
Diagnostic and Prognostic Protein Biomarkers of β-Cell Function in Type 2 Diabetes and Their Modulation with Glucose Normalization


Abstract
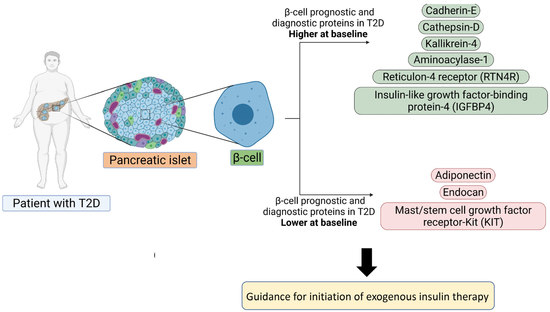
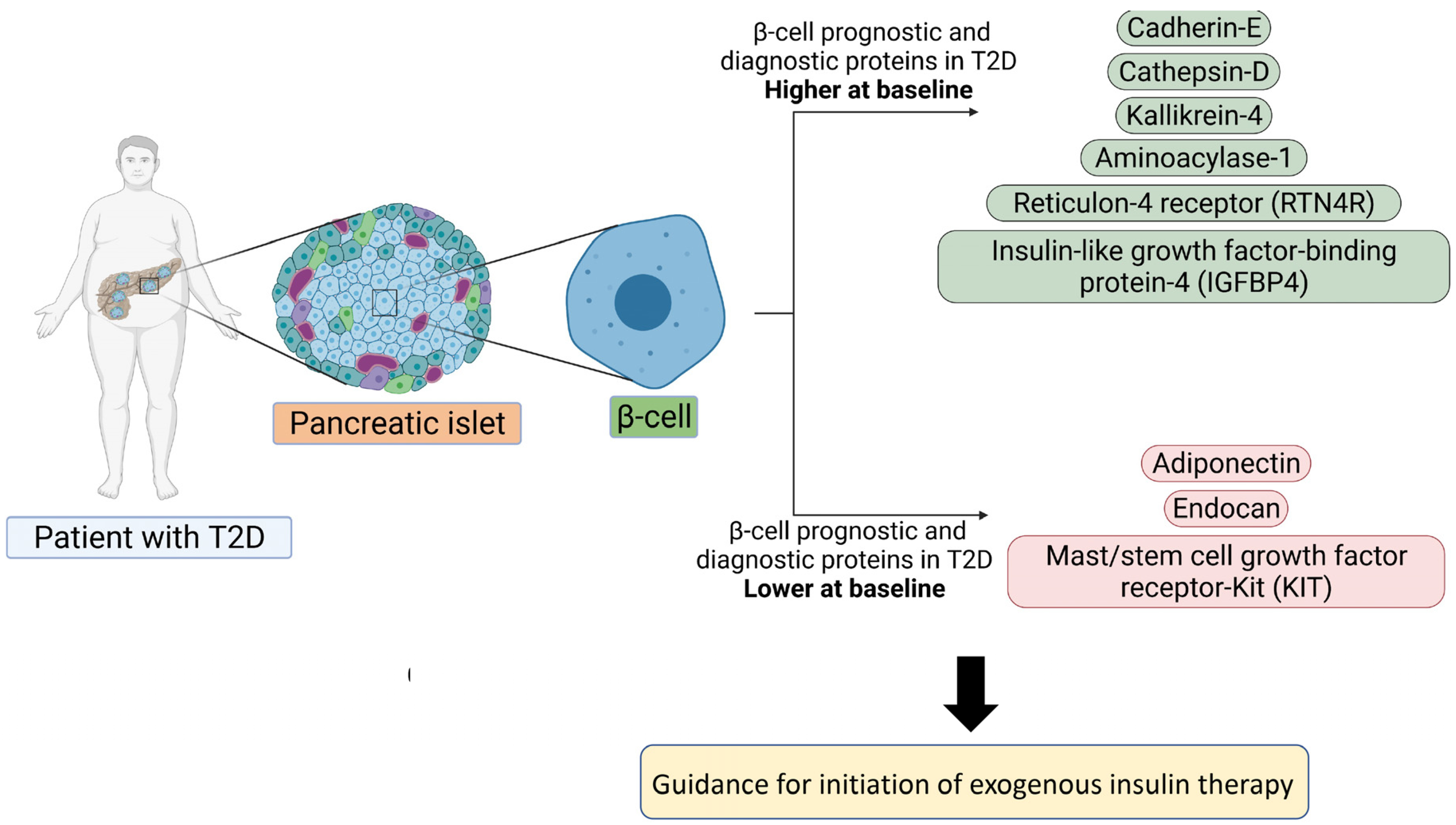
:
1. Introduction
2. Research Design and Methods
2.1. Study Design
2.2. Hyperinsulinemic Clamp
2.3. Blood Sample Preparation and Biochemical Marker Analyses
2.4. SOMA-Scan Assay
2.5. Data Processing and Analyses
2.6. Statistical Analysis
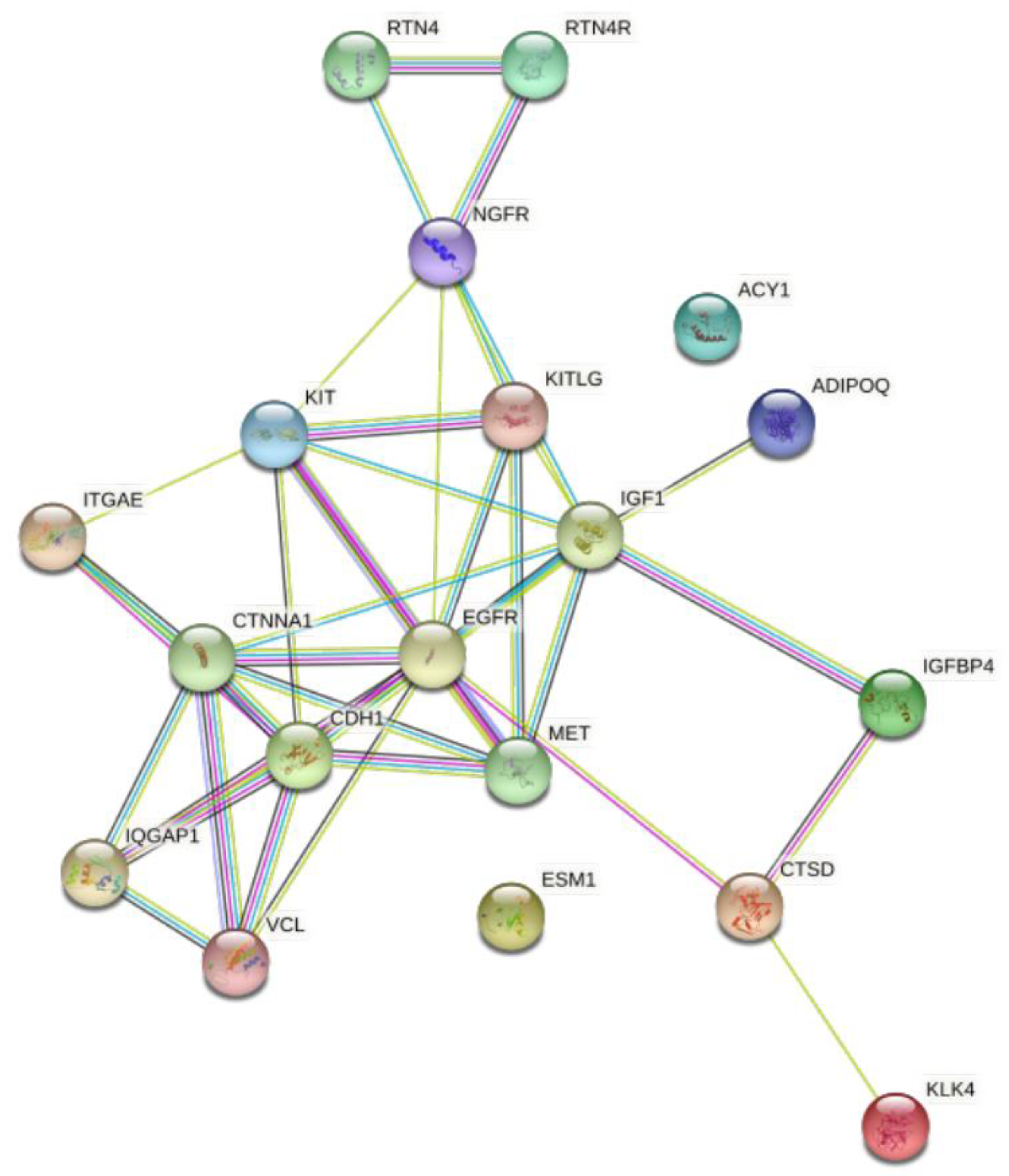
2.7. Protein-Protein Interaction; STRING Analysis
3. Results
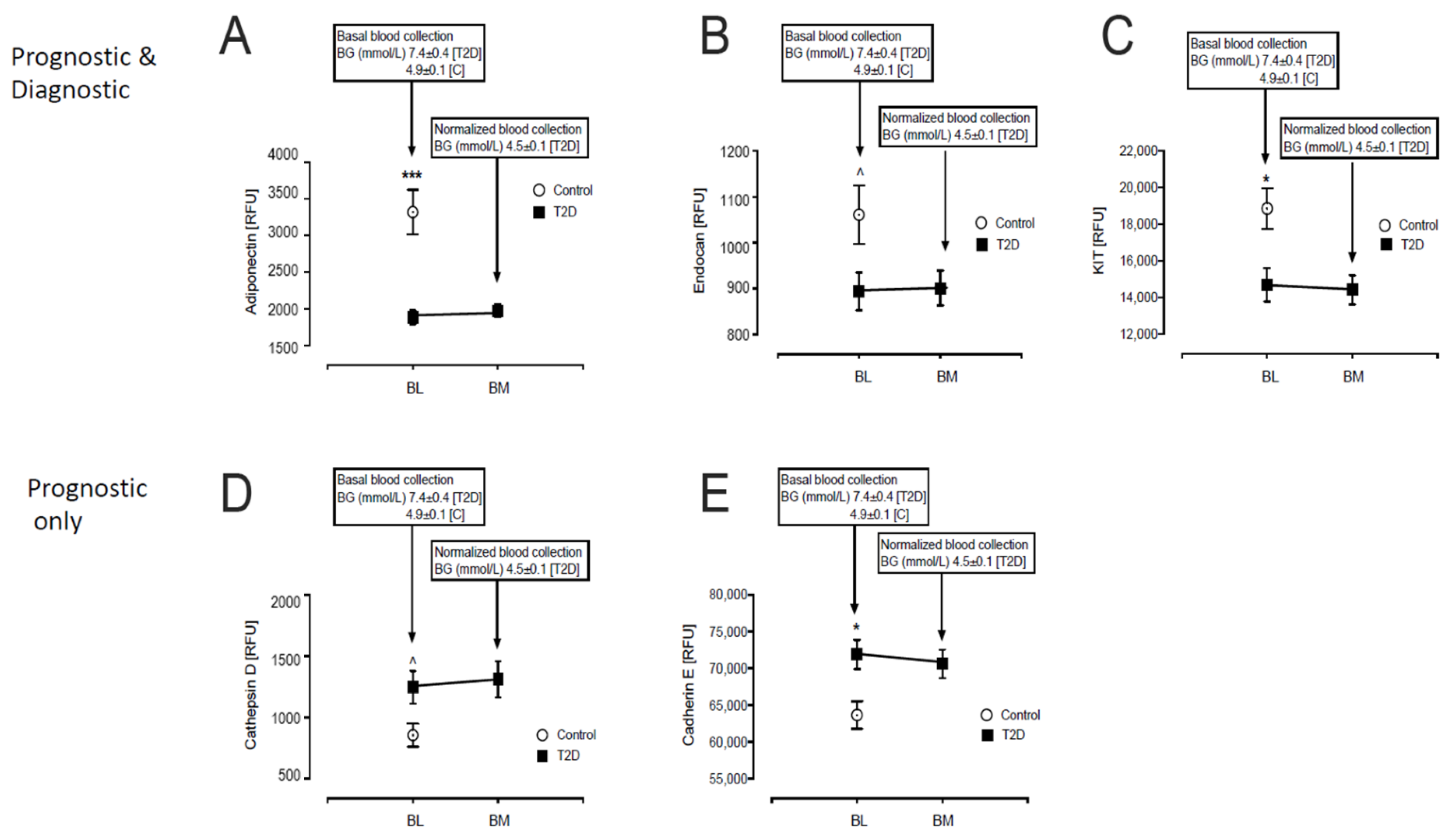
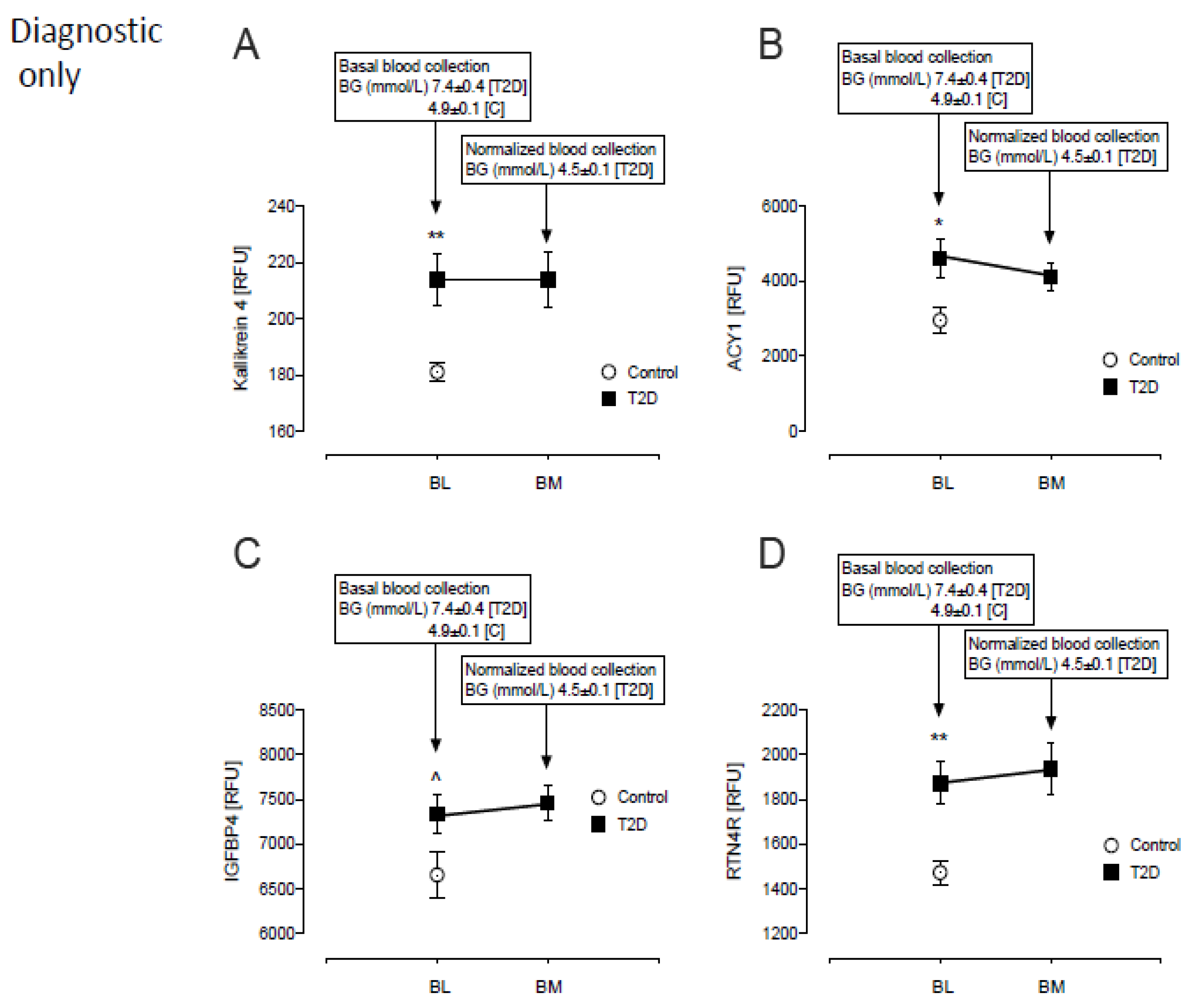
3.1. Baseline Differences between T2D and Control in Levels of Plasma Proteins Reported to Predict β-Cell Function
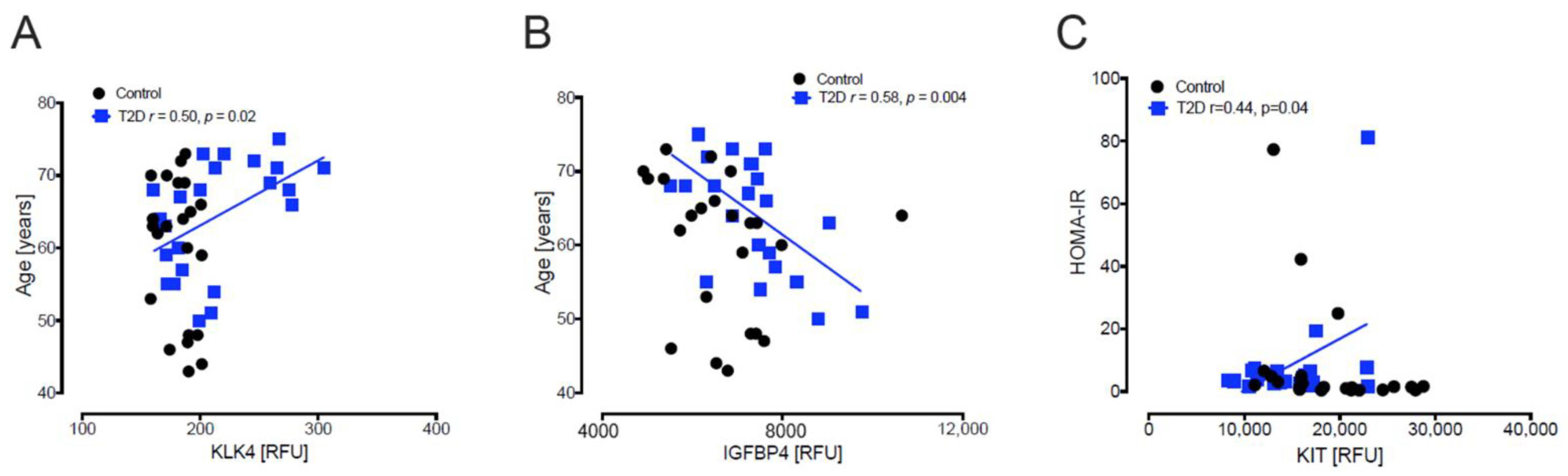
3.2. Baseline Correlations of Age, Body Mass Index, Homeostatic Model Assessment for Insulin Resistance and Homeostatic Model Assessment of β-Cell Function with Levels of Plasma Proteins Reported to Predict β-Cell Function
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Defronzo, R.A. Banting Lecture. From the triumvirate to the ominous octet: A new paradigm for the treatment of type 2 diabetes mellitus. Diabetes 2009, 58, 773–795. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Butler, A.E.; Janson, J.; Bonner-Weir, S.; Ritzel, R.; Rizza, R.A.; Butler, P.C. Beta-cell deficit and increased beta-cell apoptosis in humans with type 2 diabetes. Diabetes 2003, 52, 102–110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cerf, M.E. Beta cell dysfunction and insulin resistance. Front. Endocrinol. (Lausanne) 2013, 4, 37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klein, S.; Gastaldelli, A.; Yki-Jarvinen, H.; Scherer, P.E. Why does obesity cause diabetes? Cell Metab. 2022, 34, 11–20. [Google Scholar] [CrossRef]
- Pillon, N.J.; Loos, R.J.F.; Marshall, S.M.; Zierath, J.R. Metabolic consequences of obesity and type 2 diabetes: Balancing genes and environment for personalized care. Cell 2021, 184, 1530–1544. [Google Scholar] [CrossRef]
- Ludvik, B.; Nolan, J.J.; Baloga, J.; Sacks, D.; Olefsky, J. Effect of obesity on insulin resistance in normal subjects and patients with NIDDM. Diabetes 1995, 44, 1121–1125. [Google Scholar] [CrossRef]
- Rahier, J.; Guiot, Y.; Goebbels, R.M.; Sempoux, C.; Henquin, J.C. Pancreatic beta-cell mass in European subjects with type 2 diabetes. Diabetes Obes. Metab. 2008, 10 (Suppl. 4), 32–42. [Google Scholar] [CrossRef]
- Pipeleers, D.; Chintinne, M.; Denys, B.; Martens, G.; Keymeulen, B.; Gorus, F. Restoring a functional beta-cell mass in diabetes. Diabetes Obes. Metab. 2008, 10 (Suppl. 4), 54–62. [Google Scholar] [CrossRef]
- Guo, S.; Dai, C.; Guo, M.; Taylor, B.; Harmon, J.S.; Sander, M.; Robertson, R.P.; Powers, A.C.; Stein, R. Inactivation of specific beta cell transcription factors in type 2 diabetes. J. Clin. Investig. 2013, 123, 3305–3316. [Google Scholar] [CrossRef] [Green Version]
- Ritzel, R.A.; Butler, A.E.; Rizza, R.A.; Veldhuis, J.D.; Butler, P.C. Relationship between beta-cell mass and fasting blood glucose concentration in humans. Diabetes Care 2006, 29, 717–718. [Google Scholar] [CrossRef] [Green Version]
- Butler, A.E.; Dhawan, S.; Hoang, J.; Cory, M.; Zeng, K.; Fritsch, H.; Meier, J.J.; Rizza, R.A.; Butler, P.C. beta-Cell Deficit in Obese Type 2 Diabetes, a Minor Role of beta-Cell Dedifferentiation and Degranulation. J. Clin. Endocrinol. Metab. 2016, 101, 523–532. [Google Scholar] [CrossRef] [Green Version]
- Moin, A.S.M.; Butler, A.E. Alterations in Beta Cell Identity in Type 1 and Type 2 Diabetes. Curr. Diabetes Rep. 2019, 19, 83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kahn, S.E.; Chen, Y.C.; Esser, N.; Taylor, A.J.; van Raalte, D.H.; Zraika, S.; Verchere, C.B. The beta Cell in Diabetes: Integrating Biomarkers With Functional Measures. Endocr. Rev. 2021, 42, 528–583. [Google Scholar] [CrossRef]
- American Diabetes, A. 6. Glycemic Targets: Standards of Medical Care in Diabetes-2019. Diabetes Care 2019, 42 (Suppl. 1), S61–S70. [Google Scholar] [CrossRef] [Green Version]
- Belongie, K.J.; Ferrannini, E.; Johnson, K.; Andrade-Gordon, P.; Hansen, M.K.; Petrie, J.R. Identification of novel biomarkers to monitor beta-cell function and enable early detection of type 2 diabetes risk. PLoS ONE 2017, 12, e0182932. [Google Scholar] [CrossRef]
- Al-Qaissi, A.; Papageorgiou, M.; Deshmukh, H.; Madden, L.A.; Rigby, A.; Kilpatrick, E.S.; Atkin, S.L.; Sathyapalan, T. Effects of acute insulin-induced hypoglycaemia on endothelial microparticles in adults with and without type 2 diabetes. Diabetes Obes. Metab. 2019, 21, 533–540. [Google Scholar] [CrossRef]
- Kahal, H.; Halama, A.; Aburima, A.; Bhagwat, A.M.; Butler, A.E.; Graumann, J.; Suhre, K.; Sathyapalan, T.; Atkin, S.L. Effect of induced hypoglycemia on inflammation and oxidative stress in type 2 diabetes and control subjects. Sci. Rep. 2020, 10, 4750. [Google Scholar] [CrossRef] [Green Version]
- Kraemer, S.; Vaught, J.D.; Bock, C.; Gold, L.; Katilius, E.; Keeney, T.R.; Kim, N.; Saccomano, N.A.; Wilcox, S.K.; Zichi, D.; et al. From SOMAmer-based biomarker discovery to diagnostic and clinical applications: A SOMAmer-based, streamlined multiplex proteomic assay. PLoS ONE 2011, 6, e26332. [Google Scholar] [CrossRef] [Green Version]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]
- Benjamini, Y.; Drai, D.; Elmer, G.; Kafkafi, N.; Golani, I. Controlling the false discovery rate in behavior genetics research. Behav. Brain Res. 2001, 125, 279–284. [Google Scholar] [CrossRef] [Green Version]
- Birkett, M.A.; Day, S.J. Internal pilot studies for estimating sample size. Stat. Med. 1994, 13, 2455–2463. [Google Scholar] [CrossRef] [PubMed]
- Moin, A.S.M.; Al-Qaissi, A.; Sathyapalan, T.; Atkin, S.L.; Butler, A.E. Glucose excursions in type 2 diabetes modulate amyloid-related proteins associated with dementia. J. Transl. Med. 2021, 19, 131. [Google Scholar] [CrossRef] [PubMed]
- Kahn, S.E.; Haffner, S.M.; Heise, M.A.; Herman, W.H.; Holman, R.R.; Jones, N.P.; Kravitz, B.G.; Lachin, J.M.; O’Neill, M.C.; Zinman, B.; et al. Glycemic durability of rosiglitazone, metformin, or glyburide monotherapy. N. Engl. J. Med. 2006, 355, 2427–2443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thamer, C.; Haap, M.; Heller, E.; Joel, L.; Braun, S.; Tschritter, O.; Haring, H.; Fritsche, A. Beta cell function, insulin resistance and plasma adiponectin concentrations are predictors for the change of postprandial glucose in non-diabetic subjects at risk for type 2 diabetes. Horm. Metab. Res. 2006, 38, 178–182. [Google Scholar] [CrossRef]
- Mather, K.J.; Funahashi, T.; Matsuzawa, Y.; Edelstein, S.; Bray, G.A.; Kahn, S.E.; Crandall, J.; Marcovina, S.; Goldstein, B.; Goldberg, R.; et al. Adiponectin, change in adiponectin, and progression to diabetes in the Diabetes Prevention Program. Diabetes 2008, 57, 980–986. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Meng, R.W.; Kunutsor, S.K.; Chowdhury, R.; Yuan, J.M.; Koh, W.P.; Pan, A. Plasma adiponectin levels and type 2 diabetes risk: A nested case-control study in a Chinese population and an updated meta-analysis. Sci. Rep. 2018, 8, 406. [Google Scholar] [CrossRef]
- Straub, L.G.; Scherer, P.E. Metabolic Messengers: Adiponectin. Nat. Metab. 2019, 1, 334–339. [Google Scholar] [CrossRef]
- Li, N.; Zhao, S.; Zhang, Z.; Zhu, Y.; Gliniak, C.M.; Vishvanath, L.; An, Y.A.; Wang, M.Y.; Deng, Y.; Zhu, Q.; et al. Adiponectin preserves metabolic fitness during aging. Elife 2021, 10, e65108. [Google Scholar] [CrossRef]
- Rodrigues, K.F.; Pietrani, N.T.; Bosco, A.A.; Sousa, L.P.; Ferreira, C.N.; Sandrim, V.C.; Gomes, K.B. Endocan: A new biomarker associated with inflammation in type 2 diabetes mellitus? Diabetes Metab. Res. Rev. 2015, 31, 479–480. [Google Scholar] [CrossRef]
- Klisić, A.; Kavarić, N.; Spasojević-Kalimanovska, V.; Kotur-Stevuljević, J.; Ninić, A. Serum endocan levels in relation to traditional and non-traditional anthropometric indices in adult population. J. Med. Biochem. 2021, 40, 41–48. [Google Scholar] [CrossRef]
- Rachdi, L.; El Ghazi, L.; Bernex, F.; Panthier, J.J.; Czernichow, P.; Scharfmann, R. Expression of the receptor tyrosine kinase KIT in mature beta-cells and in the pancreas in development. Diabetes 2001, 50, 2021–2028. [Google Scholar] [CrossRef] [Green Version]
- Masini, M.; Bugliani, M.; Lupi, R.; del Guerra, S.; Boggi, U.; Filipponi, F.; Marselli, L.; Masiello, P.; Marchetti, P. Autophagy in human type 2 diabetes pancreatic beta cells. Diabetologia 2009, 52, 1083–1086. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.; Chen, B.; Zhang, X.; Tan, L.; Wang, D.W. Increased Cathepsin D Correlates with Clinical Parameters in Newly Diagnosed Type 2 Diabetes. Dis. Markers 2017, 2017, 5286408. [Google Scholar] [CrossRef] [Green Version]
- Nowak, C.; Sundstrom, J.; Gustafsson, S.; Giedraitis, V.; Lind, L.; Ingelsson, E.; Fall, T. Protein Biomarkers for Insulin Resistance and Type 2 Diabetes Risk in Two Large Community Cohorts. Diabetes 2016, 65, 276–284. [Google Scholar] [CrossRef] [Green Version]
- Wakae-Takada, N.; Xuan, S.; Watanabe, K.; Meda, P.; Leibel, R.L. Molecular basis for the regulation of islet beta cell mass in mice: The role of E-cadherin. Diabetologia 2013, 56, 856–866. [Google Scholar] [CrossRef] [Green Version]
- Mandelbaum, A.D.; Melkman-Zehavi, T.; Oren, R.; Kredo-Russo, S.; Nir, T.; Dor, Y.; Hornstein, E. Dysregulation of Dicer1 in beta cells impairs islet architecture and glucose metabolism. Exp. Diabetes Res. 2012, 2012, 470302. [Google Scholar] [CrossRef] [Green Version]
- Carvell, M.J.; Marsh, P.J.; Persaud, S.J.; Jones, P.M. E-cadherin interactions regulate beta-cell proliferation in islet-like structures. Cell. Physiol. Biochem. 2007, 20, 617–626. [Google Scholar] [CrossRef]
- Parnaud, G.; Lavallard, V.; Bedat, B.; Matthey-Doret, D.; Morel, P.; Berney, T.; Bosco, D. Cadherin engagement improves insulin secretion of single human β-cells. Diabetes 2015, 64, 887–896. [Google Scholar] [CrossRef] [Green Version]
- Veveris-Lowe, T.L.; Lawrence, M.G.; Collard, R.L.; Bui, L.; Herington, A.C.; Nicol, D.L.; Clements, J.A. Kallikrein 4 (hK4) and prostate-specific antigen (PSA) are associated with the loss of E-cadherin and an epithelial-mesenchymal transition (EMT)-like effect in prostate cancer cells. Endocr. Relat. Cancer 2005, 12, 631–643. [Google Scholar] [CrossRef] [Green Version]
- Pullen, T.J.; Huising, M.O.; Rutter, G.A. Analysis of Purified Pancreatic Islet Beta and Alpha Cell Transcriptomes Reveals 11beta-Hydroxysteroid Dehydrogenase (Hsd11b1) as a Novel Disallowed Gene. Front. Genet. 2017, 8, 41. [Google Scholar] [CrossRef]
- Rutter, G.A.; Georgiadou, E.; Martinez-Sanchez, A.; Pullen, T.J. Metabolic and functional specialisations of the pancreatic beta cell: Gene disallowance, mitochondrial metabolism and intercellular connectivity. Diabetologia 2020, 63, 1990–1998. [Google Scholar] [CrossRef] [PubMed]
- Jozsef, L.; Tashiro, K.; Kuo, A.; Park, E.J.; Skoura, A.; Albinsson, S.; Rivera-Molina, F.; Harrison, K.D.; Iwakiri, Y.; Toomre, D.; et al. Reticulon 4 is necessary for endoplasmic reticulum tubulation, STIM1-Orai1 coupling, and store-operated calcium entry. J. Biol. Chem. 2014, 289, 9380–9395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hardy, O.T.; Hohmeier, H.E.; Becker, T.C.; Manduchi, E.; Doliba, N.M.; Gupta, R.K.; White, P.; Stoeckert, C.J., Jr.; Matschinsky, F.M.; Newgard, C.B.; et al. Functional genomics of the beta-cell: Short-chain 3-hydroxyacyl-coenzyme A dehydrogenase regulates insulin secretion independent of K+ currents. Mol. Endocrinol. 2007, 21, 765–773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nichols, G.A.; Koo, Y.H.; Shah, S.N. Delay of insulin addition to oral combination therapy despite inadequate glycemic control: Delay of insulin therapy. J. Gen. Intern. Med. 2007, 22, 453–458. [Google Scholar] [CrossRef] [Green Version]
- Peyrot, M.; Rubin, R.R.; Khunti, K. Addressing barriers to initiation of insulin in patients with type 2 diabetes. Prim. Care Diabetes 2010, 4 (Suppl. 1), S11–S18. [Google Scholar] [CrossRef]
- Candia, J.; Cheung, F.; Kotliarov, Y.; Fantoni, G.; Sellers, B.; Griesman, T.; Huang, J.; Stuccio, S.; Zingone, A.; Ryan, B.M.; et al. Assessment of Variability in the SOMAscan Assay. Sci. Rep. 2017, 7, 14248. [Google Scholar] [CrossRef] [Green Version]
- Moin, A.S.M.; Al-Qaissi, A.; Sathyapalan, T.; Atkin, S.L.; Butler, A.E. Type 2 Diabetes Coagulopathy Proteins May Conflict with Biomarkers Reflective of COVID-19 Severity. Front. Endocrinol. 2021, 12, 773. [Google Scholar] [CrossRef]
- Moin, A.S.M.; Al-Qaissi, A.; Sathyapalan, T.; Atkin, S.L.; Butler, A.E. Platelet Protein-Related Abnormalities in Response to Acute Hypoglycemia in Type 2 Diabetes. Front. Endocrinol. 2021, 12, 321. [Google Scholar] [CrossRef]
- Yu, L.-R.; Sun, J.; Daniels, J.R.; Cao, Z.; Schnackenberg, L.; Choudhury, D.; Palevsky, P.M.; Ma, J.Z.; Beger, R.D.; Portilla, D. Aptamer-Based Proteomics Identifies Mortality-Associated Serum Biomarkers in Dialysis-Dependent AKI Patients. Kidney Int. Rep. 2018, 3, 1202–1213. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Baseline | Type 2 Diabetes (n = 23) | Controls (n = 23) | p-Value |
|---|---|---|---|
| Age (years) | 64 ± 8 (66) | 60 ± 10 (63) | 0.15 |
| Sex (M/F) | 12/11 | 11/12 | 0.77 |
| BMI (kg/m2) | 32 ± 4 (32) | 28 ± 3 (27) | 0.001 |
| Duration of diabetes (years) | 4.5 ± 2.2 (5.0) | N/A | |
| HbA1c (mmol/mol) | 51.2 ± 11.4 (50.0) | 37.2 ± 2.2 (37.0) | <0.0001 |
| HbA1c (%) | 6.8 ± 1.0 (6.7) | 5.6 ± 0.2 (5.5) | <0.0001 |
| Fasting plasma glucose (mmol/L) | 7.6 ± 0.4 | 4.9 ± 0.1 | <0.0001 |
| Total cholesterol (mmol/L) | 4.2 ± 1.0 (4.1) | 4.8 ± 0.67 (4.9) | 0.02 |
| Triglyceride (mmol/L) | 1.7 ± 0.7 (1.5) | 1.34 ± 0.6 (1.3) | 0.06 |
| HDL-cholesterol (mmol/L) | 1.1 ± 0.3 (1.1) | 1.5 ± 0.4 (1.4) | 0.002 |
| LDL-cholesterol (mmol/L) | 2.27 ± 0.8 (2.1) | 2.7 ± 0.7 (2.8) | 0.06 |
| CRP (mg/L) | 3.0 ± 2.7 (1.9) | 5.1 ± 10.3 (2.1) | 0.33 |
| Insulin (IU/mL) | 13.7 ± 7.6 | 21.6 ± 44.2 | 0.41 |
| HOMA-IR | 4.8 ± 3.8 | 4.8 ± 9.6 | 0.99 |
| HOMA-B | 173 ± 467 | 384 ± 762 | 0.26 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moin, A.S.M.; Sathyapalan, T.; Atkin, S.L.; Butler, A.E. Diagnostic and Prognostic Protein Biomarkers of β-Cell Function in Type 2 Diabetes and Their Modulation with Glucose Normalization. Metabolites 2022, 12, 196. https://doi.org/10.3390/metabo12030196
Moin ASM, Sathyapalan T, Atkin SL, Butler AE. Diagnostic and Prognostic Protein Biomarkers of β-Cell Function in Type 2 Diabetes and Their Modulation with Glucose Normalization. Metabolites. 2022; 12(3):196. https://doi.org/10.3390/metabo12030196
Chicago/Turabian StyleMoin, Abu Saleh Md, Thozhukat Sathyapalan, Stephen L. Atkin, and Alexandra E. Butler. 2022. "Diagnostic and Prognostic Protein Biomarkers of β-Cell Function in Type 2 Diabetes and Their Modulation with Glucose Normalization" Metabolites 12, no. 3: 196. https://doi.org/10.3390/metabo12030196