
Genomics-Based Reconstruction and Predictive Profiling of Amino Acid Biosynthesis in the Human Gut Microbiome


Abstract
:1. Introduction
2. Materials and Methods
2.1. HGM Genomic Collection
2.2. Metabolic Reconstruction
2.3. Functional Profiling of 16S Metagenomics Datasets
2.4. Comparison of Predicted Functional Profiles with the State-of-the-Art PICRUSt2 Approach
2.5. Functional Profiling of Shotgun Metagenomes
3. Results
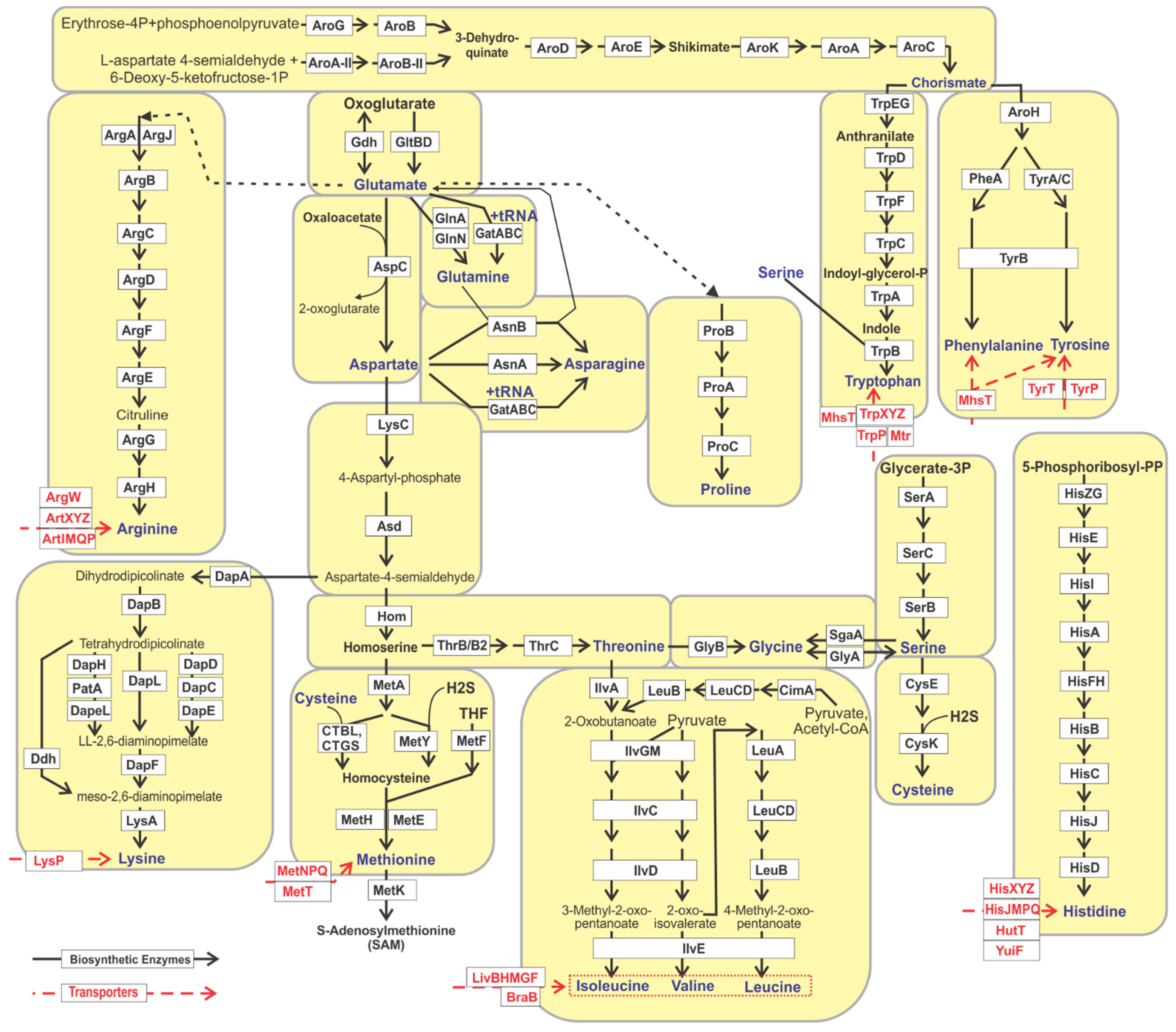
3.1. Genomic Reconstruction of Amino Acid Biosynthetic Pathways
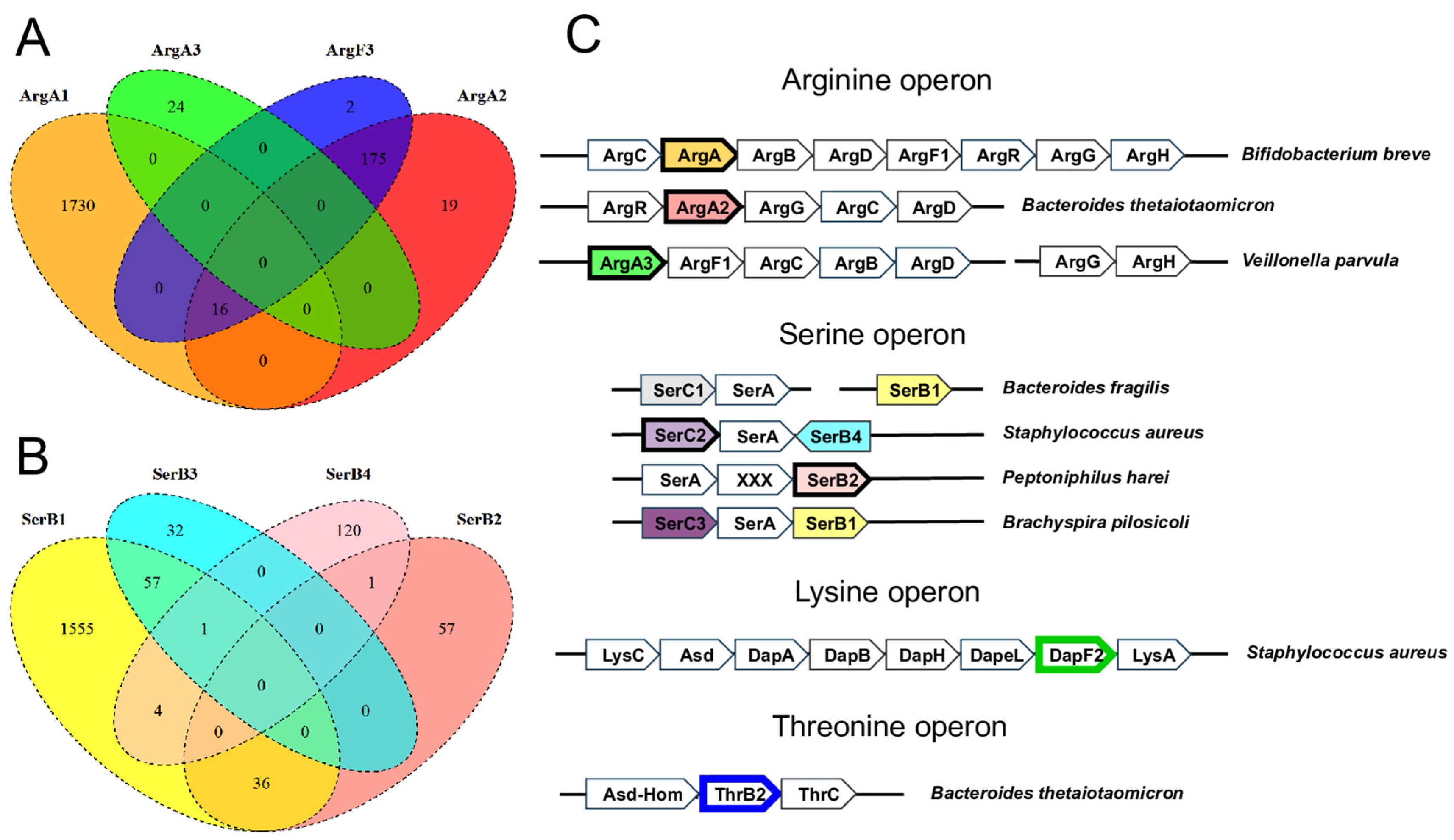
3.2. Non-Orthologous Gene Displacements in Biosynthetic Pathways
3.3. Incomplete Pathway Variants and Salvage of Amino Acid Precursors
3.4. Predicted Amino Acid Synthesis Phenotypes and Growth Requirements
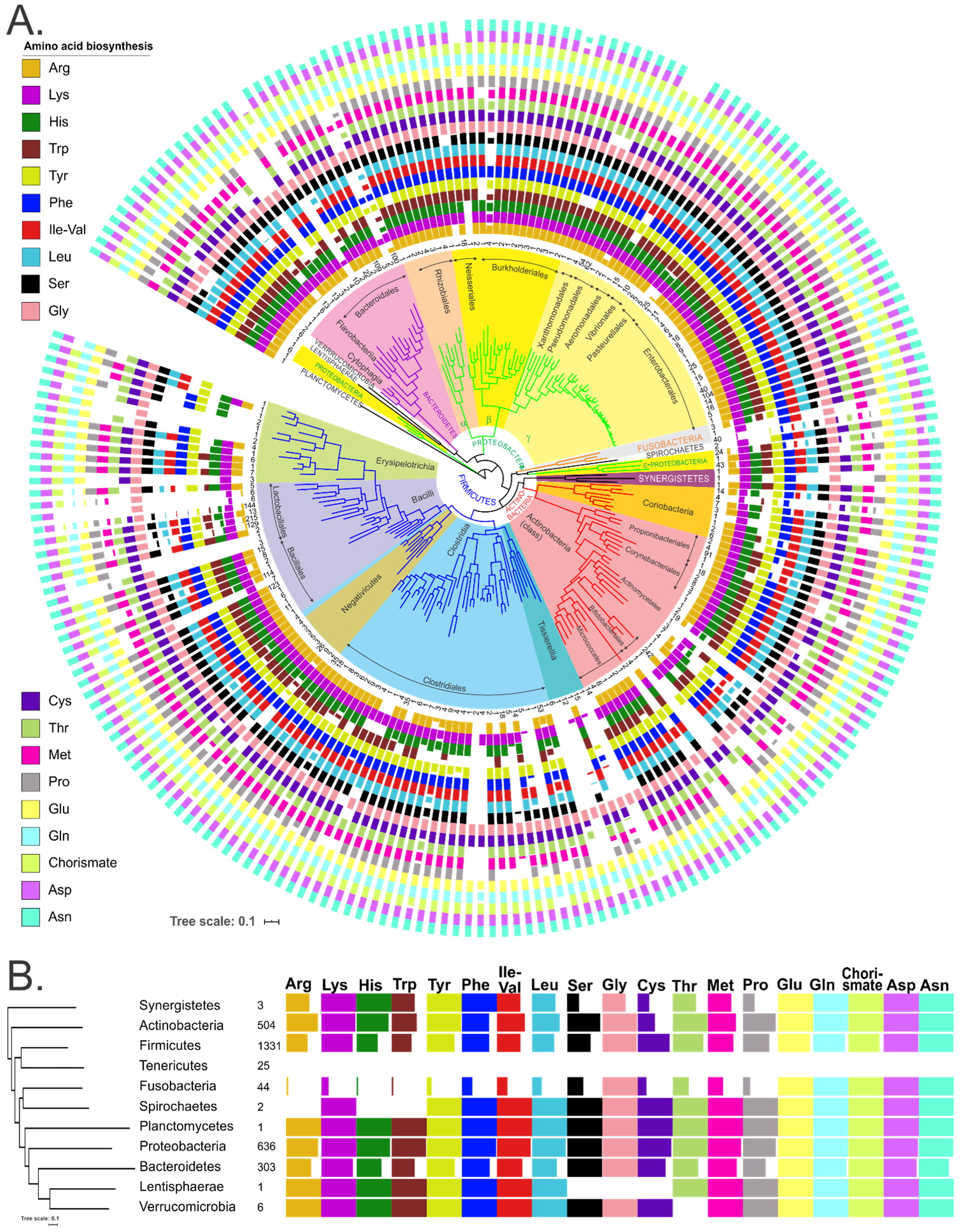
3.5. Phylogenetic Variability of Binary Amino Acid Synthesis Phenotypes
3.6. Profiling of Amino Acid Metabolic Potential of the Human Gut Microbiome
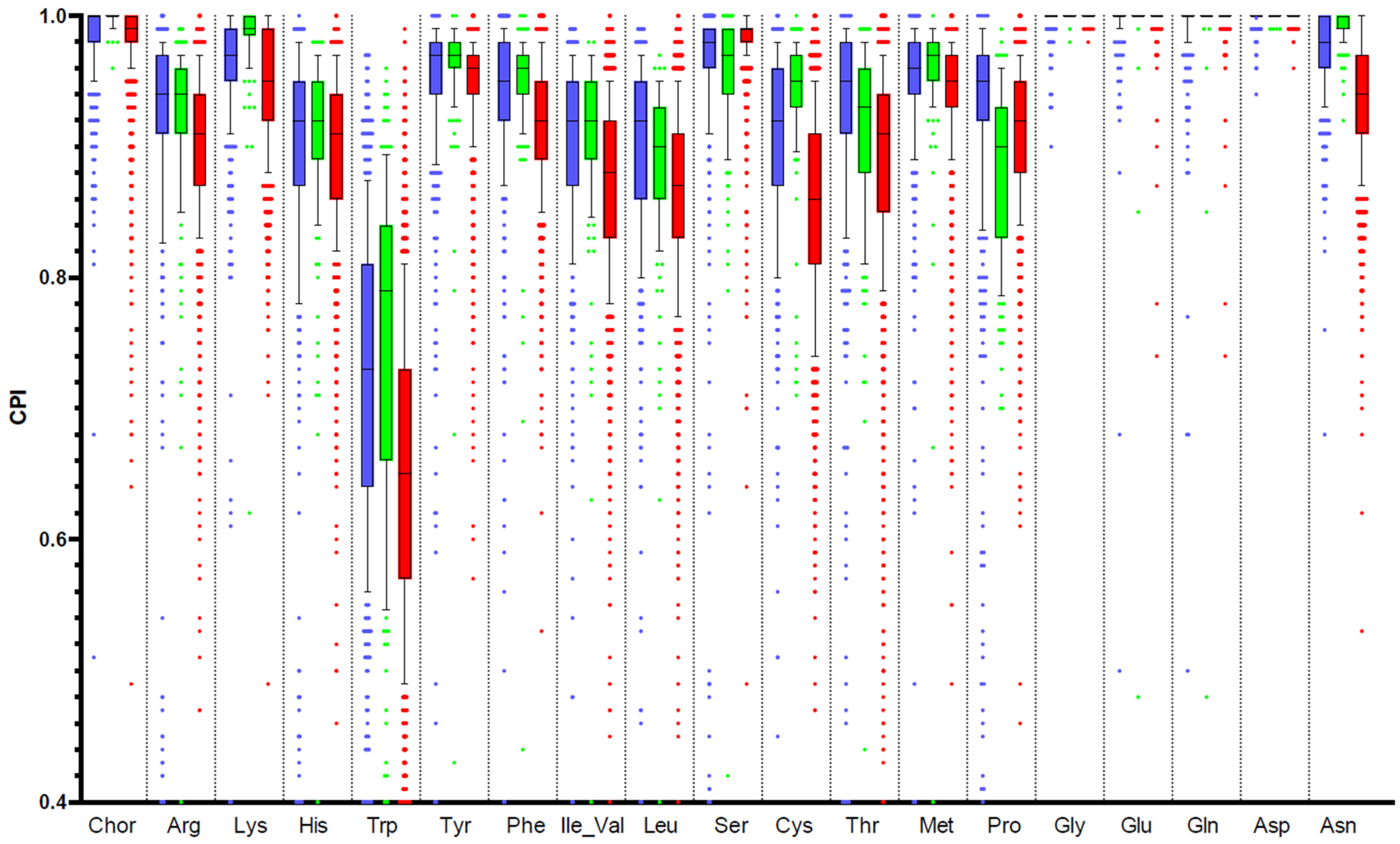
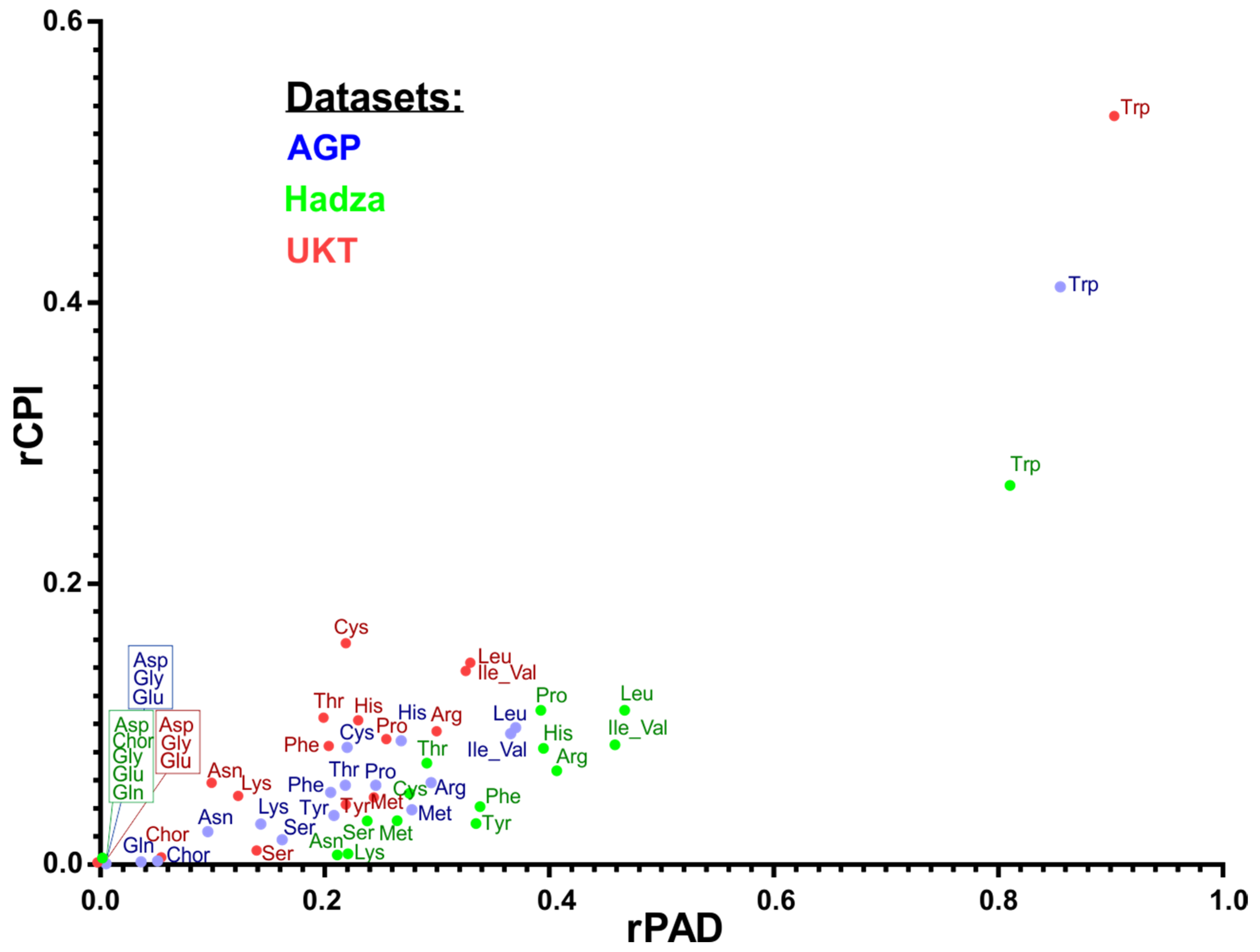
3.7. Comparison of Amino Acid Production Phenotypes and Pathway Abundances
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Depken, D.E. Gut Microbiota; Impacts on Human Health and Wellbeing. In Good Health and, Well-Being; Springer: Berlin/Heidelberg, Germany, 2019; pp. 1–9. [Google Scholar]
- Wang, B.; Yao, M.; Lv, Z.; Ling, Z.; Li, L. The Human Microbiota in Health and Disease. Engineering 2017, 3, 71–82. [Google Scholar] [CrossRef]
- Valdes, A.M.; Walter, J.; Segal, E.; Spector, T.D. Role of the gut microbiota in nutrition and health. BMJ 2018, 361, k2179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oliphant, K.; Allen-Vercoe, E. Macronutrient metabolism by the human gut microbiome: Major fermentation by-products and their impact on host health. Microbiome 2019, 7, 91. [Google Scholar] [CrossRef]
- MacDonald, T.T.; Gordon, J.N. Bacterial regulation of intestinal immune responses. Gastroenterol. Clin. N. Am. 2005, 34, 401–412. [Google Scholar] [CrossRef]
- Peterson, C.T.; Rodionov, D.A.; Osterman, A.L.; Peterson, S.N. B Vitamins and Their Role in Immune Regulation and Cancer. Nutrients 2020, 12, 3380. [Google Scholar] [CrossRef] [PubMed]
- Ding, R.X.; Goh, W.R.; Wu, R.N.; Yue, X.Q.; Luo, X.; Khine, W.W.T.; Wu, J.R.; Lee, Y.K. Revisit gut microbiota and its impact on human health and disease. J. Food Drug Anal. 2019, 27, 623–631. [Google Scholar] [CrossRef] [PubMed]
- Davila, A.M.; Blachier, F.; Gotteland, M.; Andriamihaja, M.; Benetti, P.H.; Sanz, Y.; Tome, D. Intestinal luminal nitrogen metabolism: Role of the gut microbiota and consequences for the host. Pharmacol. Res. 2013, 68, 95–107. [Google Scholar] [CrossRef]
- Almeida, A.; Nayfach, S.; Boland, M.; Strozzi, F.; Beracochea, M.; Shi, Z.J.; Pollard, K.S.; Sakharova, E.; Parks, D.H.; Hugenholtz, P.; et al. A unified catalog of 204,938 reference genomes from the human gut microbiome. Nat. Biotechnol. 2021, 39, 105–114. [Google Scholar] [CrossRef]
- Mee, M.T.; Collins, J.J.; Church, G.M.; Wang, H.H. Syntrophic exchange in synthetic microbial communities. Proc. Natl. Acad. Sci. USA 2014, 111, E2149–E2156. [Google Scholar] [CrossRef] [Green Version]
- Soto-Martin, E.C.; Warnke, I.; Farquharson, F.M.; Christodoulou, M.; Horgan, G.; Derrien, M.; Faurie, J.M.; Flint, H.J.; Duncan, S.H.; Louis, P. Vitamin Biosynthesis by Human Gut Butyrate-Producing Bacteria and Cross-Feeding in Synthetic Microbial Communities. mBio 2020, 11, e00886-20. [Google Scholar] [CrossRef]
- Seif, Y.; Choudhary, K.S.; Hefner, Y.; Anand, A.; Yang, L.; Palsson, B.O. Metabolic and genetic basis for auxotrophies in Gram-negative species. Proc. Natl. Acad. Sci. USA 2020, 117, 6264–6273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rajilic-Stojanovic, M.; de Vos, W.M. The first 1000 cultured species of the human gastrointestinal microbiota. FEMS Microbiol. Rev. 2014, 38, 996–1047. [Google Scholar] [CrossRef] [PubMed]
- Forster, S.C.; Kumar, N.; Anonye, B.O.; Almeida, A.; Viciani, E.; Stares, M.D.; Dunn, M.; Mkandawire, T.T.; Zhu, A.; Shao, Y.; et al. A human gut bacterial genome and culture collection for improved metagenomic analyses. Nat. Biotechnol. 2019, 37, 186–192. [Google Scholar] [CrossRef] [PubMed]
- Poyet, M.; Groussin, M.; Gibbons, S.M.; Avila-Pacheco, J.; Jiang, X.; Kearney, S.M.; Perrotta, A.R.; Berdy, B.; Zhao, S.; Lieberman, T.D.; et al. A library of human gut bacterial isolates paired with longitudinal multiomics data enables mechanistic microbiome research. Nat. Med. 2019, 25, 1442–1452. [Google Scholar] [CrossRef] [PubMed]
- Zou, Y.; Xue, W.; Luo, G.; Deng, Z.; Qin, P.; Guo, R.; Sun, H.; Xia, Y.; Liang, S.; Dai, Y.; et al. 1520 reference genomes from cultivated human gut bacteria enable functional microbiome analyses. Nat. Biotechnol. 2019, 37, 179–185. [Google Scholar] [CrossRef]
- Overbeek, R.; Begley, T.; Butler, R.M.; Choudhuri, J.V.; Chuang, H.Y.; Cohoon, M.; de Crecy-Lagard, V.; Diaz, N.; Disz, T.; Edwards, R.; et al. The subsystems approach to genome annotation and its use in the project to annotate 1000 genomes. Nucleic Acids Res. 2005, 33, 5691–5702. [Google Scholar] [CrossRef] [Green Version]
- Overbeek, R.; Olson, R.; Pusch, G.D.; Olsen, G.J.; Davis, J.J.; Disz, T.; Edwards, R.A.; Gerdes, S.; Parrello, B.; Shukla, M.; et al. The SEED and the Rapid Annotation of microbial genomes using Subsystems Technology (RAST). Nucleic Acids Res. 2014, 42, D206–D214. [Google Scholar] [CrossRef]
- Osterman, A.; Overbeek, R. Missing genes in metabolic pathways: A comparative genomics approach. Curr. Opin. Chem. Biol. 2003, 7, 238–251. [Google Scholar] [CrossRef]
- Rodionova, I.A.; Li, X.; Plymale, A.E.; Motamedchaboki, K.; Konopka, A.E.; Romine, M.F.; Fredrickson, J.K.; Osterman, A.L.; Rodionov, D.A. Genomic distribution of B-vitamin auxotrophy and uptake transporters in environmental bacteria from the Chloroflexi phylum. Environ. Microbiol. Rep. 2015, 7, 204–210. [Google Scholar] [CrossRef]
- Leyn, S.A.; Maezato, Y.; Romine, M.F.; Rodionov, D.A. Genomic Reconstruction of Carbohydrate Utilization Capacities in Microbial-Mat Derived Consortia. Front. Microbiol. 2017, 8, 1304. [Google Scholar] [CrossRef] [Green Version]
- Rodionov, D.A.; Yang, C.; Li, X.; Rodionova, I.A.; Wang, Y.; Obraztsova, A.Y.; Zagnitko, O.P.; Overbeek, R.; Romine, M.F.; Reed, S.; et al. Genomic encyclopedia of sugar utilization pathways in the Shewanella genus. BMC Genom. 2010, 11, 494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leyn, S.A.; Suvorova, I.A.; Kholina, T.D.; Sherstneva, S.S.; Novichkov, P.S.; Gelfand, M.S.; Rodionov, D.A. Comparative genomics of transcriptional regulation of methionine metabolism in Proteobacteria. PLoS ONE 2014, 9, e113714. [Google Scholar] [CrossRef] [PubMed]
- Rodionov, D.A.; Vitreschak, A.G.; Mironov, A.A.; Gelfand, M.S. Regulation of lysine biosynthesis and transport genes in bacteria: Yet another RNA riboswitch? Nucleic Acids Res. 2003, 31, 6748–6757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodionov, D.A.; Arzamasov, A.A.; Khoroshkin, M.S.; Iablokov, S.N.; Leyn, S.A.; Peterson, S.N.; Novichkov, P.S.; Osterman, A.L. Micronutrient Requirements and Sharing Capabilities of the Human Gut Microbiome. Front. Microbiol. 2019, 10, 1316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, V.; Rodionov, D.A.; Leyn, S.A.; Tran, D.; Iablokov, S.N.; Ding, H.; Peterson, D.A.; Osterman, A.L.; Peterson, S.N. B-Vitamin Sharing Promotes Stability of Gut Microbial Communities. Front. Microbiol. 2019, 10, 1485. [Google Scholar] [CrossRef] [PubMed]
- Blanton, L.V.; Charbonneau, M.R.; Salih, T.; Barratt, M.J.; Venkatesh, S.; Ilkaveya, O.; Subramanian, S.; Manary, M.J.; Trehan, I.; Jorgensen, J.M.; et al. Gut bacteria that prevent growth impairments transmitted by microbiota from malnourished children. Science 2016, 351, aad3311. [Google Scholar] [CrossRef] [Green Version]
- Feng, L.; Raman, A.S.; Hibberd, M.C.; Cheng, J.; Griffin, N.W.; Peng, Y.; Leyn, S.A.; Rodionov, D.A.; Osterman, A.L.; Gordon, J.I. Identifying determinants of bacterial fitness in a model of human gut microbial succession. Proc. Natl. Acad. Sci. USA 2020, 117, 2622–2633. [Google Scholar] [CrossRef] [Green Version]
- Gehrig, J.G.; Venkatesh, S.; Chang, H.; Hibberd, M.C.; Kung, V.L.; Cheng, J.; Chen, R.Y.; Subramanian, S.; Cowardin, C.A.; Meier, M.F.; et al. Effects of microbiota-directed therapeutic foods in gnotobiotic mice, gnotobiotic piglets, and undernourished children. Science 2019, 365, 139. [Google Scholar] [CrossRef] [Green Version]
- Raman, A.S.; Gehrig, J.L.; Venkatesh, S.; Chang, H.W.; Hibberd, M.C.; Subramanian, S.; Kang, G.; Bessong, P.O.; Lima, A.A.M.; Kosek, M.N.; et al. A sparse covarying unit that describes healthy and impaired human gut microbiota development. Science 2019, 365, eaau4735. [Google Scholar] [CrossRef] [Green Version]
- Iablokov, S.N.; Klimenko, N.S.; Efimova, D.A.; Shashkova, T.; Novichkov, P.S.; Rodionov, D.A.; Tyakht, A.V. Metabolic Phenotypes as Potential Biomarkers for Linking Gut Microbiome With Inflammatory Bowel Diseases. Front. Mol. Biosci. 2021, 7, 603740. [Google Scholar] [CrossRef]
- Wattam, A.R.; Davis, J.J.; Assaf, R.; Boisvert, S.; Brettin, T.; Bun, C.; Conrad, N.; Dietrich, E.M.; Disz, T.; Gabbard, J.L.; et al. Improvements to PATRIC, the all-bacterial Bioinformatics Database and Analysis Resource Center. Nucleic Acids Res. 2017, 45, D535–D542. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. MUSCLE: A multiple sequence alignment method with reduced time and space complexity. BMC Bioinform. 2004, 5, 113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stamatakis, A. Using RAxML to Infer Phylogenies. Curr. Protoc. Bioinform. 2015, 51, 11–14. [Google Scholar] [CrossRef] [PubMed]
- Letunic, I.; Bork, P. Interactive tree of life (iTOL) v3: An online tool for the display and annotation of phylogenetic and other trees. Nucleic Acids Res. 2016, 44, W242–W245. [Google Scholar] [CrossRef]
- Overbeek, R.; Bartels, D.; Vonstein, V.; Meyer, F. Annotation of bacterial and archaeal genomes: Improving accuracy and consistency. Chem. Rev. 2007, 107, 3431–3447. [Google Scholar] [CrossRef]
- Osterman, A.L.; Overbeek, R.; Rodionov, D.A. The Use of Subsystems to Encode Biosynthesis of Vitamins and Cofactors. In Comprehensive Natural Products II: Chemistry and Biology; Mander, L.N., Liu, H., Eds.; Elsevier Ltd.: Kidlington, UK, 2010; Volume 7, pp. 141–159. [Google Scholar]
- Kanehisa, M.; Goto, S.; Sato, Y.; Kawashima, M.; Furumichi, M.; Tanabe, M. Data, information, knowledge and principle: Back to metabolism in KEGG. Nucleic Acids Res. 2014, 42, D199–D205. [Google Scholar] [CrossRef] [Green Version]
- Elbourne, L.D.; Tetu, S.G.; Hassan, K.A.; Paulsen, I.T. TransportDB 2.0: A database for exploring membrane transporters in sequenced genomes from all domains of life. Nucleic Acids Res. 2017, 45, D320–D324. [Google Scholar] [CrossRef]
- Boutet, E.; Lieberherr, D.; Tognolli, M.; Schneider, M.; Bansal, P.; Bridge, A.J.; Poux, S.; Bougueleret, L.; Xenarios, I. UniProtKB/Swiss-Prot, the manually annotated section of the UniProt KnowledgeBase: How to use the entry view. Methods Mol. Biol. 2016, 1374, 23–54. [Google Scholar] [CrossRef]
- Price, M.N.; Arkin, A.P. PaperBLAST: Text mining papers for information about homologs. MSystems 2017, 2, e00039-17. [Google Scholar] [CrossRef] [Green Version]
- Rodionov, D.A. Comparative genomic reconstruction of transcriptional regulatory networks in bacteria. Chem. Rev. 2007, 107, 3467–3497. [Google Scholar] [CrossRef]
- Novichkov, P.S.; Kazakov, A.E.; Ravcheev, D.A.; Leyn, S.A.; Kovaleva, G.Y.; Sutormin, R.A.; Kazanov, M.D.; Riehl, W.; Arkin, A.P.; Dubchak, I.; et al. RegPrecise 3.0--a resource for genome-scale exploration of transcriptional regulation in bacteria. BMC Genom. 2013, 14, 745. [Google Scholar] [CrossRef] [PubMed]
- Romine, M.F.; Rodionov, D.A.; Maezato, Y.; Osterman, A.L.; Nelson, W.C. Underlying mechanisms for syntrophic metabolism of essential enzyme cofactors in microbial communities. ISME J. 2017, 11, 1434–1446. [Google Scholar] [CrossRef] [PubMed]
- Sun, E.I.; Leyn, S.A.; Kazanov, M.D.; Saier, M.H., Jr.; Novichkov, P.S.; Rodionov, D.A. Comparative genomics of metabolic capacities of regulons controlled by cis-regulatory RNA motifs in bacteria. BMC Genom. 2013, 14, 597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, M.; McNulty, N.P.; Rodionov, D.A.; Khoroshkin, M.S.; Griffin, N.W.; Cheng, J.; Latreille, P.; Kerstetter, R.A.; Terrapon, N.; Henrissat, B.; et al. Genetic determinants of in vivo fitness and diet responsiveness in multiple human gut Bacteroides. Science 2015, 350, aac5992. [Google Scholar] [CrossRef] [Green Version]
- Iablokov, S.N.; Novichkov, P.S.; Osterman, A.L.; Rodionov, D.A. Binary Metabolic Phenotypes and Phenotype Diversity Metrics for the Functional Characterization of Microbial Communities. Front. Microbiol. 2021, 12, 653314. [Google Scholar] [CrossRef]
- McDonald, D.; Hyde, E.; Debelius, J.W.; Morton, J.T.; Gonzalez, A.; Ackermann, G.; Aksenov, A.A.; Behsaz, B.; Brennan, C.; Chen, Y.; et al. American Gut: An Open Platform for Citizen Science Microbiome Research. MSystems 2018, 3, e00031-18. [Google Scholar] [CrossRef] [Green Version]
- Goodrich, J.K.; Davenport, E.R.; Beaumont, M.; Jackson, M.A.; Knight, R.; Ober, C.; Spector, T.D.; Bell, J.T.; Clark, A.G.; Ley, R.E. Genetic Determinants of the Gut Microbiome in UK Twins. Cell Host Microbe 2016, 19, 731–743. [Google Scholar] [CrossRef] [Green Version]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef]
- Schnorr, S.L.; Candela, M.; Rampelli, S.; Centanni, M.; Consolandi, C.; Basaglia, G.; Turroni, S.; Biagi, E.; Peano, C.; Severgnini, M.; et al. Gut microbiome of the Hadza hunter-gatherers. Nat. Commun. 2014, 5, 3654. [Google Scholar] [CrossRef]
- Langille, M.G.; Zaneveld, J.; Caporaso, J.G.; McDonald, D.; Knights, D.; Reyes, J.A.; Clemente, J.C.; Burkepile, D.E.; Vega Thurber, R.L.; Knight, R.; et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat. Biotechnol. 2013, 31, 814–821. [Google Scholar] [CrossRef]
- Douglas, G.M.; Maffei, V.J.; Zaneveld, J.R.; Yurgel, S.N.; Brown, J.R.; Taylor, C.M.; Huttenhower, C.; Langille, M.G.I. PICRUSt2 for prediction of metagenome functions. Nat. Biotechnol. 2020, 38, 685–688. [Google Scholar] [CrossRef] [PubMed]
- Caspi, R.; Altman, T.; Dreher, K.; Fulcher, C.A.; Subhraveti, P.; Keseler, I.M.; Kothari, A.; Krummenacker, M.; Latendresse, M.; Mueller, L.A.; et al. The MetaCyc database of metabolic pathways and enzymes and the BioCyc collection of pathway/genome databases. Nucleic Acids Res. 2012, 40, D742–D753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, Y.; Doak, T.G. A parsimony approach to biological pathway reconstruction/inference for genomes and metagenomes. PLoS Comput. Biol. 2009, 5, e1000465. [Google Scholar] [CrossRef] [Green Version]
- Lloyd-Price, J.; Arze, C.; Ananthakrishnan, A.N.; Schirmer, M.; Avila-Pacheco, J.; Poon, T.W.; Andrews, E.; Ajami, N.J.; Bonham, K.S.; Brislawn, C.J.; et al. Multi-omics of the gut microbial ecosystem in inflammatory bowel diseases. Nature 2019, 569, 655–662. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wood, D.E.; Lu, J.; Langmead, B. Improved metagenomic analysis with Kraken 2. Genome Biol. 2019, 20, 257. [Google Scholar] [CrossRef] [Green Version]
- Li, D.; Luo, R.; Liu, C.M.; Leung, C.M.; Ting, H.F.; Sadakane, K.; Yamashita, H.; Lam, T.W. MEGAHIT v1.0: A fast and scalable metagenome assembler driven by advanced methodologies and community practices. Methods 2016, 102, 3–11. [Google Scholar] [CrossRef]
- Seemann, T. Prokka: Rapid prokaryotic genome annotation. Bioinformatics 2014, 30, 2068–2069. [Google Scholar] [CrossRef]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 2015, 12, 59–60. [Google Scholar] [CrossRef]
- Patwardhan, M.N.; Wenger, C.D.; Davis, E.S.; Phanstiel, D.H. Bedtoolsr: An R package for genomic data analysis and manipulation. J. Open Source Softw. 2019, 4, 1742. [Google Scholar] [CrossRef] [Green Version]
- Robinson, M.D.; Oshlack, A. A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol. 2010, 11, R25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qin, J.; Li, R.; Raes, J.; Arumugam, M.; Burgdorf, K.S.; Manichanh, C.; Nielsen, T.; Pons, N.; Levenez, F.; Yamada, T.; et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature 2010, 464, 59–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Human Microbiome Jumpstart Reference Strains, C.; Nelson, K.E.; Weinstock, G.M.; Highlander, S.K.; Worley, K.C.; Creasy, H.H.; Wortman, J.R.; Rusch, D.B.; Mitreva, M.; Sodergren, E.; et al. A catalog of reference genomes from the human microbiome. Science 2010, 328, 994–999. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Curnow, A.W.; Hong, K.; Yuan, R.; Kim, S.; Martins, O.; Winkler, W.; Henkin, T.M.; Soll, D. Glu-tRNAGln amidotransferase: A novel heterotrimeric enzyme required for correct decoding of glutamine codons during translation. Proc. Natl. Acad. Sci. USA 1997, 94, 11819–11826. [Google Scholar] [CrossRef] [Green Version]
- Sheppard, K.; Akochy, P.M.; Salazar, J.C.; Soll, D. The Helicobacter pylori amidotransferase GatCAB is equally efficient in glutamine-dependent transamidation of Asp-tRNAAsn and Glu-tRNAGln. J. Biol. Chem. 2007, 282, 11866–11873. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.; Labedan, B.; Glansdorff, N. Surprising arginine biosynthesis: A reappraisal of the enzymology and evolution of the pathway in microorganisms. Microbiol. Mol. Biol. Rev. 2007, 71, 36–47. [Google Scholar] [CrossRef] [Green Version]
- Shi, D.; Morizono, H.; Cabrera-Luque, J.; Yu, X.; Roth, L.; Malamy, M.H.; Allewell, N.M.; Tuchman, M. Structure and catalytic mechanism of a novel N-succinyl-L-ornithine transcarbamylase in arginine biosynthesis of Bacteroides fragilis. J. Biol. Chem. 2006, 281, 20623–20631. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Shiver, A.L.; Price, M.N.; Carlson, H.K.; Trotter, V.V.; Chen, Y.; Escalante, V.; Ray, J.; Hern, K.E.; Petzold, C.J.; et al. Functional genetics of human gut commensal Bacteroides thetaiotaomicron reveals metabolic requirements for growth across environments. Cell Rep. 2021, 34, 108789. [Google Scholar] [CrossRef]
- Klemke, F.; Baier, A.; Knoop, H.; Kern, R.; Jablonsky, J.; Beyer, G.; Volkmer, T.; Steuer, R.; Lockau, W.; Hagemann, M. Identification of the light-independent phosphoserine pathway as an additional source of serine in the cyanobacterium Synechocystis sp. PCC 6803. Microbiology 2015, 161, 1050–1060. [Google Scholar] [CrossRef]
- Kuznetsova, E.; Proudfoot, M.; Gonzalez, C.F.; Brown, G.; Omelchenko, M.V.; Borozan, I.; Carmel, L.; Wolf, Y.I.; Mori, H.; Savchenko, A.V.; et al. Genome-wide analysis of substrate specificities of the Escherichia coli haloacid dehalogenase-like phosphatase family. J. Biol. Chem. 2006, 281, 36149–36161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, H.; Pandya, C.; Liu, C.; Al-Obaidi, N.F.; Wang, M.; Zheng, L.; Toews Keating, S.; Aono, M.; Love, J.D.; Evans, B.; et al. Panoramic view of a superfamily of phosphatases through substrate profiling. Proc. Natl. Acad. Sci. USA 2015, 112, E1974–E1983. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiba, Y.; Horita, S.; Ohtsuka, J.; Arai, H.; Nagata, K.; Igarashi, Y.; Tanokura, M.; Ishii, M. Structural units important for activity of a novel-type phosphoserine phosphatase from Hydrogenobacter thermophilus TK-6 revealed by crystal structure analysis. J. Biol. Chem. 2013, 288, 11448–11458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van der Oost, J.; Huynen, M.A.; Verhees, C.H. Molecular characterization of phosphoglycerate mutase in archaea. FEMS Microbiol. Lett. 2002, 212, 111–120. [Google Scholar] [CrossRef]
- Price, M.N.; Deutschbauer, A.M.; Arkin, A.P. GapMind: Automated Annotation of Amino Acid Biosynthesis. MSystems 2020, 5. [Google Scholar] [CrossRef]
- Ferrario, C.; Duranti, S.; Milani, C.; Mancabelli, L.; Lugli, G.A.; Turroni, F.; Mangifesta, M.; Viappiani, A.; Ossiprandi, M.C.; van Sinderen, D.; et al. Exploring Amino Acid Auxotrophy in Bifidobacterium bifidum PRL2010. Front. Microbiol. 2015, 6, 1331. [Google Scholar] [CrossRef]
- Diether, N.E.; Willing, B.P. Microbial Fermentation of Dietary Protein: An Important Factor in Diet(-)Microbe(-)Host Interaction. Microorganisms 2019, 7, 19. [Google Scholar] [CrossRef] [Green Version]
- Sommer, F.; Backhed, F. The gut microbiota--masters of host development and physiology. Nat. Rev. Microbiol. 2013, 11, 227–238. [Google Scholar] [CrossRef]
- Price, M.N.; Zane, G.M.; Kuehl, J.V.; Melnyk, R.A.; Wall, J.D.; Deutschbauer, A.M.; Arkin, A.P. Filling gaps in bacterial amino acid biosynthesis pathways with high-throughput genetics. PLoS Genet. 2018, 14, e1007147. [Google Scholar] [CrossRef] [Green Version]
- Priya, V.K.; Sarkar, S.; Sinha, S. Evolution of tryptophan biosynthetic pathway in microbial genomes: A comparative genetic study. Syst. Synth. Biol. 2014, 8, 59–72. [Google Scholar] [CrossRef] [Green Version]
- Akashi, H.; Gojobori, T. Metabolic efficiency and amino acid composition in the proteomes of Escherichia coli and Bacillus subtilis. Proc. Natl. Acad. Sci. USA 2002, 99, 3695–3700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, Y.; Chong, J.; Shen, S.; Chammas, J.B.; Chalifour, L.; Xia, J. TrpNet: Understanding Tryptophan Metabolism across Gut Microbiome. Metabolites 2021, 12, 10. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Amino Acid | Pathway Signature and Biosynthetic a.a. Dependencies 1 | PV 2 | BP 3 | No. Gen. 4 | Growth Requirements 5 |
|---|---|---|---|---|---|
| Proline | ProA, ProB | P | 1 | 2275 | -- |
| - | A | 0 | 581 | Pro | |
| Threonine | Hom, ThrB, ThrC | P | 1 | 2483 | -- |
| Hom, ThrC | P* | 1 | 50 | (missing ThrB) | |
| - | A | 0 | 323 | Thr | |
| Glycine | [GlyA/SgaA + Serine] or [GlyB + Threonine] | P | 1 | 2182 | -- |
| - | A | 0 | 58 | Gly | |
| GlyA/SgaA (no Serine) | P1 | 1 | 395 | Ser, Gly | |
| GlyB (no Threonine) | P2 | 1 | 25 | Thr, Gly | |
| [GlyA/SgaA (no Serine)] and [GlyB + (no Threonine)] | P3 | 1 | 23 | Thr, Ser, Gly | |
| Serine | SerA, SerC, SerB | P | 1 | 1863 | -- |
| SerA, SerC | P* | 1 | 450 | (missing SerB) | |
| - | A | 0 | 543 | Ser | |
| Leucine & Isoleucine & Valine | IlvA, IlvG, (IlvM), IlvC, IlvD, (IlvE), LeuA, LeuB, LeuC, LeuD | P1 | 1 | 1912 | -- |
| CimA, IlvG, (IlvM), IlvC, IlvD, (IlvE), LeuA, LeuB, LeuC, LeuD | P2 | 1 | 317 | -- | |
| IlvG, (IlvM), IlvC, IlvD, (IlvE), LeuA, LeuB, LeuC, LeuD | P* | 1 | 17 | (missing IlvA/CimA) | |
| IlvA, IlvG, (IlvM), IlvC, IlvD, (IlvE) | P3 | 1/0 | 60 | Leu | |
| LeuA, LeuB, LeuC, LeuD, IlvE | A* | 0 | 20 | Ile, Val | |
| - | A | 0 | 699 | Leu, Ile, Val | |
| Cysteine | CysE, CysK + Serine | P | 1 | 1919 | -- |
| - | A | 0 | 526 | Cys | |
| CysE, CysK (no Serine) | P1 | 1 | 411 | Ser, Cys | |
| Methionine | Hom, MetA, [CTBL, CTGS]/MetY, [MetH/MetE], (MetF), MetK | P | 1 | 2252 | -- |
| [MetH/MetE], (MetF), MetK | A1 | 0 | 83 | Met | |
| Hom, MetA, [CTBL, CTGS]/MetY, MetK | A2 | 0 | 25 | Met (missing MetH/E) | |
| MetK | A | 0 | 496 | Met | |
| Lysine | LysC, Asd, DapA, DapB, DapH, (PatA), DapeL, DapF, LysA | P1 | 1 | 860 | -- |
| LysC, Asd, DapA, DapB, DapD, (DapC), DapE, DapF, LysA | P2 | 1 | 1051 | -- | |
| LysC, Asd, DapA, DapB, DapL, DapF, LysA | P3 | 1 | 546 | -- | |
| LysC, Asd, DapA, DapB, Ddh, LysA | P4 | 1 | 643 | -- | |
| LysC, Asd, DapA, DapB, DapF, LysA | P* | 1 | 38 | (no amination pathway) | |
| LysA | A1 | 0 | 14 | Lys, DAP | |
| - | A | 0 | 248 | Lys | |
| Histidine | HisG, (HisZ), (HisE), HisI, HisA, HisH, HisF, HisB, HisD, (HisN), (HisC) | P | 1 | 2097 | -- |
| - | A | 0 | 759 | His | |
| Tyrosine & Phenylalanine | PheA, TyrA/TyrC, (AroH), (TyrB) | FY | 1 | 2257 | -- |
| PheA, (AroH), (TyrB) | FA | 1/0 | 40 | Tyr | |
| TyrA/TyrC, (AroH), (TyrB) | AY | 0/1 | 179 | Phe | |
| - | AA | 0 | 380 | Tyr, Phe | |
| Tryptophan | TrpA, TrpB, TrpC, (TrpD), TrpF, TrpEG | P | 1 | 1810 | -- |
| TrpA, TrpB, TrpC, (TrpD), TrpF, TrpEG (no Serine) | P1 | 1 | 140 | Ser | |
| TrpA, TrpB | A1 | 0 | 14 | Trp, indole precursors | |
| TrpA, TrpB, TrpC | A2 | 0 | 88 | Trp, indole precursors | |
| TrpA, TrpB, TrpC, TrpD, TrpF | A3 | 0 | 38 | Trp, anthranilate | |
| - | A | 0 | 766 | Trp | |
| Arginine | (ArgA/ArgJ), (ArgB), ArgC, ArgD, ArgF, (ArgE), ArgG, ArgH | P | 1 | 2061 | -- |
| ArgG, ArgH | A1 | 0 | 251 | Arg, citrulline | |
| ArgA, ArgB, ArgC, ArgD, (ArgF), (ArgE) | A2 | 0 | 5 | Arg | |
| - | A | 0 | 539 | Arg | |
| Chorismate | [AroG, AroB]/[AroA-II, AroB-II], AroD, AroE, AroK, AroA, AroC | P | 1 | 2525 | -- |
| AroD, AroE, AroK, AroA, AroC | P1 | 1 | 14 | (missing AroG/AroB) | |
| AroG, AroB, AroD, AroE, AroK, (AroC) | P2 | 1 | 37 | (missing AroA) | |
| AroG, AroB, AroD, AroE, AroA, AroC | P3 | 1 | 69 | (missing AroK) | |
| AroG, AroB, AroD, AroK, AroA, AroC | P4 | 1 | 30 | (missing AroE) | |
| AroK, AroA, AroC | As | 0 | 13 | Chorismate, shikimate | |
| - | A | 0 | 169 | Chorismate | |
| Aspartate & Asparagine | AspC, AsnA/AsnB, (GatABC) | DN | 1 | 1852 | -- |
| AspC, GatABC | DAG ^ | 1 | 899 | -- | |
| AspC | DA | 1/0 | 28 | Asn | |
| (AsnB/AsnA), (GatABC) | AA | 0 | 77 | Asp, Asn | |
| Glutamate & Glutamine | GltBD/Gdh, GlnA, (GatABC) | EQ | 1 | 2663 | -- |
| GltBD/Gdh, GatABC | EAG ^ | 1 | 17 | -- | |
| GltBD/Gdh | EA | 1/0 | 20 | Gln | |
| (GlnA), (GatABC) | AA | 0 | 156 | Gln, Glu |
| Pathway | Enzyme | Predicted Functional Role | Occurrence 1 | Evidence 2 | Example ID 3 |
|---|---|---|---|---|---|
| Arginine | ArgA2 | N-succinylglutamate synthase (EC 2.3.1.-) | 7.4% | CO, CL, CR, CF | Q8A1A5 |
| ArgA3 | N-acetylglutamate synthase (EC 2.3.1.1) | 0.8% | CO, CL | W3Y6L2 | |
| Serine | SerC2 | Phosphoserine aminotransferase (EC 2.6.1.52) | 4.0% | CO, CF, CL | Q2FXK2 |
| SerC3 | Phosphoserine aminotransferase (EC 2.6.1.52) | 6.1% | CF, CL | A5I0W7 | |
| SerB2 | Phosphoserine phosphatase (EC 3.1.3.3) | 3.6% | CO, CF | C4IFQ5 | |
| Threonine | ThrB2 | Homoserine kinase (EC 2.7.1.39) | 10.6% | CO, CL | Q5LHR7 |
| Lysine | DapF2 | Diaminopimelate epimerase (EC 5.1.1.7) | 4.4% | CO, CL, CR | W1W731 |
| RPA vs. CPI | RPA vs. MetaCyc | MetCyc Pathway Name and Annotation |
|---|---|---|
| 0.86 | 0.54 | ARGSYNBSUB-PWY: L-arginine biosynthesis II (acetyl cycle) |
| 0.59 | ARGSYN-PWY: L-arginine biosynthesis I (via L-ornithine) | |
| 0.31 | PWY-5154: L-arginine biosynthesis III (via N-acetyl-L-citrulline) | |
| 0.60 | PWY-7400: L-arginine biosynthesis IV (archaebacteria) | |
| 0.81 | 0.92 | HISTSYN-PWY: L-histidine biosynthesis |
| 0.60 | 0.64 | ILEUSYN-PWY: L-isoleucine biosynthesis I (from threonine) |
| 0.71 | PWY-5101: L-isoleucine biosynthesis II | |
| 0.69 | PWY-5103: L-isoleucine biosynthesis III | |
| 0.56 | PWY-5104: L-isoleucine biosynthesis IV | |
| 0.67 | 0.70 | LEUSYN-PWY: L-leucine biosynthesis |
| 0.69 | 0.27 | DAPLYSINESYN-PWY: L-lysine biosynthesis I |
| −0.05 | PWY-2941: L-lysine biosynthesis II | |
| 0.71 | PWY-2942: L-lysine biosynthesis III | |
| 0.72 | PWY-5097: L-lysine biosynthesis VI | |
| 0.71 | −0.16 | HOMOSER-METSYN-PWY: L-methionine biosynthesis I |
| −0.17 | HSERMETANA-PWY: L-methionine biosynthesis III | |
| 0.74 | 0.86 | TRPSYN-PWY: L-tryptophan biosynthesis |
| 0.60 | 0.64 | VALSYN-PWY: L-valine biosynthesis |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ashniev, G.A.; Petrov, S.N.; Iablokov, S.N.; Rodionov, D.A. Genomics-Based Reconstruction and Predictive Profiling of Amino Acid Biosynthesis in the Human Gut Microbiome. Microorganisms 2022, 10, 740. https://doi.org/10.3390/microorganisms10040740
Ashniev GA, Petrov SN, Iablokov SN, Rodionov DA. Genomics-Based Reconstruction and Predictive Profiling of Amino Acid Biosynthesis in the Human Gut Microbiome. Microorganisms. 2022; 10(4):740. https://doi.org/10.3390/microorganisms10040740
Chicago/Turabian StyleAshniev, German A., Sergey N. Petrov, Stanislav N. Iablokov, and Dmitry A. Rodionov. 2022. "Genomics-Based Reconstruction and Predictive Profiling of Amino Acid Biosynthesis in the Human Gut Microbiome" Microorganisms 10, no. 4: 740. https://doi.org/10.3390/microorganisms10040740