling Cascade in Human Leukemia HL-60 Cells. J. Cell. Biochem. 2015, 116, 985–997. [Google Scholar] [CrossRef] [PubMed]
- Meng, N.; Mu, X.; Lv, X.; Wang, L.; Li, N.; Gong, Y. Autophagy represses fascaplysin-induced apoptosis and angiogenesis inhibition via ROS and p8 in vascular endothelia cells. Biomed. Pharmacother. 2019, 114, 108866. [Google Scholar] [CrossRef] [PubMed]
- Oh, T.-I.; Lee, J.H.; Kim, S.; Nam, T.-J.; Kim, Y.-S.; Kim, B.M.; Yim, W.J.; Lim, J.-H. Fascaplysin Sensitizes Anti-Cancer Effects of Drugs Targeting AKT and AMPK. Molecules 2017, 23, 42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, S.; Guru, S.K.; Manda, S.; Kumar, A.; Mintoo, M.J.; Prasad, V.D.; Sharma, P.R.; Mondhe, D.M.; Bharate, S.B.; Bhushan, S. A marine sponge alkaloid derivative 4-chloro fascaplysin inhibits tumor growth and VEGF mediated angiogenesis by disrupting PI3K/Akt/mTOR signaling cascade. Chem. Interact. 2017, 275, 47–60. [Google Scholar] [CrossRef] [PubMed]
- Kuzmich, A.S.; Fedorov, S.N.; Shastina, V.V.; Shubina, L.K.; Radchenko, O.S.; Balaneva, N.N.; Zhidkov, M.E.; Park, J.-I.; Kwak, J.Y.; Stonik, V.A. The anticancer activity of 3- and 10-bromofascaplysins is mediated by caspase-8, -9, -3-dependent apoptosis. Bioorgan. Med. Chem. 2010, 18, 3834–3840. [Google Scholar] [CrossRef]
- Lyakhova, I.A.; Bryukhovetsky, I.S.; Kudryavtsev, I.V.; Khotimchenko, Y.S.; Zhidkov, M.E.; Kantemirov, A.V. Antitumor Activity of Fascaplysin Derivatives on Glioblastoma Model In Vitro. Bull. Exp. Biol. Med. 2018, 164, 666–672. [Google Scholar] [CrossRef]
- Segraves, N.L.; Robinson, S.J.; Garcia, .D.; Said, S.A.; Fu, X.; Schmitz, F.J.; Pietraszkiewicz, H.; Valeriote, A.F.A.; Crews, P. Comparison of Fascaplysin and Related Alkaloids: A Study of Structures, Cytotoxicities, and Sources. J. Nat. Prod. 2004, 67, 783–792. [Google Scholar] [CrossRef]
- Zhidkov, M.E.; Smirnova, P.A.; Tryapkin, O.A.; Kantemirov, A.V.; Khudyakova, Y.V.; Malyarenko, O.S.; Ermakova, S.P.; Grigorchuk, V.P.; Kaune, M.; Von Amsberg, G.; et al. Total Syntheses and Preliminary Biological Evaluation of Brominated Fascaplysin and Reticulatine Alkaloids and Their Analogues. Mar. Drugs 2019, 17, 496. [Google Scholar] [CrossRef] [Green Version]
- Spirin, P.; Shyrokova, E.; Lebedev, T.; Vagapova, E.; Smirnova, P.; Kantemirov, A.; Dyshlovoy, S.A.; von Amsberg, G.; Zhidkov, M.; Prassolov, V. Cytotoxic Marine Alkaloid 3,10-Dibromofascaplysin Induces Apoptosis and Synergizes with Cytarabine Resulting in Leukemia Cell Death. Mar. Drugs 2021, 19, 489. [Google Scholar] [CrossRef]
- Dyshlovoy, S.A.; Kaune, M.; Hauschild, J.; Kriegs, M.; Hoffer, K.; Busenbender, T.; Smirnova, P.A.; Zhidkov, M.E.; Poverennaya, E.V.; Oh-Hohenhorst, S.J.; et al. Efficacy and Mechanism of Action of Marine Alkaloid 3,10-Dibromofascaplysin in Drug-Resistant Prostate Cancer Cells. Mar. Drugs 2020, 18, 609. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2018. CA Cancer J. Clin. 2018, 68, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Singh, M.; Jha, R.; Melamed, J.; Shapiro, E.; Hayward, S.W.; Lee, P. Stromal Androgen Receptor in Prostate Development and Cancer. Am. J. Pathol. 2014, 184, 2598–2607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arnold, J.T.; Isaacs, J.T. Mechanisms involved in the progression of androgen-independent prostate cancers: It is not only the cancer cell’s fault. Endocr. Relat. Cancer 2002, 9, 61–73. [Google Scholar] [CrossRef]
- Gribble, G.W.; Pelcman, B. Total syntheses of the marine sponge pigments fascaplysin and homofascaplysin B and C. J. Org. Chem. 1992, 57, 3636–3642. [Google Scholar] [CrossRef]
- Rocca, P.; Marsais, F.; Godard, A.; Quéguiner, G. A short synthesis of the antimicrobial marine sponge pigment fascaplysin. Tetrahedron Lett. 1993, 34, 7917–7918. [Google Scholar] [CrossRef]
- Molina, P.; Fresneda, P.M.; García-Zafra, S.; Almendros, P. Iminophosphorane-mediated syntheses of the fascaplysin alkaloid of marine origin and nitramarine. Tetrahedron Lett. 1994, 35, 8851–8854. [Google Scholar] [CrossRef]
- Radchenko, O.S.; Novikov, V.L.; Elyakov, G.B. A simple and practical approach to the synthesis of the marine sponge pigment fascaplysin and related compounds. Tetrahedron Lett. 1997, 38, 5339–5342. [Google Scholar] [CrossRef]
- Waldmann, H.; Eberhardt, L.; Wittstein, K.; Kumar, K. Silver catalyzed cascade synthesis of alkaloid ring systems: Concise total synthesis of fascaplysin, homofascaplysin C and analogues. Chem. Commun. 2010, 46, 4622–4624. [Google Scholar] [CrossRef]
- Zhidkov, M.E.; Baranova, O.V.; Kravchenko, N.S.; Dubovitskii, S.V. A new method for the synthesis of the marine alkaloid fascaplysin. Tetrahedron Lett. 2010, 51, 6498–6499. [Google Scholar] [CrossRef]
- Bharate, S.B.; Manda, S.; Joshi, P.; Singh, B.; Vishwakarma, R.A. Total synthesis and anti-cholinesterase activity of marine-derived bis-indole alkaloid fascaplysin. MedChemComm 2012, 3, 1098–1103. [Google Scholar] [CrossRef]
- Zhidkov, M.E.; Kaminskii, V.A. A new method for the synthesis of the marine alkaloid fascaplysin based on the microwave-assisted Minisci reaction. Tetrahedron Lett. 2013, 54, 3530–3532. [Google Scholar] [CrossRef]
- Zhu, Y.-P.; Liu, M.-C.; Cai, Q.; Jia, F.-C.; Wu, A.-X. A Cascade Coupling Strategy for One-Pot Total Synthesis of β-Carboline and Isoquinoline-Containing Natural Products and Derivatives. Chem. A Eur. J. 2013, 19, 10132–10137. [Google Scholar] [CrossRef] [PubMed]
- Zhidkov, M.E.; Kantemirov, A.V.; Koisevnikov, A.V.; Andin, A.N.; Kuzmich, A.S. Syntheses of the marine alkaloids 6-oxofascaplysin, fascaplysin and their derivatives. Tetrahedron Lett. 2018, 59, 708–711. [Google Scholar] [CrossRef]
- Zhidkov, M.E.; Sidorova, M.A.; Lyakhova, I.A. One-step transformation of the marine alkaloid fascaplysin into homofascaplysins B and B-1. The first syntheses of 3-bromohomofascaplysin B and 3–bromohomofascaplysin B-1. Tetrahedron Lett. 2018, 59, 1417–1420. [Google Scholar] [CrossRef]
- Fretz, H.; Ucci-Stoll, K.; Hug, P.; Schoepfer, J.; Lang, M. Investigations on the reactivity of fascaplysin. Part I. Aromatic elec-trophilic substitutions occur at position 9. Helv. Chim. Acta 2001, 83, 3065–3068. [Google Scholar] [CrossRef]
- Zhidkov, M.E.; Baranova, O.V.; Balaneva, N.N.; Fedorov, S.N.; Radchenko, O.S.; Dubovitskii, S.V. The first syntheses of 3-bromofascaplysin, 10-bromofascaplysin and 3,10-dibromofascaplysin—marine alkaloids from Fascaplysinopsis reticulata and Didemnum sp. by application of a simple and effective approach to the pyrido[1,2-a:3,4-b′]diindole system. Tetrahedron Lett. 2007, 48, 7998–8000. [Google Scholar] [CrossRef]
- Hirsch, H.; Boocock, J.R.B.; Hickinbottom, W.J.; Hurst, S.J.; Bruce, J.M.; Zuckerman, J.J.; Green, M.; Mallion, K.B.; Mann, F.G.; Tong, B.P.; et al. Notes. J. Chem. Soc. 1963, 1318–1343. [Google Scholar] [CrossRef]
- Kim, H.-J.; Park, Y.I.; Dong, M.-S. Comparison of prostate cancer cell lines for androgen receptor-mediated reporter gene assays. Toxicol. In Vitro 2006, 20, 1159–1167. [Google Scholar] [CrossRef]
- Rai, Y.; Pathak, R.; Kumari, N.; Sah, D.K.; Pandey, S.; Kalra, N.; Soni, R.; Dwarakanath, B.S.; Bhatt, A.N. Mitochondrial biogenesis and metabolic hyperactivation limits the application of MTT assay in the estimation of radiation induced growth inhibition. Sci. Rep. 2018, 8, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Dyshlovoy, S.A.; Pelageev, D.N.; Hauschild, J.; Borisova, K.L.; Kaune, M.; Krisp, C.; Venz, S.; Sabutskii, Y.E.; Khmelevskaya, E.A.; Busenbender, T.; et al. Successful Targeting of the Warburg Effect in Prostate Cancer by Glucose-Conjugated 1,4-Naphthoquinones. Cancers 2019, 11, 1690. [Google Scholar] [CrossRef] [Green Version]
- Dyshlovoy, S.A.; Pelageev, D.N.; Jakob, L.S.; Borisova, K.L.; Hauschild, J.; Busenbender, T.; Kaune, M.; Khmelevskaya, E.A.; Graefen, M.; Bokemeyer, C.; et al. Activity of New Synthetic (2-Chloroethylthio)-1,4-naphthoquinones in Prostate Cancer Cells. Pharmaceuticals 2021, 14, 949. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | X1 | R | X2 |
|---|---|---|---|
 | |||
| 1 | - | - | - |
| 3 | - | - | 3-Br |
| 4 | - | - | 2-Br |
| 5 | 10-Br | - | - |
| 6 | 9-Br | - | - |
| 7 | 9-I | - | - |
| 8 | 10-Br | - | 3-Br |
| 9 | 8-Br | - | 3-Br |
| 10 | 9-Br | - | 2-Br |
| 11 | 9-I | - | 2-Br |
| 12 | 9-Cl | 2-Br | |
| 13 | 9, 11-Br | - | 2-Br |
| 14 | 9, 11-Cl | - | 2-Br |
| 15 | 9-Br | - | 3-Br |
| 16 | - | 7-Ph | - |
| 17 | - | 7-Et | - |
| 18 | - | 7-Me | - |
| 19 | - | 6-Me | - |
| 20 | 6-Ph | ||
| Experiment. | Heating (°C) or MW | Time, min | Reactant Ratio | Reaction Results | ||
|---|---|---|---|---|---|---|
| 1, mmol | I2, equiv. | DMSO, mL | ||||
| 1 | 120 | 280 | 0.011 | 18.8 | 1 | mixture of 1 and 7 |
| 2 | 120 | 240 | 0.007 | 28 | 0.5 | mixture of 1 and 7 |
| 3 | 120 | 120 | 0.007 | 28 | 1 | decomposition of the mixture |
| 4 | MW, 50 W | 18 | 0.04 | 5.3 | 1 | mixture of 1 and 7 (50/50) |
| 5 | MW, 50 W | 28 | 0.04 | 5.3 | 1 | 15 min—7 found, 28 min—decomposition of the mixture |
| 6 | MW, 45 W | 30 | 0.04 | 5.3 | 1 (autoclave) | mixture of 1 and 7 (minor) |
| 7 | MW, 45 W | 30 | 0.04 | 5.3 | 2 | no reaction |
| 8 | MW, 45 W | 30 | 0.01 | 18.8 | 1 | mixture of 1 and 7 (minor) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhidkov, M.E.; Kaune, M.; Kantemirov, A.V.; Smirnova, P.A.; Spirin, P.V.; Sidorova, M.A.; Stadnik, S.A.; Shyrokova, E.Y.; Kaluzhny, D.N.; Tryapkin, O.A.; et al. Study of Structure–Activity Relationships of the Marine Alkaloid Fascaplysin and Its Derivatives as Potent Anticancer Agents. Mar. Drugs 2022, 20, 185. https://doi.org/10.3390/md20030185
Zhidkov ME, Kaune M, Kantemirov AV, Smirnova PA, Spirin PV, Sidorova MA, Stadnik SA, Shyrokova EY, Kaluzhny DN, Tryapkin OA, et al. Study of Structure–Activity Relationships of the Marine Alkaloid Fascaplysin and Its Derivatives as Potent Anticancer Agents. Marine Drugs. 2022; 20(3):185. https://doi.org/10.3390/md20030185
Chicago/Turabian StyleZhidkov, Maxim E., Moritz Kaune, Alexey V. Kantemirov, Polina A. Smirnova, Pavel V. Spirin, Maria A. Sidorova, Sergey A. Stadnik, Elena Y. Shyrokova, Dmitry N. Kaluzhny, Oleg A. Tryapkin, and et al. 2022. "Study of Structure–Activity Relationships of the Marine Alkaloid Fascaplysin and Its Derivatives as Potent Anticancer Agents" Marine Drugs 20, no. 3: 185. https://doi.org/10.3390/md20030185