Effect of Expression of Human Glucosylceramidase 2 Isoforms on Lipid Profiles in COS-7 Cells

 ,
,  ,
, 
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
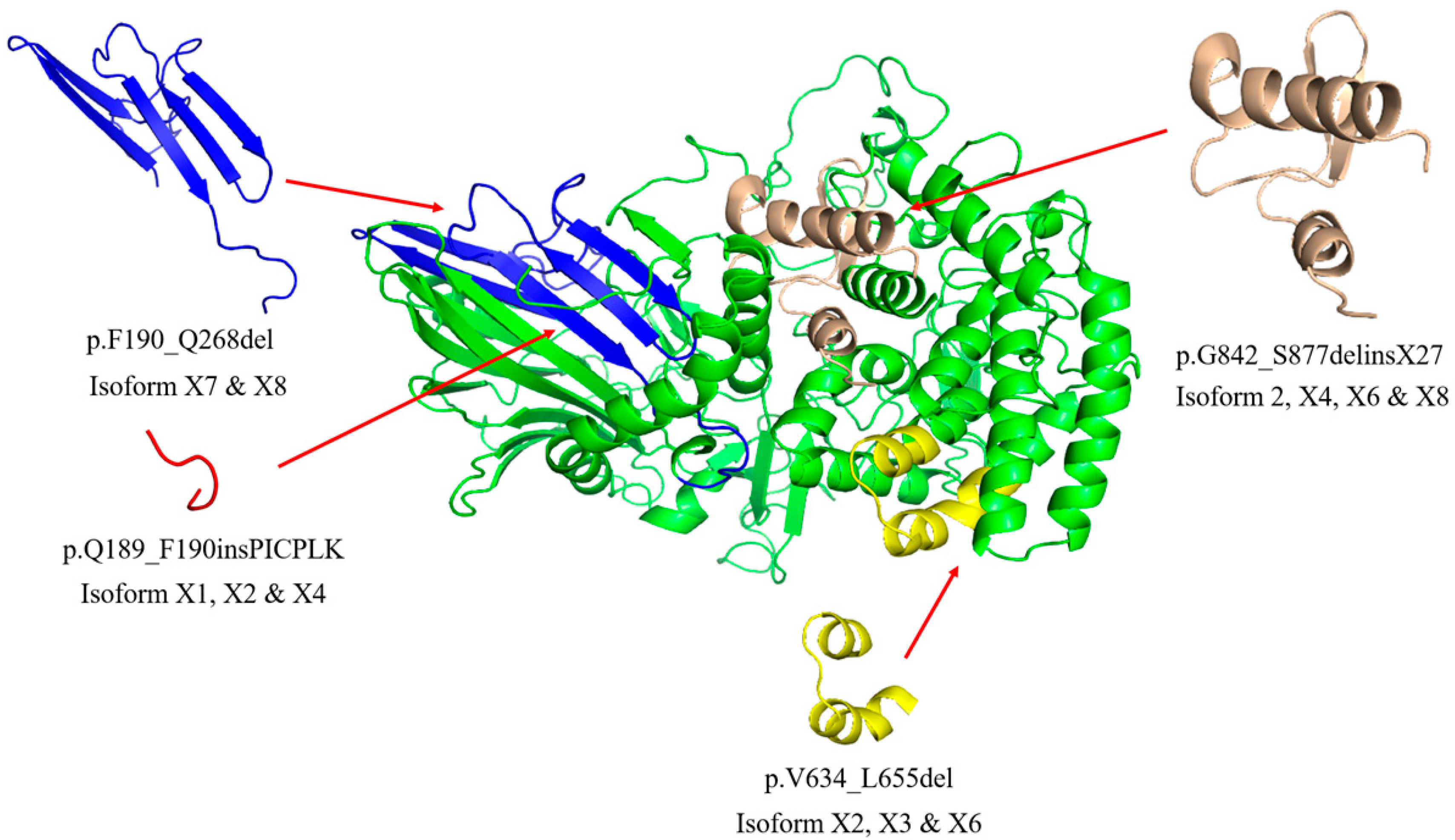
2.1. Sequence and Structure Analysis of GBA2 Isoform
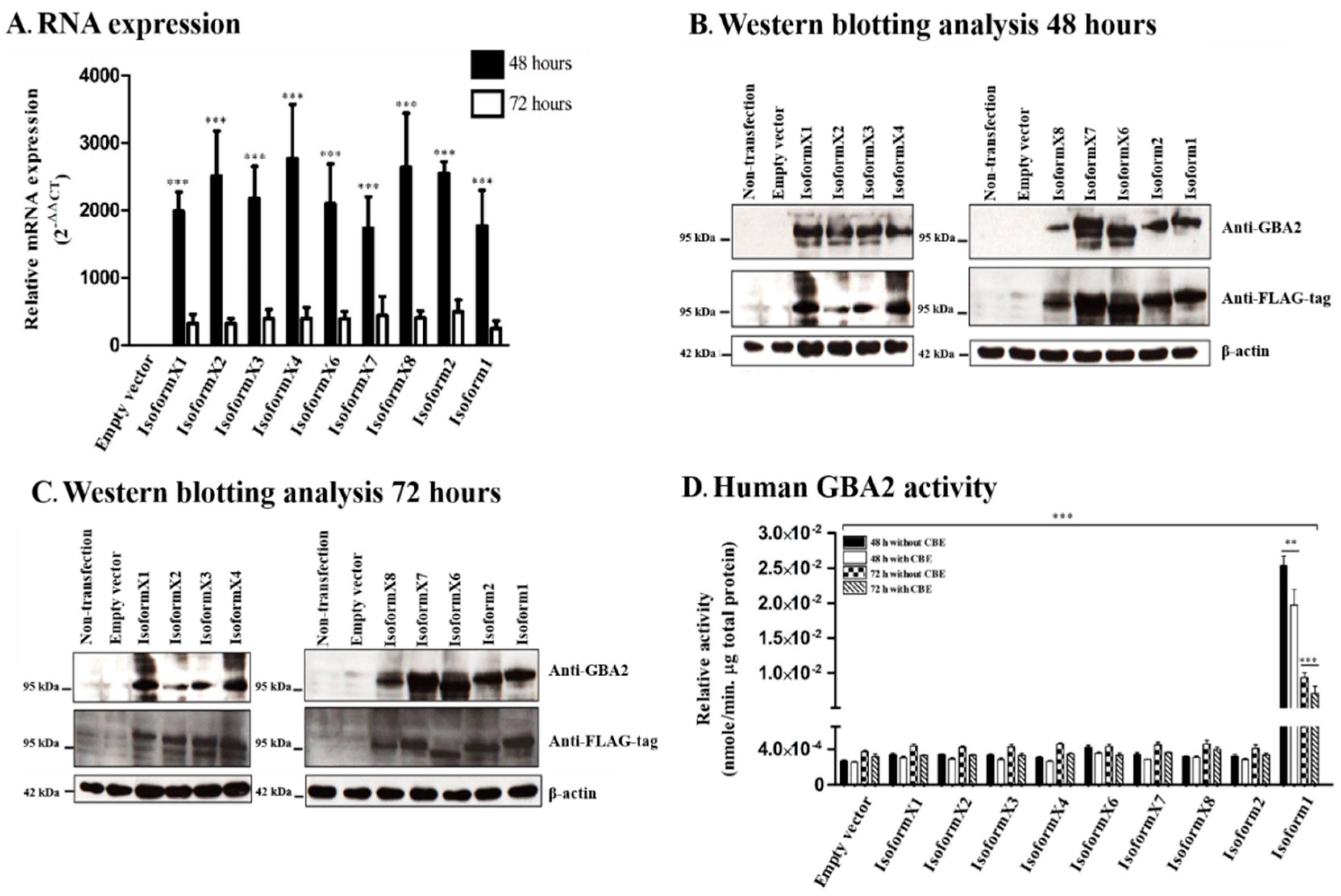
2.2. GBA2 Isoform Expression and Activity of Human GBA2 Isoforms in Transfected COS-7 Cells
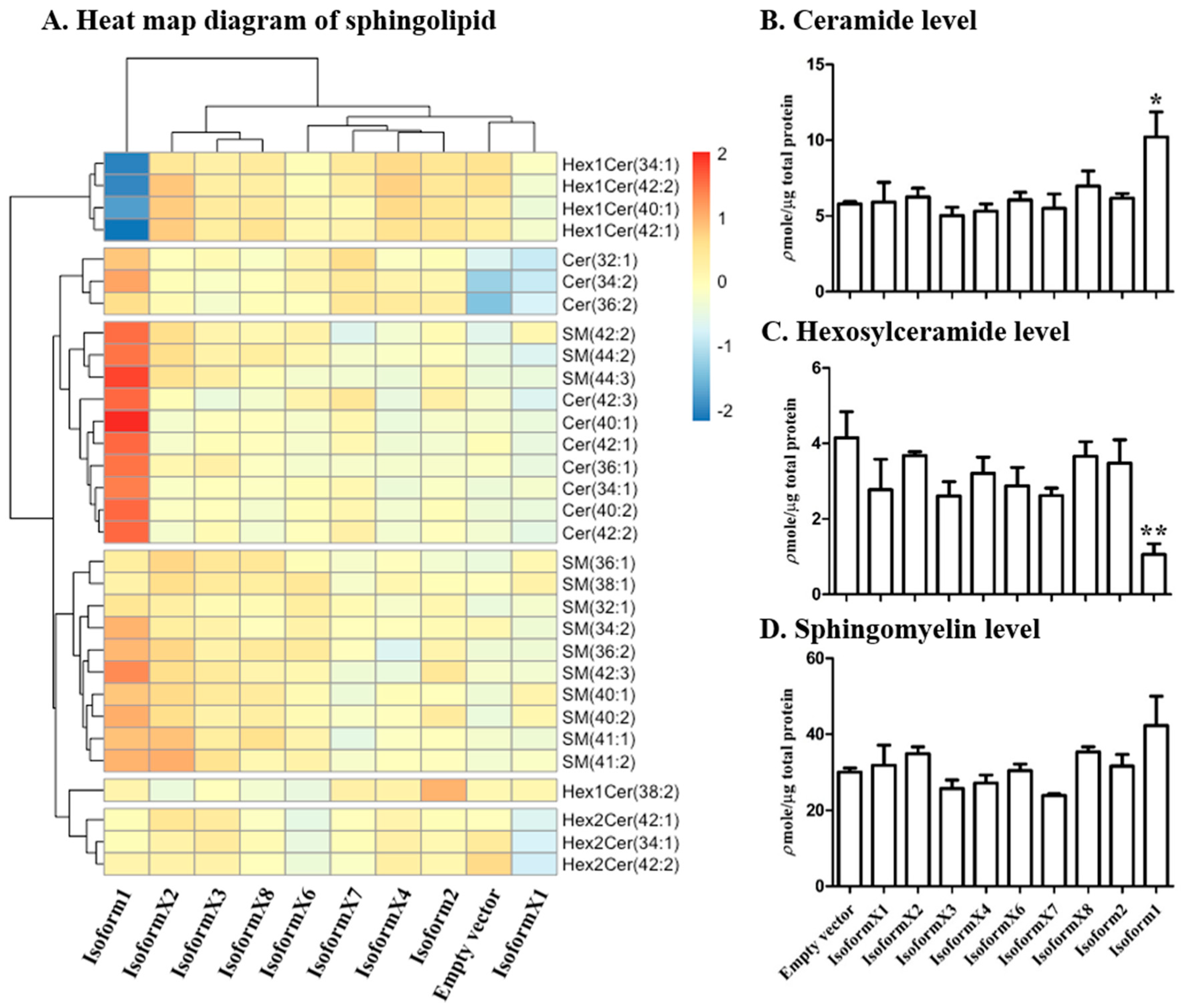
2.3. Analysis of Sphingolipid Levels in COS-7 Cells Overexpressing GBA2 Isoforms
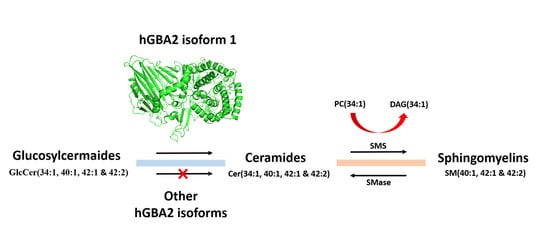
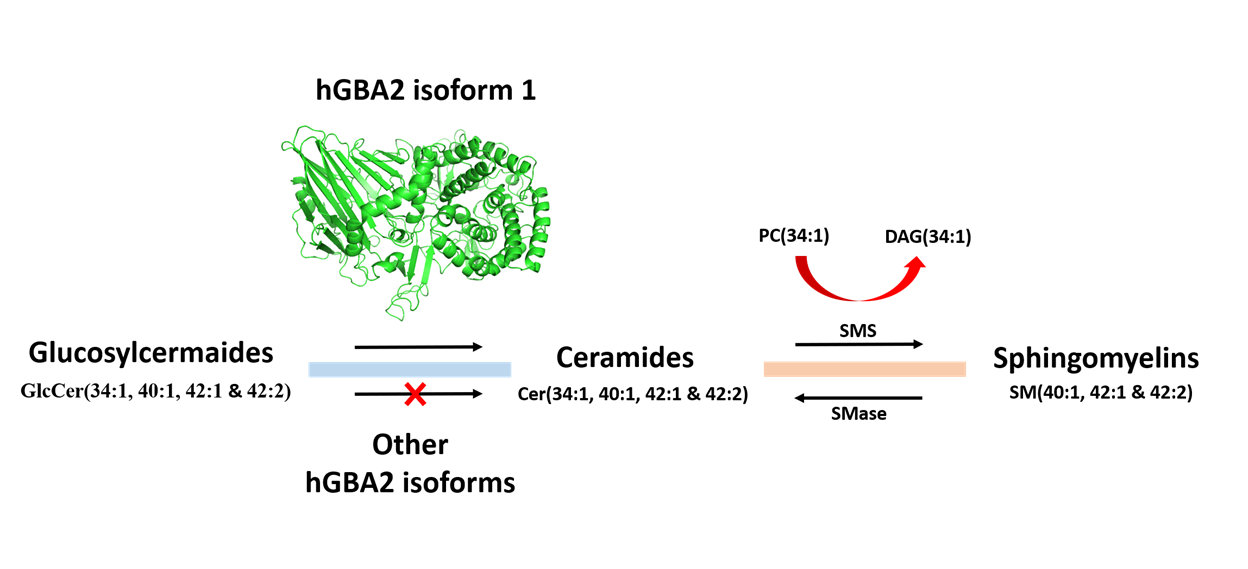
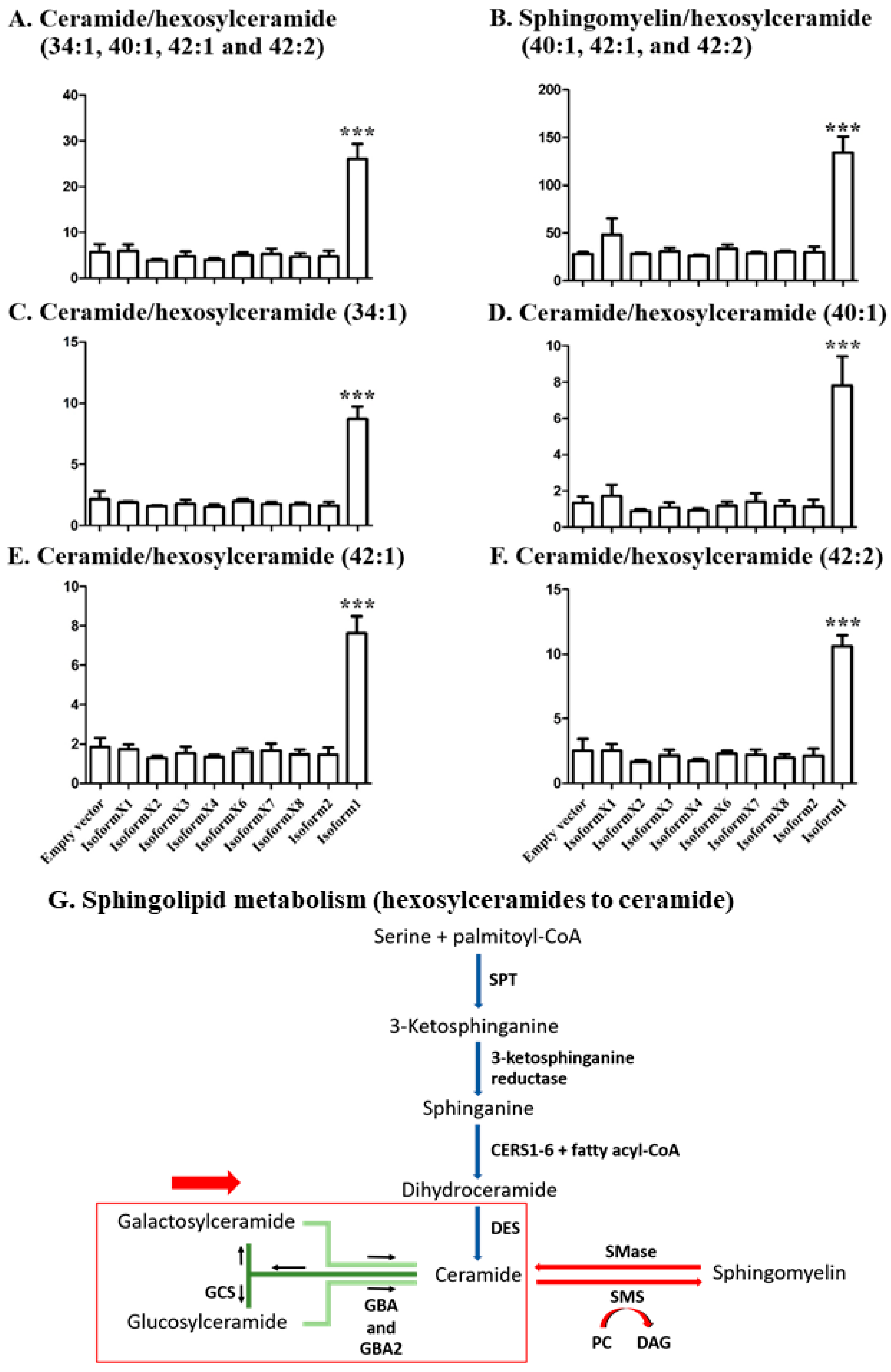
2.4. Analysis of Sphingolipid Ratios Related to the Direction of Sphingolipid Metabolic Flow
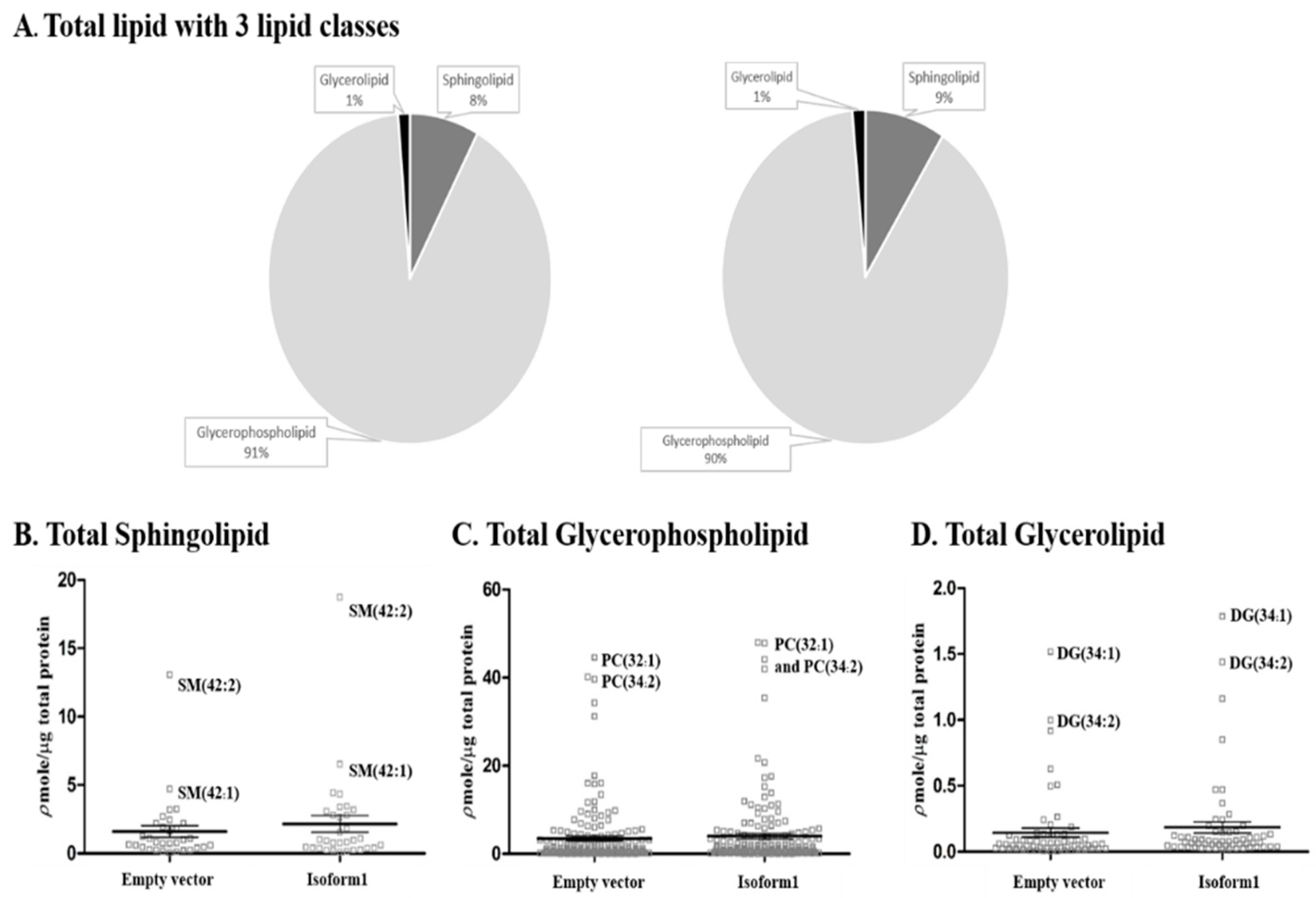
2.5. Analysis of Total Lipid Composition
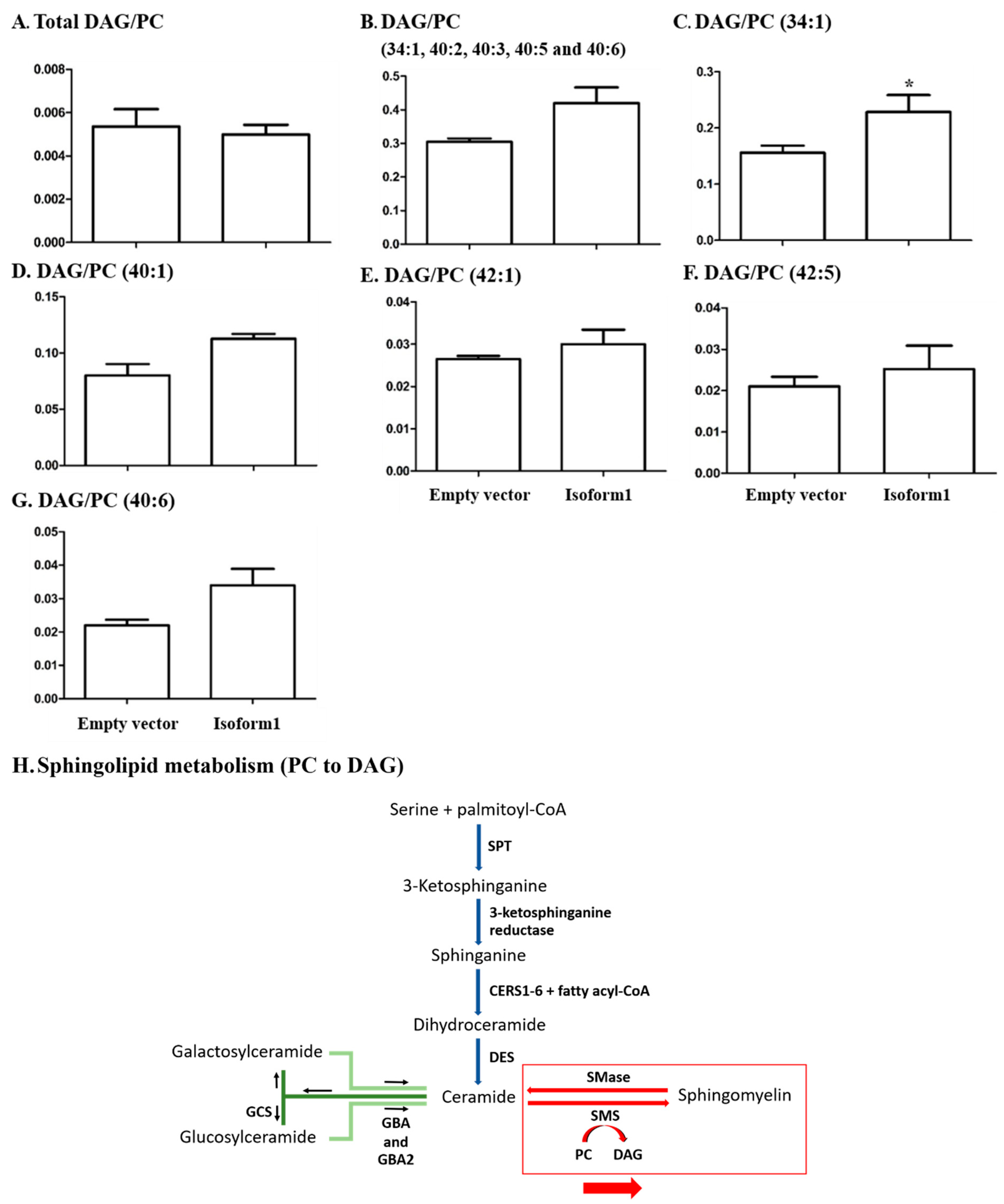
2.6. Analysis of Glycerophospholipids/Glycerolipids Involved in Sphingolipid Metabolism
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Cell Culture and Transfection
4.3. RNA Extraction and Quantitative RT-PCR
4.4. Protein Collection and Western Blotting Analysis
4.5. Measurement of GBA2 Enzyme Activity on MUG
4.6. Lipid Extraction and Lipid Measurement
4.7. Sequence and Structure Analysis
4.8. Data and Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Sakamoto, W.; Coant, N.; Canals, D.; Obeid, L.M.; Hannun, Y.A. Functions of neutral ceramidase in the Golgi apparatus. J. Lipid Res. 2018, 59, 2116–2125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, H.; Summers, S.A.; Birnbaum, M.J.; Pittman, R.N. Inhibition of Akt kinase by cell-permeable ceramide and its implications for ceramide-induced apoptosis. J. Biol. Chem. 1998, 273, 16568–16575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Radin, N.S. Designing anticancer drugs via the achilles heel: Ceramide, allylic ketones, and mitochondria. Bioorg. Med.Chem. 2003, 11, 2123–2142. [Google Scholar] [CrossRef]
- Summers, S.A. Ceramides in insulin resistance and lipotoxicity. Prog. Lipid Res. 2006, 45, 42–72. [Google Scholar] [CrossRef]
- Zeidan, Y.H.; Hannun, Y.A. Translational aspects of sphingolipid metabolism. Trends Mol. Med. 2007, 13, 327–336. [Google Scholar] [CrossRef]
- Hannun, Y.A.; Obeid, L.M. Principles of bioactive lipid signaling: Lessons from sphingolipids. Nat. Rev. Mol. Cell Biol. 2008, 9, 139–150. [Google Scholar] [CrossRef]
- Abdul-Hammed, M.; Breiden, B.; Schwarzmann, G.; Sandhoff, K. Lipids regulate the hydrolysis of membrane bound glucosylceramide by lysosomal β-glucocerebrosidase. J. Lipid Res. 2017, 58, 563–577. [Google Scholar] [CrossRef] [Green Version]
- Bielawski, J.; Szulc, Z.M.; Hannun, Y.A.; Bielawska, A. Simultaneous quantitative analysis of bioactive sphingolipids by high-performance liquid chromatography-tandem mass spectrometry. Methods 2006, 39, 82–91. [Google Scholar] [CrossRef]
- Tracey, T.J.; Steyn, F.J.; Wolvetang, E.J.; Ngo, S.T. Neuronal lipid metabolism: Multiple pathways driving functional outcomes in health and disease. Front. Mol. Neurosci. 2018, 11, 10. [Google Scholar] [CrossRef] [Green Version]
- D’Angelo, G.; Polishchuk, E.; Di Tullio, G.; Santoro, M.; Di Campli, A.; Godi, A.; van der Spoel, A.C. Glycosphingolipid synthesis requires FAPP2 transfer of glucosylceramide. Nature 2007, 449, 62–67. [Google Scholar] [CrossRef]
- Xu, Y.; Sun, Y.; Ran, H.; Quinn, B.; Witte, D.; Grabowski, G.A. Accumulation and distribution of α-synuclein and ubiquitin in the CNS of Gaucher disease mouse models. Mol. Genet. Metab. 2011, 102, 436–447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarmientos, F.; Schwarzmann, G.; Sandhoff, K. Specificity of human glucosylceramide β-glucosidase towards synthetic glucosylsphingolipids inserted into liposomes: Kinetic studies in a detergent-free assay system. Eur. J. Biochem. 1986, 160, 527–535. [Google Scholar] [CrossRef] [PubMed]
- Wilkening, G.; Linke, T.; Sandhoff, K. Lysosomal degradation on vesicular membrane surfaces Enhanced glucosylceramidase degradation by lysosomal anionic lipids and activators. J. Biol. Chem. 1998, 273, 30271–30278. [Google Scholar] [CrossRef] [Green Version]
- Akiyama, H.; Kobayashi, S.; Hirabayashi, Y.; Murakami-Murofushi, K. Cholesterol glucosylation is catalyzed by transglucosylation reaction of β-glucosidase 1. Biochem. Biophys. Res. Comm. 2013, 441, 838–843. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, E.; Schüle, R.; Smets, K.; Rastetter, A.; Boukhris, A.; Loureiro, J.L.; Gonzalez, M.A.; Mundwiller, E.; Deconinck, T.; Wessner, M.; et al. Loss of function of glucocerebrosidase GBA2 is responsible for motor neuron defects in hereditary spastic paraplegia. Am. J. Hum. Genet. 2013, 92, 238–244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hammer, M.B.; Eleuch-Fayache, G.; Schottlaender, L.V.; Nehdi, H.; Gibbs, J.R.; Arepalli, S.K.; Chong, S.B.; Hernandez, D.G.; Sailer, A.; Liu, G.; et al. Mutations in GBA2 cause autosomal-recessive cerebellar ataxia with spasticity. Am. J. Hum. Genet. 2013, 92, 245–251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Weely, S.; Brandsma, M.; Strijland, A.; Tager, J.M.; Aerts, J.M. Demonstration of the existence of a second, non-lysosomal glucocerebrosidase that is not deficient in Gaucher disease. Biochim. Biophys. Acta 1993, 1181, 55–62. [Google Scholar] [CrossRef]
- Matern, H.; Boermans, H.; Lottspeich, F.; Matern, S. Molecular cloning and expression of human bile acid β-glucosidase. Biol. Chem. 2001, 276, 37929–37933. [Google Scholar]
- Matern, H.; Heinemann, H.; Legler, G.; Matern, S. Purification and characterization of a microsomal bile acid β-glucosidase from human liver. Biol. Chem. 1997, 272, 11261–11267. [Google Scholar] [CrossRef] [Green Version]
- Yildiz, Y.; Matern, H.; Thompson, B.; Allegood, J.C.; Warren, R.L.; Ramirez, D.M.; Hammer, R.E.; Hamra, F.K.; Matern, S.; Russell, D.W. Mutation of β-glucosidase 2 causes glycolipid storage disease and impaired male fertility. J. Clin. Investig. 2006, 116, 2985–2994. [Google Scholar] [CrossRef] [Green Version]
- Burke, D.G.; Rahim, A.A.; Waddington, S.N.; Karlsson, S.; Enquist, I.; Bhatia, K.; Mehta, A.; Vellodi, A.; Heales, S. Increased glucocerebrosidase (GBA) 2 activity in GBA1 deficient mice brains and in Gaucher leucocytes. J. Inheritig. Metab. Dis. 2013, 36, 869–872. [Google Scholar] [CrossRef]
- Boot, R.G.; Verhoek, M.; Donker-Koopman, W.; Strijland, A.; Van Marle, J.; Overkleeft, H.S.; Wennekes, T.; Aerts, J.M.F.G. Identification of the non-lysosomal glucosylceramidase as β-glucosidase 2. J. Biol. Chem. 2007, 282, 1305–1312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marques, A.R.M.; Mirzaian, M.; Akiyama, H.; Wisse, P.; Ferraz, M.J.; Gaspar, P.; Ghauharali-van der Vlugt, K.; Meijer, R.; Giraldo, P.; Alfonso, P.; et al. Glucosylated cholesterol in mammalian cells and tissues: Formation and degradation by multiple cellular β-glucosidases. J. Lipid Res. 2016, 57, 451–463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akiyama, H.; Ide, M.; Nagatsuka, Y.; Sayano, T.; Nakanishi, E.; Uemura, N.; Yuyama, K.; Yamaguchi, Y.; Kamiguchi, H.; Takahashi, R.; et al. Glucocerebrosidases catalyze a transgalactosylation reaction that yields a newly-identified brain sterol metabolite, galactosylated cholesterol. Biol. Chem. 2020, 295, 5257–5277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Körschen, H.G.; Yildiz, Y.; Raju, D.N.; Schonauer, S.; Bönigk, W.; Jansen, V.; Kremmer, E.; Kaupp, U.B.; Wachten, D. The non-lysosomal β-glucosidase GBA2 is a non-integral membrane-associated protein at the endoplasmic reticulum (ER) and Golgi. J. Biol. Chem. 2013, 288, 3381–3393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mistry, P.K.; Liu, J.; Sun, L.; Chuang, W.L.; Yuen, T.; Yang, R.; Lu, P.; Zhang, K.; Li, J.; Keutzer, J.; et al. Glucocerebrosidase 2 gene deletion rescues type 1 Gaucher disease. Proc. Natl. Acad. Sci. USA 2014, 111, 4934–4939. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marques, A.R.; Aten, J.; Ottenhoff, R.; van Roomen, C.P.; Moro, D.H.; Claessen, N.; Veloz, M.F.V.; Zhou, K.; Lin, Z.; Mirzaian, M. Reducing GBA2 activity ameliorates neuropathology in Niemann-Pick type C mice. PLoS ONE 2015, 10, e0135889. [Google Scholar] [CrossRef] [Green Version]
- La Cognata, V.; D’Agata, V.; Cavalcanti, F.; Cavallaro, S. Splicing: Is there an alternative contribution to Parkinson’s disease? Neurogenetics 2015, 16, 245–263. [Google Scholar] [CrossRef] [Green Version]
- Ward, A.J.; Cooper, T.A. The pathobiology of splicing. J. Pathol. 2010, 220, 152–163. [Google Scholar] [CrossRef]
- Charoenwattanasatien, R.; Pengthaisong, S.; Breen, I.; Mutoh, R.; Sansenya, S.; Hua, Y.; Tankrathok, A.; Wu, L.; Songsiriritthigul, C.; Tanaka, H.; et al. Bacterial β-glucosidase reveals the structural and functional basis of genetic defects in human glucocerebrosidase 2 (GBA2). ACS Chem. Biol. 2016, 11, 1891–1900. [Google Scholar] [CrossRef]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; de Beer, T.A.P.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL homology modeling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sultana, S.; Reichbauer, J.; Schüle, R.; Mochel, F.; Synofzik, M.; van der Spoel, A.C. Lack of enzyme activity in GBA2 mutants associated with hereditary spastic paraplegia/cerebellar ataxia (SPG46). Biochem. Biophys. Biochem. Biophys. Res. Commun. 2015, 465, 35–40. [Google Scholar] [CrossRef] [PubMed]
- Ogura, K.; Tai, T. Molecular cloning and characterization of galactosylceramide expression factor-1 (GEF-1). Neurochem. Res. 2002, 27, 779–784. [Google Scholar] [CrossRef]
- Ridley, C.M.; Thur, K.E.; Shanahan, J.; Thillaiappan, N.B.; Shen, A.; Uhl, K.; Walden, C.M.; Rahim, A.A.; Waddington, S.N.; Platt, F.M.; et al. β-Glucosidase 2 (GBA2) activity and imino sugar pharmacology. J. Biol. Chem. 2013, 288, 26052–26066. [Google Scholar] [CrossRef] [Green Version]
- Schonauer, S.; Körschen, H.G.; Penno, A.; Rennhack, A.; Breiden, B.; Sandhoff, K.; Gutbrod, K.; Dörmann, P.; Raju, D.N.; Haberkant, P.; et al. Identification of a feedback loop involving β-glucosidase 2 and its product sphingosine sheds light on the molecular mechanisms in Gaucher disease. J. Biol. Chem. 2017, 292, 6177–6189. [Google Scholar] [CrossRef] [Green Version]
- Sorli, S.C.; Colié, S.; Albinet, V.; Dubrac, A.; Touriol, C.; Guilbaud, N.; Bedia, C.; Fabriàs, G.; Casas, J.; Ségui, B.; et al. The nonlysosomal β-glucosidase GBA2 promotes endoplasmic reticulum stress and impairs tumorigenicity of human melanoma cells. FASEB J. 2013, 27, 489–498. [Google Scholar] [CrossRef]
- Haugarvoll, K.; Johansson, S.; Rodriguez, C.E.; Boman, H.; Haukanes, B.I.; Brul, O.; Roque, F.; Jonassen, I.; Blomqvist, M.; Telstad, W.; et al. GBA2 mutations cause a Marinesco-Sjogren-like syndrome. PLoS ONE 2017, 12, e0169309. [Google Scholar] [CrossRef] [Green Version]
- Sultana, S.; Stewart, J.; van der Spoel, A.C. Truncated mutants of beta-glucosidase 2 (GBA2) are localized in the mitochondrial matrix and cause mitochondrial fragmentation. PLoS ONE 2020, 15, e0233856. [Google Scholar] [CrossRef]
- Votsi, C.; Zamba-Papanicolaou, E.; Middleton, L.T.; Pantzaris, M.; Christodoulou, K. A Novel GBA2 Gene Missense Mutation in Spastic Ataxia. Ann. Hum. Genet. 2014, 78, 13–22. [Google Scholar] [CrossRef]
- Raju, D.; Schonauer, S.; Hamzeh, H.; Flynn, K.C.; Bradke, F.; vom Dorp, K.; Dörmann, P.; Yildiz, Y.; Trötschel, C.; Poetsch, A.; et al. Accumulation of glucosylceramide in the absence of the beta-glucosidase GBA2 alters cytoskeletal dynamics. PLoS Genet. 2015, 11, e1005063. [Google Scholar] [CrossRef] [Green Version]
- Nastase, A.; Lupo, A.; Laszlo, V.; Damotte, D.; Dima, S.; Canny, E.; Alifano, M.; Popescu, I.; Klepetko, W.; Grigoroiu, M.; et al. Platinum Drug Sensitivity Polymorphisms in Stage III Non-small Cell Lung Cancer with Invasion of Mediastinal Lymph Nodes. Cancer Genom. Proteom. 2020, 17, 587–595. [Google Scholar] [CrossRef] [PubMed]
- Malekkou, A.; Sevastou, I.; Mavrikiou, G.; Georgiou, T.; Vilageliu, L.; Moraitou, M.; Michelakakis, H.; Prokopiou, C.; Drousiotou, A. A novel mutation deep within intron 7 of the GBA gene causes Gaucher disease. Mol. Genet. Genom. Med. 2020, 8, 1090. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kakouri, A.C.; Votsi, C.; Tomazou, M.; Minadakis, G.; Karatzas, E.; Christodoulou, K.; Spyrou, G.M. Analyzing gene expression profiles from ataxia and spasticity phenotypes to reveal spastic ataxia related pathways. Int. J. Mol. Sci. 2020, 21, 6722. [Google Scholar] [CrossRef] [PubMed]
- Malekkou, A.; Samarani, M.; Drousiotou, A.; Votsi, C.; Sonnino, S.; Pantzaris, M.; Chiricozzi, E.; Zamba-Papanicolaou, E.; Aureli, M.; Loberto, N.; et al. Biochemical Characterization of the GBA2 c.1780G>C Missense Mutation in Lymphoblastoid Cells from Patients with Spastic Ataxia. Int. J. Mol. Sci. 2018, 19, 3099. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bouscary, A.; Quessada, C.; Mosbach, A.; Callizot, N.; Spedding, M.; Loeffler, J.P.; Henriques, A. Ambroxol hydrochloride improves motor functions and extends survival in a mouse model of familial amyotrophic lateral sclerosis. Front. Pharmacol. 2019, 10, 883. [Google Scholar] [CrossRef] [Green Version]
- Lydic, T.A.; Busik, J.V.; Reid, G.E. A monophasic extraction strategy for the simultaneous lipidome analysis of polar and nonpolar retina lipids. J. Lipid Res. 2014, 55, 1797–1809. [Google Scholar] [CrossRef] [Green Version]
- Lydic, T.A.; Townsend, S.; Adda, C.G.; Collins, C.; Mathivanan, S.; Reid, G.E. Rapid and comprehensive ‘shotgun’lipidome profiling of colorectal cancer cell derived exosomes. Methods 2015, 87, 83–95. [Google Scholar] [CrossRef] [Green Version]
- Couttas, T.A.; Rustam, Y.H.; Song, H.; Qi, Y.; Teo, J.D.; Chen, J.; Reid, G.E.; Don, A.S. A novel function of sphingosine kinase 2 in the metabolism of sphinga-4, 14-diene lipids. Metabolites 2020, 10, 236. [Google Scholar] [CrossRef]
- Lee, R.G.; Gao, J.; Siira, S.J.; Shearwood, A.M.; Ermer, J.A.; Hofferek, V.; Mathews, J.C.; Zheng, M.; Reid, G.E.; Rackham, O.; et al. Cardiolipin is required for membrane docking of mitochondrial ribosomes and protein synthesis. J. Cell Sci. 2020, 133, 14. [Google Scholar] [CrossRef]
- Shapiro, S.S.; Wilk, M.B. An analysis of variance test for normality (complete samples). Biometrika 1965, 52, 591–611. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jatooratthawichot, P.; Talabnin, C.; Ngiwsara, L.; Rustam, Y.H.; Svasti, J.; Reid, G.E.; Ketudat Cairns, J.R. Effect of Expression of Human Glucosylceramidase 2 Isoforms on Lipid Profiles in COS-7 Cells. Metabolites 2020, 10, 488. https://doi.org/10.3390/metabo10120488
Jatooratthawichot P, Talabnin C, Ngiwsara L, Rustam YH, Svasti J, Reid GE, Ketudat Cairns JR. Effect of Expression of Human Glucosylceramidase 2 Isoforms on Lipid Profiles in COS-7 Cells. Metabolites. 2020; 10(12):488. https://doi.org/10.3390/metabo10120488
Chicago/Turabian StyleJatooratthawichot, Peeranat, Chutima Talabnin, Lukana Ngiwsara, Yepy Hardi Rustam, Jisnuson Svasti, Gavin E. Reid, and James R. Ketudat Cairns. 2020. "Effect of Expression of Human Glucosylceramidase 2 Isoforms on Lipid Profiles in COS-7 Cells" Metabolites 10, no. 12: 488. https://doi.org/10.3390/metabo10120488