

Mechanism for Utilization of the Populus-Derived Metabolite Salicin by a Pseudomonas—Rahnella Co-Culture
 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Bacterial Strains and Culture Medium
2.2. Primer Design, DNA Isolation and Microbial Quantification
2.3. Gas Chromatography-Mass Spectrometry (GC-MS) Metabolome Analysis
2.4. Sample Preparation for Proteome Measurements
2.5. Liquid Chromatography/Tandem Mass Spectrometry (LC MS/MS) Analysis
2.6. Proteomics Data Analysis
2.7. Bioinformatic Analysis
3. Results
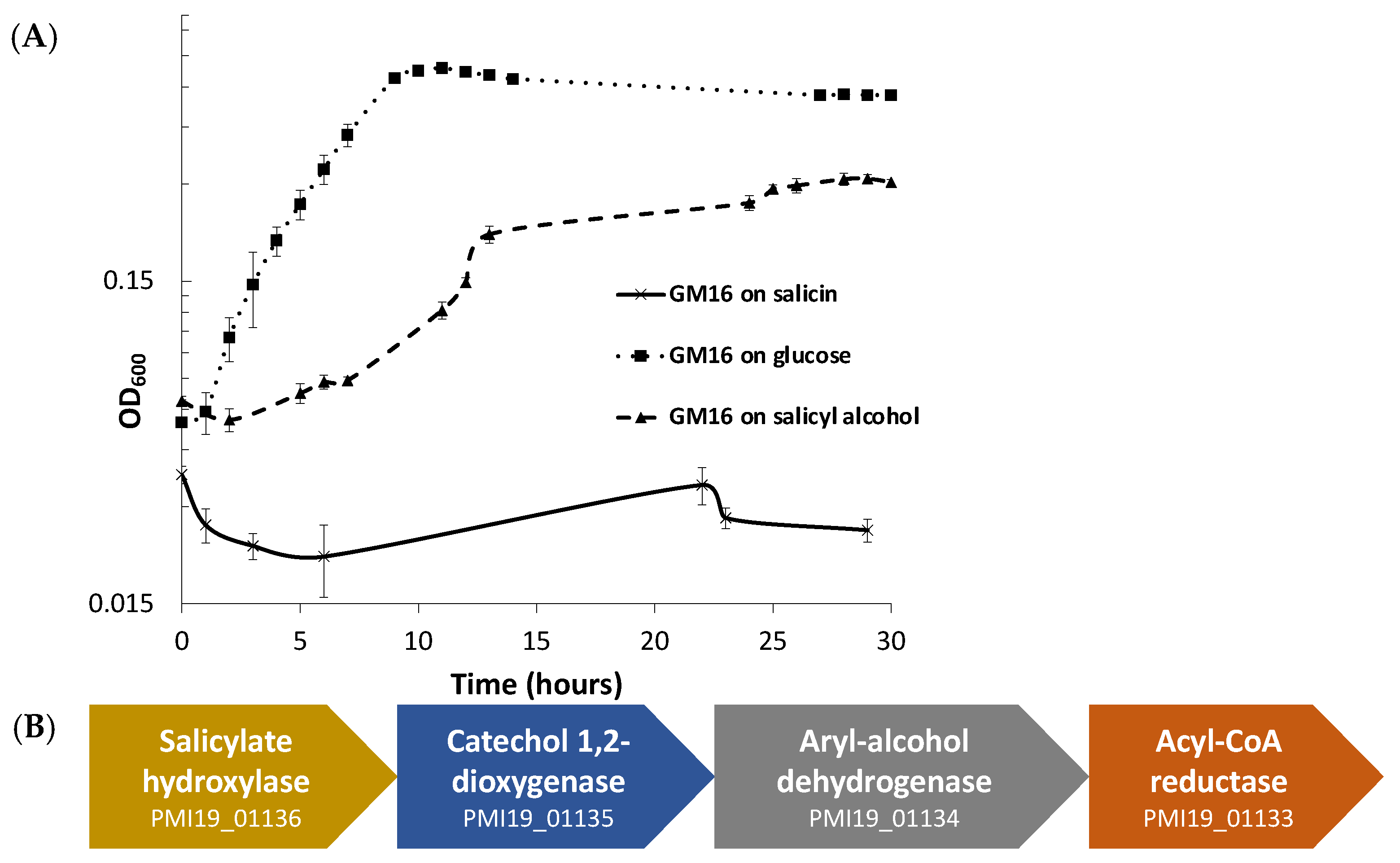
3.1. Growth and Bioinformatic Analyses of P. fluorescens GM16 Salicyl Alcohol and Salicin Metabolism
3.2. Proteomics Analysis of Salicyl Alcohol Grown Cells
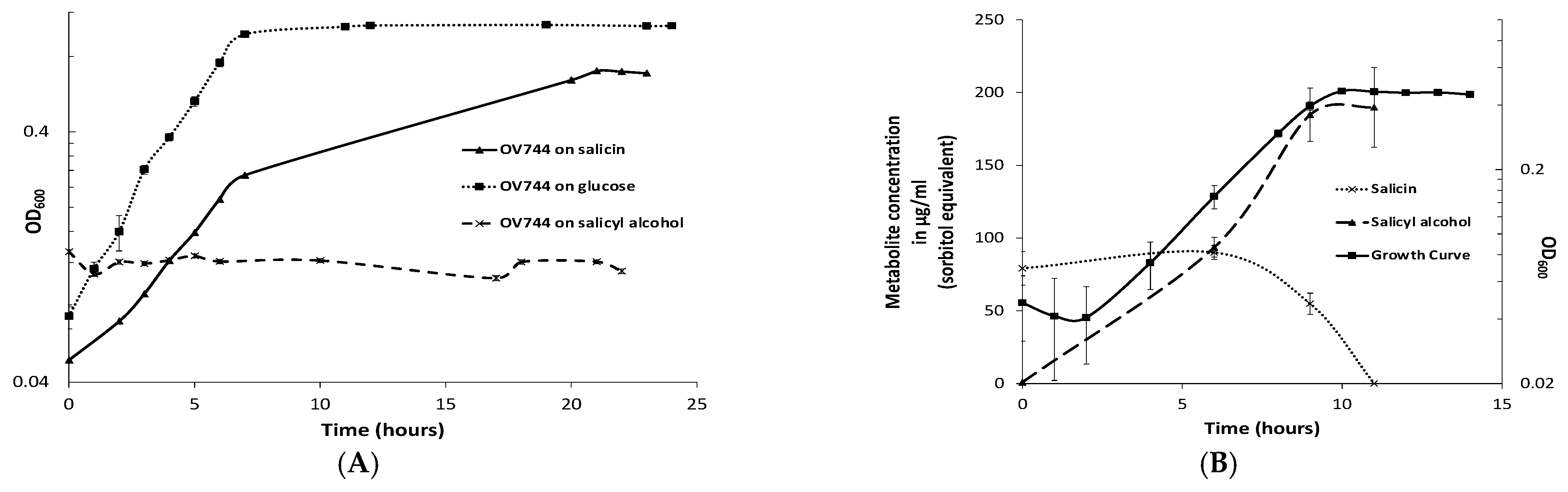
3.3. Characterization of R. aquatilis OV744 Growth on Salicin
3.4. Proteomics and Metabolomic Investigation of Salicin Utilization
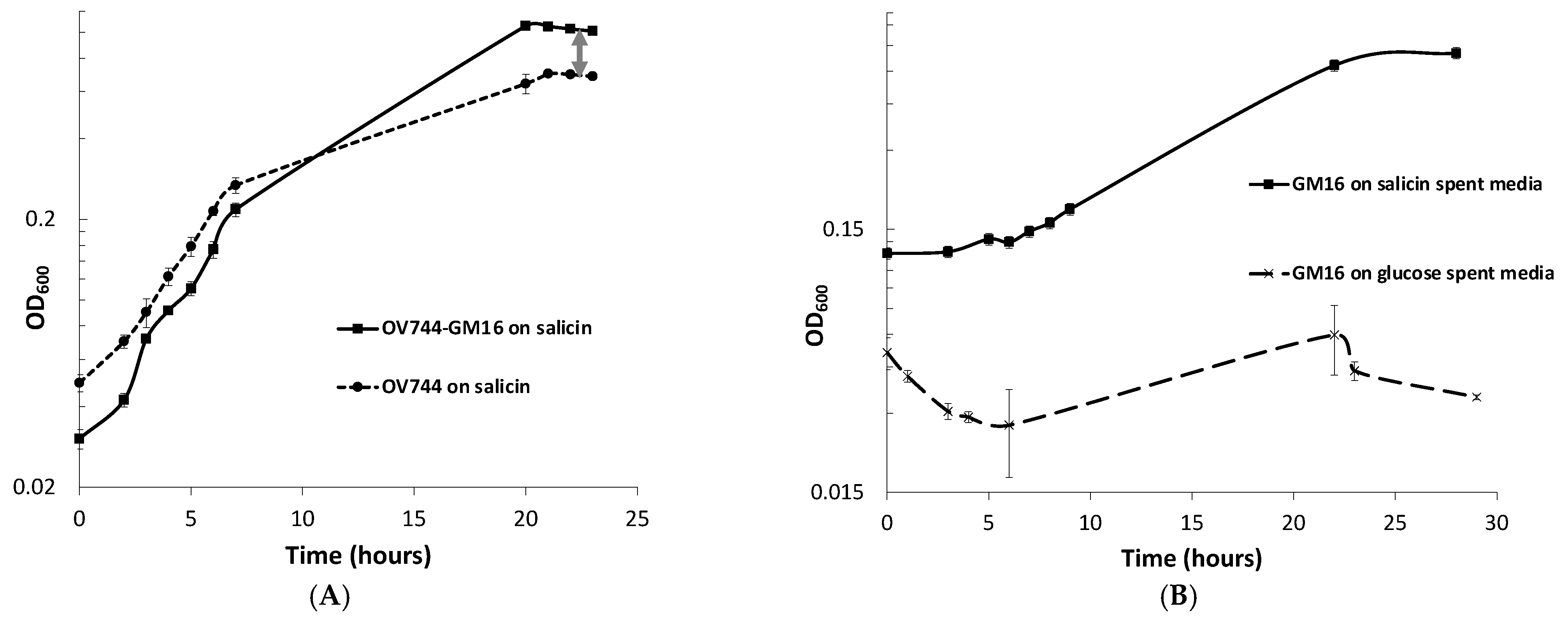
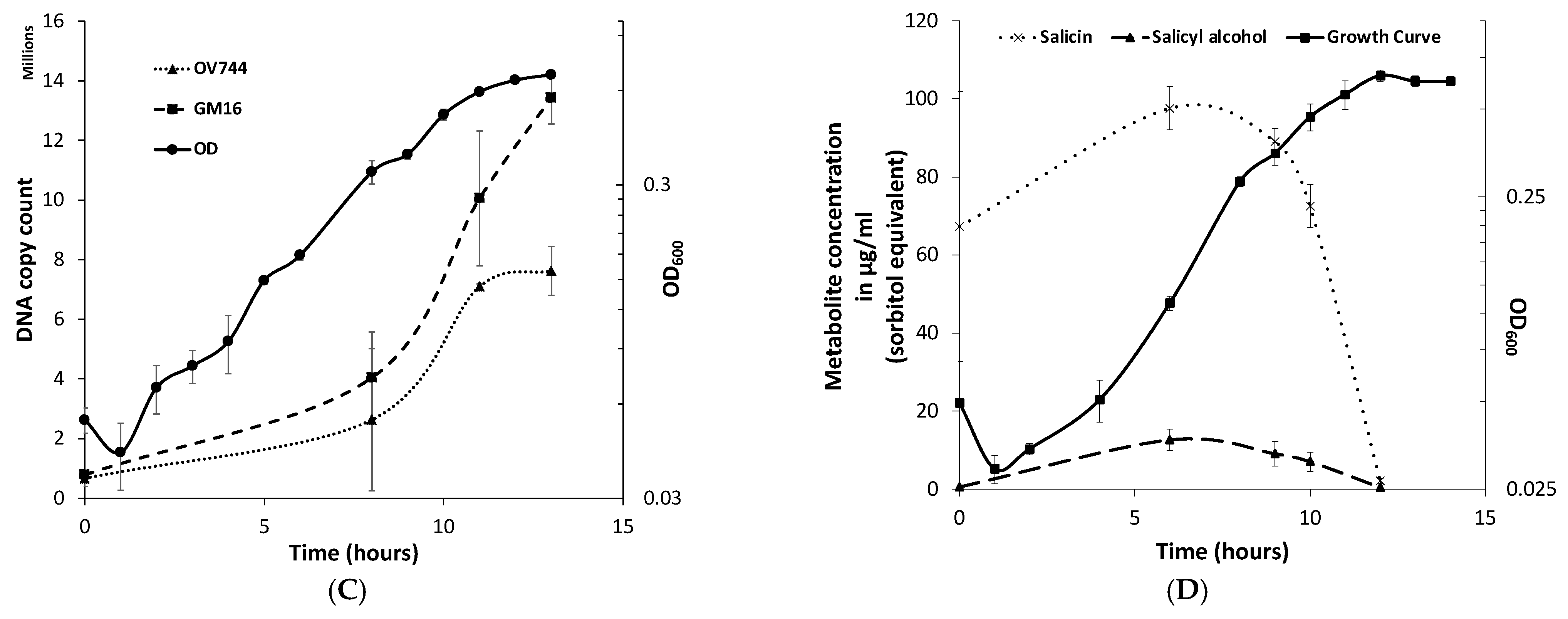
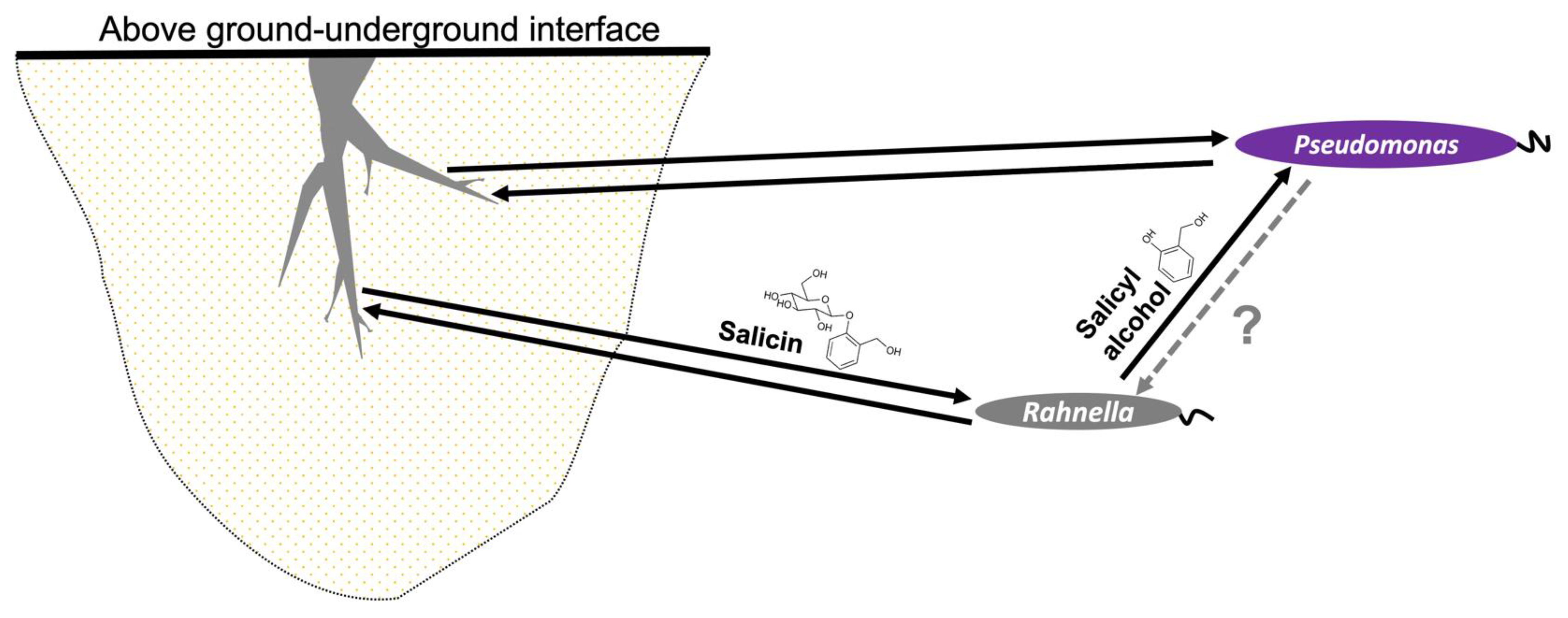
3.5. Analysis of R. aquatilis OV744 and Pseudomonas sp. Co-Culture Unidirectional Cross-Feeding Mechanism
3.6. Metabolomics and Proteomics Data Analysis of Metabolic Cross-Feeding Interaction between R. aquatilis OV744 and P. fluorescens GM16
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jun, S.-R.; Wassenaar, T.M.; Nookaew, I.; Hauser, L.; Wanchai, V.; Land, M.; Timm, C.M.; Lu, T.-Y.S.; Schadt, C.W.; Doktycz, M.J. Diversity of Pseudomonas genomes, including Populus-associated isolates, as revealed by comparative genome analysis. Appl. Environ. Microbiol. 2016, 82, 375–383. [Google Scholar] [CrossRef] [Green Version]
- Cregger, M.A.; Carper, D.L.; Christel, S.; Doktycz, M.J.; Labbé, J.; Michener, J.K.; Dove, N.C.; Johnston, E.R.; Moore, J.A.; Vélez, J.M. Plant–microbe interactions: From genes to ecosystems using Populus as a model system. Phytobiomes J. 2021, 5, 29–38. [Google Scholar] [CrossRef]
- Carper, D.L.; Weston, D.J.; Barde, A.; Timm, C.M.; Lu, T.-Y.; Burdick, L.H.; Jawdy, S.S.; Klingeman, D.M.; Robeson, M.S.; Veach, A.M. Cultivating the Bacterial Microbiota of Populus Roots. Msystems 2021, 6, e01306–e01320. [Google Scholar] [CrossRef] [PubMed]
- Henning, J.A.; Weston, D.J.; Pelletier, D.A.; Timm, C.M.; Jawdy, S.S.; Classen, A.T. Root bacterial endophytes alter plant phenotype, but not physiology. PeerJ 2016, 4, e2606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henning, J.A.; Weston, D.J.; Pelletier, D.A.; Timm, C.M.; Jawdy, S.S.; Classen, A.T. Relatively rare root endophytic bacteria drive plant resource allocation patterns and tissue nutrient concentration in unpredictable ways. Am. J. Bot. 2019, 106, 1423–1434. [Google Scholar] [CrossRef] [PubMed]
- Timm, C.M.; Campbell, A.G.; Utturkar, S.M.; Jun, S.R.; Parales, R.E.; Tan, W.A.; Robeson, M.S.; Lu, T.Y.; Jawdy, S.; Brown, S.D.; et al. Metabolic functions of Pseudomonas fluorescens strains from Populus deltoides depend on rhizosphere or endosphere isolation compartment. Front. Microbiol. 2015, 6, 1118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diaz, E.; Jimenez, J.I.; Nogales, J. Aerobic degradation of aromatic compounds. Curr. Opin. Biotechnol. 2013, 24, 431–442. [Google Scholar] [CrossRef] [Green Version]
- Donaldson, J.R.; Stevens, M.T.; Barnhill, H.R.; Lindroth, R.L. Age-related shifts in leaf chemistry of clonal aspen (Populus tremuloides). J. Chem. Ecol. 2006, 32, 1415–1429. [Google Scholar] [CrossRef] [Green Version]
- Boeckler, G.A.; Gershenzon, J.; Unsicker, S.B. Phenolic glycosides of the Salicaceae and their role as anti-herbivore defenses. Phytochemistry 2011, 72, 1497–1509. [Google Scholar] [CrossRef]
- Boeckler, G.A.; Paetz, C.; Feibicke, P.; Gershenzon, J.; Unsicker, S.B. Metabolism of poplar salicinoids by the generalist herbivore Lymantria dispar (Lepidoptera). Insect Biochem. Mol. Biol. 2016, 78, 39–49. [Google Scholar] [CrossRef]
- Mason, C.J.; Couture, J.J.; Raffa, K.F. Plant-associated bacteria degrade defense chemicals and reduce their adverse effects on an insect defoliator. Oecologia 2014, 175, 901–910. [Google Scholar] [CrossRef] [PubMed]
- Mason, C.J.; Lowe-Power, T.M.; Rubert-Nason, K.F.; Lindroth, R.L.; Raffa, K.F. Interactions between bacteria and aspen defense chemicals at the phyllosphere–herbivore interface. J. Chem. Ecol. 2016, 42, 193–201. [Google Scholar] [CrossRef] [PubMed]
- Karn, S.K.; Chakrabarty, S.; Reddy, M. Pentachlorophenol degradation by Pseudomonas stutzeri CL7 in the secondary sludge of pulp and paper mill. J. Environ. Sci. 2010, 22, 1608–1612. [Google Scholar] [CrossRef]
- Patel, M.; Patel, H.M.; Vohra, N.; Dave, S. Complete genome sequencing and comparative genome characterization of the lignocellulosic biomass degrading bacterium Pseudomonas stutzeri MP4687 from cattle rumen. Biotechnol. Rep. 2020, 28, e00530. [Google Scholar] [CrossRef] [PubMed]
- Raghunand, T.R.; Mahadevan, S. The β-glucoside genes of Klebsiella aerogenes: Conservation and divergence in relation to the cryptic bgl genes of Escherichia coli. FEMS Microbiol. Lett. 2003, 223, 267–274. [Google Scholar] [CrossRef] [Green Version]
- Pinto, S.S.; Diogo, H.P. Calorimetric studies on the phenolic glycoside D(−)-salicin. J. Pharm. Sci. 2008, 97, 5354–5362. [Google Scholar] [CrossRef]
- Paliwal, V.; Raju, S.C.; Modak, A.; Phale, P.S.; Purohit, H.J. Pseudomonas putida CSV86: A candidate genome for genetic bioaugmentation. PLoS ONE 2014, 9, e84000. [Google Scholar] [CrossRef]
- Shrivastava, R.; Basu, A.; Phale, P.S. Purification and characterization of benzyl alcohol-and benzaldehyde-dehydrogenase from Pseudomonas putida CSV86. Arch. Microbiol. 2011, 193, 553–563. [Google Scholar] [CrossRef]
- Katagiri, M.; Takemori, S.; Nakazawa, K.; Suzuki, H.; Akagi, K. Benzylalcohol dehydrogenase, a new alcohol dehydrogenase from Pseudomonas sp. Biochim. Et Biophys. Acta (BBA)-Enzymol. 1967, 139, 173–176. [Google Scholar] [CrossRef]
- Basu, A.; Dixit, S.; Phale, P. Metabolism of benzyl alcohol via catechol ortho-pathway in methylnaphthalene-degrading Pseudomonas putida CSV86. Appl. Microbiol. Biotechnol. 2003, 62, 579–585. [Google Scholar] [CrossRef]
- Schaefler, S.; Malamy, A. Taxonomic investigations on expressed and cryptic phospho-β-glucosidases in Enterobacteriaceae. J. Bacteriol. 1969, 99, 422–433. [Google Scholar] [CrossRef] [Green Version]
- el Hassouni, M.; Chippaux, M.; Barras, F. Analysis of the Erwinia chrysanthemi arb genes, which mediate metabolism of aromatic beta-glucosides. J. Bacteriol. 1990, 172, 6261–6267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schnetz, K.; Toloczyki, C.; Rak, B. Beta-glucoside (bgl) operon of Escherichia coli K-12: Nucleotide sequence, genetic organization, and possible evolutionary relationship to regulatory components of two Bacillus subtilis genes. J. Bacteriol. 1987, 169, 2579–2590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schnetz, K.; Rak, B. Regulation of the bgl operon of Escherichia coli by transcriptional antitermination. EMBO J. 1988, 7, 3271–3277. [Google Scholar] [CrossRef]
- Stubbendieck, R.M.; Vargas-Bautista, C.; Straight, P.D. Bacterial Communities: Interactions to Scale. Front. Microbiol. 2016, 7, 1234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Weert, S.; Vermeiren, H.; Mulders, I.H.; Kuiper, I.; Hendrickx, N.; Bloemberg, G.V.; Vanderleyden, J.; De Mot, R.; Lugtenberg, B.J. Flagella-driven chemotaxis towards exudate components is an important trait for tomato root colonization by Pseudomonas fluorescens. Mol. Plant-Microbe Interact. 2002, 15, 1173–1180. [Google Scholar] [CrossRef] [Green Version]
- Compant, S.; Reiter, B.; Sessitsch, A.; Nowak, J.; Clement, C.; Ait Barka, E. Endophytic colonization of Vitis vinifera L. by plant growth-promoting bacterium Burkholderia sp. strain PsJN. Appl. Environ. Microbiol. 2005, 71, 1685–1693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dennis, P.G.; Miller, A.J.; Hirsch, P.R. Are root exudates more important than other sources of rhizodeposits in structuring rhizosphere bacterial communities? FEMS Microbiol. Ecol. 2010, 72, 313–327. [Google Scholar] [CrossRef] [Green Version]
- Hibbing, M.E.; Fuqua, C.; Parsek, M.R.; Peterson, S.B. Bacterial competition: Surviving and thriving in the microbial jungle. Nat. Rev. Microbiol. 2010, 8, 15–25. [Google Scholar] [CrossRef] [Green Version]
- Freilich, S.; Zarecki, R.; Eilam, O.; Segal, E.S.; Henry, C.S.; Kupiec, M.; Gophna, U.; Sharan, R.; Ruppin, E. Competitive and cooperative metabolic interactions in bacterial communities. Nat. Commun. 2011, 2, 589. [Google Scholar] [CrossRef]
- Kemen, E. Microbe-microbe interactions determine oomycete and fungal host colonization. Curr. Opin. Plant Biol. 2014, 20, 75–81. [Google Scholar] [CrossRef] [PubMed]
- D’Souza, G.; Shitut, S.; Preussger, D.; Yousif, G.; Waschina, S.; Kost, C. Ecology and evolution of metabolic cross-feeding interactions in bacteria. Nat. Prod. Rep. 2018, 35, 455–488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- West, S.A.; Griffin, A.S.; Gardner, A.; Diggle, S.P. Social evolution theory for microorganisms. Nat. Rev. Microbiol. 2006, 4, 597–607. [Google Scholar] [CrossRef] [PubMed]
- Ponomarova, O.; Gabrielli, N.; Sévin, D.C.; Mülleder, M.; Zirngibl, K.; Bulyha, K.; Andrejev, S.; Kafkia, E.; Typas, A.; Sauer, U. Yeast creates a niche for symbiotic lactic acid bacteria through nitrogen overflow. Cell Syst. 2017, 5, 345–357.e346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Egan, M.; Motherway, M.O.C.; Kilcoyne, M.; Kane, M.; Joshi, L.; Ventura, M.; van Sinderen, D. Cross-feeding by Bifidobacterium breve UCC2003 during co-cultivation with Bifidobacterium bifidum PRL2010 in a mucin-based medium. BMC Microbiol. 2014, 14, 282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blair, P.M.; Land, M.L.; Piatek, M.J.; Jacobson, D.A.; Lu, T.-Y.S.; Doktycz, M.J.; Pelletier, D.A. Exploration of the biosynthetic potential of the Populus microbiome. mSystems 2018, 3, e00045-18. [Google Scholar] [CrossRef] [Green Version]
- Brown, S.D.; Utturkar, S.M.; Klingeman, D.M.; Johnson, C.M.; Martin, S.L.; Land, M.L.; Lu, T.-Y.S.; Schadt, C.W.; Doktycz, M.J.; Pelletier, D.A. Twenty-one genome sequences from Pseudomonas species and 19 genome sequences from diverse bacteria isolated from the rhizosphere and endosphere of Populus deltoides. J. Bacteriol. 2012, 194, 5991–5993. [Google Scholar] [CrossRef] [Green Version]
- Sayers, E.W.; Beck, J.; Bolton, E.E.; Bourexis, D.; Brister, J.R.; Canese, K.; Comeau, D.C.; Funk, K.; Kim, S.; Klimke, W. Database resources of the national center for biotechnology information. Nucleic Acids Res. 2021, 49, D10. [Google Scholar] [CrossRef]
- Chen, I.-M.A.; Chu, K.; Palaniappan, K.; Ratner, A.; Huang, J.; Huntemann, M.; Hajek, P.; Ritter, S.J.; Webb, C.; Wu, D. The IMG/M data management and analysis system v. 7: Content updates and new features. Nucleic Acids Res. 2023, 51, D723–D732. [Google Scholar] [CrossRef]
- Neidhardt, F.C.; Bloch, P.L.; Smith, D.F. Culture medium for enterobacteria. J. Bacteriol. 1974, 119, 736–747. [Google Scholar] [CrossRef]
- Timm, C.M.; Pelletier, D.A.; Jawdy, S.S.; Gunter, L.E.; Henning, J.A.; Engle, N.; Aufrecht, J.; Gee, E.; Nookaew, I.; Yang, Z.; et al. Two Poplar-Associated Bacterial Isolates Induce Additive Favorable Responses in a Constructed Plant-Microbiome System. Front. Plant Sci. 2016, 7, 497. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tschaplinski, T.J.; Standaert, R.F.; Engle, N.L.; Martin, M.Z.; Sangha, A.K.; Parks, J.M.; Smith, J.C.; Samuel, R.; Jiang, N.; Pu, Y. Down-regulation of the caffeic acid O-methyltransferase gene in switchgrass reveals a novel monolignol analog. Biotechnol. Biofuels 2012, 5, 71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thompson, M.R.; Chourey, K.; Froelich, J.M.; Erickson, B.K.; VerBerkmoes, N.C.; Hettich, R.L. Experimental approach for deep proteome measurements from small-scale microbial biomass samples. Anal. Chem. 2008, 80, 9517–9525. [Google Scholar] [CrossRef] [PubMed]
- Washburn, M.P.; Wolters, D.; Yates III, J.R. Large-scale analysis of the yeast proteome by multidimensional protein identification technology. Nat. Biotechnol. 2001, 19, 242. [Google Scholar] [CrossRef] [PubMed]
- Wolters, D.A.; Washburn, M.P.; Yates, J.R. An automated multidimensional protein identification technology for shotgun proteomics. Anal. Chem. 2001, 73, 5683–5690. [Google Scholar] [CrossRef] [PubMed]
- Tabb, D.L.; Fernando, C.G.; Chambers, M.C. MyriMatch: Highly accurate tandem mass spectral peptide identification by multivariate hypergeometric analysis. J. Proteome Res. 2007, 6, 654–661. [Google Scholar] [CrossRef] [Green Version]
- Ma, Z.-Q.; Dasari, S.; Chambers, M.C.; Litton, M.D.; Sobecki, S.M.; Zimmerman, L.J.; Halvey, P.J.; Schilling, B.; Drake, P.M.; Gibson, B.W. IDPicker 2.0: Improved protein assembly with high discrimination peptide identification filtering. J. Proteome Res. 2009, 8, 3872–3881. [Google Scholar] [CrossRef] [Green Version]
- Moore, R.E.; Young, M.K.; Lee, T.D. Qscore: An algorithm for evaluating SEQUEST database search results. J. Am. Soc. Mass Spectrom. 2002, 13, 378–386. [Google Scholar] [CrossRef] [Green Version]
- Elias, J.E.; Gygi, S.P. Target-decoy search strategy for increased confidence in large-scale protein identifications by mass spectrometry. Nat. Methods 2007, 4, 207. [Google Scholar] [CrossRef]
- Team, R.C. R: A Language and Environment for Statistical Computing. 2014. Available online: https://www.R-project.org (accessed on 30 December 2022).
- Liu, H.; Sadygov, R.G.; Yates, J.R. A model for random sampling and estimation of relative protein abundance in shotgun proteomics. Anal. Chem. 2004, 76, 4193–4201. [Google Scholar] [CrossRef]
- Zhang, Y.; Wen, Z.; Washburn, M.P.; Florens, L. Refinements to label free proteome quantitation: How to deal with peptides shared by multiple proteins. Anal. Chem. 2010, 82, 2272–2281. [Google Scholar] [CrossRef] [PubMed]
- Zybailov, B.; Mosley, A.L.; Sardiu, M.E.; Coleman, M.K.; Florens, L.; Washburn, M.P. Statistical analysis of membrane proteome expression changes in Saccharomyces c erevisiae. J. Proteome Res. 2006, 5, 2339–2347. [Google Scholar] [CrossRef] [PubMed]
- Hong, F.; Breitling, R.; McEntee, C.W.; Wittner, B.S.; Nemhauser, J.L.; Chory, J. RankProd: A bioconductor package for detecting differentially expressed genes in meta-analysis. Bioinformatics 2006, 22, 2825–2827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Estenson, K.; Hurst, G.B.; Standaert, R.F.; Bible, A.N.; Garcia, D.; Chourey, K.; Doktycz, M.J.; Morrell-Falvey, J.L. Characterization of Indole-3-acetic acid biosynthesis and the effects of this phytohormone on the proteome of the plant-associated microbe Pantoea sp. YR343. J. Proteome Res. 2018, 17, 1361–1374. [Google Scholar] [CrossRef] [PubMed]
- Chai, J.; Kora, G.; Ahn, T.-H.; Hyatt, D.; Pan, C. Functional phylogenomics analysis of bacteria and archaea using consistent genome annotation with UniFam. BMC Evol. Biol. 2014, 14, 207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ogata, H.; Goto, S.; Sato, K.; Fujibuchi, W.; Bono, H.; Kanehisa, M. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 1999, 27, 29–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ross, W.D. Fungi associated with root diseases of aspen in Wyoming. Can. J. Bot. 1976, 54, 734–744. [Google Scholar] [CrossRef]
- Andersen, D.C.; MacMahon, J.A. Population dynamics and bioenergetics of a fossorial herbivore, Thomomys talpoides (Rodentia: Geomyidae), in a spruce-fir sere. Ecol. Monogr. 1981, 51, 179–202. [Google Scholar] [CrossRef]
- Wallner, W.E.; Wagner, D.L.; Parker, B.L.; Tobi, D.L. Bioecology of the conifer swift moth, Korscheltellus gracilis, a root feeder associated with spruce-fir decline. In Forest Insect Guilds: Patterns of Interaction with Host Trees, Abakan, Siberia, 13–17 August 1989; Baranchikov, Y.N., Mattson, W.J., Hain, F.P., Payne, T.L., Eds.; USSR Gen. Tech. Rep. NE-153; US Department of Agriculture, Forest Service, Northeastern Forest Experiment Station: Radnor, PA, USA, 1991; Volume 153, pp. 199–204. [Google Scholar]
- Coyle, D.; Mattson, W.; Raffa, K. Invasive root feeding insects in natural forest ecosystems of North America. In Root Feeders: An Ecosystem Perspective; CAB International: Wallingford, UK, 2008; pp. 134–151. [Google Scholar]
- Stevens, M.T.; Gusse, A.C.; Lindroth, R.L. Root Chemistry in Populus tremuloides: Effects of Soil Nutrients, Defoliation, and Genotype. J. Chem. Ecol. 2014, 40, 31–38. [Google Scholar] [CrossRef]
- Randriamanana, T.R.; Nybakken, L.; Lavola, A.; Aphalo, P.J.; Nissinen, K.; Julkunen-Tiitto, R. Sex-related differences in growth and carbon allocation to defence in Populus tremula as explained by current plant defence theories. Tree Physiol. 2014, 34, 471–487. [Google Scholar] [CrossRef]
- Ruuhola, T. Dynamics of Salicylates in Willows and Its Relation to Herbivory; University of Joensuu: Joensuu, Finland, 2001. [Google Scholar]
- Ruuhola, T.; Julkunen-Tiitto, R.; Vainiotalo, P. In vitro degradation of willow salicylates. J. Chem. Ecol. 2003, 29, 1083–1097. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Daza, M.C.; Pulido-Mateos, E.C.; Lupien-Meilleur, J.; Guyonnet, D.; Desjardins, Y.; Roy, D. Polyphenol-mediated gut microbiota modulation: Toward prebiotics and further. Front. Nutr. 2021, 8, 689456. [Google Scholar] [CrossRef] [PubMed]
- Lyu, D.; Msimbira, L.A.; Nazari, M.; Antar, M.; Pagé, A.; Shah, A.; Monjezi, N.; Zajonc, J.; Tanney, C.A.S.; Backer, R.; et al. The coevolution of plants and microbes underpins sustainable agriculture. Microorganisms 2021, 9, 1036. [Google Scholar] [CrossRef] [PubMed]
- Pang, Z.; Chen, J.; Wang, T.; Gao, C.; Li, Z.; Guo, L.; Xu, J.; Cheng, Y. Linking plant secondary metabolites and plant microbiomes: A review. Front. Plant Sci. 2021, 12, 621276. [Google Scholar] [CrossRef] [PubMed]
- Zangoui, P.; Vashishtha, K.; Mahadevan, S. Evolution of aromatic beta-glucoside utilization by successive mutational steps in Escherichia coli. J. Bacteriol. 2015, 197, 710–716. [Google Scholar] [CrossRef] [Green Version]
- Masika, P.; Sultana, N.; Afolayan, A.; Houghton, P. Isolation of two antibacterial compounds from the bark of Salix capensis. South Afr. J. Bot. 2005, 71, 441–443. [Google Scholar] [CrossRef] [Green Version]
- Cameron, R.K.; Zaton, K. Intercellular salicylic acid accumulation is important for age-related resistance in Arabidopsis to Pseudomonas syringae. Physiol. Mol. Plant Pathol. 2004, 65, 197–209. [Google Scholar] [CrossRef]
- Terada, H. Uncouplers of oxidative phosphorylation. Environ. Health Perspect. 1990, 87, 213–218. [Google Scholar] [CrossRef]
- Gross, J.; Schumacher, K.; Schmidtberg, H.; Vilcinskas, A. Protected by fumigants: Beetle perfumes in antimicrobial defense. J. Chem. Ecol. 2008, 34, 179. [Google Scholar] [CrossRef]
- Hussain, H.; Badawy, A.; Elshazly, A.; Elsayed, A.; Krohn, K.; Riaz, M.; Schulz, B. Chemical constituents and antimicrobial activity of Salix subserrata. Rec. Nat. Prod. 2011, 5, 133. [Google Scholar]
- Appel, H.M. Phenolics in ecological interactions: The importance of oxidation. J. Chem. Ecol. 1993, 19, 1521–1552. [Google Scholar] [CrossRef] [PubMed]
- Pourcel, L.; Routaboul, J.-M.; Cheynier, V.; Lepiniec, L.; Debeaujon, I. Flavonoid oxidation in plants: From biochemical properties to physiological functions. Trends Plant Sci. 2007, 12, 29–36. [Google Scholar] [CrossRef] [PubMed]
- Bittner, S. When quinones meet amino acids: Chemical, physical and biological consequences. Amino Acids 2006, 30, 205–224. [Google Scholar] [CrossRef] [PubMed]
- Friedman, M. Food browning and its prevention: An overview. J. Agric. Food Chem. 1996, 44, 631–653. [Google Scholar] [CrossRef]
- Jiménez, J.I.; Nogales, J.; García, J.L.; Díaz, E. A Genomic View of the Catabolism of Aromatic Compounds in Pseudomonas. In Handbook of Hydrocarbon and Lipid Microbiology; Springer: Berlin/Heidelberg, Germany, 2010; pp. 1297–1325. [Google Scholar] [CrossRef]
- Garcia, V.; Godoy, P.; Daniels, C.; Hurtado, A.; Ramos, J.L.; Segura, A. Functional analysis of new transporters involved in stress tolerance in Pseudomonas putida DOT-T1E. Environ. Microbiol. Rep. 2010, 2, 389–395. [Google Scholar] [CrossRef]
- Fillet, S.; Daniels, C.; Pini, C.; Krell, T.; Duque, E.; Bernal, P.; Segura, A.; Lu, D.; Zhang, X.; Ramos, J.-L. Transcriptional control of the main aromatic hydrocarbon efflux pump in Pseudomonas. Environ. Microbiol. Rep. 2012, 4, 158–167. [Google Scholar] [CrossRef]
- Segura, A.; Molina, L.; Fillet, S.; Krell, T.; Bernal, P.; Munoz-Rojas, J.; Ramos, J.-L. Solvent tolerance in Gram-negative bacteria. Curr. Opin. Biotechnol. 2012, 23, 415–421. [Google Scholar] [CrossRef]
- Ramos, J.L.; Sol Cuenca, M.; Molina-Santiago, C.; Segura, A.; Duque, E.; Gomez-Garcia, M.R.; Udaondo, Z.; Roca, A. Mechanisms of solvent resistance mediated by interplay of cellular factors in Pseudomonas putida. FEMS Microbiol. Rev. 2015, 39, 555–566. [Google Scholar] [CrossRef] [Green Version]
- Ramos-Gonzalez, M.I.; Godoy, P.; Alaminos, M.; Ben-Bassat, A.; Ramos, J.L. Physiological characterization of Pseudomonas putida DOT-T1E tolerance to p-hydroxybenzoate. Appl. Env. Microbiol. 2001, 67, 4338–4341. [Google Scholar] [CrossRef] [Green Version]
- Jagmann, N.; Brachvogel, H.P.; Philipp, B. Parasitic growth of Pseudomonas aeruginosa in co-culture with the chitinolytic bacterium Aeromonas hydrophila. Environ. Microbiol. 2010, 12, 1787–1802. [Google Scholar] [CrossRef]
- Mellefont, L.; McMeekin, T.; Ross, T. Effect of relative inoculum concentration on Listeria monocytogenes growth in co-culture. Int. J. Food Microbiol. 2008, 121, 157–168. [Google Scholar] [CrossRef] [PubMed]
- Buchanan, R.L.; Bagi, L.K. Microbial competition: Effect of culture conditions on the suppression of Listeria monocytogenes Scott A by Carnobacterium piscicola. J. Food Prot. 1997, 60, 254–261. [Google Scholar] [CrossRef] [PubMed]
- Morris, J.J.; Lenski, R.E.; Zinser, E.R. The Black Queen Hypothesis: Evolution of dependencies through adaptive gene loss. MBio 2012, 3, e00036-12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sonowal, R.; Nandimath, K.; Kulkarni, S.S.; Koushika, S.P.; Nanjundiah, V.; Mahadevan, S. Hydrolysis of aromatic beta-glucosides by non-pathogenic bacteria confers a chemical weapon against predators. Proc. Biol. Sci. 2013, 280, 20130721. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Locus Tag | Annotation | pfp ≤ 0.05 a | log2[SalOH:Glu] b |
|---|---|---|---|
| PMI19_01133 | acyl-CoA reductase | Yes | 6.2 |
| PMI19_01134 | aryl-alcohol dehydrogenase | Yes | 6.1 |
| PMI19_01136 | salicylate hydroxylase | Yes | (up) |
| PMI19_01135 | catechol 1,2-dioxygenase | Yes | (up) |
| PMI19_01184 | catechol 1,2-dioxygenase | Yes | 6.8 |
| PMI19_03945 | catechol 1,2-dioxygenase | No | (up) |
| PMI19_01186 | muconate cycloisomerase | Yes | (up) |
| PMI19_03943 | muconate cycloisomerase | - | ND |
| PMI19_01185 | muconolactone delta-isomerase | Yes | (up) |
| PMI19_03944 | muconolactone delta-isomerase | - | ND |
| PMI19_04396 | 3-oxoadipate enol-lactonase | Yes | (up) |
| PMI19_04846 | 3-oxoadipate enol-lactonase | - | ND |
| PMI19_01175 | 3-oxoadipate CoA-transferase, alpha subunit | Yes | (up) |
| PMI19_01176 | 3-oxoadipate CoA-transferase, beta subunit | No | (up) |
| PMI19_00175 | acetyl-CoA acyltransferase | - | ND |
| PMI19_01217 | 3-ketoacyl-CoA thiolase | No | 0.25 |
| PMI19_03256 | 3-ketoacyl-CoA thiolase | No | 0.73 |
| PMI19_04401 | 3-oxoadipyl-CoA thiolase | Yes | 4.6 |
| Locus Tag | Annotation | pfp ≤ 0.05 a | log2[Sal:Glu] b |
|---|---|---|---|
| EX29DRAFT_02289 | PTS system beta-glucoside-specific IIA component, Glc family (TC 4.A.1.2.2)/PTS system beta-glucoside-specific IIB component, Glc family (TC 4.A.1.2.2)/PTS system beta-glucoside-specific IIC component, Glc family (TC 4.A.1.2.2) | Yes | (up) |
| EX29DRAFT_02290 | 6-phospho-beta-glucosidase | Yes | (up) |
| EX29DRAFT_02291 | transcriptional antiterminator, BglG family | - | ND |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dahal, S.; Hurst, G.B.; Chourey, K.; Engle, N.L.; Burdick, L.H.; Morrell-Falvey, J.L.; Tschaplinski, T.J.; Doktycz, M.J.; Pelletier, D.A. Mechanism for Utilization of the Populus-Derived Metabolite Salicin by a Pseudomonas—Rahnella Co-Culture. Metabolites 2023, 13, 140. https://doi.org/10.3390/metabo13020140
Dahal S, Hurst GB, Chourey K, Engle NL, Burdick LH, Morrell-Falvey JL, Tschaplinski TJ, Doktycz MJ, Pelletier DA. Mechanism for Utilization of the Populus-Derived Metabolite Salicin by a Pseudomonas—Rahnella Co-Culture. Metabolites. 2023; 13(2):140. https://doi.org/10.3390/metabo13020140
Chicago/Turabian StyleDahal, Sanjeev, Gregory B. Hurst, Karuna Chourey, Nancy L. Engle, Leah H. Burdick, Jennifer L. Morrell-Falvey, Timothy J. Tschaplinski, Mitchel J. Doktycz, and Dale A. Pelletier. 2023. "Mechanism for Utilization of the Populus-Derived Metabolite Salicin by a Pseudomonas—Rahnella Co-Culture" Metabolites 13, no. 2: 140. https://doi.org/10.3390/metabo13020140