
RNA Sequencing Reveals Widespread Transcription of Natural Antisense RNAs in Entamoeba Species

 , , , , ,
, , , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Entamoeba Species and In Vitro Culture
2.2. RNA Preparation
2.3. Library Construction, Sequencing and Read Processing
2.4. Reference Genomes
2.5. Data Repositories
2.6. De Novo Assembly of Transcriptomes
2.7. Analyses of Small RNAs
2.8. TSS Identification and Annotation
2.9. PAS Identification and Annotation
2.10. Motif Enrichment
2.11. Orthologs, Core and Pan-Genome Identification
2.12. Differential Expression Analysis
2.13. eggNOG Annotations and GO Terms Enrichment
3. Results
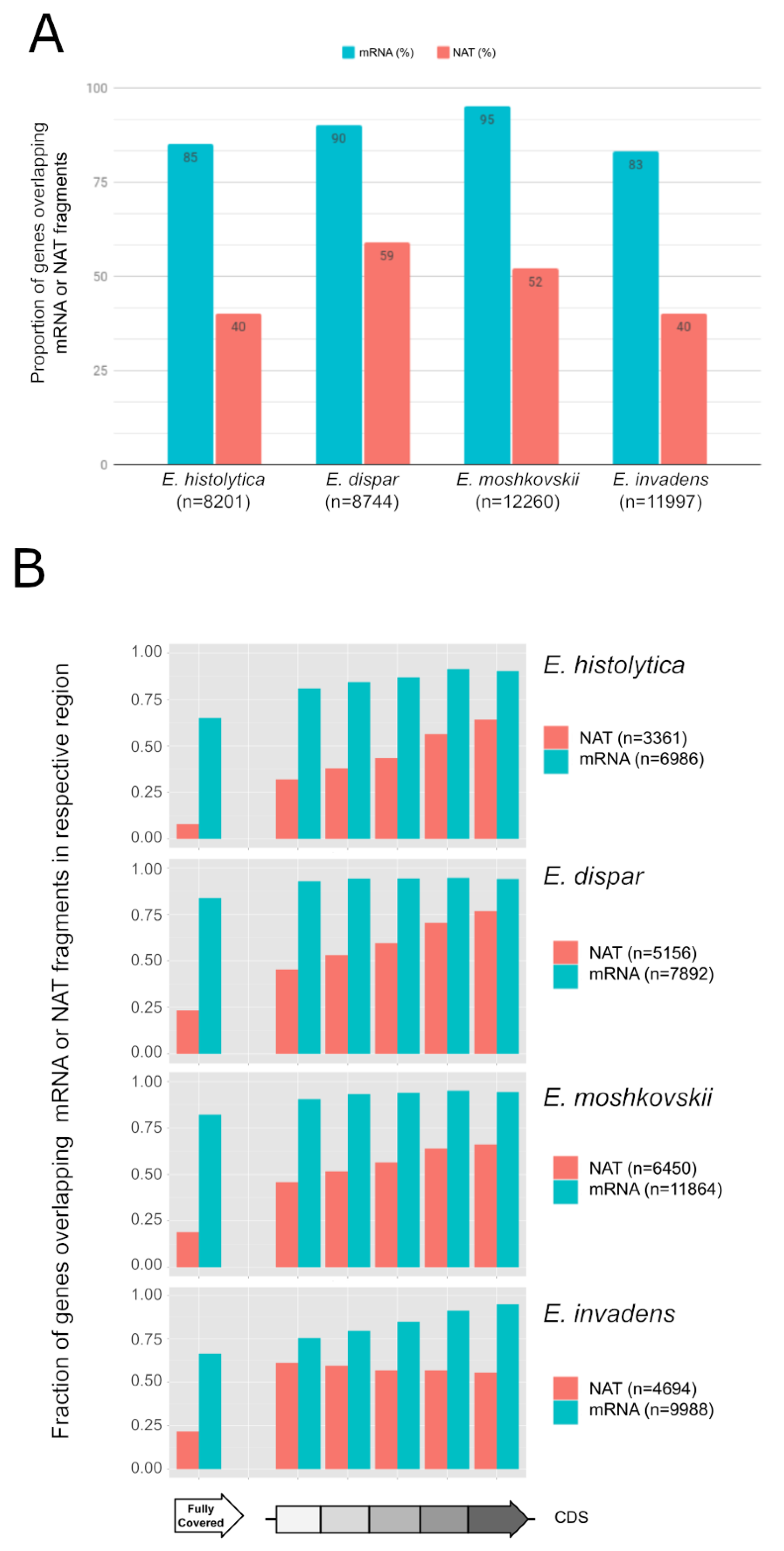
3.1. Widespread NAT in Entamoeba Species
3.2. Small RNAs and NATs Are Independent Entities of Non-Coding RNAs
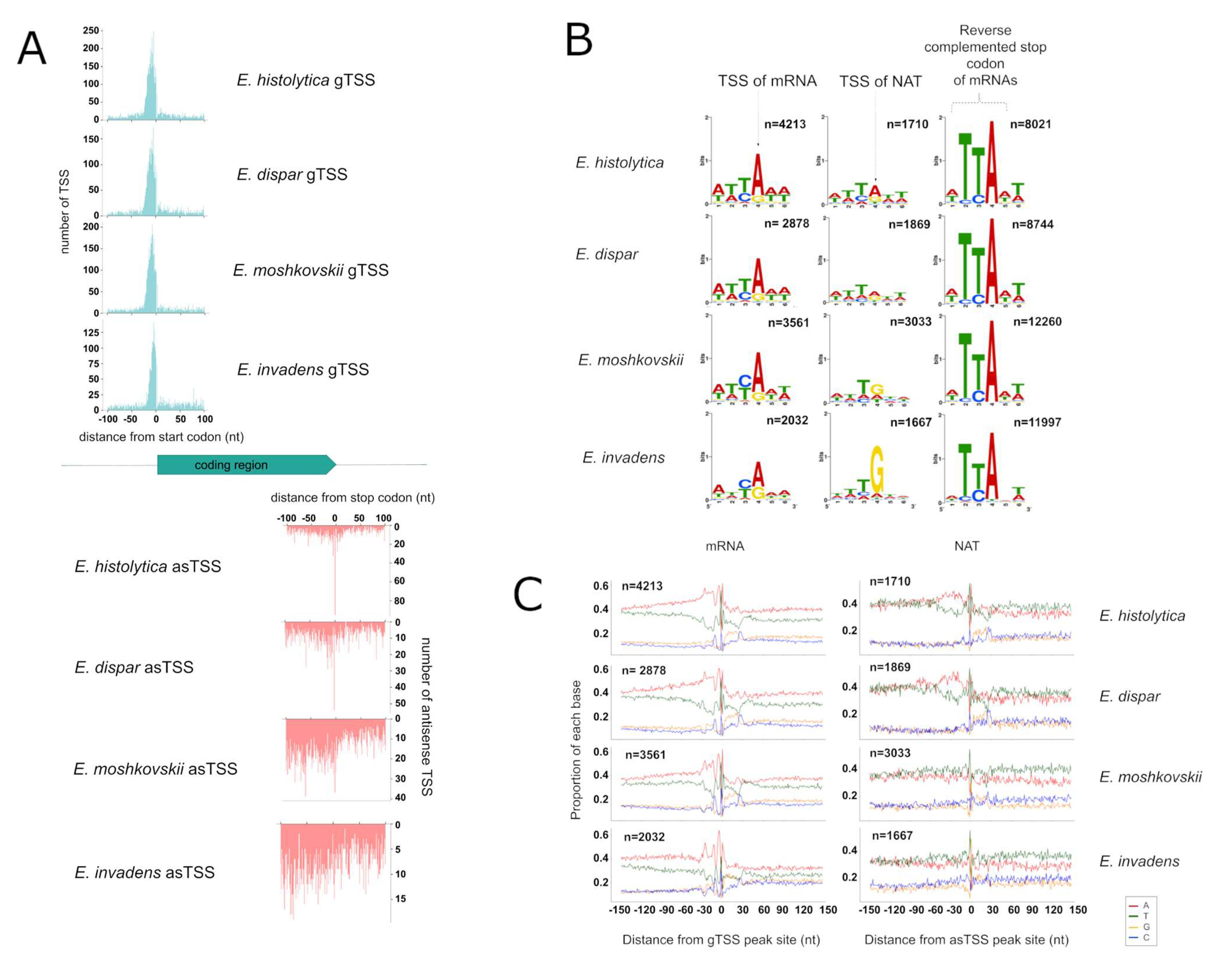
3.3. Identification of TSS for mRNA and NATs
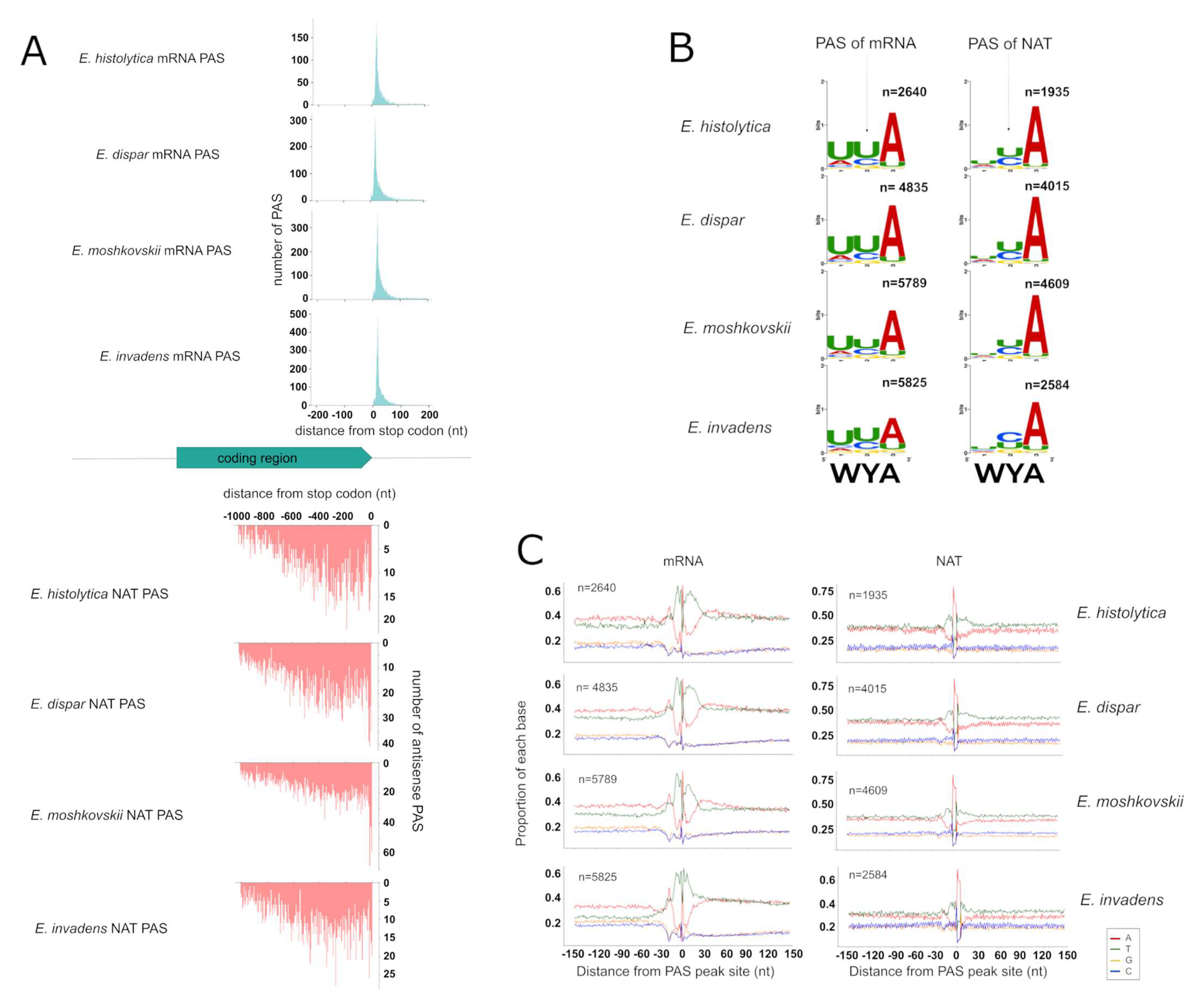
3.4. Identification of PAS for mRNAs and NATs
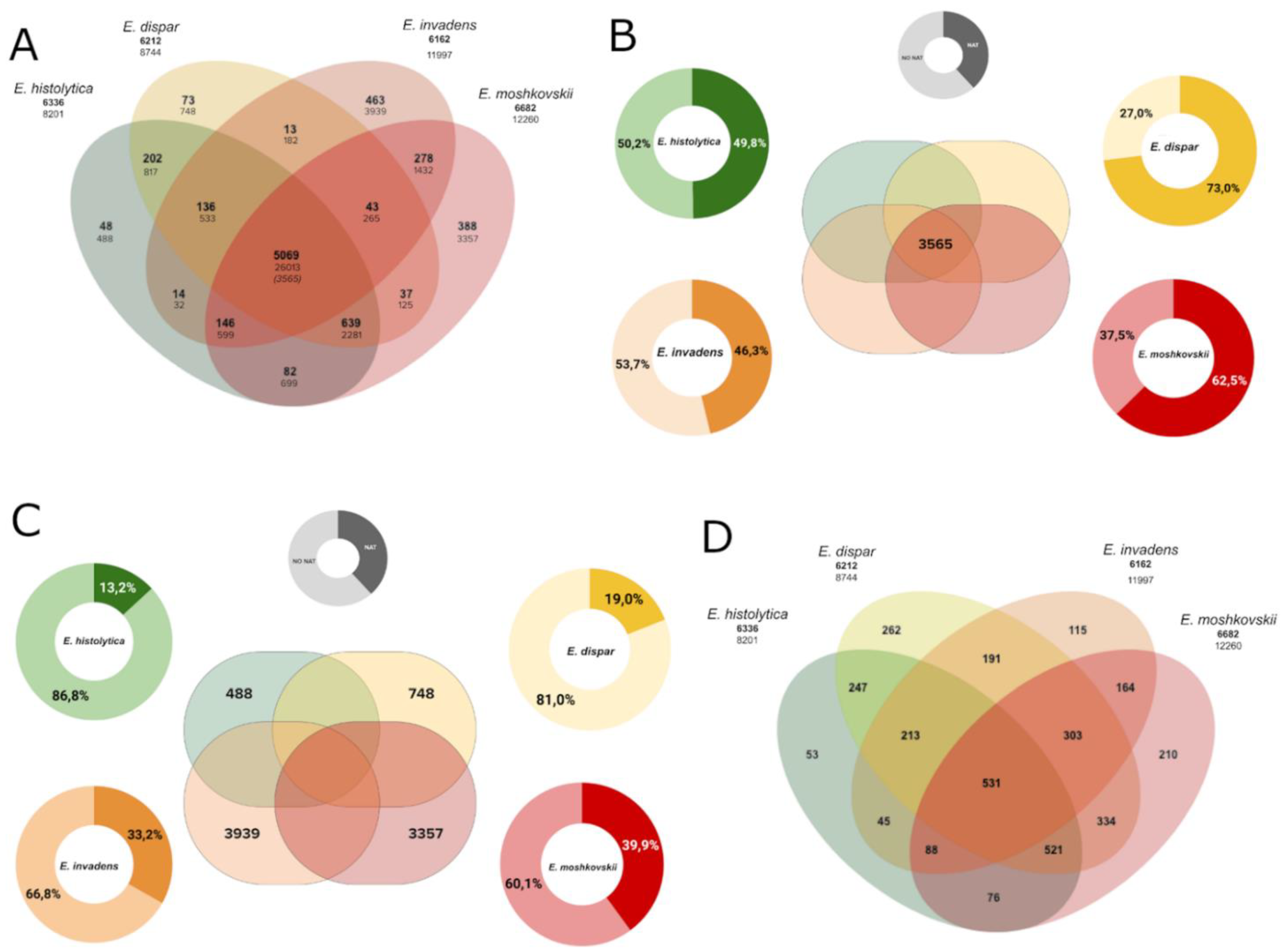
3.5. NAT in the Core-Genome and Species-Specific Genes
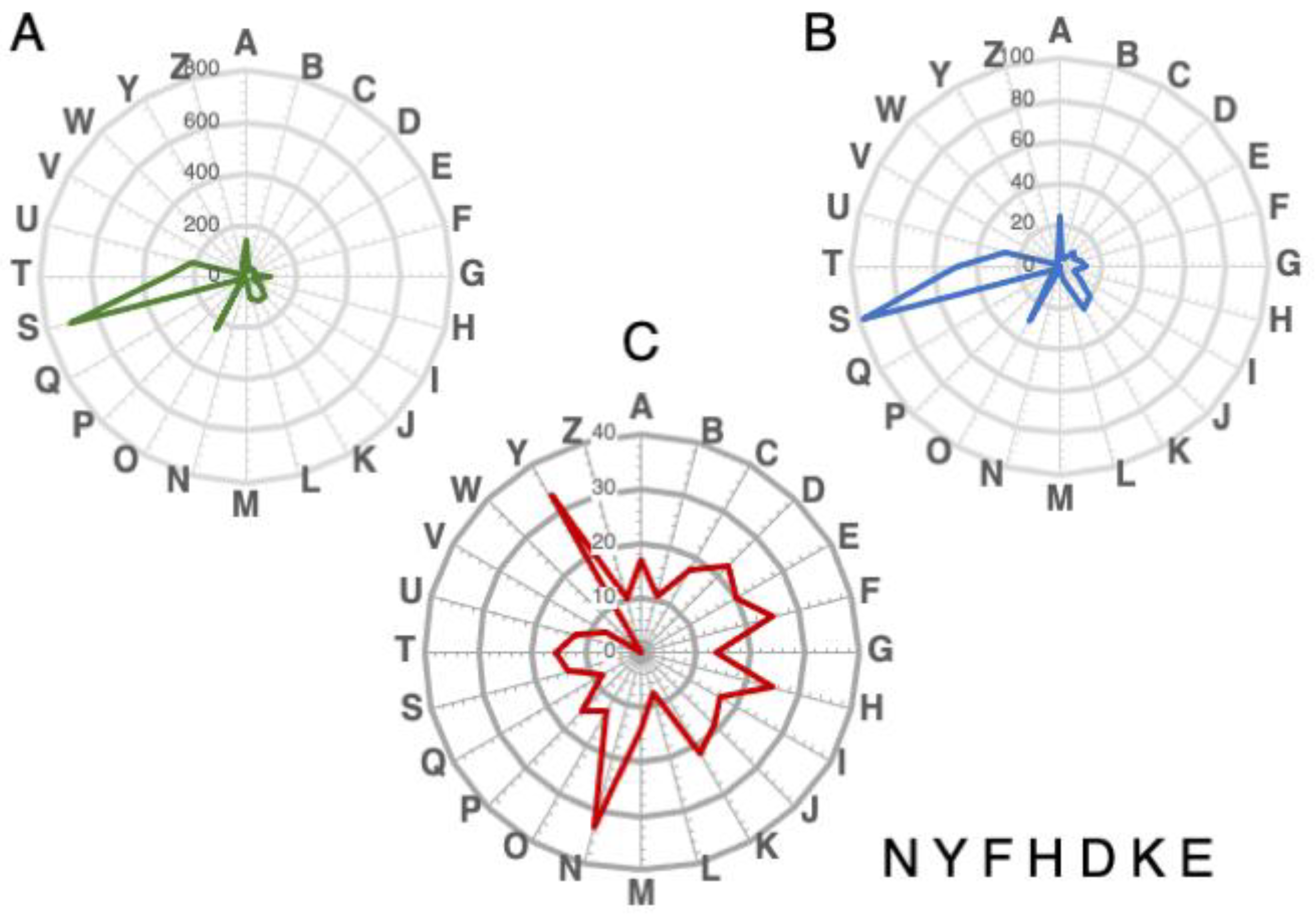
3.6. Functional Annotations of NAT-Associated Genes Conserved in All Species
3.7. Expression of NATs in Syntenic Blocks
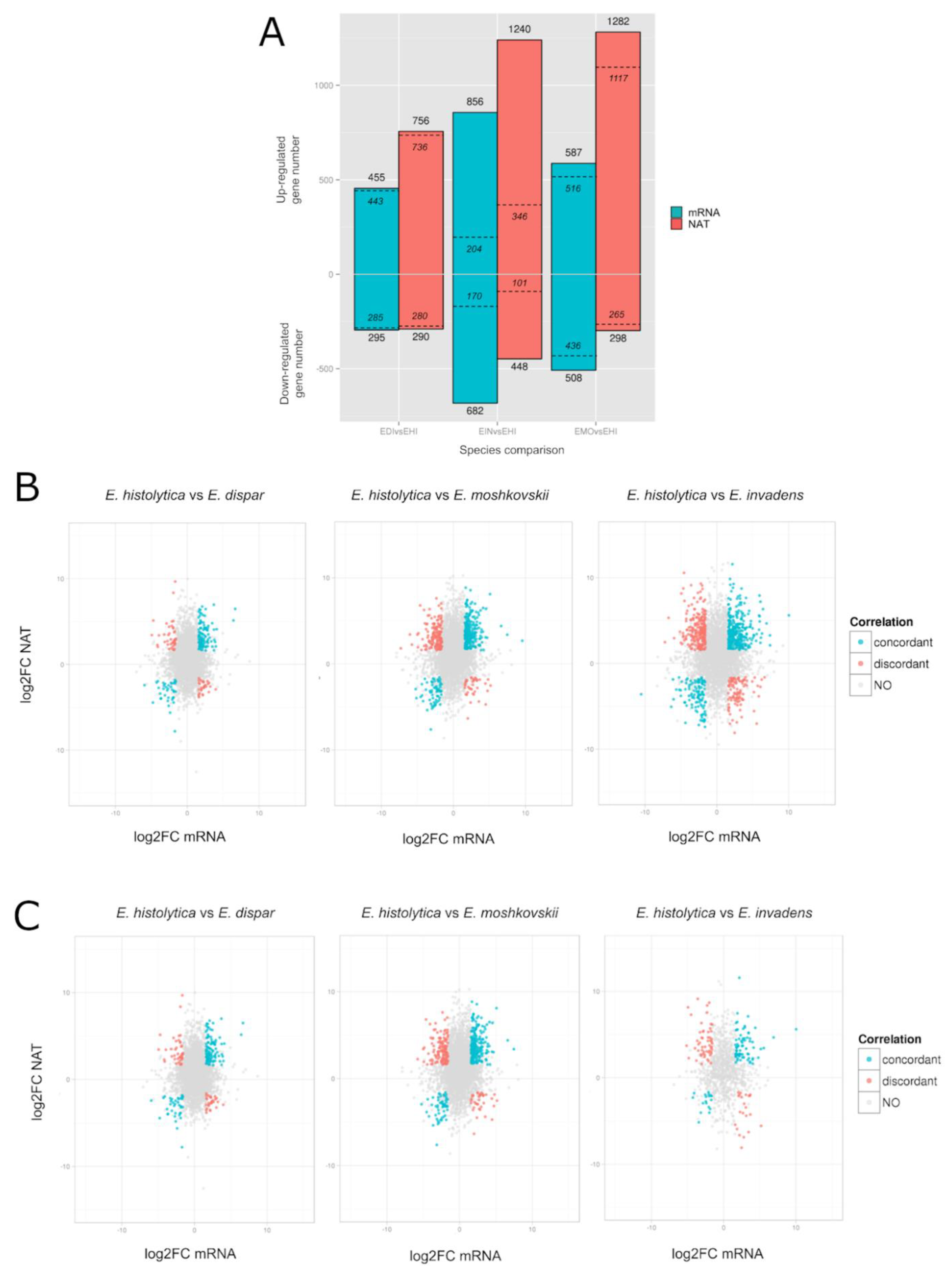
3.8. Differential Gene Expression Profile of Entamoeba Histolytica Antisense RNA Transcription
3.9. Functional Annotations of E. histolytica Syntenic Genes with Differentially Expressed NATs
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Stensvold, C.R.; Lebbad, M.; Victory, E.L.; Verweij, J.J.; Tannich, E.; Alfellani, M.; Legarraga, P.; Clark, C.G. Increased sampling reveals novel lineages of Entamoeba: Consequences of genetic diversity and host specificity for taxonomy and molecular detection. Protist 2011, 162, 525–541. [Google Scholar] [CrossRef] [Green Version]
- Stensvold, C.R. Pinning down the role of common luminal intestinal parasitic protists in human health and disease-status and challenges. Parasitology 2019, 146, 695–701. [Google Scholar] [CrossRef]
- Lokmer, A.; Aflalo, S.; Amougou, N.; Lafosse, S.; Froment, A.; Tabe, F.E.; Poyet, M.; Groussin, M.; Said-Mohamed, R.; Ségurel, L. Response of the human gut and saliva microbiome to urbanization in Cameroon. Sci. Rep. 2020, 10, 2856. [Google Scholar] [CrossRef] [Green Version]
- Marie, C.; Petri, W.A. Regulation of virulence of Entamoeba histolytica. Annu Rev. Microbiol 2014, 68, 493–520. [Google Scholar] [CrossRef]
- Cui, Z.; Li, J.; Chen, Y.; Zhang, L. Molecular epidemiology, evolution, and phylogeny of Entamoeba spp. Infect. Genet. Evol. 2019, 75, 104018. [Google Scholar] [CrossRef]
- Clark, C.G.; Diamond, L.S. The Laredo strain and other ‘Entamoeba histolytica-like’ amoebae are Entamoeba moshkovskii. Mol. Biochem. Parasitol. 1991, 46, 11–18. [Google Scholar] [CrossRef]
- Heredia, R.D.; Fonseca, J.A.; López, M.C. Entamoeba moshkovskii perspectives of a new agent to be considered in the diagnosis of amebiasis. Acta Trop. 2012, 123, 139–145. [Google Scholar] [CrossRef] [PubMed]
- Eichinger, D. Encystation in parasitic protozoa. Curr. Opin. Microbiol. 2001, 4, 421–426. [Google Scholar] [CrossRef]
- Chia, M.-Y.; Jeng, C.-R.; Hsiao, S.-H.; Lee, A.-H.; Chen, C.-Y.; Pang, V.F. Entamoeba invadens myositis in a common water monitor lizard (Varanus salvator). Vet. Pathol. 2009, 46, 673–676. [Google Scholar] [CrossRef]
- Loftus, B.; Anderson, I.; Davies, R.; Alsmark, U.C.M.; Samuelson, J.; Amedeo, P.; Roncaglia, P.; Berriman, M.; Hirt, R.P.; Mann, B.J.; et al. The genome of the protist parasite Entamoeba histolytica. Nature 2005, 433, 865–868. [Google Scholar] [CrossRef]
- Clark, C.; Alsmark, U.; Tazreiter, M.; Saito-Nakano, Y.; Ali, V.; Marion, S.; Weber, C.; Mukherjee, C.; Bruchhaus, I.; Tannich, E.; et al. Structure and content of the Entamoeba histolytica genome. Adv. Parasitol. 2007, 65, 51–190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lorenzi, H.A.; Puiu, D.; Miller, J.R.; Brinkac, L.M.; Amedeo, P.; Hall, N.; Caler, E.V. New assembly, reannotation and analysis of the Entamoeba histolytica genome reveal new genomic features and protein content information. PLoS Negl. Trop. Dis. 2010, 4, e716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aurrecoechea, C.; Barreto, A.; Brestelli, J.; Brunk, B.P.; Caler, E.V.; Fischer, S.; Gajria, B.; Gao, X.; Gingle, A.; Grant, G.R.; et al. AmoebaDB and MicrosporidiaDB: Functional genomic resources for Amoebozoa and Microsporidia species. Nucleic Acids Res. 2011, 39, D612–D619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilson, I.W.; Weedall, G.D.; Lorenzi, H.; Howcroft, T.; Hon, C.-C.; Deloger, M.; Guillén, N.; Paterson, S.; Clark, C.G.; Hall, N. Genetic Diversity and Gene Family Expansions in Members of the Genus Entamoeba. Genome Biol. Evol. 2019, 11, 688–705. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Samuelson, J.; Clark, C.G.; Eichinger, D.; Paul, J.; Van Dellen, K.; Hall, N.; Anderson, I.; Loftus, B. Gene discovery in the Entamoeba invadens genome. Mol. Biochem. Parasitol. 2003, 129, 23–31. [Google Scholar] [CrossRef]
- Ehrenkaufer, G.M.; Weedall, G.D.; Williams, D.; Lorenzi, H.A.; Caler, E.; Hall, N.; Singh, U. The genome and transcriptome of the enteric parasite Entamoeba invadens, a model for encystation. Genome Biol. 2013, 14, R77. [Google Scholar] [CrossRef] [Green Version]
- Zaki, M.; Meelu, P.; Sun, W.; Clark, C.G. Simultaneous differentiation and typing of Entamoeba histolytica and Entamoeba dispar. J. Clin. Microbiol 2002, 40, 1271–1276. [Google Scholar] [CrossRef] [Green Version]
- Willhoeft, U.; Buss, H.; Tannich, E. Genetic differences between Entamoeba histolytica and Entamoeba dispar. Arch. Med. Res. 2000, 31, S254. [Google Scholar] [CrossRef]
- Lorenzi, H.; Thiagarajan, M.; Haas, B.; Wortman, J.; Hall, N.; Caler, E. Genome wide survey, discovery and evolution of repetitive elements in three Entamoeba species. BMC Genom. 2008, 9, 595. [Google Scholar] [CrossRef] [Green Version]
- Kumari, V.; Sharma, R.; Yadav, V.P.; Gupta, A.K.; Bhattacharya, A.; Bhattacharya, S. Differential distribution of a SINE element in the Entamoeba histolytica and Entamoeba dispar genomes: Role of the LINE-encoded endonuclease. BMC Genom. 2011, 12, 267. [Google Scholar] [CrossRef] [Green Version]
- Pritham, E.J.; Feschotte, C.; Wessler, S.R. Unexpected diversity and differential success of DNA transposons in four species of Entamoeba protozoans. Mol. Biol. Evol. 2005, 22, 1751–1763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clark, C.G.; Ali IK, M.; Zaki, M.; Loftus, B.J.; Hall, N. Unique organisation of tRNA genes in Entamoeba histolytica. Mol. Biochem. Parasitol. 2006, 146, 24–29. [Google Scholar] [CrossRef]
- Tawari, B.; Ali, I.K.M.; Scott, C.; Quail, M.A.; Berriman, M.; Hall, N.; Clark, C.G. Patterns of evolution in the unique tRNA gene arrays of the genus Entamoeba. Mol. Biol. Evol. 2008, 25, 187–198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hackney, J.A.; Ehrenkaufer, G.M.; Singh, U. Identification of putative transcriptional regulatory networks in Entamoeba histolytica using Bayesian inference. Nucleic Acids Res. 2007, 35, 2141–2152. [Google Scholar] [CrossRef] [PubMed]
- Ehrenkaufer, G.M.; Haque, R.; Hackney, J.A.; Eichinger, D.J.; Singh, U. Identification of developmentally regulated genes in Entamoeba histolytica: Insights into mechanisms of stage conversion in a protozoan parasite. Cell. Microbiol. 2007, 9, 1426–1444. [Google Scholar] [CrossRef]
- De Cádiz, A.E.; Jeelani, G.; Nakada-Tsukui, K.; Caler, E.; Nozaki, T. Transcriptome analysis of encystation in Entamoeba invadens. PLoS ONE 2013, 8, e74840. [Google Scholar] [CrossRef]
- Manna, D.; Ehrenkaufer, G.M.; Singh, U. Regulation of gene expression in the protozoan parasite Entamoeba invadens: Identification of core promoter elements and promoters with stage-specific expression patterns. Int. J. Parasitol. 2014, 44, 837–845. [Google Scholar] [CrossRef] [Green Version]
- Naiyer, S.; Kaur, D.; Ahamad, J.; Singh, S.S.; Singh, Y.P.; Thakur, V.; Bhattacharya, A.; Bhattacharya, S. Transcriptomic analysis reveals novel downstream regulatory motifs and highly transcribed virulence factor genes of Entamoeba histolytica. BMC Genom. 2019, 20, 206. [Google Scholar] [CrossRef]
- Zhang, H.; Ehrenkaufer, G.M.; Pompey, J.M.; Hackney, J.A.; Singh, U. Small RNAs with 5′-polyphosphate termini associate with a Piwi-related protein and regulate gene expression in the single-celled eukaryote Entamoeba histolytica. PLoS Pathog. 2008, 4, e1000219. [Google Scholar] [CrossRef]
- Zhang, H.; Ehrenkaufer, G.M.; Manna, D.; Hall, N.; Singh, U. High Throughput Sequencing of Entamoeba 27nt Small RNA Population Reveals Role in Permanent Gene Silencing But No Effect on Regulating Gene Expression Changes during Stage Conversion, Oxidative, or Heat Shock Stress. PLoS ONE 2015, 10, e0134481. [Google Scholar] [CrossRef]
- Suresh, S.; Ehrenkaufer, G.; Zhang, H.; Singh, U. Development of RNA Interference Trigger-Mediated Gene Silencing in Entamoeba invadens. Infect. Immun 2016, 84, 964–975. [Google Scholar] [CrossRef] [Green Version]
- Mornico, D.; Hon, C.-C.; Koutero, M.; Weber, C.; Coppee, J.-Y.; Dillies, M.-A.; Guillen, N. Genomic determinants for initiation and length of natural antisense transcripts in Entamoeba histolytica. Sci. Rep. 2020, 10, 20190. [Google Scholar] [CrossRef]
- Zamorano, A.; López-Camarillo, C.; Orozco, E.; Weber, C.; Guillen, N.; Marchat, L.A. In silico analysis of EST and genomic sequences allowed the prediction of cis-regulatory elements for Entamoeba histolytica mRNA polyadenylation. Comput. Biol. Chem. 2008, 32, 256–263. [Google Scholar] [CrossRef] [PubMed]
- Hon, C.-C.; Weber, C.; Sismeiro, O.; Proux, C.; Koutero, M.; Deloger, M.; Das, S.; Agrahari, M.; Dillies, M.-A.; Jagla, B.; et al. Quantification of stochastic noise of splicing and polyadenylation in Entamoeba histolytica. Nucleic Acids Res. 2013, 41, 1936–1952. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mar-Aguilar, F.; Treviño, V.; Salinas-Hernández, J.E.; Taméz-Guerrero, M.M.; Barrón-González, M.P.; Morales-Rubio, E.; Treviño-Neávez, J.; Verduzco-Martínez, J.A.; Morales-Vallarta, M.R.; Resendez-Pérez, D. Identification and characterization of microRNAs from Entamoeba histolytica HM1-IMSS. PLoS ONE 2013, 8, e68202. [Google Scholar] [CrossRef] [PubMed]
- Saha, A.; Bhattacharya, S.; Bhattacharya, A. Serum stress responsive gene EhslncRNA of Entamoeba histolytica is a novel long noncoding RNA. Sci. Rep. 2016, 6, 27476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clark, C.G.; Diamond, L.S. Methods for cultivation of luminal parasitic protists of clinical importance. Clin. Microbiol. Rev. 2002, 15, 329–341. [Google Scholar] [CrossRef] [Green Version]
- Criscuolo, A.; Brisse, S. AlienTrimmer: A tool to quickly and accurately trim off multiple short contaminant sequences from high-throughput sequencing reads. Genomics 2013, 102, 500–506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dobin, A.; Gingeras, T.R. Mapping RNA-seq Reads with STAR. Curr. Protoc. Bioinform. 2015, 51, 11.14.1–11.14.19. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [Green Version]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [Green Version]
- Wu, T.D.; Watanabe, C.K. GMAP: A genomic mapping and alignment program for mRNA and EST sequences. Bioinformatics 2005, 21, 1859–1875. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liao, Y.; Smyth, G.K.; Shi, W. featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Ehrenkaufer, G.M.; Hall, N.; Singh, U. Identification of oligo-adenylated small RNAs in the parasite Entamoeba and a potential role for small RNA control. BMC Genom. 2020, 21, 879. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Trapnell, C.; Pop, M.; Salzberg, S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009, 10, R25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quinlan, A.R. BEDTools: The Swiss-Army Tool for Genome Feature Analysis. Curr. Protoc. Bioinform. 2014, 47, 11.12.1–11.12.34. [Google Scholar] [CrossRef]
- Bullard, J.H.; Purdom, E.; Hansen, K.D.; Dudoit, S. Evaluation of statistical methods for normalization and differential expression in mRNA-Seq experiments. BMC Bioinform. 2010, 11, 94. [Google Scholar] [CrossRef] [Green Version]
- Dugar, G.; Herbig, A.; Förstner, K.U.; Heidrich, N.; Reinhardt, R.; Nieselt, K.; Sharma, C.M. High-resolution transcriptome maps reveal strain-specific regulatory features of multiple Campylobacter jejuni isolates. PLoS Genet. 2013, 9, e1003495. [Google Scholar] [CrossRef] [PubMed]
- Bailey, T.L. DREME: Motif discovery in transcription factor ChIP-seq data. Bioinformatics 2011, 27, 1653–1659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Emms, D.M.; Kelly, S. OrthoFinder: Phylogenetic orthology inference for comparative genomics. Genome Biol. 2019, 20, 238. [Google Scholar] [CrossRef] [Green Version]
- Wagner, G.P.; Kin, K.; Lynch, V.J. Measurement of mRNA abundance using RNA-seq data: RPKM measure is inconsistent among samples. Theory Biosci. 2012, 131, 281–285. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Varet, H.; Brillet-Guéguen, L.; Coppée, J.-Y.; Dillies, M.-A. SARTools: A DESeq2- and EdgeR-Based R Pipeline for Comprehensive Differential Analysis of RNA-Seq Data. PLoS ONE 2016, 11, e0157022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2019. [Google Scholar]
- Wickham, H. ggplot2: Elegant Graphics for Data Analysis; Springer: Berlin/Heidelberg, Germany, 2016. [Google Scholar]
- Huerta-Cepas, J.; Szklarczyk, D.; Heller, D.; Hernández-Plaza, A.; Forslund, S.K.; Cook, H.V.; Mende, D.R.; Letunic, I.; Rattei, T.; Jensen, L.J.; et al. eggNOG 5.0: A hierarchical, functionally and phylogenetically annotated orthology resource based on 5090 organisms and 2502 viruses. Nucleic Acids Res. 2019, 47, D309–D314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Ehrenkaufer, G.M.; Hall, N.; Singh, U. Small RNA pyrosequencing in the protozoan parasite Entamoeba histolytica reveals strain-specific small RNAs that target virulence genes. BMC Genom. 2013, 14, 53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- North, J.A.; Wildenthal, J.A.; Erb, T.J.; Evans, B.S.; Byerly, K.M.; Gerlt, J.A.; Tabita, F.R. A bifunctional salvage pathway for two distinct S-adenosylmethionine by-products that is widespread in bacteria, including pathogenic Escherichia coli. Mol. Microbiol 2020, 113, 923–937. [Google Scholar] [CrossRef] [PubMed]
- Nakada-Tsukui, K.; Saito-Nakano, Y.; Ali, V.; Nozaki, T. A retromerlike complex is a novel Rab7 effector that is involved in the transport of the virulence factor cysteine protease in the enteric protozoan parasite Entamoeba histolytica. Mol. Biol. Cell 2005, 16, 5294–5303. [Google Scholar] [CrossRef] [Green Version]
- Tsuchiya, H.; Ohtake, F.; Arai, N.; Kaiho, A.; Yasuda, S.; Tanaka, K.; Saeki, Y. In Vivo Ubiquitin Linkage-type Analysis Reveals that the Cdc48-Rad23/Dsk2 Axis Contributes to K48-Linked Chain Specificity of the Proteasome. Mol. Cell 2017, 66, 488–502. [Google Scholar] [CrossRef] [Green Version]
- Bodnar, N.; Kim, K.H.; Ji, Z.; Wales, T.E.; Svetlov, V.; Nudler, E.; Engen, J.R.; Walz, T.; Rapoport, T.A. Structure of the Cdc48 ATPase with its ubiquitin-binding cofactor Ufd1-Npl4. Nat. Struct. Mol. Biol. 2018, 25, 616–622. [Google Scholar] [CrossRef]
- Bays, N.W.; Hampton, R.Y. Cdc48-Ufd1-Npl4: Stuck in the middle with Ub. Curr. Biol. 2002, 12, R366–R371. [Google Scholar] [CrossRef] [Green Version]
- Fu, M.; Blackshear, P.J. RNA-binding proteins in immune regulation: A focus on CCCH zinc finger proteins. Nat. Rev. Immunol. 2017, 17, 130–143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kastenmayer, J.P.; Green, P.J. Novel features of the XRN-family in Arabidopsis: Evidence that AtXRN4, one of several orthologs of nuclear Xrn2p/Rat1p, functions in the cytoplasm. Proc. Natl. Acad. Sci. USA 2000, 97, 13985–13990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weber, C.; Koutero, M.; Dillies, M.-A.; Varet, H.; Lopez-Camarillo, C.; Coppée, J.Y.; Hon, C.-C.; Guillén, N. Extensive transcriptome analysis correlates the plasticity of Entamoeba histolytica pathogenesis to rapid phenotype changes depending on the environment. Sci. Rep. 2016, 6, 35852. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wight, M.; Werner, A. The functions of natural antisense transcripts. Essays Biochem. 2013, 54, 91–101. [Google Scholar] [CrossRef] [PubMed]
- Nakada-Tsukui, K.; Watanabe, N.; Maehama, T.; Nozaki, T. Phosphatidylinositol Kinases and Phosphatases in Entamoeba histolytica. Front. Cell Infect. Microbiol. 2019, 9, 150. [Google Scholar] [CrossRef]
- Watanabe, N.; Nakada-Tsukui, K.; Maehama, T.; Nozaki, T. Dynamism of PI4-Phosphate during Interactions with Human Erythrocytes in Entamoeba histolytica. Microorganisms 2020, 8, 1050. [Google Scholar] [CrossRef]
- Saito-Nakano, Y.; Wahyuni, R.; Nakada-Tsukui, K.; Tomii, K.; Nozaki, T. Rab7D small GTPase is involved in phago-, trogocytosis and cytoskeletal reorganization in the enteric protozoan Entamoeba histolytica. Cell Microbiol. 2021, 23, e13267. [Google Scholar] [CrossRef]
- Weedall, G.D.; Clark, C.G.; Koldkjaer, P.; Kay, S.; Bruchhaus, I.; Tannich, E.; Paterson, S.; Hall, N. Genomic diversity of the human intestinal parasite Entamoeba histolytica. Genome Biol. 2012, 13, R38. [Google Scholar] [CrossRef] [Green Version]
- Wanowska, E.; Kubiak, M.R.; Rosikiewicz, W.; Makałowska, I.; Szcześniak, M.W. Natural antisense transcripts in diseases: From modes of action to targeted therapies. Wiley Interdiscip. Rev. RNA 2018, 9. [Google Scholar] [CrossRef] [Green Version]
- Zinad, H.S.; Natasya, I.; Werner, A. Natural Antisense Transcripts at the Interface between Host Genome and Mobile Genetic Elements. Front. Microbiol. 2017, 8, 2292. [Google Scholar] [CrossRef]
- Siegel, T.N.; Hon, C.-C.; Zhang, Q.; Lopez-Rubio, J.-J.; Scheidig-Benatar, C.; Martins, R.M.; Sismeiro, O.; Coppée, J.-Y.; Scherf, A. Strand-specific RNA-Seq reveals widespread and developmentally regulated transcription of natural antisense transcripts in Plasmodium falciparum. BMC Genom. 2014, 15, 150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Radke, J.R.; Behnke, M.S.; Mackey, A.J.; Radke, J.B.; Roos, D.S.; White, M.W. The transcriptome of Toxoplasma gondii. BMC Biol. 2005, 3, 26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elmendorf, H.G.; Singer, S.M.; Nash, T.E. The abundance of sterile transcripts in Giardia lamblia. Nucleic Acids Res. 2001, 29, 4674–4683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Militello, K.T.; Refour, P.; Comeaux, C.A.; Duraisingh, M.T. Antisense RNA and RNAi in protozoan parasites: Working hard or hardly working? Mol. Biochem. Parasitol. 2008, 157, 117–126. [Google Scholar] [CrossRef]
- Fahim, A.; Afrin, F.; Wen, G.; Ananvoranich, S. Characterization of natural antisense transcripts arisen from the locus encoding Toxoplasma gondii ubiquitin-like protease. Mol. Biochem. Parasitol. 2020, 240, 111334. [Google Scholar] [CrossRef]
- Donaldson, M.E.; Ostrowski, L.A.; Goulet, K.M.; Saville, B.J. Transcriptome analysis of smut fungi reveals widespread intergenic transcription and conserved antisense transcript expression. BMC Genom. 2017, 18, 340. [Google Scholar] [CrossRef]
- Donaldson, M.E.; Saville, B.J. Natural antisense transcripts in fungi. Mol. Microbiol. 2012, 85, 405–417. [Google Scholar] [CrossRef]
- Zhang, H.; Alramini, H.; Tran, V.; Singh, U. Nucleus-localized antisense small RNAs with 5′-polyphosphate termini regulate long term transcriptional gene silencing in Entamoeba histolytica G3 strain. J. Biol. Chem. 2011, 286, 44467–44479. [Google Scholar] [CrossRef] [Green Version]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mornico, D.; Hon, C.-C.; Koutero, M.; Weber, C.; Coppée, J.-Y.; Clark, C.G.; Dillies, M.-A.; Guillen, N. RNA Sequencing Reveals Widespread Transcription of Natural Antisense RNAs in Entamoeba Species. Microorganisms 2022, 10, 396. https://doi.org/10.3390/microorganisms10020396
Mornico D, Hon C-C, Koutero M, Weber C, Coppée J-Y, Clark CG, Dillies M-A, Guillen N. RNA Sequencing Reveals Widespread Transcription of Natural Antisense RNAs in Entamoeba Species. Microorganisms. 2022; 10(2):396. https://doi.org/10.3390/microorganisms10020396
Chicago/Turabian StyleMornico, Damien, Chung-Chau Hon, Mikael Koutero, Christian Weber, Jean-Yves Coppée, C Graham Clark, Marie-Agnes Dillies, and Nancy Guillen. 2022. "RNA Sequencing Reveals Widespread Transcription of Natural Antisense RNAs in Entamoeba Species" Microorganisms 10, no. 2: 396. https://doi.org/10.3390/microorganisms10020396