
High-Resolution Melting PCR as Rapid Genotyping Tool for Brucella Species


EU-RL Brucellosis, Bacterial Zoonoses Unit, Animal Health Laboratory, University Paris-Est, ANSES, 94700 Maisons-Alfort, France
*
Author to whom correspondence should be addressed.
†
These authors contributed equally to this work.
Microorganisms 2022, 10(2), 336; https://doi.org/10.3390/microorganisms10020336
Submission received: 21 December 2021
/
Revised: 26 January 2022
/
Accepted: 28 January 2022
/
Published: 1 February 2022
(This article belongs to the Special Issue Emerging Themes in Brucella and Brucellosis)
Abstract
:Brucella sp. are the causative agents of brucellosis. One of the main characteristics of the Brucella genus concerns its very high genetic homogeneity. To date, classical bacteriology typing is still considered as the gold standard assay for direct diagnosis of Brucella. Molecular approaches are routinely used for the identification of Brucella at the genus level. However, genotyping is more complex, and to date, no method exists to quickly assign a strain into species and biovar levels, and new approaches are required. Next generation sequencing (NGS) opened a new era into the diagnosis of bacterial diseases. In this study, we designed a high-resolution melting (HRM) method for the rapid screening of DNA and direct assignment into one of the 12 species of the Brucella genus. This method is based on 17 relevant single nucleotide polymorphisms (SNPs), identified and selected from a whole genome SNP (wgSNP) analysis based on 988 genomes (complete and drafts). These markers were tested against the collection of the European Reference Laboratory (EU-RL) for brucellosis (1440 DNAs extracted from Brucella strains). The results confirmed the reliability of the panel of 17 SNP markers, allowing the differentiation of each species of Brucella together with biovars 1, 2, and 3 of B. suis and vaccine strain Rev1 (B. melitensis) within 3 h, which is a considerable gain of time for brucellosis diagnosis. Therefore, this genotyping tool provides a new and quick alternative for Brucella identification based on SNPs with the HRM-PCR assay.
1. Introduction
Brucellosis, also known as ‘undulant fever’, is a zoonotic disease caused by Gram-negative bacteria of the Brucella genus [1]. Brucellae are a group of facultative intracellular Alphaproteobacteria that usually infect domesticated animals and humans, but also wildlife [1,2,3,4]. To date, the genus is composed of 12 species [5], mostly characterized by phenotypical and biochemical preferences: the six classical species B. abortus (bovine), B. melitensis (caprine and ovine), B. suis (porcine), B. canis (canine), B. ovis (ovine), B. neotomae (desert woodrat) [4,6,7,8], and six new species B. ceti (dolphins, porpoises and whales) [9], B. pinnipedialis (seals) [9], B. microti (common vole, frogs, wild boar) [10,11,12,13], B. inopinata (natural host not clearly identified, incidental association with breast implant) [14], B. papionis (baboon) [15] and B. vulpis [16]. The genus is therefore in constant evolution and new and atypical hosts are regularly identified, such as Brucella isolated in frogs [17,18] or more recently Brucella DNA detected in bats [19].
The population structure of the Brucella genus is highly clonal, with all Brucella species sharing sequence similarity values from 98% to 100% [20]. This particular genetic homogeneity explains the challenge of Brucella genotyping. Originally, the genus was classified as a single species subdivided into biovars [21]. In contrast to other bacteria, the ribosomal RNA gene sequences provide little information about internal separation within the genus as many members possess identical 16S rRNA sequences [22]. However, species and biovars can be differentiated using traditional microbiological tests, serological and phenotypic traits that match with the preferred host specificity [2,6]. These methods are considered tedious and hazardous for people in direct contact with live bacteria. The diagnosis of bacteria belonging to the Brucella genus was always difficult, especially concerning the biotyping of isolates and the gold standard approach is still the bacteriology [6,23]. Several specific high-resolution molecular methods are currently available for species identification, such as DNA-based methods (fragment analysis and sequencing) [2,20,24], restriction fragment length polymorphism PCR (RFLP) [25], multiple locus variable number of tandem repeat analysis (MLVA) [26,27], multilocus sequence typing (MLST) [28] or multilocus sequence analysis (MLSA) [29], SNP typing [3], average nucleotide identity (ANI) [30], and more recently the matrix-assisted laser desorption ionization-time of flight mass spectrometry analysis (MALDI-TOF) [31]. These molecular methods allow the distinction between most species of the Brucella genus, but to date, there is no perfect molecular method allowing to quickly and correctly assign a strain into the Brucella genus at species and biovar levels.
The WGS and the use of draft whole genome sequences in replacement of MLST and large-scale SNP analyses provide a powerful alternative to study the genome diversity [20]. Until now, hundreds of strains were sequenced and analyzed to produce some draft or complete genomes [32]. Some comparative studies of Brucella genomes demonstrated the high-resolution power of this kind of approach [2,3,33,34,35], as it was already established for other clonal bacterial species [36,37,38]. To date, more than 600 complete or draft Brucella genomes are available on NCBI and/or PATRIC. Based on these whole genome comparisons, it is possible to identify some specific phylogenetic variants that can be used within a genotyping tool, such as the HRM-PCR [37,39,40]. Previous studies on Brucella identification using HRM-PCR are reported, but not designed for the identification of all currently described Brucella species and associated biovars [41,42].
In this paper, we compared 988 Brucella genomes to identify 12,730 SNPs on the Brucella core-genome. A set of 17 fixed SNPs was identified in silico and used to develop a quick and accurate genotyping tool based on the HRM-PCR. This set of SNPs was tested and validated on a total of 1440 DNAs from different Brucella species. This genotyping tool will be used as routine tool in the network of EU Reference Laboratories for a quick Brucella identification.
2. Materials and Methods
2.1. wgSNP Analyses
Complete and draft genomes belonging to the Brucella genus were retrieved from NCBI (https://www.ncbi.nlm.nih.gov/genome/?term=brucella, 27 January 2022) and PAthosystems Resource Integration Center (PATRIC) (https://www.patricbrc.org/search/?keyword(brucella), 27 January 2022) [43]. A total of 988 genomes sequences (410 B. abortus, 370 B. melitensis, 79 B. suis, 49 B. sp., 35 B. canis, 17 B. ovis, 10 B. ceti, 6 B. pinnipedialis, 4 B. neotomae, 3 B. microti, 2 B. vulpis, 2 B. papionis and 1 B. inopinata) were used in this study (Supplementary File S1).
The genome of B. melitensis strain 16M was used as the reference genome for all the analyses. Chimeric genomes of chromosomes 1 and 2 were generated to compare complete and drafts genomes. Sequence analyses were performed in BioNumerics 7.6.2 (Applied Maths, BioMérieux, Marcy–l’Étoile, France). Synthetic sequencing reads of 250 bp and 50× coverage were generated in silico for all the sequences using ART [44]. The generated reads were mapped against the reference chimeric genome using BWA in BioNumerics. A set of SNPs was deduced for each genome sequence data using BioNumerics wgSNP module. Data were filtered by different ways: inter-SNP distance (minimum 10 bp between SNPs), repetitive elements (VNTR, rRNA), absolute coverage of 20× for each SNPs, unreliable bases (remove positions with at least one unreliable base, i.e., Ns), ambiguous bases (remove positions with at least one ambiguous base, i.e., IUPAC code) and gaps are removed. The matrix of filtered SNPs is used to generate a phylogenetic tree, using a maximum-likelihood approach (BioNumerics 7.6.2 and MEGA X), allowing phylogenetic analyses. The tree was rooted using strains of ‘atypical’ Brucella, as in a recent study [5]. Based on this phylogenetic tree, fixed variants specific to species and biovars are extracted and are used to develop the HRM-PCR assay.
2.2. DNA
DNA used in this study was extracted from pure bacterial suspension, either using phenol-chloroform for reference strains or High Pure PCR Template Preparation Kit (Roche®) for field strains. In total, 29 reference strains and 1411 field strains were used in this study.
2.3. HRM-PCR
High-resolution melting (HRM) is a post-PCR technique that determines with high precision the melt profile of PCR products. A new generation dye is incorporated into a double-stranded DNA. Using a slow constant increase in temperature, fluorescence acquisition allows the distinction between two different populations of amplicons. The method can be used to interrogate small number of SNPs. The filtered SNP matrix is composed of relevant variants. Species- or biovar-specific SNPs, i.e., having the specific variant, which is different from all other sequences, are extracted from this matrix. Once the fixed variants are identified, the 100 bp SNP flanking region is extracted, and primers are designed (Table 1) using Primer3plus (http://www.bioinformatics.nl/cgi-bin/primer3plus/primer3plus.cgi, 27 January 2022). A maximum amplicon size of 100 bp is expected, but most of the time, smaller amplicons are targeted.
Amplification was performed on the ViiA7™ Real-Time PCR System (Life Technologies, Carlsbad, CA, USA) using the LightCycler® 480 High Resolution Melting Master Mix (Roche Diagnostics). The reaction mixture consisted of 0.2 μM of each primer, 1×LightCycler® 480 HRM master mix and 2.5 mM MgCl2 in a 10-μL final volume. The following parameters were used: 10 min at 95 °C were followed by 40 cycles consisting of 10 s at 95 °C, 10 s at 58 °C and 20 s at 72 °C. Samples were next heated to 95 °C for 30 s, cooled down to 65 °C for 1 min and heated from 65 °C to 94 °C at a rate of 1 °C/s with 25 acquisitions/°C. For each HRM assay, a positive expected allele sample is always added, to compare the tested DNAs. The presence of non-specific amplicons was tested with water sample. The melting temperature and melting curves profiles were analyzed by the QuantStudio™ Real-Time PCR Software (version 1.2).
2.4. Assessment of HRM-PCR Performances
The HRM-PCR assay was designed to identify the species and biovars after the confirmation of the Brucella genus. The assay was challenged on intra-Brucella inclusivity and exclusivity for each couple of primers. For the inclusivity test (i.e., belonging to a specific Brucella species), all primers were tested on field DNAs from various Brucella species (previously identified by bacteriology). For the exclusivity (i.e., belonging to another Brucella species), all primers were tested on DNAs from other Brucella species that the previously identified species.
The reference strains for each species are described in Supplementary File S2. For the inclusivity tests, all field DNAs belonging to the expected species were included (from 0 to 589 DNAs, Table 2). For the exclusivity tests, a randomly chosen panel was tested for each couple of primers (from 42 to 297 DNAs, Table 3), to avoid testing more than thousands of DNAs for each couple of primers.
3. Results
3.1. SNP-Based Phylogeny of the Brucella Genus
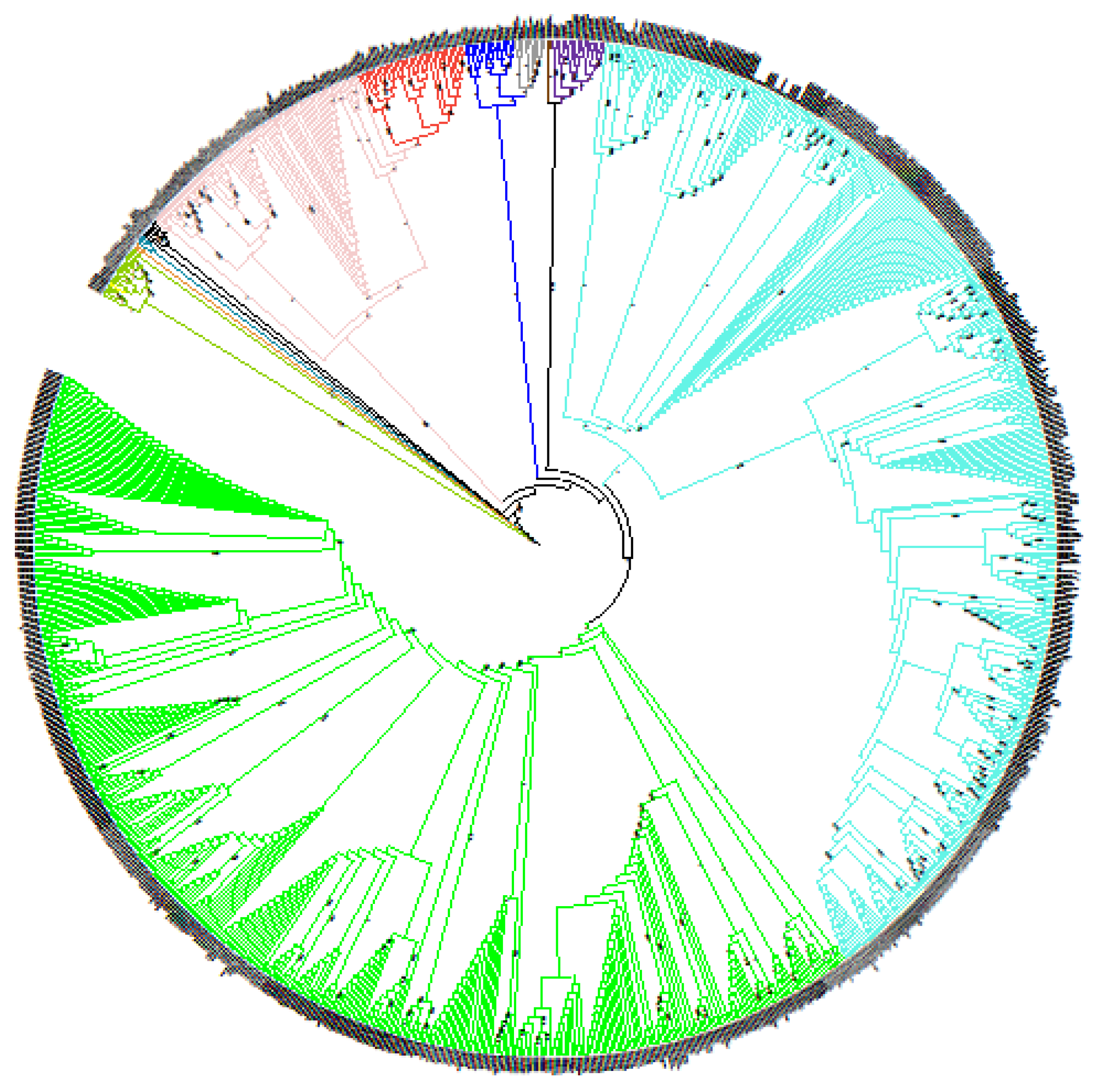
A global comparison of complete and draft genomes of the Brucella genus generated by NGS was performed. Genome-wide comparison of 988 genomes yielded 12,730 SNPs after filtering allowing reconstruction of the Brucella genus phylogeny (Figure 1). This phylogenetic tree highlighted that all Brucella species were clearly separated into different clusters, i.e., each species can be defined by specific branches, thus providing a species-specific panel of SNPs. This phylogenetic tree was rooted with the clade of ‘atypical’ Brucella strains (including B. inopinata and strains isolated in bullfrogs) and as expected, Brucella species were split into two groups: the first one regrouping ‘classical species’ and the second group containing ‘atypical’ or early-diverging Brucella strains [5,45]. The most basal strain from the classical species was B. microti CCM 4915, whereas other lineages radiated quickly, indicating that all species diverged almost simultaneously, as previously reported [5,45].
3.2. Development of HRM-PCR Scheme
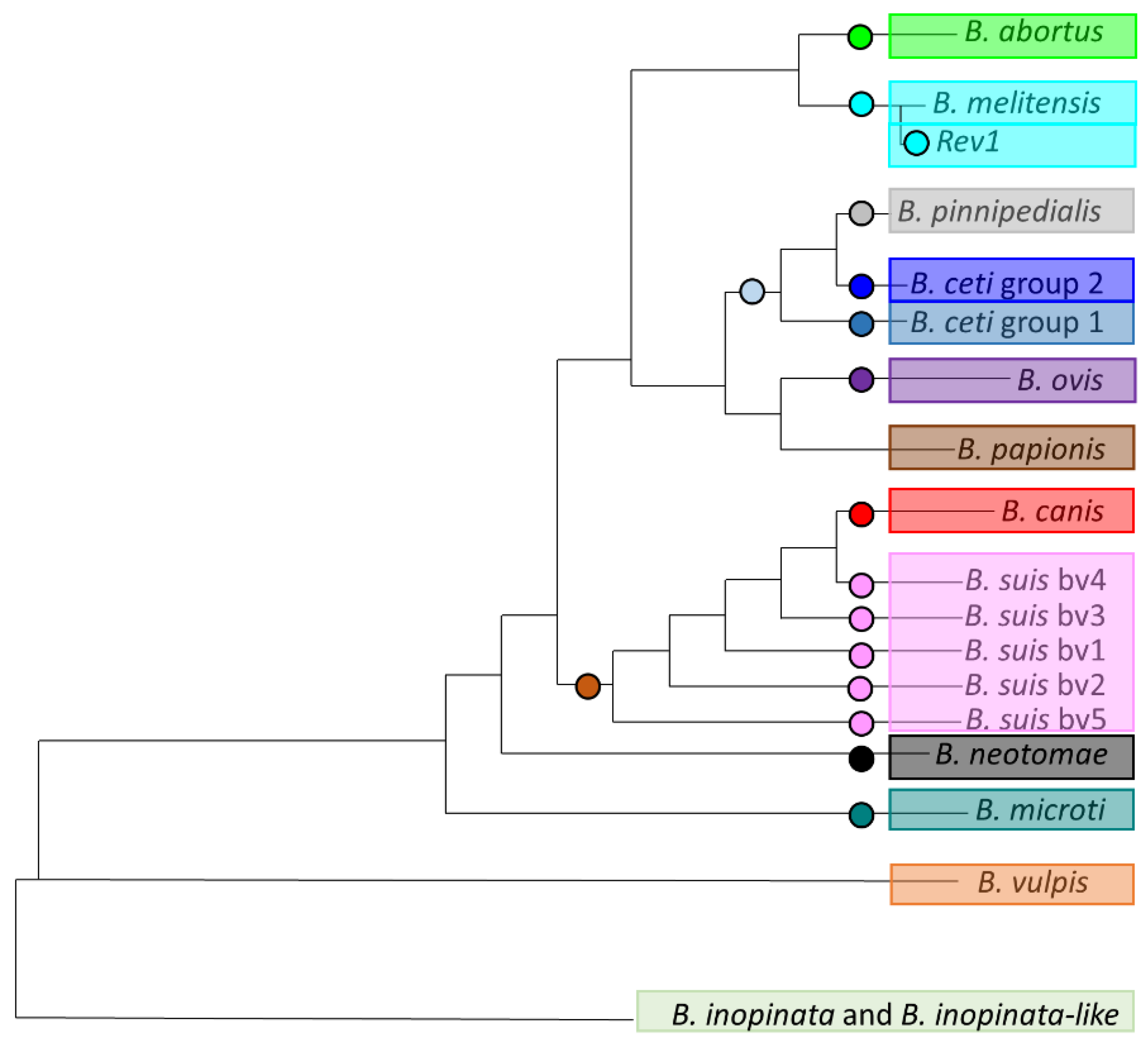
Once this phylogeny was well supported by NGS data, extracting fixed variants (SNPs) was the main crucial step to design HRM assays (Figure 2). A panel of 17 SNPs was thus selected, and specific primers were designed (Figure 2, Table 1). These SNPs were validated in silico for genotyping and the expected alternate alleles for different populations are indicated in Table 1.
3.3. Assessment of HRM-PCR Performances
These diagnostic assays were successfully validated on DNA extracted from reference strains for each species. A total of 29 reference DNAs were used and results were concordant in 100% of tests (inclusivity and exclusivity tests, Table 2 and Table 3). Results in Table 2 for reference DNA can be counted twice, as B. suis biovars DNA are included into the B. suis/canis primer test, explaining a higher number of DNA tested than 29. Finally, the developed HRM-PCR scheme was applied on the complete DNA collection of EU-RL. For the field collection, 1411 DNAs were tested, and results were concordant with previous bacteriological typing and multiplex PCR (Bruce-ladder and Suis-ladder) in 100% of cases (inclusivity and exclusivity tests, Table 2 and Table 3).
4. Discussion
Comparative whole-genome sequencing is a powerful and reliable way for characterization of any bacterial pathogen, especially for highly clonal bacteria like B. anthracis, Brucella sp., Francisella tularensis or Mycobacterium tuberculosis [37,38,45,46,47]. Cost, time, and biosecurity are important issues for all diagnostic assays. Concerning brucellosis, at least 10 days are required for bacteriology and classical phenotyping. In this aim, HRM-PCR is a very attractive assay, developed from WGS analyses, low-cost (only a couple of primers is required, approximately 0.5 € per test) and rapid.
This study confirms that HRM-PCR can be applied to Brucella for a quick assignment of a DNA into a species. The results are very relevant for reference DNAs with 100% of results satisfying both inclusivity and exclusivity tests. Concerning field DNA, results are satisfying with 100% of concordant assignment for inclusivity and exclusivity tests. One important point about HRM-PCR concerns quality of DNA. In this study, a concentration between 10 and 100 fg was determined as the minimal concentration for DNA. This concentration was estimated using Qubit dsDNA High Sensitivity kit and serial dilutions of the DNA were performed and tested in PCR HRM. The last dilution detected was estimated at the minimal concentration of DNA required. This new HRM-PCR scheme is validated on reference strains (100% inclusivity and exclusivity) and on field samples (100% inclusivity and exclusivity). Relatively rare species like B. vulpis, B. papionis, or B. inopinata were not included in this scheme as they are not adequately represented in laboratories’ collection. Previous studies based on HRM-PCR allow the identification of five [41] to six species [42]. One of the main difficulties of the HRM-PCR remains the multiplexing of targets. A previous study allowed testing five targets in one multiplex reaction [41]. In this study, we decided to focus on the detection of all Brucella species without multiplexing, including identification of B. suis biovars.
Using classic bacteriological typing takes almost 10 days to identify a strain. After DNA extraction, when using Bruce-ladder and Suis-ladder, results can be expected within 24 h. MALDI-TOF can provide results within a very short time and is mainly used for the identification of the genus, with remaining lacks of accuracy at species level [48]. Recent improvements were reported for Brucella canis species [49]. Nevertheless, the main bottleneck of MALDI-TOF is related to the database required to obtain a correct identification. The database should be well designed and enriched with spectra from different origins to correctly reach the species level. As regards other molecular methods, multiplex PCR (Bruce and Suis ladder) allows for the identification of species, vaccine strains, and biovars from B. suis, but the results are not obtained as fast as with HRM-PCR. With the panel of 17 markers designed in this study, it is possible to assign a DNA extracted from a strain into nine Brucella species (including all five biovars of B. suis and vaccine strain B. melitensis Rev1) in less than 3 h, which is an important gain in time in case of outbreak investigations. Moreover, even if DNA from reference strains is not available in all laboratories, the possibility to use synthetic amplicons (by oligonucleotides synthesis) as reference amplicon for each couple of primers is another advantage for this method. With the access to a thermocycler allowing HRM analysis, only the primers for the 17 SNPs assay, the synthetic amplicon used as reference profile and the DNA extracted from the suspected strain are necessary to identify precisely the species/biovars involved.
5. Conclusions
In this study, we propose a quick and easy-to-use way to assign whole genome data to a species using a molecular approach, directly developed from WGS data and wgSNP, the HRM-PCR. This genotyping tool developed in this study with the related 17 SNP panel dedicated to Brucella species and biovars is a very powerful approach to quickly identify a strain and assign it to the correct species. This method will be used in routine in EU-RL and can be applied mostly for brucellosis reference laboratories in the world.
This SNP scheme will probably be improved in the future, as laboratories produce more and more data and share it with the scientific community. Moreover, the use of HRM-PCR directly on DNA extracted from specimens is a promising outlook, as preamplification step combined with HRM showed promising results. This will be the next step for future analyses, ‘filling the gaps’ to combat brucellosis.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/microorganisms10020336/s1, Supplementary File S1: List of genomes sequences used in this study; Supplementary File S2: list of reference DNA used in this study.
Author Contributions
Conceptualization, G.G. and V.M.; data curation, G.G. and L.P.; formal analysis, G.G. and L.P.; funding acquisition, C.P.; methodology, G.G.; supervision, V.M. and C.P.; validation, G.G. and L.P.; writing—original draft, G.G.; writing—review and editing, G.G., V.M. and C.P. All authors have read and agreed to the published version of the manuscript.
Funding
We are very thankful to the DG SANTE—DG for Health and Food Safety, European Commission for the support and funding in the framework of EU Reference Laboratory for Brucellosis (grant agreement no. SI2.801943).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Scholz, H.C.; Banai, M.; Cloeckaert, A.; Kampfer, P.; Whatmore, A.M. Brucella. In Bergey’s Manual of Systematics of Archaea and Bacteria; John Wiley and Sons: Hoboken, NJ, USA, 2018; pp. 1–38. [Google Scholar]
- Foster, J.; Beckstrom-Sternberg, S.M.; Pearson, T.; Beckstrom-Sternberg, J.S.; Chain, P.S.G.; Roberto, F.F.; Hnath, J.; Brettin, T.; Keim, P. Whole-Genome-Based Phylogeny and Divergence of the Genus Brucella. J. Bacteriol. 2009, 191, 2864–2870. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foster, J.T.; Price, L.B.; Beckstrom-Sternberg, S.M.; Pearson, T.; Brown, W.D.; Kiesling, D.M.; Allen, C.A.; Liu, C.M.; Beckstrom-Sternberg, J.; Roberto, F.F.; et al. Genotyping of Brucella species using clade specific SNPs. BMC Microbiol. 2012, 12, 110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corbel, M.J.; WHO. Brucellosis in Humans and Animals; WHO Press: Geneva, Switzerland, 2006. [Google Scholar]
- Whatmore, A.M.; Foster, J.T. Emerging diversity and ongoing expansion of the genus Brucella. Infect. Genet. Evol. 2021, 92, 104865. [Google Scholar] [CrossRef] [PubMed]
- OIE. Brucellosis (infection with B. abortus, B. melitensis and B. suis). In OIE Terrestrial Manual; OIE: Paris, France, 2016.suis). In OIE Terrestrial Manual; OIE: Paris, France, 2016. [Google Scholar]
- Moreno, E.; Cloeckaert, A.; Moriyón, I. Brucella evolution and taxonomy. Vet. Microbiol. 2002, 90, 209–227. [Google Scholar] [CrossRef]
- Osterman, B.; Moriyon, I. International Committee on Systematics of Prokaryotes; Subcommittee on the taxonomy of Brucella. Int. J. Syst. Evol. Microbiol. 2006, 56, 1173–1175. [Google Scholar] [CrossRef] [Green Version]
- Foster, G.; Osterman, B.S.; Godfroid, J.; Jacques, I.; Cloeckaert, A. Brucella ceti sp. nov. and Brucella pinnipedialis sp. nov. for Brucella strains with cetaceans and seals as their preferred hosts. Int. J. Syst. Evol. Microbiol. 2007, 57, 2688–2693. [Google Scholar] [CrossRef] [Green Version]
- Audic, S.; Lescot, M.; Claverie, J.-M.; Scholz, H.C. Brucella microti: The genome sequence of an emerging pathogen. BMC Genom. 2009, 10, 352. [Google Scholar] [CrossRef] [Green Version]
- Jaý, M.; Girault, G.; Perrot, L.; Taunay, B.; Vuilmet, T.; Rossignol, F.; Pitel, P.-H.; Picard, E.; Ponsart, C.; Mick, V. Phenotypic and Molecular Characterization of Brucella microti-Like Bacteria from a Domestic Marsh Frog (Pelophylax ridibundus). Front. Vet. Sci. 2018, 5, 283. [Google Scholar] [CrossRef]
- Jaÿ, M.; Freddi, L.; Mick, V.; Durand, B.; Girault, G.; Perrot, L.; Taunay, B.; Vuilmet, T.; Azam, D.; Ponsart, C.; et al. Brucella microti-like prevalence in French farms producing frogs. Transbound. Emerg. Dis. 2019, 67, 617–625. [Google Scholar] [CrossRef]
- Rónai, Z.; Kreizinger, Z.; Dán, A.; Drees, K.P.; Foster, J.T.; Bányai, K.; Marton, S.; Szeredi, L.; Jánosi, S.; Gyuranecz, M. First isolation and characterization of Brucella microti from wild boar. BMC Vet. Res. 2015, 11, 147. [Google Scholar] [CrossRef] [Green Version]
- Scholz, H.C.; Nöckler, K.; Göllner, C.; Bahn, P.; Vergnaud, G.; Tomaso, H.; Al Dahouk, S.; Kampfer, P.; Cloeckaert, A.; Maquart, M.; et al. Brucella inopinata sp. nov., isolated from a breast implant infection. Int. J. Syst. Evol. Microbiol. 2010, 60, 801–808. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whatmore, A.M.; Davison, N.; Cloeckaert, A.; Al Dahouk, S.; Zygmunt, M.; Brew, S.D.; Perrett, L.L.; Koylass, M.S.; Vergnaud, G.; Quance, C.; et al. Brucella papionis sp. nov., isolated from baboons (Papio spp.). Int. J. Syst. Evol. Microbiol. 2014, 64, 4120–4128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scholz, H.C.; Revilla-Fernandez, S.; Al Dahouk, S.; Hammerl, J.A.; Zygmunt, M.S.; Cloeckaert, A.; Koylass, M.; Whatmore, A.M.; Blom, J.; Vergnaud, G.; et al. Brucella vulpis sp. nov., isolated from mandibular lymph nodes of red foxes (Vulpes vulpes). Int. J. Syst. Evol. Microbiol. 2016, 66, 2090–2098. [Google Scholar] [CrossRef] [PubMed]
- Scholz, H.C.; Mühldorfer, K.; Shilton, C.; Benedict, S.; Whatmore, A.; Blom, J.; Eisenberg, T. The Change of a Medically Important Genus: Worldwide Occurrence of Genetically Diverse Novel Brucella Species in Exotic Frogs. PLoS ONE 2016, 11, e0168872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eisenberg, T.; Hamann, H.-P.; Kaim, U.; Schlez, K.; Seeger, H.; Schauerte, N.; Melzer, F.; Tomaso, H.; Scholz, H.C.; Koylass, M.S.; et al. Isolation of potentially novel Brucella spp. from frogs. Appl. Environ. Microbiol. 2012, 78, 3753–3755. [Google Scholar] [CrossRef] [Green Version]
- Bai, Y.; Urushadze, L.; Osikowicz, L.; McKee, C.; Kuzmin, I.; Kandaurov, A.; Babuadze, G.; Natradze, I.; Imnadze, P.; Kosoy, M. Molecular Survey of Bacterial Zoonotic Agents in Bats from the Country of Georgia (Caucasus). PLoS ONE 2017, 12, e0171175. [Google Scholar] [CrossRef]
- Scholz, H.; Vergnaud, G. Molecular characterisation of Brucella species. Rev. Sci. Tech. Off. Int. Epiz. 2013, 32, 149–162. [Google Scholar] [CrossRef] [Green Version]
- Verger, J.-M.; Grimont, F.; Grimont, P.A.D.; Grayon, M. Brucella, a Monospecific Genus as Shown by Deoxyribonucleic Acid Hybridization. Int. J. Syst. Bacteriol. 1985, 35, 292–295. [Google Scholar] [CrossRef]
- Gee, J.E.; De, B.K.; Levett, P.N.; Whitney, A.M.; Novak, R.T.; Popovic, T. Use of 16S rRNA gene sequencing for rapid confirmatory identification of Brucella isolates. J. Clin. Microbiol. 2004, 42, 3649–3654. [Google Scholar] [CrossRef] [Green Version]
- Alton, G.G.; Jones, L.M.; Pietz, D.E. Laboratory Techniques in Brucellosis; WHO: Geneva, Switzerland, 1975. [Google Scholar]
- Yu, W.L.; Nielsen, K. Review of Detection of Brucella sp. by Polymerase Chain Reaction. Croat. Med. J. 2010, 51, 306–313. [Google Scholar] [CrossRef] [Green Version]
- Al Dahouk, S.; Tomaso, H.; Prenger-Berninghoff, E.; Splettstoesser, W.D.; Scholz, H.C.; Neubauer, H. Identification of Bruicella Species and Biotypes using Polymerase Chain Reaction-Restriction Fragment Length Polymorphism (PCR-RFLP). Crit. Rev. Microbiol. 2005, 31, 191–196. [Google Scholar] [CrossRef] [PubMed]
- Le Flèche, P.; Jacques, I.; Grayon, M.; Al Dahouk, S.; Bouchon, P.; Denoeud, F.; Nöckler, K.; Neubauer, H.; Guilloteau, L.A.; Vergnaud, G. Evaluation and selection of tandem repeat loci for a Brucella MLVA typing assay. BMC Microbiol. 2006, 6, 9–14. [Google Scholar] [CrossRef] [PubMed]
- Al Dahouk, S.; Le Flèche, P.; Nöckler, K.; Jacques, I.; Grayon, M.; Scholz, H.C.; Tomaso, H.; Vergnaud, G.; Neubauer, H. Evaluation of Brucella MLVA typing for human brucellosis. J. Microbiol. Methods 2007, 69, 137–145. [Google Scholar] [CrossRef] [PubMed]
- Whatmore, A.M.; Perrett, L.L.; MacMillan, A.P. Characterisation of the genetic diversity of Brucella by multilocus sequencing. BMC Microbiol. 2007, 7, 34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whatmore, A.M.; Koylass, M.S.; Muchowski, J.; Edwards-Smallbone, J.; Gopaul, K.K.; Perrett, L.L. Extended Multilocus Sequence Analysis to Describe the Global Population Structure of the Genus Brucella: Phylogeography and Relationship to Biovars. Front. Microbiol. 2016, 7, 2049. [Google Scholar] [CrossRef] [PubMed]
- Konstantinidis, K.T.; Tiedje, J.M. Genomic insights that advance the species definition for prokaryotes. Proc. Natl. Acad. Sci. USA 2005, 102, 2567–2572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mesureur, J.; Ranaldi, S.; Monnin, V.; Girard, V.; Arend, S.; Welker, M.; O’Callaghan, D.; Lavigne, J.-P.; Keriel, A. A Simple and Safe Protocol for Preparing Brucella Samples for Matrix-Assisted Laser Desorption Ionization-Time of Flight Mass Spectrometry Analysis. J. Clin. Microbiol. 2016, 54, 449–452. [Google Scholar] [CrossRef] [Green Version]
- O’Callaghan, D.; Whatmore, A. Brucella genomics as we enter the multi-genome era. Briefings Funct. Genom. 2011, 10, 334–341. [Google Scholar] [CrossRef] [Green Version]
- Sankarasubramanian, J.; Vishnu, U.S.; Gunasekaran, P.; Rajendhran, J. A genome-wide SNP-based phylogenetic analysis distinguishes different biovars of Brucella suis. Infect. Genet. Evol. 2016, 41, 213–217. [Google Scholar] [CrossRef]
- Tan, K.-K.; Tan, Y.-C.; Chang, L.-Y.; Lee, K.W.; Nore, S.S.; Yee, W.-Y.; Isa, M.N.M.; Jafar, F.L.; Hoh, C.-C.; AbuBakar, S. Full genome SNP-based phylogenetic analysis reveals the origin and global spread of Brucella melitensis. BMC Genom. 2015, 16, 93. [Google Scholar] [CrossRef] [Green Version]
- Holzapfel, M.; Girault, G.; Keriel, A.; Ponsart, C.; O’Callaghan, D.; Mick, V. Comparative Genomics and in vitro Infection of Field Clonal Isolates of Brucella melitensis Biovar 3 Did Not Identify Signature of Host Adaptation. Front. Microbiol. 2018, 9, 2505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vergnaud, G.; Girault, G.; Thierry, S.; Pourcel, C.; Madani, N.; Blouin, Y. Comparison of French and Worldwide Bacillus anthracis Strains Favors a Recent, Post-Columbian Origin of the Predominant North-American Clade. PLoS ONE 2016, 11, e0146216. [Google Scholar] [CrossRef] [Green Version]
- Girault, G.; Blouin, Y.; Vergnaud, G.; Derzelle, S. High-throughput sequencing of Bacillus anthracis in France: Investigating genome diversity and population structure using whole- genome SNP discovery. BMC Genom. 2014, 15, 288. [Google Scholar] [CrossRef] [Green Version]
- Blouin, Y.; Hauck, Y.; Soler, C.; Fabre, M.; Vong, R.; Dehan, C.; Cazajous, G.; Massoure, P.-L.; Kraemer, P.; Jenkins, A.; et al. Significance of the identification in the Horn of Africa of an exceptionally deep branching Mycobacterium tuberculosis clade. PLoS ONE 2012, 7, e52841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Girault, G.; Wattiau, P.; Saqib, M.; Martin, B.; Vorimore, F.; Singha, H.; Engelsma, M.; Roest, H.; Spicic, S.; Grunow, R.; et al. High-resolution melting PCR analysis for rapid genotyping of Burkholderia mallei. Infect. Genet. Evol. 2018, 63, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Girault, G.; Thierry, S.; Cherchame, E.; Derzelle, S. Application of High-Throughput Sequencing: Discovery of Informative SNPs to Subtype Bacillus anthracis. Adv. Biosci. Biotechnol. 2014, 5, 669–677. [Google Scholar] [CrossRef] [Green Version]
- Gopaul, K.K.; Sells, J.; Lee, R.; Beckstrom-Sternberg, S.M.; Foster, J.T.; Whatmore, A.M. Development and assessment of multiplex high resolution melting assay as a tool for rapid single-tube identification of five Brucella species. BMC Res. Notes 2014, 7, 903. [Google Scholar] [CrossRef] [Green Version]
- Winchell, J.M.; Wolff, B.J.; Tiller, R.; Bowen, M.D.; Hoffmaster, A.R. Rapid identification and discrimination of Brucella isolates by use of real-time PCR and high-resolution melt analysis. J. Clin. Microbiol. 2010, 48, 697–702. [Google Scholar] [CrossRef] [Green Version]
- Wattam, A.R.; Abraham, D.; Dalay, O.; Disz, T.L.; Driscoll, T.; Gabbard, J.L.; Gillespie, J.J.; Gough, R.; Hix, D.; Kenyon, R.; et al. PATRIC, the bacterial bioinformatics database and analysis resource. Nucleic Acids Res. 2014, 42, D581–D591. [Google Scholar] [CrossRef] [Green Version]
- Huang, W.; Li, L.; Myers, J.R.; Marth, G.T. ART: A next-generation sequencing read simulator. Bioinformatics 2012, 28, 593–594. [Google Scholar] [CrossRef] [Green Version]
- Wattam, A.R.; Foster, J.T.; Mane, S.P.; Beckstrom-Sternberg, S.M.; Beckstrom-Sternberg, J.M.; Dickerman, A.W.; Keim, P.; Pearson, T.; Shukla, M.; Ward, D.V.; et al. Comparative Phylogenomics and Evolution of the Brucellae Reveal a Path to Virulence. J. Bacteriol. 2014, 196, 920–930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dwibedi, C.; Birdsell, D.; Lärkeryd, A.; Myrtennäs, K.; Öhrman, C.; Nilsson, E.; Karlsson, E.; Hochhalter, C.; Rivera, A.; Maltinsky, S.; et al. Long-range dispersal moved Francisella tularensis into Western Europe from the East. Microb. Genom. 2016, 2, e000100. [Google Scholar] [CrossRef] [PubMed]
- Kevin, M.; Girault, G.; Caspar, Y.; Cherfa, M.A.; Mendy, C.; Tomaso, H.; Gavier-Widen, D.; Escudero, R.; Maurin, M.; Durand, B.; et al. Phylogeography and Genetic Diversity of Francisella tularensis subsp. holarctica in France (1947–2018). Front. Microbiol. 2020, 11, 287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mesureur, J.; Arend, S.; Cellière, B.; Courault, P.; Cotte-Pattat, P.-J.; Totty, H.; Deol, P.; Mick, V.; Girard, V.; Touchberry, J.; et al. A MALDI-TOF MS database with broad genus coverage for species-level identification of Brucella. PLoS Neglected Trop. Dis. 2018, 12, e0006874. [Google Scholar] [CrossRef] [PubMed]
- da Silva, D.A.V.; Brendebach, H.; Grutzke, J.; Dieckmann, R.; Soares, R.M.; de Lima, J.T.R.; Keid, L.B.; Hofreuter, D.; Al Dahouk, S. MALDI-TOF MS and genomic analysis can make the difference in the clarification of canine brucellosis outbreaks. Sci. Rep. 2020, 10, 19256. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Global phylogeny of Brucella genus. Evolutionary history was inferred by using Maximum Likelihood method and Kimura 2-parameter model. Initial tree(s) for heuristic search was obtained automatically by applying Maximum Parsimony method. Tree is not drawn to scale (branch lengths does not correspond to number of substitutions per site). This analysis involved 988 genome sequences. There was a total of 12,730 positions in final dataset. Evolutionary analyses were conducted in MEGA X. All species are color-coded: green for B. abortus, light blue for B. melitensis, grey for B. pinnipedialis, dark blue for B. ceti, purple for B. ovis, brown for B. papionis, red for B. canis, pink for B. suis, black for B. neotomae, light green for B. sp. dark green for B. microti, yellow for B. inopinata and orange for B. vulpis. Bootstrap values are indicated at branch nodes (between 0 and 1).
Figure 1.
Global phylogeny of Brucella genus. Evolutionary history was inferred by using Maximum Likelihood method and Kimura 2-parameter model. Initial tree(s) for heuristic search was obtained automatically by applying Maximum Parsimony method. Tree is not drawn to scale (branch lengths does not correspond to number of substitutions per site). This analysis involved 988 genome sequences. There was a total of 12,730 positions in final dataset. Evolutionary analyses were conducted in MEGA X. All species are color-coded: green for B. abortus, light blue for B. melitensis, grey for B. pinnipedialis, dark blue for B. ceti, purple for B. ovis, brown for B. papionis, red for B. canis, pink for B. suis, black for B. neotomae, light green for B. sp. dark green for B. microti, yellow for B. inopinata and orange for B. vulpis. Bootstrap values are indicated at branch nodes (between 0 and 1).

Figure 2.
Position of 17 selected single nucleotide polymorphisms (SNPs) for high-resolution melting (HRM)-PCR on phylogenetic tree of Brucella. This figure represents a simplified phylogeny of Brucella genus. Each circle represent a fixed SNP selected on branch for identification of related species or biovar.
Figure 2.
Position of 17 selected single nucleotide polymorphisms (SNPs) for high-resolution melting (HRM)-PCR on phylogenetic tree of Brucella. This figure represents a simplified phylogeny of Brucella genus. Each circle represent a fixed SNP selected on branch for identification of related species or biovar.


{kind=link}
{kind=link}
Table 1.
SNPs used for HRM-PCR and related information.
| Species Targeted | Genomic Position (16M ref) | Forward Primer (5′-3′) | Forward Primer Coordinates (16M ref) | Reverse Primer (5′-3′) | Reverse Primer Coordinates (16M ref) | Locus Tag | Locus Tag Information | Amplicon Size (bp) | Targeted Allele (Related to Column 1) | Other Allele (Related to All Other Brucella) |
|---|---|---|---|---|---|---|---|---|---|---|
| B. abortus | 609866 | acgaagaagcgatctcgatg | 609832-609851 | aggaaaggccgatgatgtaa | 609905-609924 | BMEI0587 | coml, competence lipoprotein | 93 | T | C |
| B. melitensis | 375209 | cggtccgggccacctttacg | 375164-375183 | ggcccggcaattgctcctga | 375225-375244 | NR | NR | 81 | C | T |
| B. suis-B. canis | 687223 | ctggcggaaaaggatttgat | 687162-687181 | aatcacgacaaaccacagca | 687232-687251 | BMEI0664 | sugar transport system permease protein | 90 | T | C |
| B. suis biovar 1 | 656162 | tgacatggaccctgttttcc | 656196-656215 | cagcgtgacactgaacatgg | 656138-656157 | BMEI0629 | hypothetical protein | 78 | G | A |
| B. suis biovar 2 | 221777 | agaccttgcgcttgaacg | 221821-221838 | gccacactgctgagttcg | 221755-221772 | BMEI0215 | (di)nucleoside polyphosphate hydrolase | 84 | T | C |
| B. suis biovar 3 | 1368151 | gtatggcggaatgcagga | 1368178-1368195 | cacaaacgccagtgaacg | 1368132-1368149 | NR | NR | 64 | A | G |
| B. suis biovar 4 | 2026823 | aagatcgccgtcgtctcg | 2026873-2026890 | ggccacaacagcctgaac | 2026801-2026818 | NR | NR | 90 | A | G |
| B. suis biovar 5 | 159143 | cttccgttgaagggcaatc | 159161-159179 | gcctcgaaaacgaaatcatc | 159085-159104 | NR | NR | 95 | C | T |
| B. canis | 937299 | gagaactgacccgatggaaa | 937238-937257 | caagggaaccgaatatctgc | 937302-937231 | NR | NR | 84 | C | T |
| B. microti | 1111504 | aactgccggatgtgaaaaag | 1111529-1111548 | aaggatcgaggcgtcataaa | 1111478-1111497 | NR | NR | 71 | C | T |
| Marine Brucella | 1237960 | gcgatttcattgcccttg | 1237892-1237909 | ttgaaatgggcttcatcca | 1237961-1237979 | NR | NR | 88 | A | G |
| B. ceti 1 | 318627 | aatgccgcaatcttcatctt | 318637-318656 | cctctgcgcgacagtttaag | 318587-318606 | NR | NR | 70 | A | C |
| B. ceti 2 | 121188 | ctcgctcccaaacactaccc | 121150-121169 | cgttcgccccttatatttga | 121220-121239 | NR | NR | 90 | C | T |
| B. pinnipedialis | 369804 | tgcgggatttcaaggataag | 369821-369840 | aagatcgccagatcgtgct | 369768-369786 | BMEI0358 | deoxyuridine 5′-triphosphate nucleotidohydrolase | 73 | T | C |
| B. ovis | 576553 | atgggctttggcggtatt | 576495-576512 | cgcccaggtagagctttg | 576558-576575 | BMEI0556 | alpha-ketoglutarate permease | 81 | T | C |
| B. neotomae | 1010822 | atggcgaattcgatgaaaag | 1010854-1010873 | tgtcttcacagacgggaatg | 1010775-1010794 | NR | NR | 99 | T | G |
| B. melitensis Rev 1 | 139509 | cttcacgccatgcttctttt | 139556-139575 | atgctcaccaccttcaacg | 139483-139501 | BMEI0141 | dihydrolipoamide succinyltransferase component (e2) of 2-oxoglutarate dehydrogenase complex | 93 | T | C |
Table 2.
Results of inclusivity tests.
| Expected SNP Profile (Inclusivity Test) | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Reference DNA | Field DNA | Total | ||||||||
| Expected Allele | Other Allele | Total | % | Expected Allele | Other Allele | Total | % | |||
| Primers | B. abortus | 11 | 0 | 11 | 100 | 96 | 0 | 96 | 100 | 107 |
| B. melitensis | 6 | 0 | 6 | 100 | 232 | 0 | 232 | 100 | 238 | |
| B. melitensis Rev1 | 2 | 0 | 2 | 100 | 6 | 0 | 6 | 100 | 8 | |
| B. suis/canis | 6 | 0 | 6 | 100 | 589 | 0 | 589 | 100 | 595 | |
| B. suis biovar 1 | 1 | 0 | 1 | 100 | 26 | 0 | 26 | 100 | 27 | |
| B. suis biovar 2 | 1 | 0 | 1 | 100 | 194 | 0 | 194 | 100 | 195 | |
| B. suis biovar 3 | 1 | 0 | 1 | 100 | 2 | 0 | 2 | 100 | 3 | |
| B. suis biovar 4 | 1 | 0 | 1 | 100 | 0 | 0 | 0 | NA | 1 | |
| B. suis biovar 5 | 1 | 0 | 1 | 100 | 0 | 0 | 0 | NA | 1 | |
| B. canis | 1 | 0 | 1 | 100 | 82 | 0 | 82 | 100 | 83 | |
| B. marine | 3 | 0 | 3 | 100 | 14 | 0 | 14 | 100 | 17 | |
| B. ceti group 1 | 1 | 0 | 1 | 100 | 8 | 0 | 8 | 100 | 9 | |
| B. ceti group 2 | 1 | 0 | 1 | 100 | 4 | 0 | 4 | 100 | 5 | |
| B. pinnipedialis | 1 | 0 | 1 | 100 | 2 | 0 | 2 | 100 | 3 | |
| B. microti | 1 | 0 | 1 | 100 | 68 | 0 | 68 | 100 | 69 | |
| B. ovis | 1 | 0 | 1 | 100 | 88 | 0 | 88 | 100 | 89 | |
| B. neotomae | 1 | 0 | 1 | 100 | 0 | 0 | 0 | NA | 1 | |
| Total DNA | 40 | 1411 | 1451 | |||||||
Table 3.
Results of exclusivity tests.
| Other SNP Profile (Exclusion Test) | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Reference DNA | Field DNA | Total | ||||||||
| Expected Allele | Other Allele | Total | % | Expected Allele | Other Allele | Total | % | |||
| Primers | B. abortus | 0 | 16 | 16 | 100 | 0 | 218 | 218 | 100 | 234 |
| B. melitensis | 0 | 21 | 21 | 100 | 0 | 66 | 66 | 100 | 87 | |
| B. melitensis Rev1 | 0 | 26 | 26 | 100 | 0 | 56 | 56 | 100 | 82 | |
| B. suis/canis | 0 | 22 | 22 | 100 | 0 | 43 | 43 | 100 | 65 | |
| B. suis biovar 1 | 0 | 26 | 26 | 100 | 0 | 297 | 297 | 100 | 323 | |
| B. suis biovar 2 | 0 | 25 | 25 | 100 | 0 | 63 | 63 | 100 | 88 | |
| B. suis biovar 3 | 0 | 28 | 28 | 100 | 0 | 94 | 94 | 100 | 122 | |
| B. suis biovar 4 | 0 | 25 | 25 | 100 | 0 | 52 | 52 | 100 | 77 | |
| B. suis biovar 5 | 0 | 25 | 25 | 100 | 0 | 54 | 54 | 100 | 79 | |
| B. canis | 0 | 26 | 26 | 100 | 0 | 64 | 64 | 100 | 90 | |
| B. marine | 0 | 24 | 24 | 100 | 0 | 57 | 57 | 100 | 81 | |
| B. ceti group 1 | 0 | 26 | 26 | 100 | 0 | 63 | 63 | 100 | 89 | |
| B. ceti group 2 | 0 | 26 | 26 | 100 | 0 | 68 | 68 | 100 | 94 | |
| B. pinnipedialis | 0 | 26 | 26 | 100 | 0 | 70 | 70 | 100 | 96 | |
| B. microti | 0 | 26 | 26 | 100 | 0 | 59 | 59 | 100 | 85 | |
| B. ovis | 0 | 28 | 28 | 100 | 0 | 42 | 42 | 100 | 70 | |
| B. neotomae | 0 | 27 | 27 | 100 | 0 | 45 | 45 | 100 | 72 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Girault, G.; Perrot, L.; Mick, V.; Ponsart, C. High-Resolution Melting PCR as Rapid Genotyping Tool for Brucella Species. Microorganisms 2022, 10, 336. https://doi.org/10.3390/microorganisms10020336
AMA Style
Girault G, Perrot L, Mick V, Ponsart C. High-Resolution Melting PCR as Rapid Genotyping Tool for Brucella Species. Microorganisms. 2022; 10(2):336. https://doi.org/10.3390/microorganisms10020336
Chicago/Turabian StyleGirault, Guillaume, Ludivine Perrot, Virginie Mick, and Claire Ponsart. 2022. "High-Resolution Melting PCR as Rapid Genotyping Tool for Brucella Species" Microorganisms 10, no. 2: 336. https://doi.org/10.3390/microorganisms10020336
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.