
Current and Future Perspectives on the Structural Identification of Small Molecules in Biological Systems

 ,
,  , ,
, ,  and
and 
Abstract
:1. Introduction
1.1. Structure Elucidation
1.2. Structure Elucidation by Nuclear Magnetic Resonance Spectroscopy (NMR)
1.3. Gas Chromatography-Mass Spectrometry (GC-MS) Based Metabolomics
1.4. Liquid Chromatography-Mass Spectrometry Based Metabolomics
1.5. Towards Standardization in Metabolomics
- Confident identifications are based upon a minimum of two different pieces of confirmatory data relative to an authentic standard.
- Putatively annotated compounds (e.g., without chemical reference standards, based upon physicochemical properties and/or spectral similarity with public/commercial spectral libraries).
- Putatively characterized compound classes (e.g., based upon characteristic physicochemical properties of a chemical class of compounds, or by spectral similarity to known compounds of a chemical class).
- Unknown compounds—although unidentified or unclassified these metabolites can still be differentiated and quantified based on spectral data.
2. Structure Elucidation by Nuclear Magnetic Resonance Spectroscopy (NMR)
2.1. NMR in Metabolomics
2.1.1. How Much Sample Is Required?
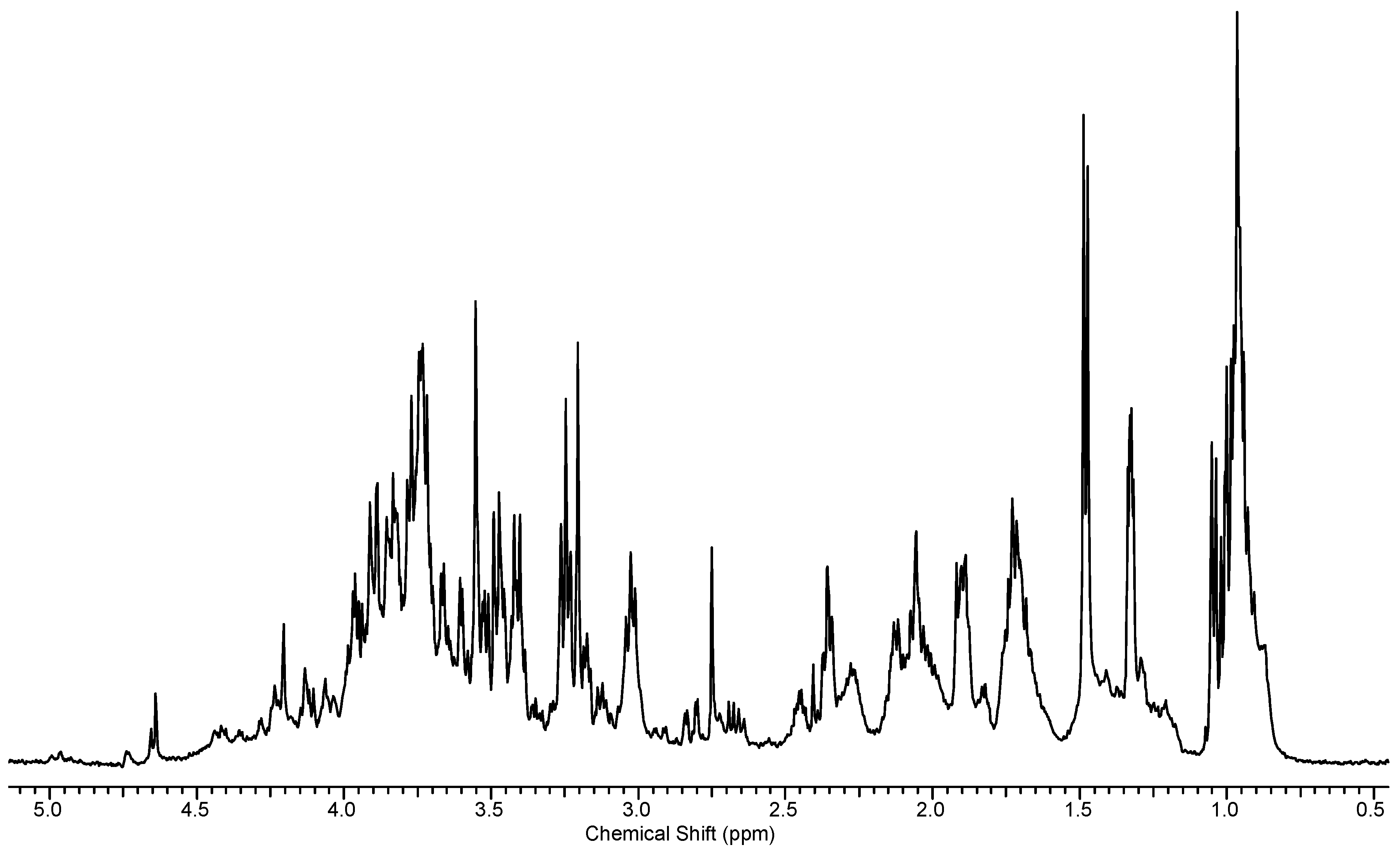
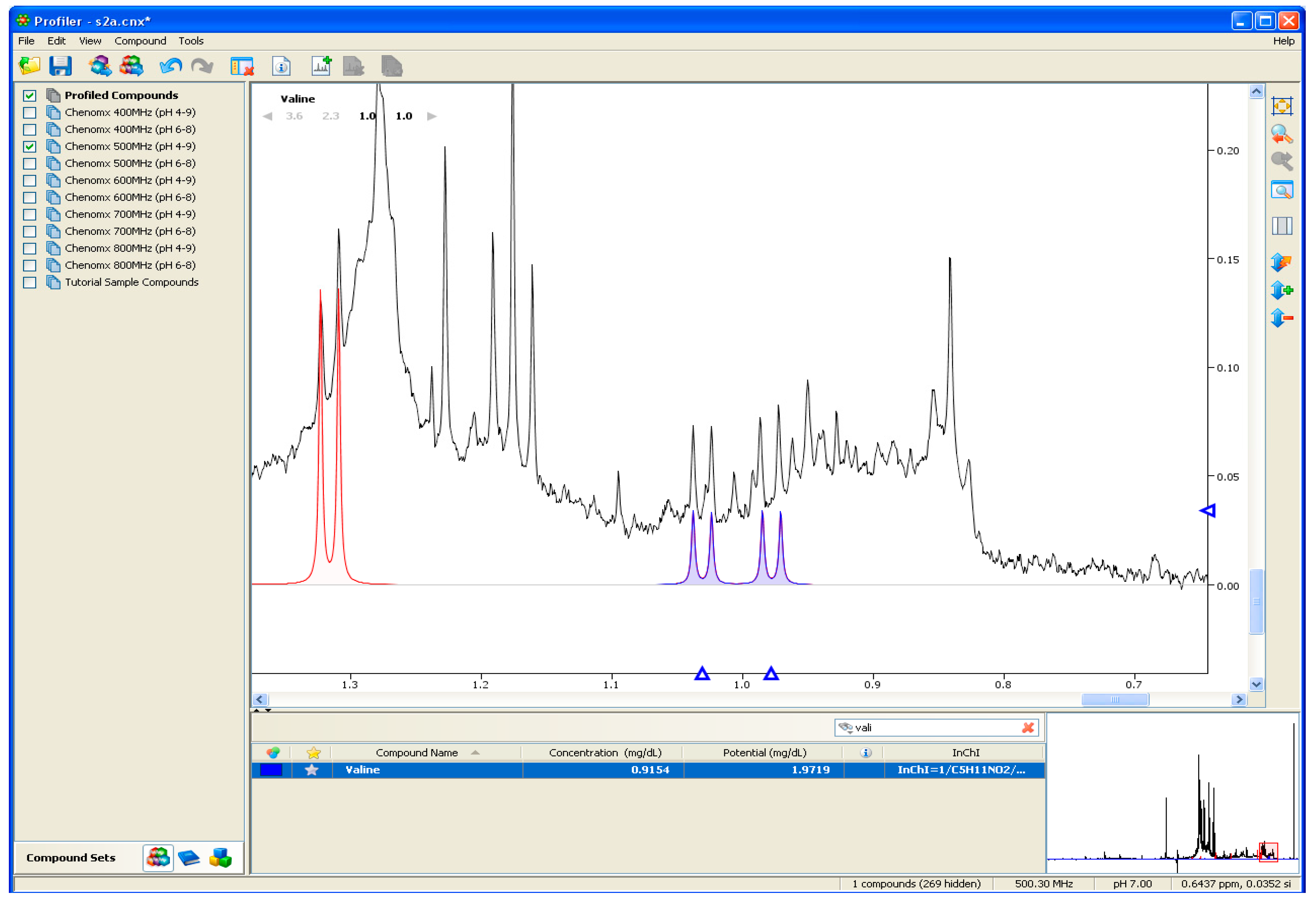
2.1.2. Assigning Peaks
2.2. Identification of Unknown Metabolites
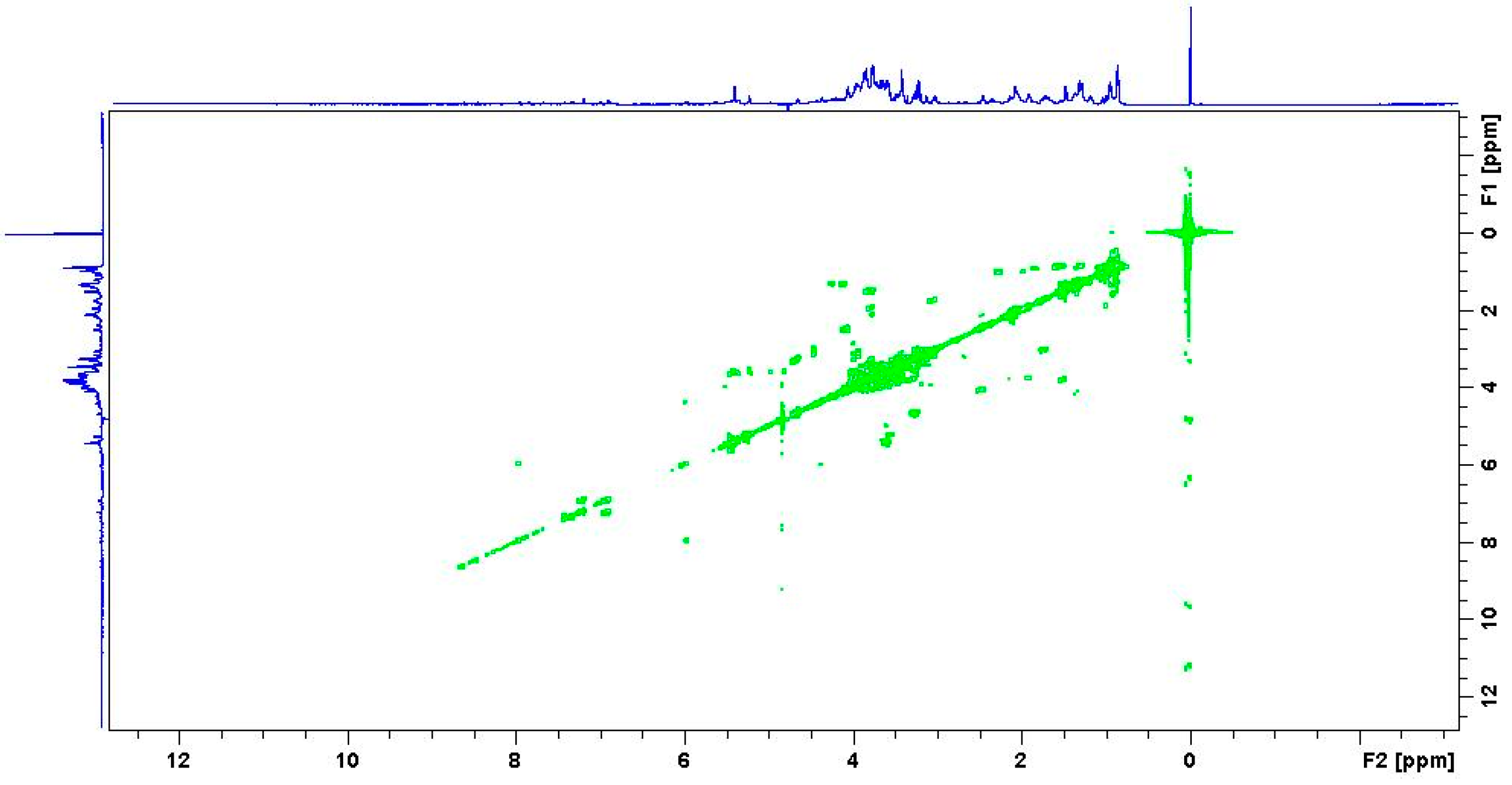
2.3. Multidimensional NMR
2.4. Computational Structure Assignment
3. Mass Spectrometry
3.1. Types of Ion Sources
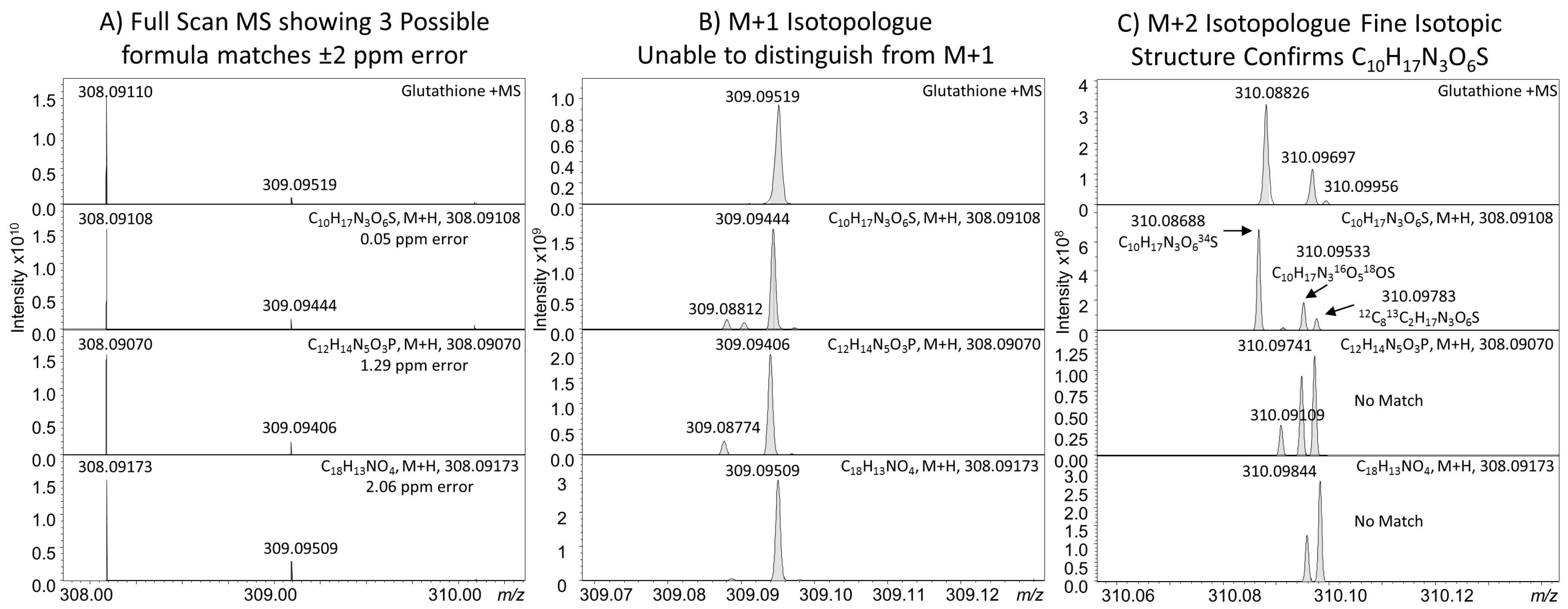
3.2. Mass Analysis
3.3. Tandem Mass Spectrometry
3.4. Chromatography-Mass Spectrometry
3.5. Ion Mobility-Mass Spectrometry
3.6. Metabolite Annotation
3.7. Organisation of MS/MS Data in Molecular Network to Enhance Annotation
4. Fourier Transform Infrared Spectroscopy (FTIR)
5. Future Perspectives
Acknowledgments
Conflicts of Interest
Abbreviations
| HSQC | Heteronuclear Single Quantum Coherence |
| COSY | Correlation spectroscopy |
| TOCSY | Total correlation spectroscopy |
| HMBC | Heteronuclear multiple-bond correlation spectroscopy |
| NOESY | Nuclear Overhauser effect spectroscopy |
| ROESY | Rotating frame nuclear Overhauser effect spectroscopy |
References
- Katz, L.; Baltz, R.H. Natural product discovery: Past, present, and future. J. Ind. Microbiol. Biotechnol. 2016, 43, 155–176. [Google Scholar] [CrossRef] [PubMed]
- David, B.; Wolfender, J.-L.; Dias, D.A. The pharmaceutical industry and natural products: Historical status and new trends. Phytochem. Rev. 2015, 14, 299–315. [Google Scholar] [CrossRef]
- Lia, D.; Misialek, J.R.; Boerwinkle, E.; Gottesman, R.F.; Sharrette, A.R.; Mosley, T.H.; Coresh, J.; Wruck, L.M.; Knopman, D.S.; Alonso, A. Plasma phospholipids and prevalence of mild cognitive impairment and/or dementia in the aric neurocognitive study (ARIC-NCS). Alzheimer Dement. Diagn. Assess. Dis. Monit. 2016, 3, 73–82. [Google Scholar] [CrossRef] [PubMed]
- González-Domínguez, R.; Rupérez, F.J.; García-Barrera, T.; Barbas, C.; Gómez-Ariza, J.L. Metabolomic-driven elucidation of serum disturbances associated with Alzheimer’s disease and mild cognitive impairment. Curr. Alzheimer Res. 2016, 13, 641–653. [Google Scholar] [CrossRef] [PubMed]
- Fiandaca, M.S.; Zhong, X.; Cheema, A.K.; Orquiza, M.H.; Chidambaram, S.; Tan, M.T.; Gresenz, C.R.; FitzGerald, K.T.; Nalls, M.A.; Singleton, A.B.; et al. Plasma 24-metabolite panel predicts preclinical transition to clinical stages of Alzheimer’s disease. Front. Neurol. 2015, 12, 237. [Google Scholar] [CrossRef] [PubMed]
- Ramirez, T.; Daneshian, M.; Kamp, H.; Bois, F.Y.; Clench, M.R.; Coen, M.; Donley, B.; Fischer, S.M.; Ekman, D.R.; Fabian, E.; et al. Metabolomics in toxicology and preclinical research. ALTEX 2013, 30, 209–225. [Google Scholar] [CrossRef] [PubMed]
- Castillo-Peinado, L.S.; Luquee de Castro, M.D. Present and foreseeable future of metabolomics in forensic analysis. Anal. Chim. Acta 2016, 925, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Breton, R.C.; Reynolds, W.F. Using nmr to identify and characterize natural products. Nat. Prod. Rep. 2013, 30, 501–524. [Google Scholar] [CrossRef] [PubMed]
- Inglese, J.; Shamu, C.E.; Guy, R.K. Reporting data from high-throughput screening of small-molecule libraries. Nat. Chem. Biol. 2007, 3, 438–441. [Google Scholar] [CrossRef] [PubMed]
- Elyashberg, M. Identification and structure elucidation by NMR spectroscopy. Trends Anal. Chem. 2015, 69, 88–97. [Google Scholar] [CrossRef]
- Gierth, P.; Codina, A.; Schumann, F.; Kovacs, H.; Kupce, E. Fast experiments for structure elucidation of small molecules: Hadamard nmr with multiple receivers. Magn. Reson. Chem. 2015, 53, 940–944. [Google Scholar] [CrossRef] [PubMed]
- Williams, R.B.; O’Neil-Johnson, M.; Williams, A.J.; Wheeler, P.; Pol, R.; Moser, A. Dereplication of natural products using minimal NMR data inputs. Org. Biomol. Chem. 2015, 13, 9957–9962. [Google Scholar] [CrossRef] [PubMed]
- Jayaseelan, K.V.; Steinbeck, C. Building blocks for automated elucidation of metabolites: Natural product-likeness for candidate ranking. BMC Bioinform. 2014, 15, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Koichi, S.; Arisaka, M.; Koshino, H.; Aoki, A.; Iwata, S.; Uno, T.; Satoh, H. Chemical structure elucidation from 13C NMR chemical shifts: Efficient data processing using bipartite matching and maximal clique algorithms. J. Chem. Inform. Model. 2014, 54, 1027–1035. [Google Scholar] [CrossRef] [PubMed]
- Plainchont, B.; Nuzillard, J.M.; Rodrigues, G.V.; Ferreira, M.J.; Scotti, M.T.; Emerenciano, V.P. New improvements in automatic structure elucidation using the LSD (logic for structure determination) and the sistemat expert systems. Nat. Prod. Commun. 2010, 5, 763–770. [Google Scholar] [PubMed]
- Available online: http://eos.Univ-reims.Fr/lsd/index_eng.html (accessed on 10 December 2016).
- Wang, T.; Shao, K.; Chu, Q.; Ren, Y.; Mu, Y.; Qu, L.; He, J.; Jin, C.; Xia, B. Automics: An integrated platform for NMR-based metabonomics spectral processing and data analysis. BMC Bioinform. 2009, 10, 1471–2105. [Google Scholar] [CrossRef] [PubMed]
- Cui, Q.; Lewis, I.A.; Hegeman, A.D.; Anderson, M.E.; Li, J.; Schulte, C.F.; Westler, W.M.; Eghbalnia, H.R.; Sussman, M.R.; Markley, J.L. Metabolite identification via the madison metabolomics consortium database. Nat. Biotechnol. 2008, 26, 162–164. [Google Scholar] [CrossRef] [PubMed]
- Bayesil. Available online: http://bayesil.ca/ (accessed on 10 December 2016).
- Metabolomics Fiehn Lab. Available online: http://fiehnlab.Ucdavis.Edu/staff/kind/metabolomics/structure_elucidation/ (accessed on 10 December 2016).
- Bax Group. Available online: https://spin.Niddk.Nih.Gov/bax/software/ (accessed on 10 December 2016).
- Kind, T.; Wohlgemuth, G.; Lee do, Y.; Lu, Y.; Palazoglu, M.; Shahbaz, S.; Fiehn, O. Fiehnlib: Mass spectral and retention index libraries for metabolomics based on quadrupole and time-of-flight gas chromatography/mass spectrometry. Anal. Chem. 2009, 15, 10038–10048. [Google Scholar] [CrossRef] [PubMed]
- Available online: http://cp.Literature.Agilent.Com/litweb/pdf/5989-8310en.Pdf (accessed on 10 December 2016).
- Kopka, J.; Schauer, N.; Krueger, S.; Birkemeyer, C.; Usadel, B.; Bergmüller, E.; Dörmann, P.; Weckwerth, W.; Gibon, Y.; Stitt, M.; et al. [email protected]: The golm metabolome database. Bioinformatics 2005, 21, 1635–1638. [Google Scholar] [CrossRef] [PubMed]
- Golm Metabolome Database. Available online: http://gmd.Mpimp-golm.Mpg.De (accessed on 10 December 2016).
- NIST. Available online: http://www.Nist.Gov (accessed on 10 December 2016).
- Available online: http://www.Sisweb.Com/software/wiley-registry.Htm (accessed on 10 December 2016).
- Roessner, U.; Luedemann, A.; Brust, D.; Fiehn, O.; Linke, T.; Willmitzer, L.; Fernie, A.R. Metabolic profiling allows comprehensive phenotyping of genetically or environmentally modified plant systems. Plant Cell 2001, 13, 11–29. [Google Scholar] [CrossRef] [PubMed]
- Boughton, B.A.; Callahan, D.L.; Silva, C.; Bowne, J.; Nahid, A.; Rupasinghe, T.; Tull, D.L.; McConville, M.J.; Bacic, A.; Roessner, U. Comprehensive profiling and quantitation of amine group containing metabolites. Anal. Chem. 2011, 83, 7523–7530. [Google Scholar] [CrossRef] [PubMed]
- Xu, F.; Zou, L.; Liu, Y.; Zhang, Z.; Ong, C.N. Enhancement of the capabilities of liquid chromatography-mass spectrometry with derivatization: General principles and applications. Mass Spectrom. Rev. 2011, 30, 1143–1172. [Google Scholar] [CrossRef] [PubMed]
- Tang, Z.; Guengerich, F.P. Dansylation of unactivated alcohols for improved mass spectral sensitivity and application to analysis of cytochrome p450 oxidation products in tissue extracts. Anal. Chem. 2010, 82, 7706–7712. [Google Scholar] [CrossRef] [PubMed]
- Dictionary of Natural Products. Available online: http://dnp.Chemnetbase.Com (accessed on 10 December 2016).
- HMDB. Available online: http://www.Hmdb.Ca (accessed on 10 December 2016).
- Scripps Center for Metabolomics. Available online: http://metlin.Scripps.Edu/ (accessed on 10 December 2016).
- MassBank. Available online: http://www.Massbank.Jp (accessed on 10 December 2016).
- Reading, E.; Munoz-Muriedas, J.; Roberts, A.D.; Dear, G.J.; Robinson, C.V.; Beaumont, C. Elucidation of drug metabolite structural isomers using molecular modeling coupled with ion mobility mass spectrometry. Anal. Chem. 2016, 88, 2273–2280. [Google Scholar] [CrossRef] [PubMed]
- Eugster, P.J.; Boccard, J.; Debrus, B.; Breant, L.; Wolfender, J.L.; Martel, S.; Carrupt, P.A. Retention time prediction for dereplication of natural products (CXHYOZ) in LC-MS metabolite profiling. Phytochemistry 2014, 108, 196–207. [Google Scholar] [CrossRef] [PubMed]
- Abate-Pella, D.; Freund, D.M.; Ma, Y.; Simon-Manso, Y.; Hollender, J.; Broeckling, C.D.; Huhman, D.V.; Krokhin, O.V.; Stoll, D.R.; Hegeman, A.D.; et al. Retention projection enables accurate calculation of liquid chromatographic retention times across labs and methods. J. Chromatogr. A 2015, 18, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Sumner, L.W.; Amberg, A.; Barrett, D.; Beale, M.H.; Beger, R.; Daykin, C.A.; Fan, T.W.-M.; Fiehn, O.; Goodacre, R.; Griffin, J.L.; et al. Proposed minimum reporting standards for chemical analysis. Metabolomics 2007, 3, 211–221. [Google Scholar] [CrossRef] [PubMed]
- Salek, R.M.; Neumann, S.; Schober, D.; Hummel, J.; Billiau, K.; Kopka, J.; Correa, E.; Reijmers, T.; Rosato, A.; Tenori, L.; et al. Coordination of standards in metabolomics (COSMOS): Facilitating integrated metabolomics data access. Metabolomics 2015, 11, 1587–1597. [Google Scholar] [CrossRef] [PubMed]
- Data producers deserve citation credit. Available online: http://www.nature.com/ng/journal/v41/n10/full/ng1009-1045.html (accessed on 10 December 2016).
- Hanson, L.G. Is quantum mechanics necessary for understanding magnetic resonance? Concepts Magn. Reson. Part A 2008, 32, 329–340. [Google Scholar] [CrossRef]
- Keeler, J. Understanding NMR Spectroscopy, 1 ed.; John Wiley and Sons: Chichester, UK, 2005. [Google Scholar]
- Levitt, M.H. Spin Dynamics: Basics of Nuclear Magnetic Resonance, 2nd ed.; Wiley: London, UK, 2008; p. 740. [Google Scholar]
- Wolfender, J.L.; Rudaz, S.; Choi, Y.H.; Kim, H.K. Plant metabolomics: From holistic data to relevant biomarkers. Curr. Med. Chem 2013, 20, 1056–1090. [Google Scholar] [CrossRef] [PubMed]
- Oliver, S.G.; Winson, M.K.; Kell, D.B.; Baganz, F. Systematic functional analysis of the yeast genome. Trends Biotechnol. 1998, 16, 373–378. [Google Scholar] [CrossRef]
- Nicholson, J.K.; Osborn, D. Kidney lesions in pelagic seabirds with high tissue levels of cadmium and mercury. J. Zool. 1983, 200, 99–118. [Google Scholar] [CrossRef]
- Jones, O.A.H.; Cheung, V.L. An introduction to metabolomics and its potential application in veterinary science. Comp. Med. 2007, 57, 436–442. [Google Scholar] [PubMed]
- Lindon, J.C.; Nicholson, J.K. Spectroscopic and statistical techniques for information recovery in metabonomics and metabolomics. Annu. Rev. Anal. Chem. 2008, 1, 45–69. [Google Scholar] [CrossRef] [PubMed]
- Salek, R.M.; Maguire, M.L.; Bentley, E.; Rubtsov, D.V.; Hough, T.; Cheeseman, M.; Nunez, D.; Sweatman, B.C.; Haselden, J.N.; Cox, R.D.; et al. A metabolomic comparison of urinary changes in type 2 diabetes in mouse, rat, and human. Physiol. Genom. 2007, 29, 99–108. [Google Scholar] [CrossRef] [PubMed]
- Heather, L.C.; Wang, X.; West, J.A.; Griffin, J.L. A practical guide to metabolomic profiling as a discovery tool for human heart disease. J. Mol. Cell. Cardiol. 2013, 55, 2–11. [Google Scholar] [CrossRef] [PubMed]
- Jones, O.A.; Hügel, H.M. Bridging the gap: Basic metabolomics methods for natural product chemistry. Methods Mol. Biol. 2013, 1055, 245–266. [Google Scholar] [PubMed]
- Bothwell, J.H.; Griffin, J.L. An introduction to biological nuclear magnetic resonance spectroscopy. Biol. Rev. Camb. Philos. Soc. 2011, 86, 493–510. [Google Scholar] [CrossRef] [PubMed]
- Molinski, T.F. NMR of natural products at the ‘nanomole-scale’. Nat. Prod. Rep. 2010, 27, 321–329. [Google Scholar] [CrossRef] [PubMed]
- Kwan, A.H.; Mobli, M.; Gooley, P.R.; King, G.F.; Mackay, J.P. Macromolecular nmr spectroscopy for the non-spectroscopist. FEBS J. 2011, 278, 687–703. [Google Scholar] [CrossRef] [PubMed]
- Rossini, A.J.; Widdifield, C.M.; Zagdoun, A.; Lelli, M.; Schwarzwalder, M.; Coperet, C.; Lesage, A.; Emsley, L. Dynamic nuclear polarization enhanced NMR spectroscopy for pharmaceutical formulations. J. Am. Chem. Soc. 2014, 136, 2324–2334. [Google Scholar] [CrossRef] [PubMed]
- Flogel, U.; Jacoby, C.; Godecke, A.; Schrader, J. In vivo 2D mapping of impaired murine cardiac energetics in no-induced heart failure. Magn. Reson. Med. 2007, 57, 50–58. [Google Scholar] [CrossRef] [PubMed]
- Duarte, I.F.; Stanley, E.G.; Holmes, E.; Lindon, J.C.; Gil, A.M.; Tang, H.; Ferdinand, R.; McKee, C.G.; Nicholson, J.K.; Vilca-Melendez, H.; et al. Metabolic assessment of human liver transplants from biopsy samples at the donor and recipient stages using high-resolution magic angle spinning 1H NMR spectroscopy. Anal. Chem. 2005, 77, 5570–5578. [Google Scholar] [CrossRef] [PubMed]
- Wishart, D.S.; Jewison, T.; Guo, A.C.; Wilson, M.; Knox, C.; Liu, Y.; Djoumbou, Y.; Mandal, R.; Aziat, F.; Dong, E.; et al. HMDB 3.0—The human metabolome database in 2013. Nucleic Acids Res. 2013, 41, D801–D807. [Google Scholar] [CrossRef] [PubMed]
- Jones, O.A.H.; Sdepanian, S.; Lofts, S.; Svendsen, C.; Spurgeon, D.J.; Maguire, M.L.; Griffin, J.L. Metabolomic analysis of soil communities can be used for pollution assessment. Environ. Sci. Technol. 2014, 33, 61–64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gomez, J.; Brezmes, J.; Mallol, R.; Rodriguez, M.A.; Vinaixa, M.; Salek, R.M.; Correig, X.; Canellas, N. Dolphin: A tool for automatic targeted metabolite profiling using 1D and 2D 1H-NMR data. Anal. Bioanal. Chem. 2014, 406, 7967–7976. [Google Scholar] [CrossRef] [PubMed]
- Ravanbakhsh, S.; Liu, P.; Bjorndahl, T.C.; Mandal, R.; Grant, J.R.; Wilson, M.; Eisner, R.; Sinelnikov, I.; Hu, X.; Luchinat, C.; et al. Accurate, fully-automated NMR spectral profiling for metabolomics. PLoS ONE 2015, 10, e0124219. [Google Scholar] [CrossRef] [PubMed]
- Gillard, J.; Frenkel, J.; Devos, V.; Sabbe, K.; Paul, C.; Rempt, M.; Inze, D.; Pohnert, G.; Vuylsteke, M.; Vyverman, W. Metabolomics enables the structure elucidation of a diatom sex pheromone. Angew. Chem. Int. Ed. Engl. 2013, 52, 854–857. [Google Scholar] [CrossRef] [PubMed]
- Nakabayashi, R.; Kusano, M.; Kobayashi, M.; Tohge, T.; Yonekura-Sakakibara, K.; Kogure, N.; Yamazaki, M.; Kitajima, M.; Saito, K.; Takayama, H. Metabolomics-oriented isolation and structure elucidation of 37 compounds including two anthocyanins from Arabidopsis thaliana. Phytochemistry 2009, 70, 1017–1029. [Google Scholar] [CrossRef] [PubMed]
- Kwan, E.E.; Huang, S.G. Structural elucidation with nmr spectroscopy: Practical strategies for organic chemists. Eur. J. Org. Chem. 2008, 2008, 2671–2688. [Google Scholar] [CrossRef]
- Spraul, M.; Freund, A.S.; Nast, R.E.; Withers, R.S.; Maas, W.E.; Corcoran, O. Advancing nmr sensitivity for LC-NMR-MS using a cryoflow probe: Application to the analysis of acetaminophen metabolites in urine. Anal. Chem. 2003, 75, 1536–1541. [Google Scholar] [CrossRef] [PubMed]
- Dias, D.; Urban, S. Phytochemical analysis of the southern australian marine alga, Plocamium mertensii using HPLC-NMR. Phytochem. Anal. 2008, 19, 453–470. [Google Scholar] [CrossRef] [PubMed]
- Urban, S.; Dias, D.A. NMR spectroscopy: Structure elucidation of cycloelatanene A: A natural product case study. In Metabolomics Tools for Natural Product Discovery: Methods and Protocols; Roessner, U., Dias, A.D., Eds.; Humana Press: Totowa, NJ, USA, 2013; pp. 99–116. [Google Scholar]
- Analytical Chemistry—A Guide to Proton Nuclear Magnetic Resonance (NMR). Available online: http://www.Compoundchem.Com/2015/02/24/proton-nmr/ (accessed on 10 December 2016).
- Analytical Chemistry—A Guide to 13-C Nuclear Magnetic Resonance (NMR). Available online: http://www.Compoundchem.Com/2015/04/07/carbon-13-nmr/ (accessed on 10 December 2016).
- Wheeler, P.; Hayward, S.; Elyashberg, M. Computer-assisted structure elucidation in routine analysis. Am. Lab. 2016, 48, 12–14. [Google Scholar]
- Bisson, J.; Simmler, C.; Chen, S.-N.; Friesen, J.B.; Lankin, D.C.; McAlpine, J.B.; Pauli, G.F. Dissemination of original NMR data enhances reproducibility and integrity in chemical research. Natl. Prod. Rep. 2016, 33, 1028–1033. [Google Scholar] [CrossRef] [PubMed]
- Kind, T.; Fiehn, O. Seven golden rules for heuristic filtering of molecular formulas obtained by accurate mass spectrometry. BMC Bioinform. 2007, 8, 105. [Google Scholar] [CrossRef] [PubMed]
- Wolfender, J.-L.; Marti, G.; Thomas, A.; Bertrand, S. Current approaches and challenges for the metabolite profiling of complex natural extracts. J. Chromatogr. A 2015, 1382, 136–164. [Google Scholar] [CrossRef] [PubMed]
- Schymanski, E.L.; Jeon, J.; Gulde, R.; Fenner, K.; Ruff, M.; Singer, H.; Hollender, J. Identifying small molecules via high resolution mass spectrometry: Communicating confidence. Environ. Sci. Technol. 2014, 48, 2097–2098. [Google Scholar] [CrossRef] [PubMed]
- Glish, G.L.; Vachet, R.W. The basics of mass spectrometry in the twenty-first century. Nat. Rev. Drug Discov. 2003, 2, 140–150. [Google Scholar] [CrossRef] [PubMed]
- Palma, P.; Famiglini, G.; Trufelli, H.; Pierini, E.; Termopoli, V.; Cappiello, A. Electron ionization in LC-MS: Recent developments and applications of the direct-EI LC-MS interface. Anal. Bioanal. Chem. 2011, 399, 2683–2693. [Google Scholar] [CrossRef] [PubMed]
- Ho, C.S.; Lam, C.W.; Chan, M.H.; Cheung, R.C.; Law, L.K.; Lit, L.C.; Ng, K.F.; Suen, M.W.; Tai, H.L. Electrospray ionisation mass spectrometry: Principles and clinical applications. Clin. Biochem. Rev. 2003, 24, 3–12. [Google Scholar] [PubMed]
- Hoffmann, E.D.; Stroobant, V. Mass Spectrometry: Principles and Applications, 3rd ed.; Wiley: New York, NY, USA, 2007; p. 502. [Google Scholar]
- Kind, T.; Fiehn, O. Advances in structure elucidation of small molecules using mass spectrometry. Bioanal. Rev. 2010, 2, 23–60. [Google Scholar] [CrossRef] [PubMed]
- Stahnke, H.; Kittlaus, S.; Kempe, G.; Alder, L. Reduction of matrix effects in liquid chromatography-electrospray ionization-mass spectrometry by dilution of the sample extracts: How much dilution is needed? Anal. Chem. 2012, 84, 1474–1482. [Google Scholar] [CrossRef] [PubMed]
- Konermann, L.; Ahadi, E.; Rodriguez, A.D.; Vahidi, S. Unraveling the mechanism of electrospray ionization. Anal. Chem. 2013, 85, 2–9. [Google Scholar] [CrossRef] [PubMed]
- Lewis, J.K.; Wei, J.; Siuzdak, G. Matrix-assisted laser desorption/ionization mass spectrometry in peptide and protein analysis. Encycl. Anal. Chem. 2006. [Google Scholar] [CrossRef]
- Shroff, R.; Rulisek, L.; Doubsky, J.; Svatos, A. Acid-base-driven matrix-assisted mass spectrometry for targeted metabolomics. Proc. Natl. Acad. Sci. USA 2009, 106, 10092–10096. [Google Scholar] [CrossRef] [PubMed]
- Gholipour, Y.; Erra-Balsells, R.; Nonami, H. Nanoparticles applied to mass spectrometry metabolomics and pesticide residue analysis. In Nanotechnology and Plant Sciences: Nanoparticles and Their Impact on Plants; Siddiqui, H.M., Al-Whaibi, H.M., Mohammad, F., Eds.; Springer: Cham, Switzerland, 2015; pp. 289–303. [Google Scholar]
- Gholipour, Y.; Giudicessi, S.L.; Nonami, H.; Erra-Balsells, R. Diamond, titanium dioxide, titanium silicon oxide, and barium strontium titanium oxide nanoparticles as matrixes for direct matrix-assisted laser desorption/ionization mass spectrometry analysis of carbohydrates in plant tissues. Anal. Chem. 2010, 82, 5518–5526. [Google Scholar] [CrossRef] [PubMed]
- Hamm, G.; Carré, V.; Poutaraud, A.; Maunit, B.; Frache, G.; Merdinoglu, D.; Muller, J.F. Determination and imaging of metabolites from vitis vinifera leaves by laser desorption/ionisation time-of-flight mass spectrometry. Rapid Commun. Mass Spectrom. 2010, 24, 335–342. [Google Scholar] [CrossRef] [PubMed]
- Wiegelmann, M.; Soltwisch, J.; Jaskolla, T.W.; Dreisewerd, K. Matching the laser wavelength to the absorption properties of matrices increases the ion yield in UV-MALDI mass spectrometry. Anal. Bioanal. Chem. 2013, 405, 6925–6932. [Google Scholar] [CrossRef] [PubMed]
- Cajka, T.; Riddellova, K.; Tomaniova, M.; Hajslova, J. Ambient mass spectrometry employing a dart ion source for metabolomic fingerprinting/profiling: A powerful tool for beer origin recognition. Metabolomics 2011, 7, 500–508. [Google Scholar] [CrossRef]
- Park, H.M.; Kim, H.J.; Jang, Y.P.; Kim, S.Y. Direct analysis in real time mass spectrometry (DART-MS) analysis of skin metabolome changes in the ultraviolet B-induced mice. Biomol. Ther. 2013, 21, 470–475. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Seo, Y.T.; Park, S.-I.; Jeong, S.H.; Kim, M.K.; Jang, Y.P. DART–TOF–MS based metabolomics study for the discrimination analysis of geographical origin of angelica gigas roots collected from Korea and China. Metabolomics 2015, 11, 64–70. [Google Scholar] [CrossRef]
- Nagao, H.; Miki, S.; Toyoda, M. Development of a miniaturized multi-turn time-of-flight mass spectrometer with a pulsed fast atom bombardment ion source. Eur. J. Mass Spectrom. 2014, 20, 215–220. [Google Scholar] [CrossRef] [PubMed]
- Miura, D.; Tsuji, Y.; Takahashi, K.; Wariishi, H.; Saito, K. A strategy for the determination of the elemental composition by fourier transform ion cyclotron resonance mass spectrometry based on isotopic peak ratios. Anal. Chem. 2010, 82, 5887–5891. [Google Scholar] [CrossRef] [PubMed]
- De Hoffmann, E. Tandem mass spectrometry: A primer. J. Mass Spectrom. 1996, 31, 129–137. [Google Scholar] [CrossRef]
- Sleno, L.; Volmer, D.A. Ion activation methods for tandem mass spectrometry. J. Mass Spectrom. 2004, 39, 1091–1112. [Google Scholar] [CrossRef] [PubMed]
- Olsen, J.V.; Macek, B.; Lange, O.; Makarov, A.; Horning, S.; Mann, M. Higher-energy C-trap dissociation for peptide modification analysis. Nat. Meth. 2007, 4, 709–712. [Google Scholar] [CrossRef] [PubMed]
- Syka, J.E.; Coon, J.J.; Schroeder, M.J.; Shabanowitz, J.; Hunt, D.F. Peptide and protein sequence analysis by electron transfer dissociation mass spectrometry. Proc. Natl. Acad. Sci. USA 2004, 101, 9528–9533. [Google Scholar] [CrossRef] [PubMed]
- Zubarev, R.A.; Horn, D.M.; Fridriksson, E.K.; Kelleher, N.L.; Kruger, N.A.; Lewis, M.A.; Carpenter, B.K.; McLafferty, F.W. Electron capture dissociation for structural characterization of multiply charged protein cations. Anal. Chem. 2000, 72, 563–573. [Google Scholar] [CrossRef] [PubMed]
- Gauthier, J.W.; Trautman, T.R.; Jacobson, D.B. Sustained off-resonance irradiation for collision-activated dissociation involving fourier transform mass spectrometry. Collision-activated dissociation technique that emulates infrared multiphoton dissociation. Anal. Chim. Acta 1991, 246, 211–225. [Google Scholar] [CrossRef]
- Thomas, M.C.; Mitchell, T.W.; Harman, D.G.; Deeley, J.M.; Nealon, J.R.; Blanksby, S.J. Ozone-induced dissociation: Elucidation of double bond position within mass-selected lipid ions. Anal. Chem. 2008, 80, 303–311. [Google Scholar] [CrossRef] [PubMed]
- Tsugawa, H.; Cajka, T.; Kind, T.; Ma, Y.; Higgins, B.; Ikeda, K.; Kanazawa, M.; VanderGheynst, J.; Fiehn, O.; Arita, M. MS-DIAL: Data-independent MS/MS deconvolution for comprehensive metabolome analysis. Nat. Methods 2015, 12, 523–526. [Google Scholar] [CrossRef] [PubMed]
- Lanucara, F.; Holman, S.W.; Gray, C.J.; Eyers, C.E. The power of ion mobility-mass spectrometry for structural characterization and the study of conformational dynamics. Nat. Chem. 2014, 6, 281–294. [Google Scholar] [CrossRef] [PubMed]
- Cumeras, R.; Figueras, E.; Davis, C.E.; Baumbach, J.I.; Gràcia, I. Review on ion mobility spectrometry. Part 1: Current instrumentation. Analyst 2015, 140, 1376–1390. [Google Scholar] [CrossRef] [PubMed]
- Silveira, J.A.; Ridgeway, M.E.; Park, M.A. High resolution trapped ion mobility spectrometery of peptides. Anal. Chem. 2014, 86, 5624–5627. [Google Scholar] [CrossRef] [PubMed]
- Adams, K.J.; Montero, D.; Aga, D.; Fernandez-Lima, F. Isomer separation of polybrominated diphenyl ether metabolites using NANOESI-TIMS-MS. Int. J. Ion Mobil. Spectrom. 2016, 19, 69–76. [Google Scholar] [CrossRef] [PubMed]
- Lipidomics Gateway. Available online: http://www.Lipidmaps.Org (accessed on 10 December 2016).
- mzCloud. Available online: http://www.Mzcloud.Org (accessed on 10 December 2016).
- Grant Lab. Available online: http://metabolomics.Pharm.Uconn.Edu/iimdb (accessed on 10 December 2016).
- LipidBlast. Available online: http://fiehnlab.Ucdavis.Edu/projects/lipidblast (accessed on 10 December 2016).
- Allen, F.; Greiner, R.; Wishart, D. Competitive fragmentation modeling of ESI-MS/MS spectra for putative metabolite identification. Metabolomics 2015, 11, 98–110. [Google Scholar] [CrossRef]
- Ridder, L.; van der Hooft, J.J.; Verhoeven, S. Automatic compound annotation from mass spectrometry data using magma. Mass Spectrom. 2014, 3, S0033. [Google Scholar] [CrossRef] [PubMed]
- Duhrkop, K.; Shen, H.; Meusel, M.; Rousu, J.; Bocker, S. Searching molecular structure databases with tandem mass spectra using CSI:Fingerid. Proc. Natl. Acad. Sci. USA 2015, 112, 12580–12585. [Google Scholar] [CrossRef] [PubMed]
- Ruttkies, C.; Schymanski, E.L.; Wolf, S.; Hollender, J.; Neumann, S. Metfrag relaunched: Incorporating strategies beyond in silico fragmentation. J. Cheminform. 2016, 8, 16–115. [Google Scholar] [CrossRef] [PubMed]
- Critical Assessment of Small Molecule Identification. Available online: http://casmi-contest.Org/ (accessed on 10 December 2016).
- Allard, P.M.; Peresse, T.; Bisson, J.; Gindro, K.; Marcourt, L.; Pham, V.C.; Roussi, F.; Litaudon, M.; Wolfender, J.L. Integration of molecular networking and in-silico MS/MS fragmentation for natural products dereplication. Anal. Chem. 2016, 88, 3317–3323. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Carver, J.J.; Phelan, V.V.; Sanchez, L.M.; Garg, N.; Peng, Y.; Nguyen, D.D.; Watrous, J.; Kapono, C.A.; Luzzatto-Knaan, T.; et al. Sharing and community curation of mass spectrometry data with global natural products social molecular networking. Nat. Biotechnol. 2016, 34, 828–837. [Google Scholar] [CrossRef] [PubMed]
- Cabral, R.S.; Allard, P.M.; Marcourt, L.; Young, M.C.; Queiroz, E.F.; Wolfender, J.L. Targeted isolation of indolopyridoquinazoline alkaloids from conchocarpus fontanesianus based on molecular networks. J. Nat. Prod. 2016, 79, 2270–2278. [Google Scholar] [CrossRef] [PubMed]
- Mariey, L.; Signolle, J.P.; Amiel, C.; Travert, J. Discrimination, classification, identification of microorganisms using FTIR spectroscopy and chemometrics. Vib. Spectrosc. 2001, 26, 151–159. [Google Scholar] [CrossRef]
- Jackson, M.; Mantsch, H.H. The use and misuse of FTIR spectroscopy in the determination of protein structure. Crit. Rev. Biochem. Mol. Biol. 1995, 30, 95–120. [Google Scholar] [CrossRef] [PubMed]
- Nurrulhidayah, A.F.; Che Man, Y.B.; Amin, I.; Arieff Salleh, R.; Farawahidah, M.Y.; Shuhaimi, M.; Khatib, A. FTIR-ATR spectroscopy based metabolite fingerprinting as a direct determination of butter adulterated with lard. Int. J. Food Prop. 2015, 18, 372–379. [Google Scholar] [CrossRef]
- Adt, I.; Toubas, D.; Pinon, J.M.; Manfait, M.; Sockalingum, G.D. FTIR spectroscopy as a potential tool to analyse structural modifications during morphogenesis of Candida albicans. Arch. Microbiol. 2006, 185, 277–285. [Google Scholar] [CrossRef] [PubMed]
- Del Bove, M.; Lattanzi, M.; Rellini, P.; Pelliccia, C.; Fatichenti, F.; Cardinali, G. Comparison of molecular and metabolomic methods as characterization tools of debaryomyces hansenii cheese isolates. Food Microbiol. 2009, 26, 453–459. [Google Scholar] [CrossRef] [PubMed]
- Roscini, L.; Corte, L.; Antonielli, L.; Rellini, P.; Fatichenti, F.; Cardinali, G. Influence of cell geometry and number of replicas in the reproducibility of whole cell FTIR analysis. Analyst 2010, 135, 2099–2105. [Google Scholar] [CrossRef] [PubMed]
- Szeghalmi, A.; Kaminskyj, S.; Gough, K.M. A synchrotron ftir microspectroscopy investigation of fungal hyphae grown under optimal and stressed conditions. Anal. Bioanal. Chem. 2007, 387, 1779–1789. [Google Scholar] [CrossRef] [PubMed]
- Corte, L.; Tiecco, M.; Roscini, L.; de Vincenzi, S.; Colabella, C.; Germani, R.; Tascini, C.; Cardinali, G. FTIR metabolomic fingerprint reveals different modes of action exerted by structural variants of n-alkyltropinium bromide surfactants on Escherichia coli and Listeria innocua cells. PLoS ONE 2015, 10, e0115275. [Google Scholar] [CrossRef] [PubMed]
- Kamnev, A.A. FTIR spectroscopic studies of bacterial cellular responses to environmental factors, plant-bacterial interactions and signalling. J. Spectrosc. 2008, 22, 83–95. [Google Scholar] [CrossRef]
- Corte, L.; Roscini, L.; Zadra, C.; Antonielli, L.; Tancini, B.; Magini, A.; Emiliani, C.; Cardinali, G. Effect of ph on potassium metabisulphite biocidic activity against yeast and human cell cultures. Food Chem. 2012, 134, 1327–1336. [Google Scholar] [CrossRef] [PubMed]
- Corte, L.; Dell’Abate, M.T.; Magini, A.; Migliore, M.; Felici, B.; Roscini, L.; Sardella, R.; Tancini, B.; Emiliani, C.; Cardinali, G. Assessment of safety and efficiency of nitrogen organic fertilizers from animal-based protein hydrolysates—A laboratory multidisciplinary approach. J. Sci. Food Agric. 2014, 94, 235–245. [Google Scholar] [CrossRef] [PubMed]
- Burke, M.; Small, D.M.; Antolasic, F.; Hughes, J.G.; Spencer, M.J.S.; Blanch, E.W.; Jones, O.A.H. Infrared spectroscopy-based metabolomic analysis for the detection of preharvest sprouting in grain. Cereal Chem. 2016, 93, 444–449. [Google Scholar] [CrossRef]
- Kummerle, M.; Scherer, S.; Seiler, H. Rapid and reliable identification of food-borne yeasts by fourier-transform infrared spectroscopy. Appl. Environ. Microbiol. 1998, 64, 2207–2214. [Google Scholar] [PubMed]
- Winson, M.K.; Goodacre, R.; Timmins, É.M.; Jones, A.; Alsberg, B.K.; Woodward, A.M.; Rowland, J.J.; Kell, D.B. Diffuse reflectance absorbance spectroscopy taking in chemometrics (drastic). A hyperspectral FT-IR-based approach to rapid screening for metabolite overproduction. Anal. Chim. Acta 1997, 348, 273–282. [Google Scholar] [CrossRef]
- Olson, E.S.; Stanley, D.C.; Gallagher, J.R. Characterization of intermediates in the microbial desulfurization of dibenzothiophene. Energy Fuels 1993, 7, 159–164. [Google Scholar] [CrossRef]
- Guitton, J.; Desage, M.; Alamercery, S.; Dutruch, L.; Dautraix, S.; Perdrix, J.P.; Brazier, J.L. Gas chromatographic-mass spectrometry and gas chromatographic-fourier transform infrared spectroscopy assay for the simultaneous identification of fentanyl metabolites. J. Chromatogr. B Biomed. Sci. Appl. 1997, 693, 59–70. [Google Scholar] [CrossRef]
- Lewis, P.D.; Lewis, K.E.; Ghosal, R.; Bayliss, S.; Lloyd, A.J.; Wills, J.; Godfrey, R.; Kloer, P.; Mur, L.A. Evaluation of FTIR spectroscopy as a diagnostic tool for lung cancer using sputum. BMC Cancer 2010, 10, 640. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Himmelreich, U.; Somorjai, R.L.; Dolenko, B.; Lee, O.C.; Daniel, H.M.; Murray, R.; Mountford, C.E.; Sorrell, T.C. Rapid identification of candida species by using nuclear magnetic resonance spectroscopy and a statistical classification strategy. Appl. Environ. Microbiol. 2003, 69, 4566–4574. [Google Scholar] [CrossRef] [PubMed]
- Psychogios, N.; Hau, D.D.; Peng, J.; Guo, A.C.; Mandal, R.; Bouatra, S.; Sinelnikov, I.; Krishnamurthy, R.; Eisner, R.; Gautam, B.; et al. The human serum metabolome. PLoS ONE 2011, 6, e16957. [Google Scholar] [CrossRef] [PubMed]
- Skogerson, K.; Runnebaum, R.; Wohlgemuth, G.; de Ropp, J.; Heymann, H.; Fiehn, O. Comparison of gas chromatography-coupled time-of-flight mass spectrometry and 1H nuclear magnetic resonance spectroscopy metabolite identification in white wines from a sensory study investigating wine body. J. Agric. Food Chem. 2009, 57, 6899–6907. [Google Scholar] [CrossRef] [PubMed]
- Bouatra, S.; Aziat, F.; Mandal, R.; Guo, A.C.; Wilson, M.R.; Knox, C.; Bjorndahl, T.C.; Krishnamurthy, R.; Saleem, F.; Liu, P.; et al. The human urine metabolome. PLoS ONE 2013, 8, e73076. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schymanski, E.L.; Singer, H.P.; Slobodnik, J.; Ipolyi, I.M.; Oswald, P.; Krauss, M.; Schulze, T.; Haglund, P.; Letzel, T.; Grosse, S.; et al. Non-target screening with high-resolution mass spectrometry: Critical review using a collaborative trial on water analysis. Anal. Bioanal. Chem. 2015, 407, 6237–6255. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Chen, D.; Wang, N.; Zhang, T.; Zhou, R.; Huan, T.; Lu, Y.; Su, X.; Xie, Q.; Li, L.; et al. Development of high-performance chemical isotope labeling LC-MS for profiling the human fecal metabolome. Anal. Chem. 2015, 87, 829–836. [Google Scholar] [CrossRef] [PubMed]
- Markley, J.L.; Bruschweiler, R.; Edison, A.S.; Eghbalnia, H.R.; Powers, R.; Raftery, D.; Wishart, D.S. The future of NMR-based metabolomics. Curr. Opin. Biotechnol. 2016, 43, 34–40. [Google Scholar] [CrossRef] [PubMed]
- Theis, T.; Ortiz, G.X., Jr.; Logan, A.W.; Claytor, K.E.; Feng, Y.; Huhn, W.P.; Blum, V.; Malcolmson, S.J.; Chekmenev, E.Y.; Wang, Q.; et al. Direct and cost-efficient hyperpolarization of long-lived nuclear spin states on universal 15N2-diazirine molecular tags. Sci. Adv. 2016, 2, e1501438. [Google Scholar] [CrossRef] [PubMed]
- Onjiko, R.M.; Moody, S.A.; Nemes, P. Single-cell mass spectrometry reveals small molecules that affect cell fates in the 16-cell embryo. Proc. Natl. Acad. Sci. USA 2015, 112, 6545–6550. [Google Scholar] [CrossRef] [PubMed]
- Da Silva, R.R.; Dorrestein, P.C.; Quinn, R.A. Illuminating the dark matter in metabolomics. Proc. Natl. Acad. Sci. USA 2015, 112, 12549–12550. [Google Scholar] [CrossRef] [PubMed]
- Markley, J.L.; Ulrich, E.L.; Berman, H.M.; Henrick, K.; Nakamura, H.; Akutsu, H. Biomagresbank (BMRB) as a partner in the worldwide protein data bank (WWPDB): New policies affecting biomolecular NMR depositions. J. Biomol. NMR 2008, 40, 153–155. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.A.; O’Maille, G.; Want, E.J.; Qin, C.; Trauger, S.A.; Brandon, T.R.; Custodio, D.E.; Abagyan, R.; Siuzdak, G. Metlin: A metabolite mass spectral database. Ther. Drug Monit. 2005, 27, 747–751. [Google Scholar] [CrossRef] [PubMed]
- Massbank of North America (MoNA). Available online: http://mona.Fiehnlab.Ucdavis.Edu (accessed on 10 December 2016).
- Allen, F.; Pon, A.; Greiner, R.; Wishart, D. Computational prediction of electron ionization mass spectra to assist in GC/MS compound identification. Anal. Chem. 2016, 88, 7689–7697. [Google Scholar] [CrossRef] [PubMed]
- Kale, N.S.; Haug, K.; Conesa, P.; Jayseelan, K.; Moreno, P.; Rocca-Serra, P.; Nainala, V.C.; Spicer, R.A.; Williams, M.; Li, X.; et al. Metabolights: An open-access database repository for metabolomics data. Curr. Protoc. Bioinform. 2016, 53. [Google Scholar] [CrossRef]
- Huan, T.; Tang, C.; Li, R.; Shi, Y.; Lin, G.; Li, L. Mycompoundid MS/MS search: Metabolite identification using a library of predicted fragment-ion-spectra of 383,830 possible human metabolites. Anal. Chem. 2015, 87, 10619–10626. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Thiessen, P.A.; Bolton, E.E.; Chen, J.; Fu, G.; Gindulyte, A.; Han, L.; He, J.; He, S.; Shoemaker, B.A.; et al. Pubchem substance and compound databases. Nucleic Acids Res. 2016, 44, D1202–D1213. [Google Scholar] [CrossRef] [PubMed]
- Allen, F.; Pon, A.; Wilson, M.; Greiner, R.; Wishart, D. CFM-ID: A web server for annotation, spectrum prediction and metabolite identification from tandem mass spectra. Nucleic Acids Res. 2014, 42, W94–W99. [Google Scholar] [CrossRef] [PubMed]
- Kind, T.; Okazaki, Y.; Saito, K.; Fiehn, O. Lipidblast templates as flexible tools for creating new in-silico tandem mass spectral libraries. Anal. Chem. 2014, 86, 11024–11027. [Google Scholar] [CrossRef] [PubMed]
- Brouard, C.; Shen, H.; Duhrkop, K.; d’Alche-Buc, F.; Bocker, S.; Rousu, J. Fast metabolite identification with input output Kernel regression. Bioinformatics 2016, 32, i28–i36. [Google Scholar] [CrossRef] [PubMed]
- Jeffryes, J.G.; Colastani, R.L.; Elbadawi-Sidhu, M.; Kind, T.; Niehaus, T.D.; Broadbelt, L.J.; Hanson, A.D.; Fiehn, O.; Tyo, K.E.; Henry, C.S. Mines: Open access databases of computationally predicted enzyme promiscuity products for untargeted metabolomics. J. Cheminform. 2015, 7, 15–87. [Google Scholar] [CrossRef] [PubMed]
- Marchant, C.A.; Briggs, K.A.; Long, A. In silico tools for sharing data and knowledge on toxicity and metabolism: Derek for windows, meteor, and vitic. Toxicol. Mech. Methods 2008, 18, 177–187. [Google Scholar] [CrossRef] [PubMed]
- Wicker, J.; Lorsbach, T.; Gutlein, M.; Schmid, E.; Latino, D.; Kramer, S.; Fenner, K. Envipath—The environmental contaminant biotransformation pathway resource. Nucleic Acids Res. 2016, 44, D502–D508. [Google Scholar] [CrossRef] [PubMed]
- Audoin, C.; Cocandeau, V.; Thomas, O.P.; Bruschini, A.; Holderith, S.; Genta-Jouve, G. Metabolome consistency: Additional parazoanthines from the mediterranean zoanthid parazoanthus axinellae. Metabolites 2014, 4, 421–432. [Google Scholar] [CrossRef] [PubMed]
- Sumner, L.W.; Lei, Z.; Nikolau, B.J.; Saito, K. Modern plant metabolomics: Advanced natural product gene discoveries, improved technologies, and future prospects. Nat. Prod. Rep. 2015, 32, 212–229. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
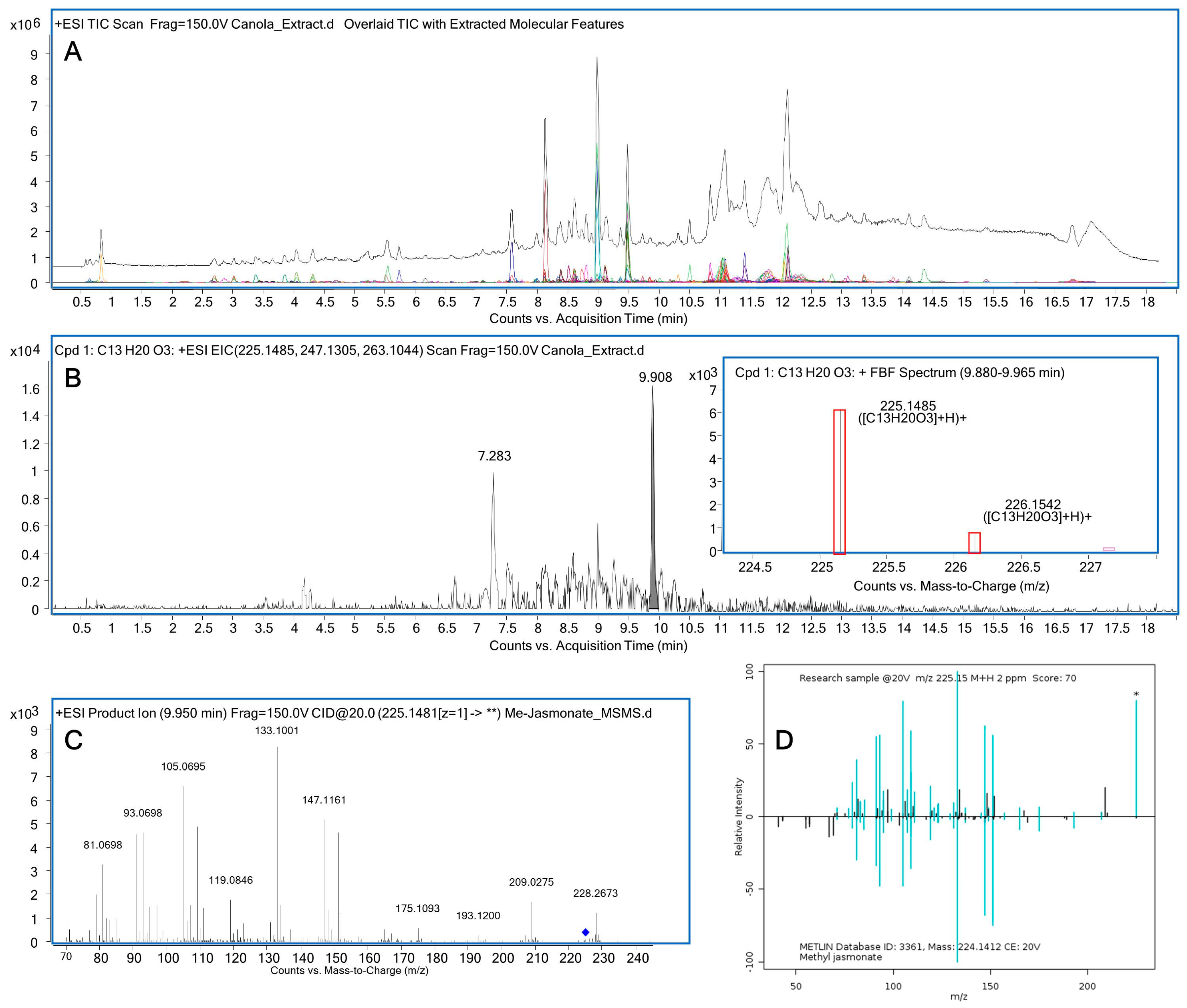{kind=link}
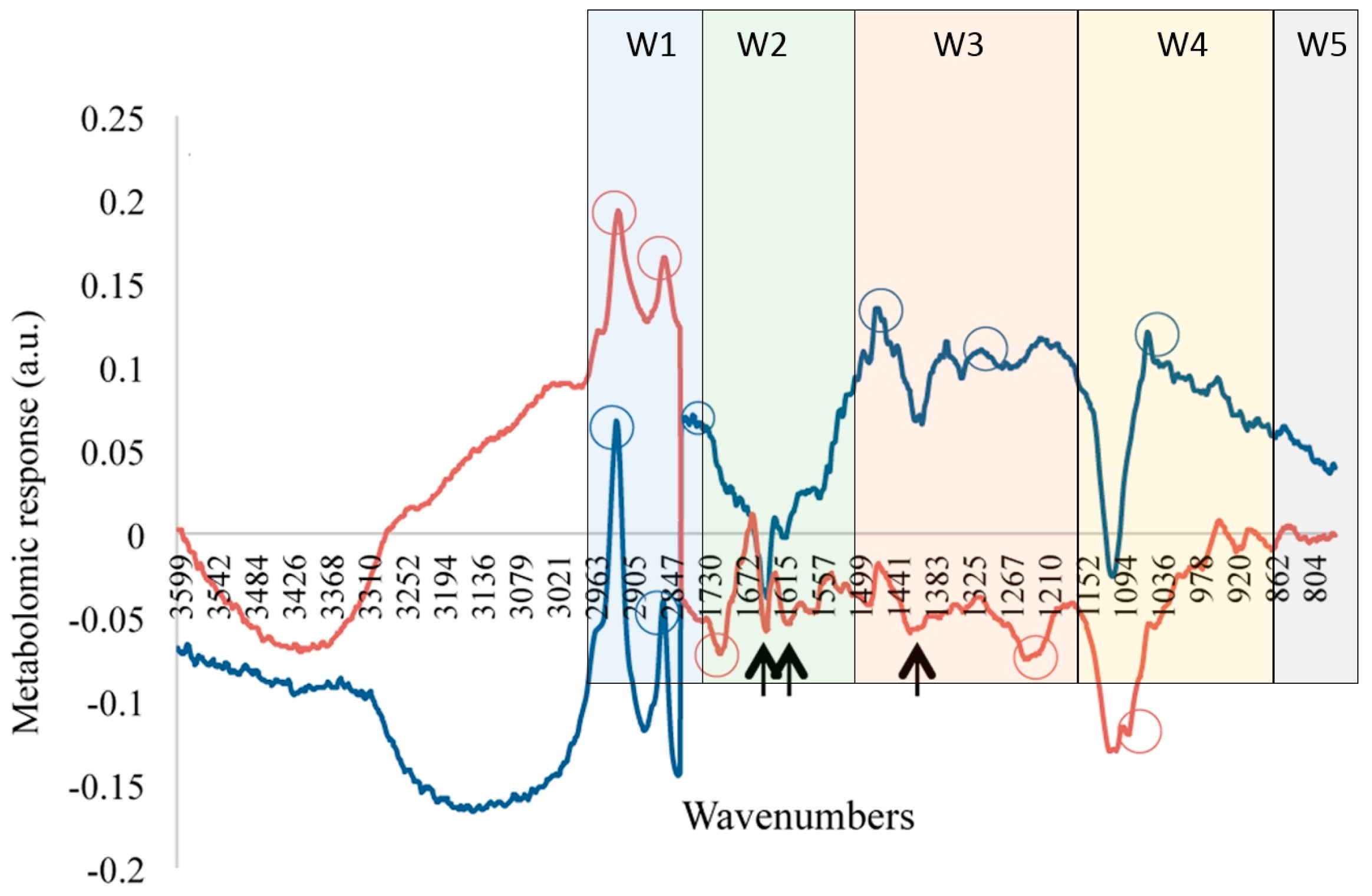{kind=link}
| Mass Analyzer | Mass Resolution | Mass Range (Da) | MS/MS | MSn | Acquisition Speed |
|---|---|---|---|---|---|
| Quadrupole | ~1000 | 50–6000 | Yes | No | Medium |
| Ion Trap | ~1000 | 50–4000 | Yes | Yes | Medium |
| TOF | 2500–40,000 | 20–500,000 | No | No | Fast |
| TOF/TOF | >20,000 | 20–500,000 | Yes | No | Fast |
| Orbitrap | >100,000 | 40–4000 | Yes | Yes | Slow |
| FT-ICR | >200,000 | 10–10,000 | Yes | Yes | Slow |
| Ion Mobility Q-TOF | 13,000/40,000 | Up to 40,000 | Yes | No | Fast |
| Does the FTIR spectra have a Carbonyl (C=O) band? Strong band at 1820–1660 cm−1 | Yes | Acid | • Look for indications that an O–H band is present (broad absorption near 3300–2500 cm−1; will overlap the C–H stretch near 3000 cm−1). |
| • Look for indications that a C–O single bond is present (1100–1300 cm−1). | |||
| • Carbonyl band (near 1725–1700 cm−1). | |||
| Ester | • Look for C–O absorption (medium intensity near 1300–1000 cm−1. There will be no O–H band (3600–3300 cm−1). | ||
| Aldehyde | • Look for aldehyde e type C–H absorption bands (two weak absorptions to the right of the C–H stretch near 2850 cm−1 and 2750 cm−1). | ||
| • Carbonyl band (near 1740–1720 cm−1). | |||
| Ketone | • The weak aldehyde C–H absorption bands will be absent. | ||
| • Carbonyl band (near 1725–1705 cm−1). | |||
| No | Alcohol | • Look for OH band (broad adsorption at 3600–3300 cm−1). | |
| • Look for C–O absorption band (near 1300–1000 cm−1). | |||
| Alkene | • Look for weak absorption near 1650 cm−1 for a double bond. | ||
| • Look for CH stretch band near 3000 cm−1. | |||
| Aromatic | • Look for the benzene double bonds (medium to strong absorptions near 1650–1450 cm−1). | ||
| • The CH stretch band is much weaker than in alkenes. | |||
| Alkane | • The main absorption will be the C–H stretch near 3000 cm−1. | ||
| • Look for another band near 1450 cm−1. | |||
| Alkyl bromide | • Look for the C–H stretch near 3000 cm−1. | ||
| • Look for another band to the right of 667 cm−1. |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dias, D.A.; Jones, O.A.H.; Beale, D.J.; Boughton, B.A.; Benheim, D.; Kouremenos, K.A.; Wolfender, J.-L.; Wishart, D.S. Current and Future Perspectives on the Structural Identification of Small Molecules in Biological Systems. Metabolites 2016, 6, 46. https://doi.org/10.3390/metabo6040046
Dias DA, Jones OAH, Beale DJ, Boughton BA, Benheim D, Kouremenos KA, Wolfender J-L, Wishart DS. Current and Future Perspectives on the Structural Identification of Small Molecules in Biological Systems. Metabolites. 2016; 6(4):46. https://doi.org/10.3390/metabo6040046
Chicago/Turabian StyleDias, Daniel A., Oliver A.H. Jones, David J. Beale, Berin A. Boughton, Devin Benheim, Konstantinos A. Kouremenos, Jean-Luc Wolfender, and David S. Wishart. 2016. "Current and Future Perspectives on the Structural Identification of Small Molecules in Biological Systems" Metabolites 6, no. 4: 46. https://doi.org/10.3390/metabo6040046