
The Impact of Simvastatin on Lipidomic Markers of Cardiovascular Risk in Human Liver Cells Is Secondary to the Modulation of Intracellular Cholesterol

 ,
, 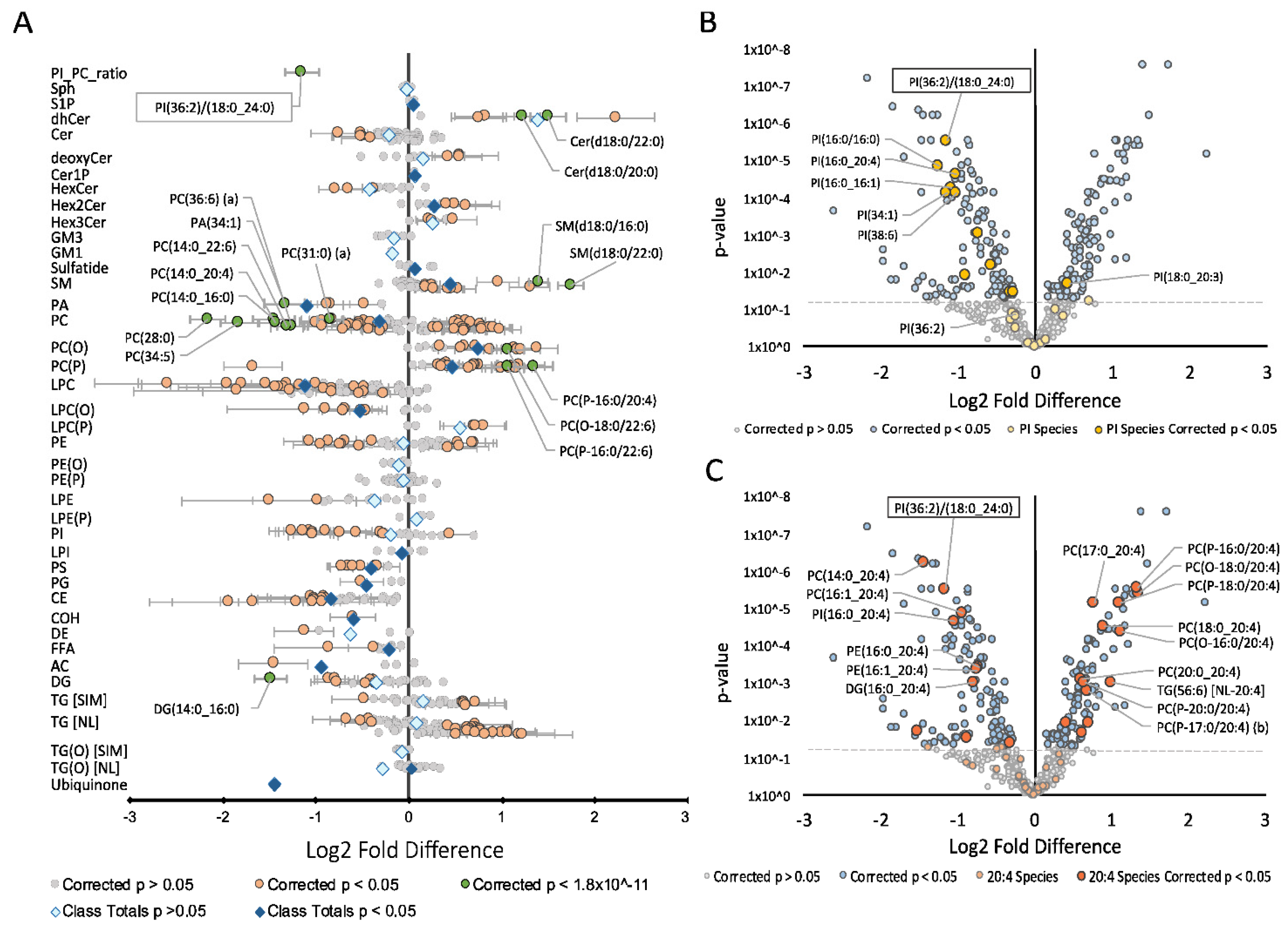{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Effect of Simvastatin on the Lipidome
2.2. Effect of Alternative Cholesterol Modulating Treatment on the Lipidome
2.3. Gene Expression
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Treatment of Cells with Cholesterol Modulators
4.3. Lipid Extraction
4.4. Liquid Chromatography Electrospray Ionisation Tandem Mass Spectrometry
4.5. Quantification of Lipid Species
4.6. RNA Isolation and Reverse Transcription-Polymerase Chain Reaction (RT-PCR) and Quantitative RT-PCR (qPCR)
4.7. Data Presentation and Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Liao, J.K.; Laufs, U. Pleiotropic Effects of Statins. Annu. Rev. Pharmacol. Toxicol. 2004, 45, 89–118. [Google Scholar] [CrossRef] [Green Version]
- Ballantyne, C.M.; Raichlen, J.S.; Nicholls, S.J.; Erbel, R.; Tardif, J.C.; Brener, S.J.; Cain, V.A.; Nissen, S.E. Effect of Rosuvastatin Therapy on Coronary Artery Stenoses Assessed by Quantitative Coronary Angiography. Circulation 2008, 117. [Google Scholar] [CrossRef] [Green Version]
- Rossouw, J.E. Lipid-lowering interventions in angiographic trials. Am. J. Cardiol. 1995, 76, 86–92. [Google Scholar] [CrossRef]
- Weng, T.C.; Kao Yang, Y.H.; Lin, S.J.; Tai, S.H. A systematic review and meta-analysis on the therapeutic equivalence of statins. J. Clin. Pharm. Ther. 2010, 35, 139–151. [Google Scholar] [CrossRef] [PubMed]
- Scandinavian Simvastatin Survival Study Group. Randomised trial of cholesterol lowering in 4444 patients with coronary heart disease: The Scandinavian Simvastatin Survival Study (4S). Lancet 1994, 344, 1383–1389. [Google Scholar] [CrossRef]
- Sacks, F.M.; Pfeffer, M.A.; Moye, L.A.; Rouleau, J.L.; Rutherford, J.D.; Cole, T.G.; Brown, L.; Warnica, J.W.; Arnold, J.M.O.; Wun, C.C.; et al. The effect of pravastatin on coronary events after myocardial infarction in patients with average cholesterol levels. Cholesterol and Recurrent Events Trial investigators. N. Engl. J. Med. 1996, 335, 1001–1009. [Google Scholar] [CrossRef]
- Long-Term Intervention with Pravastatin in Ischaemic Disease (LIPID) Study Group. Prevention of cardiovascular events and death with pravastatin in patients with coronary heart disease and a broad range of initial cholesterol levels. N. Engl. J. Med. 1998, 339, 1349–1357. [Google Scholar] [CrossRef] [Green Version]
- Shepherd, J.; Cobbe, S.M.; Ford, I.; Isles, C.G.; Lorimer, A.R.; Macfarlane, P.W.; McKillop, J.H.; Packard, C.J. Prevention of Coronary Heart Disease with Pravastatin in Men with Hypercholesterolemia. West of Scotland coronary prevention study group. N. Engl. J. Med. 1995, 333, 1301–1307. [Google Scholar] [CrossRef]
- Kureishi, Y.; Luo, Z.; Shiojima, I.; Bialik, A.; Fulton, D.; Lefer, D.J.; Sessa, W.C.; Walsh, K. The HMG-CoA reductase inhibitor simvastatin activates the protein kinase Akt and promotes angiogenesis in normocholesterolemic animals. Nat. Med. 2001, 7, 129. [Google Scholar] [CrossRef]
- Rikitake, Y.; Kawashima, S.; Takeshita, S.; Yamashita, T.; Azumi, H.; Yasuhara, M.; Nishi, H.; Inoue, N.; Yokoyama, M. Anti-oxidative properties of fluvastatin, an HMG-CoA reductase inhibitor, contribute to prevention of atherosclerosis in cholesterol-fed rabbits. Atherosclerosis 2001, 154, 87–96. [Google Scholar] [CrossRef]
- Martínez-González, J.; Raposo, B.; Rodríguez, C.; Badimon, L. 3-hydroxy-3-methylglutaryl coenzyme a reductase inhibition prevents endothelial NO synthase downregulation by atherogenic levels of native LDLs: Balance between transcriptional and posttranscriptional regulation. Arterioscler. Thromb. Vasc. Biol. 2001, 21, 804–809. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jayawardana, K.S.; Mundra, P.A.; Giles, C.; Barlow, C.K.; Nestel, P.J.; Barnes, E.H.; Kirby, A.; Thompson, P.; Sullivan, D.R.; Alshehry, Z.H.; et al. Changes in plasma lipids predict pravastatin efficacy in secondary prevention. JCI Insight 2019, 4, e128438. [Google Scholar] [CrossRef]
- Kapur, N.K.; Musunuru, K. Clinical efficacy and safety of statins in managing cardiovascular risk. Vasc. Health Risk Manag. 2008, 4, 341–353. [Google Scholar] [CrossRef] [Green Version]
- Lydic, A.T.; Goo, Y.H. Lipidomics unveils the complexity of the lipidome in metabolic diseases. Clin. Transl. Med. 2018, 7. [Google Scholar] [CrossRef]
- Gaposchkin, D.P.; Zoeller, R.A. Plasmalogen status influences docosahexaenoic acid levels in a macrophage cell line. Insights using ether lipid-deficient variants. J. Lipid Res. 1999, 40, 495–503. [Google Scholar] [CrossRef]
- Ford, D.A.; Gross, R.W. Plasmenylethanolamine is the major storage depot for arachidonic acid in rabbit vascular smooth muscle and is rapidly hydrolyzed after angiotensin II stimulation. Proc. Natl. Acad. Sci. USA 1989, 86, 3479–3483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blank, M.L.; Smith, L.Z.; Lee, Y.J.; Snyder, F. Effects of eicosapentaenoic and docosahexaenoic acid supplements on phospholipid composition and plasmalogen biosynthesis in P388D1 cells. Arch. Biochem. Biophys. 1989, 269, 603–611. [Google Scholar] [CrossRef]
- Das, U.N. Essential fatty acids as possible mediators of the actions of statins. Prostaglandins Leukot. Essent. Fat. Acids 2001, 65, 37–40. [Google Scholar] [CrossRef]
- Das, U.N. Beneficial actions of statins in the reduction of atrial fibrillation and stabilization and regression of coronary plaques: But how and why? Circ. J. 2011, 75, 224–225. [Google Scholar] [CrossRef] [Green Version]
- Hrboticky, N.; Tang, L.; Zimmer, B.; Lux, I.; Weber, P.C. Lovastatin increases arachidonic acid levels and stimulates thromboxane synthesis in human liver and monocytic cell lines. J. Clin. Investig. 1994, 93, 195–203. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.H.Y.; Lemaitre, R.N.; Imamura, F.; King, I.B.; Song, X.; Spiegelman, D.; Siscovick, D.S.; Mozaffarian, D. Fatty acids in the de novo lipogenesis pathway and risk of coronary heart disease. The Cardiovascular Health Study. Am. J. Clin. Nutr. 2011, 94, 431–438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borradaile, N.M.; Han, X.; Harp, J.D.; Gale, S.E.; Ory, D.S.; Schaffer, J.E. Disruption of endoplasmic reticulum structure and integrity in lipotoxic cell death. J. Lipid Res. 2006, 47, 2726–2737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sainsbury, C.A.; Sattar, N.; Connell, J.M.; Hillier, C.; Petrie, J.R. Non-esterified fatty acids impair endothelium-dependent vasodilation in rat mesenteric resistance vessels. Clin. Sci. 2004, 107, 625–629. [Google Scholar] [CrossRef] [Green Version]
- Schwartz, E.A.; Zhang, W.Y.; Karnik, S.K.; Borwege, S.; Anand, V.R.; Laine, P.S.; Su, Y.; Reaven, P.D. Nutrient modification of the innate immune response: A novel mechanism by which saturated fatty acids greatly amplify monocyte inflammation. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 802–808. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- González-Périz, A.; Horrill, O.R.; Ferré, N.; Gronert, K.; Dong, B.; Morán-Salvador, E.; Titos, E.; Martínez-Clemente, M.; López-Parra, M.; Arroyo, V.; et al. Obesity-induced insulin resistance and hepatic steatosis are alleviated by omega-3 fatty acids: A role for resolvins and protectins. FASEB J. 2009, 23, 1946–1957. [Google Scholar] [CrossRef] [Green Version]
- Christian, A.E.; Haynes, M.P.; Phillips, M.C.; Rothblat, G.H. Use of cyclodextrins for manipulating cellular cholesterol content. J. Lipid Res. 1997, 38, 2264–2272. [Google Scholar] [CrossRef]
- Hua, X.; Sakai, J.; Ho, Y.K.; Goldstein, J.L.; Brown, M.S. Hairpin Orientation of Sterol Regulatory Element-binding Protein-2 in Cell Membranes as Determined by Protease Protection. J. Biol. Chem. 1995, 270, 29422–29427. [Google Scholar] [CrossRef] [Green Version]
- Feig, J.E.; Shang, Y.; Rotllan, N.; Vengrenyuk, Y.; Wu, C.; Shamir, R.; Torra, I.P.; Fernandez-Hernando, C.; Fisher, E.A.; Garabedian, M.J. Statins Promote the Regression of Atherosclerosis via Activation of the CCR7-Dependent Emigration Pathway in Macrophages. PLoS ONE 2011, 6, e28534. [Google Scholar] [CrossRef]
- Moon, A.Y.; Hammer, R.E.; Horton, J.D. Deletion of ELOVL5 leads to fatty liver through activation of SREBP-1c in mice. J. Lipid Res. 2009, 50, 412–423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- RiséL, P.; Ghezzi, S.; Carissimi, R.; Mastromauro, F.; Petroni, A.; Galli, C. Delta5 Desaturase mRNA levels are increased by simvastatin via SREBP-1 at early stages, not via PPARα, in THP-1 cell. Eur. J. Pharmacol. 2008, 571, 97–105. [Google Scholar] [CrossRef]
- Jang, J.E.; Park, H.S.; Yoo, H.J.; Baek, I.J.; Yoon, J.E.; Ko, M.S.; Kim, A.R.; Kim, H.S.; Park, H.S.; Lee, S.E.; et al. Protective role of endogenous plasmalogens against hepatic steatosis and steatohepatitis. Hepatology 2017, 66, 416–431. [Google Scholar] [CrossRef] [Green Version]
- Brown, R.E.; Jarvis, K.L.; Hyland, K.J. Protein measurement using bicinchoninic acid: Elimination of interfering substances. Anal. Biochem. 1989, 180, 136–139. [Google Scholar] [CrossRef]
- Weir, J.M.; Wong, G.; Barlow, C.K.; Greeve, M.A.; Kowalczyk, A.; Almasy, L.; Comuzzie, A.G.; Mahaney, M.C.; Jowett, J.B.; Shaw, J.; et al. Plasma lipid profiling in a large population-based cohort. J. Lipid Res. 2013, 54, 2898–2908. [Google Scholar] [CrossRef] [Green Version]
- Huynh, K.; Barlow, C.K.; Jayawardana, K.S.; Weir, J.M.; Mellett, N.A.; Cinel, M.; Magliano, D.J.; Shaw, J.E.; Drew, B.G.; Meikle, P.J. High-Throughput Plasma Lipidomics: Detailed Mapping of the Associations with Cardiometabolic Risk Factors. Cell Chem. Biol. 2019, 26, 71–84.e4. [Google Scholar] [CrossRef] [PubMed]
- Benjamini, Y.; Hochberg, Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. Ser. B (Methodol.) 1995, 57, 289–300. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schooneveldt, Y.L.; Giles, C.; Keating, M.F.; Mellett, N.A.; Jurrjens, A.W.; Paul, S.; Calkin, A.C.; Meikle, P.J. The Impact of Simvastatin on Lipidomic Markers of Cardiovascular Risk in Human Liver Cells Is Secondary to the Modulation of Intracellular Cholesterol. Metabolites 2021, 11, 340. https://doi.org/10.3390/metabo11060340
Schooneveldt YL, Giles C, Keating MF, Mellett NA, Jurrjens AW, Paul S, Calkin AC, Meikle PJ. The Impact of Simvastatin on Lipidomic Markers of Cardiovascular Risk in Human Liver Cells Is Secondary to the Modulation of Intracellular Cholesterol. Metabolites. 2021; 11(6):340. https://doi.org/10.3390/metabo11060340
Chicago/Turabian StyleSchooneveldt, Yvette L., Corey Giles, Michael F. Keating, Natalie A. Mellett, Aaron W. Jurrjens, Sudip Paul, Anna C. Calkin, and Peter J. Meikle. 2021. "The Impact of Simvastatin on Lipidomic Markers of Cardiovascular Risk in Human Liver Cells Is Secondary to the Modulation of Intracellular Cholesterol" Metabolites 11, no. 6: 340. https://doi.org/10.3390/metabo11060340