
Molecular Regulatory Mechanisms Drive Emergent Pathogenetic Properties of Neisseria gonorrhoeae


1
Department of Immunology, Graduate School of Biomedical Sciences, Tufts University School of Medicine, Boston, MA 02111, USA
2
Pacific Northwest National Laboratory, Richland, WA 99354, USA
*
Author to whom correspondence should be addressed.
Microorganisms 2022, 10(5), 922; https://doi.org/10.3390/microorganisms10050922
Submission received: 28 March 2022
/
Revised: 25 April 2022
/
Accepted: 26 April 2022
/
Published: 28 April 2022
(This article belongs to the Special Issue Regulation of Gene Expression in Response to Environmental Changes in Bacterial Pathogens)
Abstract
:Neisseria gonorrhoeae is the causative agent of the sexually transmitted infection (STI) gonorrhea, with an estimated 87 million annual cases worldwide. N. gonorrhoeae predominantly colonizes the male and female genital tract (FGT). In the FGT, N. gonorrhoeae confronts fluctuating levels of nutrients and oxidative and non-oxidative antimicrobial defenses of the immune system, as well as the resident microbiome. One mechanism utilized by N. gonorrhoeae to adapt to this dynamic FGT niche is to modulate gene expression primarily through DNA-binding transcriptional regulators. Here, we describe the major N. gonorrhoeae transcriptional regulators, genes under their control, and how these regulatory processes lead to pathogenic properties of N. gonorrhoeae during natural infection. We also discuss the current knowledge of the structure, function, and diversity of the FGT microbiome and its influence on gonococcal survival and transcriptional responses orchestrated by its DNA-binding regulators. We conclude with recent multi-omics data and modeling tools and their application to FGT microbiome dynamics. Understanding the strategies utilized by N. gonorrhoeae to regulate gene expression and their impact on the emergent characteristics of this pathogen during infection has the potential to identify new effective strategies to both treat and prevent gonorrhea.
1. Introduction
The Gram-negative bacterium Neisseria gonorrhoeae is the causative agent of the sexually transmitted infection (STI) gonorrhea. The geographical distribution of gonorrhea is very diverse. The estimates of gonorrhea cases in adults aged 15 to 49 varied considerably across the different WHO regions. According to WHO statistics, the WHO African region had the highest incidence with 41 cases per 1000 women and 50 cases per 1000 men, whereas the WHO European region had the lowest incidence with seven cases per 1000 women and 11 per 1000 men. A variety of factors contribute to this variation, including sexual orientation, socioeconomic status, demographics, geographical location, cultural practices, and educational background [1,2]. In 2019, 616,392 gonococcal infection cases were reported to the Centers for Disease Control and Prevention (CDC), making it the second most common reportable disease in the United States [3]. A purulent discharge, composed of polymorphonuclear leukocytes (PMNs), exfoliated epithelial cells, and N. gonorrhoeae, is a hallmark of gonorrhea in men [4,5,6]. However, subjects infected with N. gonorrhoeae are often asymptomatic, an outcome observed more frequently in women than in men [7,8,9,10]. Asymptomatic infections in women are of concern since women are unaware they are infected and do not seek treatment, resulting in prolonged, untreated gonococcal infection. Untreated infections can lead to severe complications, including pelvic inflammatory disease (PID), infertility, and ectopic pregnancy, along with the possibility of further transmission, a public health concern [7,11]. Most gonococcal infections can be successfully treated with oral antibiotic intervention. However, strains of N. gonorrhoeae resistant to multiple antibiotics have emerged worldwide [12,13,14,15,16,17,18]. As a result of prolonged and untreated infection and antibiotic-resistant gonococcal strains, gonorrhea remains a significant STI. As such, we need new strategies to combat this infection.
The male and female urogenital tracts are the primary sites of N. gonorrhoeae infection, although infections at extragenital sites, including the rectum and pharynx, are sometimes detected [1,19]. The female genital tract (FGT) and its resident microbiome have been proposed to act as a barrier to pathogenic infections [20]. The mucosal immune system of the vagina primarily constitutes an epithelial cell mechanical barrier and innate immune cells, including macrophages and natural killer cells [21,22,23]. Recognition of mucosal pathogens by these immune cells results in the induction and secretion of chemokines, cytokines, innate immune molecules, and the recruitment of PMNs to the site of infection. Innate immune molecules secreted by immune and non-immune cells include antimicrobial peptides (AMPs) that target and disrupt bacterial cell walls, proteins that sequester nutrients essential for pathogens, and reactive oxygen species (ROS) primarily produced by PMNs [21,24,25]. In addition, macrophages, dendritic cells (DCs), and other antigen-presenting cells (APCs) stimulate adaptive immune responses, leading to the initiation of humoral and cellular immunity [21,25]. N. gonorrhoeae adapts to this complex FGT mucosal niche through different mechanisms, including regulatory mechanisms that calibrate gene expression in response to different environmental stimuli, such as fluctuating levels of nutrients, antimicrobial compounds, and antibodies directed against various N. gonorrhoeae surface antigens. Control of genes involved in these processes is mediated through different mechanisms, such as phase variation and transcriptional regulation through DNA-binding transcriptional regulators and nucleoid-associated proteins (NAP) [26,27,28,29,30,31]. Among other mechanisms, these gene regulatory processes enable N. gonorrhoeae to survive in the FGT environment, promoting its pathogenesis [29].
2. DNA-Binding Transcriptional Regulators
One mechanism for regulation of gene expression in N. gonorrhoeae is achieved through DNA-binding regulators that respond to various stress factors, such as ROS, antibiotics, and nutrient scarcity. This mechanism facilitates the adaptation of N. gonorrhoeae to the dynamic environment of the host. Here we describe a subset of the ~34 putative transcriptional regulators identified in N. gonorrhoeae based on homology searches, beginning with those responding to varying iron levels at the site of the infection [29].
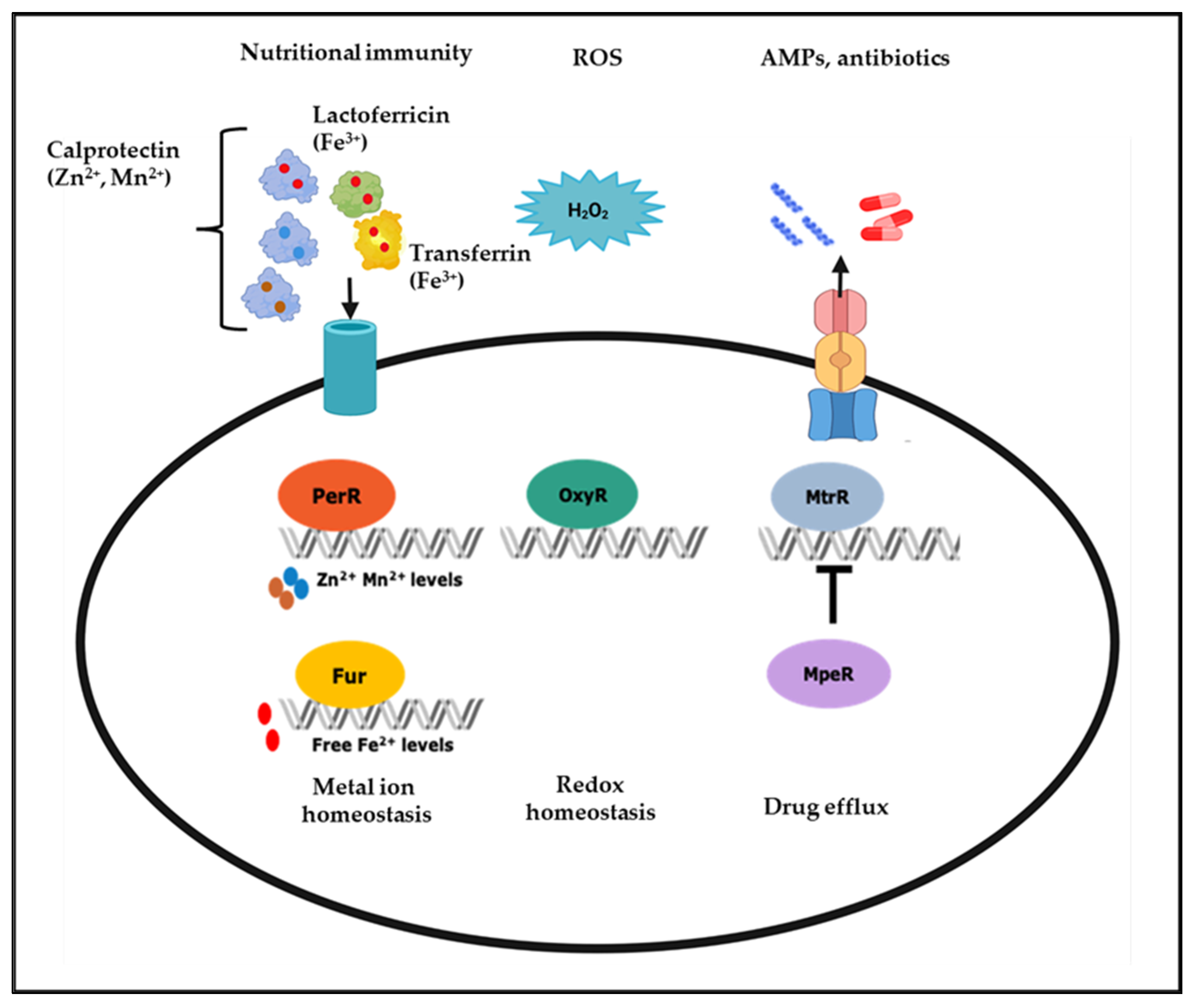
Iron plays a vital role in various bacterial cellular processes, including DNA replication, transcription, metabolism, and responses to oxidative stress, making it essential for the optimal growth of pathogens [32,33]. Nutritional immunity defense strategies of the human host include depriving pathogens of iron to prevent their survival and pathogenesis [33,34]. This strategy involves maintaining low free iron levels using iron storage proteins including lactoferrin in mucosal tissues and transferrin in plasma, lymph, and cerebrospinal fluid [35,36,37,38]. In the vagina, lactoferrin levels increase in response to bacterial infections and during the mid-follicular phase of the menstrual cycle as estrogen levels rise, further sequestering iron. However, iron from heme becomes more available to bacteria during the menses [35,36]. Despite the importance of iron in numerous biological processes, excess iron may cause ROS generation via the Fenton reaction; therefore, it is crucial for the pathogen to control cellular iron homeostasis during infection [39,40]. In the mucosal niche, gonococci maintain iron homeostasis through a complex iron-responsive network controlled by multiple transcriptional regulators, including the Ferric Uptake Regulator (Fur) and Mtr Protein Efflux Regulator (MpeR) (Figure 1) [29,37,41,42,43,44].
Fur is a global transcriptional iron-response regulator found in Gram-negative and positive bacteria [45]. The classic mechanism of Fur regulation involves Fur binding directly to DNA sequences to inhibit transcription. Without iron, Fur exists as an inactive monomer. However, increasing ferrous iron levels or other divalent cations leads to Fur dimerization and binding to the promoter region of target genes. This interaction blocks subsequent binding by RNA polymerase and leads to decreased transcription of target genes [45,46,47,48]. Fur can also bind to DNA in the absence of iron, a process known as Apo-Fur mediated regulation but this is less common than iron-driven Fur activation [48,49]. In addition to acting as a repressor, Fur can act as a direct activator of gene expression via binding directly to promoter regions, facilitating RNA polymerase binding, and leading to increased transcription of target genes [41,45,50,51]. As a direct regulator of iron homeostasis in N. gonorrhoeae, Fur regulates the expression of the genes encoding TonB and TonB-dependent transporters FbpA, TbpA, and TbpB that extract iron from ferritin and transferrin in the host, respectively [52,53,54]. Fur can also indirectly regulate genes by repressing a repressor, with the targets of such repressors transcribed and translated at higher rates. In this indirect role, Fur targets the expression of non-coding RNAs such as regulatory small RNAs (sRNAs), as is the case for the Fur-repressed sRNA NrrF, which controls transcription of the sdhC/A genes. Fur-mediated repression of NrrF results in increased translation of sdhC/A transcripts; thus, the expression of functional SdhC/A proteins is indirectly activated by Fur [45,55,56,57].
Fur acts as a global transcriptional regulator, regulating the expression of other transcriptional regulators, such as MpeR, another iron response N. gonorrhoeae regulator. Under iron-replete conditions, Fur negatively regulates the expression of mpeR in an iron-dependent manner [41]. MpeR has two putative α-helix–turn–α-helix (HTH) DNA-binding motifs that share homology with the arabinose operon regulatory protein (AraC) family transcriptional regulators [41,58,59,60]. AraC family regulators participate in multiple cellular processes, including oxidative stress, carbon metabolism, and pathogenesis [59,61]. Like Fur, MpeR functions as a transcriptional activator and repressor. For example, MpeR regulates the mtrCDE efflux pump operon by repressing the expression of mtrR, the gene encoding the mtrCDE efflux pump operon repressor (Figure 1). In contrast to its role as a repressor, MpeR has been shown to activate iron-response genes, including fetA, pointing to its role in maintaining the iron homeostasis [42]. The binding of MpeR to the fetA promoter has been demonstrated by electrophoretic mobility shift assay (EMSA), but its specific DNA-binding motif has yet to be determined experimentally [42,60]. High-throughput transcriptomic studies of the N. gonorrhoeae’s MpeR and H2O2 regulon have demonstrated that mpeR gene expression increases in response to H2O2 and that MpeR regulates oxidative stress, energy metabolism, and transport genes’ expression [60,62,63]. MpeR also has a role in gonococcal interactions with innate immune cells. N. gonorrhoeae upregulates the expression of iron response genes, including mpeR, in response to nutritional immune defenses of monocytes [37,64]. Transcriptomic analysis of N. gonorrhoeae in cervicovaginal lavage specimens from infected women revealed that mpeR is expressed during natural infection by this pathogen [43]. Collectively, these studies show that MpeR responds to different environmental conditions relevant to natural infection, including iron-limitation and ROS (H2O2), suggesting it is critical in the survival and pathogenesis of N. gonorrhoeae during natural infection.
In addition to iron, other trace metals, including zinc and manganese, play a role in the interactions of N. gonorrhoeae with the host immune and non-immune cells [29]. Calprotectin, produced by epithelial cells and PMNs, is a zinc and manganese chelator that sequesters these trace metals from pathogens [65]. N. gonorrhoeae can extract zinc and manganese from calprotectin via the respective surface-expressed metal transport proteins MntABC and TdfH, whose expression is controlled by the transcriptional regulators Zinc Uptake Regulation Protein (Zur) and Peroxide Responsive Regulator (PerR) [29,66,67]. Furthermore, zinc is also needed for redox homeostasis and is a cofactor of another DNA-binding protein, the redox-responsive regulator NmlR that represses alcohol dehydrogenase (adhC) and multi-copper oxidase encoding gene copA [68].
Unlike iron which could contribute to ROS generation, manganese protects N. gonorrhoeae against oxidative stress by catalyzing ROS removal [66,69,70]. Due to the role of iron and manganese in oxidative stress responses, there is a coordination between iron and manganese homeostasis and ROS defenses [71,72,73]. For example, in N. gonorrhoeae, oxidative stress response genes, such as katA, are regulated by multiple oxidative-stress response regulators, including OxyR and PerR, as well as iron-response regulator Fur [29,41,62,66,74]. OxyR in N. gonorrhoeae negatively regulates the expression of katA, with the OxyR regulon consisting of only two additional genes: peroxiredoxin (prx) and glutathione reductase (gor) [74,75]. This repression of katA is contrary to the OxyR in Escherichia coli and Salmonella typhimurium which regulate katA positively [74,75,76]. Another transcriptional regulator involved in the oxidative stress-response is LexA, which represses the expression of three genes, including itself and the DNA-repair enzyme recN. Oxidation of a cysteine residue in the LexA protein causes its detachment from DNA, resulting in the de-repression of the three genes in its regulon [77].
As a facultative anaerobe, N. gonorrhoeae can be cultured from the female genital tract with obligate anaerobes, and antibodies to proteins required for anaerobiosis have been detected in sera of women with gonorrhea [78,79]. In the absence of oxygen, N. gonorrhoeae can use nitrite or nitric oxide as a terminal electron acceptor via the activity of a truncated denitrification pathway composed of a nitrite reductase (aniA) and a nitric oxide reductase (norB). The expression of genes related to nitrogen respiration is complex as it involves multiple pathways and many proteins that require sensitive transcriptional control. These proteins sense specific molecules and bind to DNA to regulate genes under their control [80,81,82,83,84]. For example, the fumarate nitrate reduction (FNR) protein drives the expression of aniA in the absence of oxygen. At the same time, further control is mediated by the ability of NsrR to repress aniA expression in the absence of nitric oxide [82]. Additional DNA-binding proteins, including Fur and an ArsR-like protein NsrR, are involved in promoting and repressing the transcription of norB, depending on the presence of iron or nitric oxide [41,83,85,86].
3. Phase Variation
Phase variation is the reversible, stochastic switching between turning on and off the expression of a gene through genetic or epigenetic mechanisms [87,88]. Genetic regulation of phase variation is dictated by changes in the DNA sequence at a specific genetic locus. Slipped-strand mispairing (SSM) is the most common genetic phase variation mechanism in Neisseria spp. and is dictated by different kinds of DNA repeats, including homopolymer tracts and tandem repeats of hundreds of bases in the genome [89,90]. SSM occurs during DNA replication or repair when DNA repeats are misaligned between the mother and daughter DNA strands. DNA repeats are then either expanded or contracted, resulting in phase-variable gene expression at the transcriptional and translational levels, depending on the DNA repeats’ position in reference to its open reading frame [26,87]. Several phase-variable genes encode surface antigens that are potential vaccine candidates [26,91]. Loss or gain in the number of cytosines in the poly-cytosine tract present close to the transcriptional start site of surface-expressed antigen fetA determines its expression levels [41,92]. Aside from the insertion of nucleotides, epigenetic regulation of phase variation is determined by the methylation status of the regulatory region of a phase-variable gene or operon rather than their DNA sequences [88,93,94]. Phase variation of DNA methyltransferases leads to coordinated global differential methylation of the bacteria’s genome that corresponds to the on and off methyltransferase variants. Each different global methylation results in the differential expression of a particular gene set called a phasevarion, i.e., phase-variable regulon. Phase variation of the N. gonorrhoeae methyltransferase ModA13 comprises 54 genes involved in oxidative stress and antibiotic resistance, including mtrF and trx [93,94,95].
4. Global Gene Co-Expression Network of N. gonorrhoeae
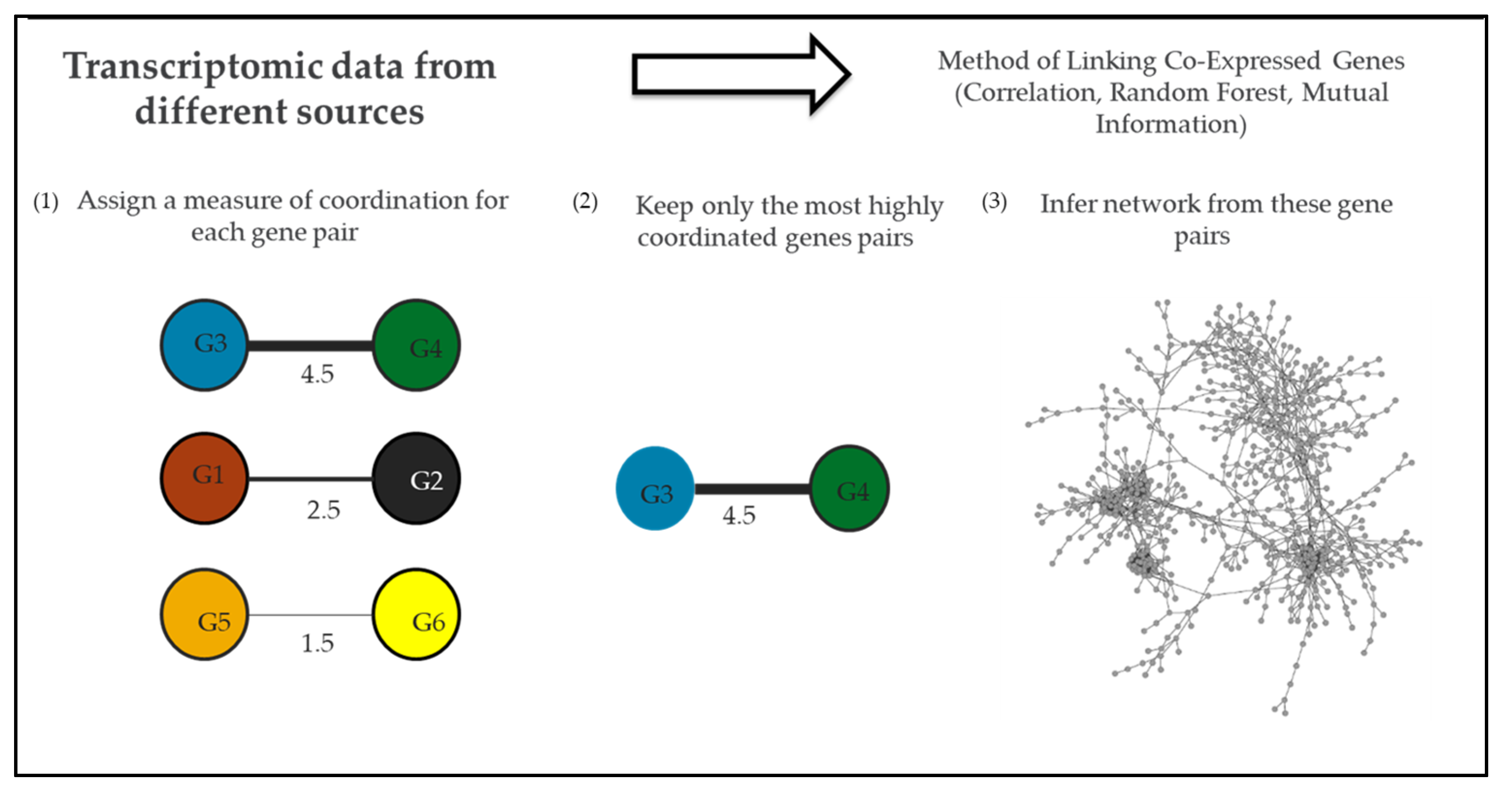
Comparative transcriptomics and pathway enrichment analysis of microarray and RNA-Seq (RNA sequencing) data have helped to define global regulons of transcriptional regulators and their associated pathways under specific experimental conditions [41,60,62,81]. However, a systems biology approach can be used to study the interactions of genes from multiple regulons when there is enough transcriptomic data that describes a system (e.g., N. gonorrhoeae) under a variety of conditions [96,97]. Recently, the research community has reached this depth of transcriptomic data for N. gonorrhoeae, opening up the possibility of gene co-expression analysis of this pathogen [31]. With a gene co-expression network approach, genes represent nodes in a network, and instances of high co-expression between individual gene pairs represent edges in a network. Edges are calculated by analyzing a gene’s expression profile across a range of conditions and linking genes with similar expression profiles (Figure 2) [98]. Using network analysis it is possible to identify new potential targets of known regulators by identifying the edges connecting regulators and their potential targets within the network [31,96]. Network studies have been applied to pathogens such as Salmonella to identify processes crucial to the infection [99].
To gain an understanding of the global gene–gene interactions within N. gonorrhoeae (including during natural infection and in vitro conditions related to natural infection such as H2O2 treatment) we constructed the first gene co-expression network for this pathogen (Figure 2) [31]. This network contained 1118 N. gonorrhoeae genes (representing 56% of the gonococcal genome) linked by 1499 edges. We then utilized this network to expand our understanding of regulatory pathways within N. gonorrhoeae, with a particular focus on Fur. Genes clustered in the network neighborhood of Fur (near Fur in the network) were isolated and further examined. A total of 173 genes were identified in this network neighborhood, including 9 of 23 known targets of Fur. The likelihood of selecting this many Fur targets through random sampling of 173 genes in a network of 1118 was less than 0.002%. We, therefore, reasoned that there might also be undiscovered Fur targets within this network neighborhood. To identify them, we took advantage of the known binding site of Fur in the 5′ UTR of target genes. In our search for known Fur binding sites within the promoter region of genes within the Fur network neighborhood, we identified 11 genes that contain binding sites for Fur but had not been previously identified as Fur targets. In agreement with their role as putative Fur targets, many of these genes are regulated by iron but they lose this regulation in a Fur-knockout strain [31,41]. The discovery of these new potential targets of Fur was only possible after this inference of a gene co-expression network that links, among other genes, regulators, and targets.
We also used a process termed guilty-by-association (GBA) in conjunction with our network to identify putative functions of unknown genes. In a gene co-expression network genes that are linked by edges are likely to participate in similar pathways and share functions. Extending this analysis means that unknown genes can be putatively identified, at least insofar as to what pathways they are involved in, by examining which known characterized genes they associate within the network. This is the basis of the GBA analysis [102]. We applied GBA analysis to our network and found that, for known genes, it assigned the correct function 85% of the time. Applying this approach to all ~700 unknown hypothetical proteins, which also include uncharacterized transcriptional regulators in the N. gonorrhoeae genome, we assigned putative functions to 313 (~44%). These functions included gene regulation, DNA metabolism, energy metabolism, general metabolism, phage associated, pilin, replication, stress, translation, and transport-related genes [31].
5. Interactions between N. gonorrhoeae and the FGT Microbiome
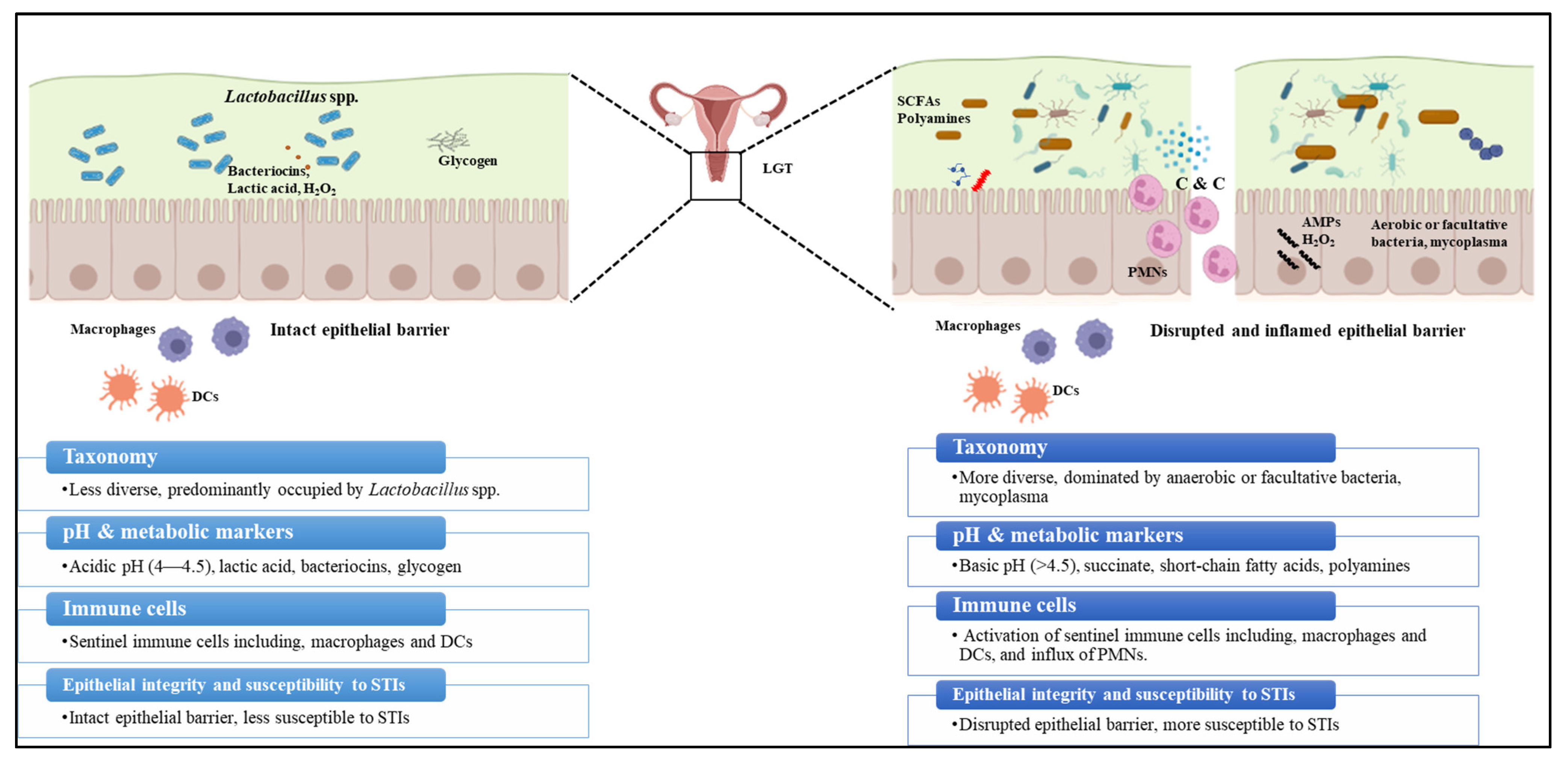
Along with the host immune response, N. gonorrhoeae interacts with the resident microbiota of the FGT mucosal niche [29,103]. This community provides additional protection against bacterial, viral, parasitic, and fungal infections. Even in healthy individuals, multiple factors including diet, environment, host genetics, and exposure to microbes during early stages of life contribute to inter- and intrapersonal variability in the microbiome [104,105]. 16S rRNA sequencing analysis is widely used for microbiota profiling; it involves clustering the composite 16S sequences of the microbiota into species, or higher taxonomic levels where species cannot be identified, based on similarities in the sequences [106]. This information is used to measure the diversity metrics alpha and beta diversity that determine the microbiota diversity. Alpha diversity is directly proportional to the number of microbial species and the evenness of their relative abundances within a sample. In contrast, beta diversity is directly proportional to taxonomical differences between samples [104,106,107]. The FGT microbiome encompasses a heterogenous ecosystem with distinct microbiota density and composition at and within the lower and the upper female genital tract (LGT and UGT) [108] The vaginal microbiome at the LGT, consisting of ~200–300 bacterial species, has the lowest species-level alpha diversity, low beta diversity at the genus level compared to the microbiome from other anatomical niches, and high beta diversity at the species level [108,109,110]. Disruption to the composition of the “core healthy microbiota” (dysbiosis) or natural changes in the microbiome due to the changes in female physiology, results in changes to the FGT ecology (Figure 3) [111,112]. In general, bacterial mass at the LGT is several orders of magnitude higher than the UGT, whereas LGT microbiota is less diverse than the UGT [108,110]. Lactobacillus iners or Lactobacillus crispatus are the predominant microbes in a healthy vagina (LGT), followed by Lactobacillus jensenii and Lactobacillus gasseri, which are known to play a crucial role in the vaginal microbiome homeostasis [105,108,113]. Non-lactobacillus genera including Gardenella, Prevotella, Peptoniphilus, Peptostreptococcus, Anaerococcus, Veillonella, Megasphaera, Leptotrichia, Sneathia, or Atopobium represent a small proportion of the healthy vaginal microbiome [108,114]. Unlike the LGT microbiota, the UGT microbiota has traditionally been overlooked because it was considered sterile except when infected with pathogenic bacteria [115]. Additionally, studies on the UGT microbiome are difficult since access to the UGT ecosystem is cumbersome, involving a transcervical procedure risking contamination with cervicovaginal microbiome or invasive methods including hysterectomy and surgical laparoscopy. Nevertheless, recent studies using 16S rRNA amplicon analysis have provided evidence of colonization of distinct microbial communities along the female reproductive tract [109,110,115,116]. Identified major genera of the endometrial microbiota among different studies include Flavobacterium, Gardnerella, Bifidobacterium, Streptococcus, Prevotella, Pseudomonas, Acinetobacter, Vagococcus, Sphingobium, and Lactobacillus. It should be noted, however, that these genera do not always appear in all studies, with the exception of Lactobacillus, which was consistently observed [109,115,117,118]. The Fallopian tube microbiome is dominated by the phyla Firmicutes (Staphylococcus sp., Enterococcus sp., and Lactobacillus sp.) followed by Pseudomonads (Pseudomonas sp. and Burkholderia sp.) [115,119,120]. Despite these previous studies, detailed characterization of the UGT microbiome’s structure, function, and diversity is still in its infancy.
Multiple epidemiological studies have shown an association between Lactobacillus spp. vaginal occupancy and protection from N. gonorrhoeae infections [121]. Lactobacillus spp. play a crucial role in maintaining vaginal microbiome homeostasis and providing a barrier against pathogenic microbes by competitive adhesion to the vaginal epithelium and producing antimicrobial products, namely lactic acid and H2O2 [122,123]. Different strains of L. crispatus isolated from the vagina of healthy premenopausal women have been demonstrated to inhibit the growth of N. gonorrhoeae in vitro primarily by maintaining an acidic pH from the lactic acid metabolism [124]. L. jensenii has been shown to block N. gonorrhoeae adherence and invasion of epithelial cells in vitro [125]. Later studies showed L. jensenii inhibits N. gonorrhoeae adherence to epithelial cells via the production of surface-associated enolase [126]. In L. crispatus, enolase and glutamine synthetase [127,128] were shown to interfere with the interaction of N. gonorrhoeae with epithelial cells in vitro [128]. In contrast to a healthy Lactobacillus population, disruption to the composition of the “core healthy” vaginal microbiota (dysbiosis) is associated with susceptibility to multiple STIs. For example, bacterial vaginosis (BV), a dysbiotic state of the vaginal microbiome, is associated with an increased risk of acquiring STIs, including gonorrhea, chlamydia, HIV, and reproductive and gynecological complications [103,108]. BV is characterized by increased microbiome diversity due to the outgrowth of the anaerobe Gardnerella vaginalis along with other anaerobic and facultative organisms including Atopobium vaginae, Bacteroides spp., Mobiluncus spp., and genital mycoplasmas, accompanied by the reduction in Lactobacillus spp. Clinical symptoms of BV include (1) vaginal pH > 4.5, (2) grayish-white vaginal discharge with a (3) distinct fishy odor due to the presence of polyamines, and (4) detection of clue cells (epithelial cells covered with Gram-negative rod cells) in the vaginal smears examined under the microscope. Limited studies have examined the mechanistic details of how N. gonorrhoeae responds to the healthy and dysbiotic vaginal ecology and how this could potentially impact treatment and vaccine development [129,130,131]. In vitro studies have shown that N. gonorrhoeae responds to acidic conditions by modulating the expression of surface-expressed proteins including Rmp, an important determinant of gonococcal vaccine efficacy, and stress response proteins Hsp63 [132,133,134]. Another study has shown N. gonorrhoeae can develop resistance to lactic acid in a polyamine-dependent manner [135].
Sexual intercourse is associated with changes in the FGT microbiome and the risk of BV in the female partner; this is partly dependent on the composition of the male genital tract (MGT) microbiome [136,137]. Like the FGT microbiome, the MGT microbiome is predominantly present in the LGT, primarily in the urethra and the coronal sack, with variation between individuals. The UGT is typically considered sterile unless infected [138]. A variety of studies have examined the urinary tract microbiome of men without STIs. They have identified considerable diversity among the microbial taxa, including Corynebacterium, Streptococcus, Staphylococcus, Propionibacterium, Sneathia, Veillonella, Prevotella, Ureaplasma, Mycoplasma, Anaerococcus, Atopobium, Aerococcus, Gemella, Enterococcus, Finegoldia, Lactobacillus, Gardnerella, Alphaproteobacteria, and Prevotella [139,140,141,142,143,144,145]. Using an approach that incorporated several machine learning classifiers, Mehta et al. have identified penile microbiota in the meatus and the glans/coronal sulcus that accurately predicted the occurrence of BV in a female partner. The following ten meatal taxa are critical for predicting BV incidence, from the most important to the least: Parvimonas, Lactobacillus iners, Fastidiosipula, Negativicoccus, Lactobacillus crispatus, Dialister, Sneathia sanguinegens, Gardnerella vaginalis, Prevotella corporis, and Corynebacterium. Notably, some of these bacteria have been associated with BV in the FGT [136]. An analysis of the urethral microbiota of healthy individuals and STI patients found that the amount of Staphylococcus spp., Streptococcus spp., and Corynebacterium spp. was an effective diagnostic indicator for distinguishing the two groups. STI patients had significantly lower levels of the above three species than clinically healthy individuals. On the other hand, the relative number of Anaerococcus spp. were significantly higher in men infected with N. gonorrhoeae than in healthy men [146].
As with gonorrhea, BV is often asymptomatic and when symptomatic, antibiotics such as metronidazole or clindamycin are commonly used to treat polymicrobial BV, although with a low success rate and subsequent recurrence of symptoms [103,108,147]. Polymicrobial etiology and variations in the BV-associated microbiome between subjects of different racial backgrounds contribute to the low success rate of metronidazole treatment. In some cases, treatment of BV with a combination of antibiotics and Lactobacillus probiotics has had a higher success rate than treatment with antibiotics alone [130]. Amstel clinical criteria and Nugent score are the most widely accepted BV diagnostic methods. Nugent score and Amstel’s clinical criteria are subjective diagnostic methods that depend on the observer’s skills to accurately assess the clinical symptoms. This limitation often leads to misdiagnosis [131,148,149]. This subjective diagnosis combined with the fact that many women with BV may be asymptomatic means that it is imperative to use tools that provide a comprehensive picture of the individual-specific BV microbiome in an unbiased manner. A recent study has demonstrated the usefulness of next-generation sequencing for the accurate diagnosis and management of recurrent BV by identifying specific microbes and drug-resistant genes contributing to the symptoms [150]. Other studies have applied metatranscriptomics to identify transcriptionally active microbes, functional pathways, and genes of the vaginal microbiome responsible for resistance to the metronidazole treatment [151,152].
6. Application of Gene Regulatory Data to Modeling of the Female Genital Tract Microbiome
Network analysis, agent-based modeling, and genome-scale metabolic modeling (GEM) are a few computational analyses used to study various aspects of the microbiome including its structure, function, and dynamics of the microbial community (Table 1). One of the major goals of microbiome modeling is to identify interactions between different microorganisms within a community together with host–microbiome interactions [153]. The inference of such networks and models, and their application to a better understanding of STIs, is based on fundamental gene expression data from N. gonorrhoeae and other microbes of the genital tract. Thus, these networks and models serve as links between the gene regulatory pathways described above and the emergent processes of infection. Most of the data collected has come from the global genital tract microbiome; therefore, results have instead focused on the community rather than on the N. gonorrhoeae. An analysis of the network provides a structure and function relationship between components of the network based on their connectivity within the bacterial community’s network. It identifies the key microbes or metabolites in the community represented by highly connected hubs/nodes. This network approach is similar to the gene co-expression networks described above. For example, GEM is a data-driven modeling approach that generates a model using experimental genomic or biochemical data [154]. In addition to providing a temporal overview of the bacterial communities, computational modeling can predict how the microbiome will evolve over time [155]. Agent-based modeling uses a stochastic simulation approach by simulating the responses of bacterial communities to perturbations in various biological entities such as an enzyme and its substrate. For example, the flux balance analysis (FBA) approach is used to predict the growth phenotype of a microbe by calculating the flux and flow of metabolites through a network [156].
So far, most modeling work relies exclusively on amplicon analysis. However, the emergent qualities of the vaginal microbiome result from functions expressed by these species. Beyond its role in the diagnosis and management of BV and identifying transcriptionally active microbes, metatranscriptomics analysis of the FGT may be used to model interactions between processes and species in this anatomical site. As a result, other studies have moved beyond amplicon analysis of species and included additional -omics analysis, including metatranscriptomics and metabolomics to better understand how these functions are linked to healthy states [157]. A recent study that examined the Multi-Omics Microbiome Study: Pregnancy Initiative MOMS-PI dataset performed paired metatranscriptomic and metagenomic analyses of 122 vaginal samples (41 from premature births and 81 from term births) and discovered that the taxa that were most transcriptionally active were also associated with premature births. L. crispatus, for example, showed some of the highest transcription levels of several genes [158]. Additionally, this study found a higher expression of genes from G. vaginalis to be associated with term birth. Another study that examined transcriptomic profiles of vaginal microbiomes from pregnant women also found these higher levels of expression from G. vaginalis [159]. The results of these studies indicate that it is the functional pathways expressed by the microbiome that are responsible for its effects on reproductive health. Microbiome and host interactions are likely to lead to these effects and future research should examine this in greater detail.
Modeling of the female genital tract metabolome has also been reported. Noecker et al. used a community-based metabolite potential (CMP) score built from metagenomic data [160]. Each of these scores predicts a community’s potential to deplete or generate each metabolite. These analyses were used to associate specific metabolites and functional pathways to either healthy vaginal microbiomes or those with BV. Metabolomic and taxonomic data from women suffering from vulvovaginal candidiasis (yeast infection) also linked metabolites and microbial taxa and found that Lactobacillus abundance was positively associated with lactate and 4-hydroxyphenylacetate, isoleucine, leucine, tryptophan, phenylalanine, and aspartate. Lactobacillus was negatively correlated with formate, acetate, 2-hydroxyisovalerate, and alanine. In contrast, other bacterial taxa were positively correlated with the metabolites that Lactobacillus was negatively correlated with; these included Gardnerella, Prevotella, Megasphaera, Atopobium, Dialister, and Clostridium. This taxon also showed a positive correlation with organic acids and amines [161]. Further analyses of these types will enable a better understanding of the relationship between taxonomic presence or absence of bacterial species and disease states.

{kind=link}
{kind=link}
{kind=link}
Table 1.
Studies carrying out predictive modeling of the female genital tract.
| Study | Input Data (Features) | Modeling | Major Inferences from the Study (Labels) |
|---|---|---|---|
| [162] | A large longitudinal study looking at more than 3620 women with high Nugent scores | Correlative | There is an association between a high Nugent score and acquisition of N. gonorrhoeae, C. trachomatis, or T. vaginalis infection |
| [163] | 16S amplicon data of vaginal swabs from women from four ethnic/racial groups | Correlative | Prediction of T. vaginalis infection is associated with high bacterial diversity and reduction in Lactobacillus spp. |
| [158] | An analysis of vaginal samples from women who have experienced preterm or term births (control) using 16S amplicons, metagenomic and metatranscriptomic sequencing was carried out | Associative model using a Mann–Whitney U test and assigning weights to these taxa using L1-regularized logistic regression | The abundance of Lactobacillus spp. No difference between pregnant and non-pregnant women Differs in preterm and full-term pregnancies Prediction of preterm birth based on selecting OTUs associated with premature birth Premature birth is significantly associated with four taxa: Sneathia amnii, BV-associated bacterium 1 (BVAB1), Prevotella cluster 2, and TM7-H1 |
| [164] | 16S amplicon timescale data of vaginal samples collected for each subject across 16 weeks | Vagina-specific dynamic microbial interaction network (MIN) | Subject-specific interaction predictions L. iners prevents growth of other Lactobacillus spp. and L. jensenii aids the growth of Gardnerella sp. Finegoldia sp. have a highly important position in the vaginal microbiome and synergistic relationships with Sneathia and Anarococcus sp. L. iners was found to promote growth of Gardnerella as well as to promote growth of Atopobium, Prevotella, Parvimonas, Sneathia, and Mobiluncus |
| [114,165] | The longitudinal study included analysis of 16S amplicon sequencing and the Nugent score for vaginal samples | Mixed effects model Dynamic Bayesian network | L. iners and Streptococcus taxa are linked to menstrual cycle Found positive relationships between L. iners and Atopobium as well as Atopobium and Gardnerella |
| MOMS-PI dataset metatranscriptomic and metagenomic analysis of 122 vaginal samples | |||
| [160] | Integrated taxonomic and metabolomic data | Community-based metabolite potential (CMP) score | Association of specific metabolites and functional pathways to either healthy vaginal microbiomes or those with BV |
| [161] | Integrated metabolomic and taxonomic data collected from healthy women and women with BV, vulvovaginal candidiasis, and Chlamydia trachomatis infection | Co-abundance network of Spearman correlation coefficient | Lactobacillus spp. abundance was positively associated with lactate and 4-hydroxyphenylacetate, isoleucine, leucine, tryptophan, phenylalanine, and aspartate Lactobacillus was negatively correlated with formate, acetate, 2-hydroxyisovalerate, and alanine In contrast, other bacterial taxa were positively correlated with the metabolites that Lactobacillus was negatively correlated with; these include Gardnerella, Prevotella, Megasphaera, Atopobium, Dialister, and Clostridium. These taxa also showed a positive correlation with organic acids and amines |
7. Conclusions
As an obligate human pathogen, N. gonorrhoeae has evolved mechanisms to colonize the FGT and co-habit with the microbiome, while also being able to adapt to the host immune response. Like other pathogens, N. gonorrhoeae has several transcriptional regulators that enable the organism to respond to and survive specific defenses of the host innate immune responses. While we have an overall understanding of how some of these transcriptional regulators function in gene regulation, there are many other uncharacterized putative transcriptional regulators that must be studied. Furthermore, the detailed analysis of gene expression during natural infection is still in its infancy. Moving forward, a major focus should be the integration of molecular-level data collected in vitro with system-level information collected during natural infection. Future studies should also focus on bridging this gap by utilizing various microbiological techniques, RNA-Seq, ChIP-Seq, and bioinformatics analysis tools, such as gene co-expression network analysis and Bayesian network analysis for causality analysis. The same challenges exist in our understanding of the interactions of N. gonorrhoeae with the microbiome during natural infection. There have been numerous epidemiological and in vitro studies emphasizing the necessity of the vaginal microbiome, especially Lactobacillus species, to protect against infecting pathogens. However, there is still a gap in our understanding of the interactions between functionally active genes, proteins, and metabolites in the microbiome, infecting pathogens, and the host. Future studies should focus on transcriptional, proteomic, and metabolic profiling to understand the impact of the microbiome on the regulation of N. gonorrhoeae’s gene expression that results in infection, and susceptibility to antibiotics and probiotic treatment. Collecting -omics data of these types will help in our overall understanding of gonococcal pathogenesis during natural infection of the human genital tract.
Author Contributions
Conceptualization, A.S., R.M. and C.G.; writing—original draft preparation, A.S., R.M. and C.G.; writing—review and editing, C.G. and R.M.; visualization, A.S., R.M. and C.G.; supervision, C.G; funding acquisition, C.G. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by NIAID 5R01AI130946-04 and 5R01AI116969-04 to C.G.
Data Availability Statement
Not applicable.
Acknowledgments
We thank Carolyn Kramer and Paola Massari for their critical review of the paper.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Unemo, M.; Seifert, H.S.; Hook, E.W.; Hawkes, S.; Ndowa, F.; Dillon, J.-A.R. Gonorrhoea. Nat. Rev. Dis. Prim. 2019, 5, 79. [Google Scholar] [CrossRef]
- Rowley, J.; Vander Hoorn, S.; Korenromp, E.; Low, N.; Unemo, M.; Abu-Raddad, L.J.; Chico, R.M.; Smolak, A.; Newman, L.; Gottlieb, S.; et al. Chlamydia, gonorrhoea, trichomoniasis and syphilis: Global prevalence and incidence estimates, 2016. Bull. World Health Organ. 2019, 97, 548–562. [Google Scholar] [CrossRef]
- Centers for Disease Control and Prevention. Sexually Transmitted Disease Surveillance 2017; U.S. Department of Health and Human Services: Atlanta, GA, USA, 2017; Available online: https://www.cdc.gov/std/stats17/2017-STD-Surveillance-Report_CDC-clearance-9.10.18.pdf (accessed on 16 March 2022).
- Apicella, M.A.; Ketterer, M.; Lee, F.K.N.; Zhou, D.; Rice, P.A.; Blake, M.S. The Pathogenesis of Gonococcal Urethritis in Men: Confocal and Immunoelectron Microscopic Analysis of Urethral Exudates from Men Infected with Neisseria gonorrhoeae. J. Infect. Dis. 1996, 173, 636–646. [Google Scholar] [CrossRef]
- Farzadegan, H.; Roth, I.L. Scanning electron microscopy and freeze-etching of gonorrhoeal urethral exudate. Sex. Transm. Infect. 1975, 51, 83–91. [Google Scholar] [CrossRef] [Green Version]
- Katherine, H.; Ram, S.; Darville, T. Chapter 219 Neisseria gonorrhoeae (Gonococcus). In Nelson Textbook of Pediatrics; Elsevier: Amsterdam, The Netherlands, 2020. [Google Scholar]
- Hook, E.W.; Holmes, K.K. Gonococcal Infections. Ann. Intern. Med. 1985, 102, 229–243. [Google Scholar] [CrossRef]
- Edwards, J.L.; Apicella, M.A. The Molecular Mechanisms Used by Neisseria gonorrhoeae To Initiate Infection Differ between Men and Women. Clin. Microbiol. Rev. 2004, 17, 965–981. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Detels, R.; Green, A.M.; Klausner, J.D.; Katzenstein, D.; Gaydos, C.; Handsfield, H.H.; Pequegnat, W.; Mayer, K.; Hartwell, T.D.; Quinn, T.C. The Incidence and Correlates of Symptomatic and Asymptomatic Chlamydia trachomatis and Neisseria gonorrhoeae Infections in Selected Populations in Five Countries. Sex Transm Dis 2011, 38, 503–509. [Google Scholar] [CrossRef] [Green Version]
- Edwards, J.L.; Butler, E.K. The Pathobiology of Neisseria gonorrhoeae Lower Female Genital Tract Infection. Front. Microbiol. 2011, 2, 102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiesenfeld, H.C.; Hillier, S.L.; Meyn, L.A.; Amortegui, A.J.; Sweet, R.L. Subclinical Pelvic Inflammatory Disease and Infertility. Obstet. Gynecol. 2012, 120, 37–43. [Google Scholar] [CrossRef]
- Wu, H.; Soler-García, A.A.; Jerse, A.E. A Strain-Specific Catalase Mutation and Mutation of the Metal-Binding Transporter Gene mntC Attenuate Neisseria gonorrhoeae In Vivo but Not by Increasing Susceptibility to Oxidative Killing by Phagocytes. Infect. Immun. 2009, 77, 1091–1102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, A.; Buono, S.; Katz, K.A.; Pandori, M.W. Clinical Neisseria gonorrhoeae Isolates in the United States with Resistance to Azithromycin Possess Mutations in All 23S rRNA Alleles and the mtrR Coding Region. Microb. Drug Resist. 2011, 17, 425–427. [Google Scholar] [CrossRef]
- Nguyen, D.; Gose, S.; Castro, L.; Chung, K.; Bernstein, K.; Samuel, M.; Bauer, H.; Pandori, M. Neisseria gonorrhoeae Strain with Reduced Susceptibilities to Extended-Spectrum Cephalosporins. Emerg. Infect. Dis. 2014, 20, 1211. [Google Scholar] [CrossRef] [Green Version]
- Su, X.-H.; Wang, B.-X.; Le, W.-J.; Liu, Y.-R.; Wan, C.; Li, S.; Alm, R.A.; Mueller, J.P.; Rice, P.A. Multidrug-Resistant Neisseria gonorrhoeae Isolates from Nanjing, China, Are Sensitive to Killing by a Novel DNA Gyrase Inhibitor, ETX0914 (AZD0914). Antimicrob. Agents Chemother. 2016, 60, 621–623. [Google Scholar] [CrossRef] [Green Version]
- Brunner, A.; Nemes-Nikodem, E.; Jeney, C.; Szabo, D.; Marschalko, M.; Karpati, S.; Ostorhazi, E. Emerging azithromycin-resistance among the Neisseria gonorrhoeae strains isolated in Hungary. Ann. Clin. Microbiol. Antimicrob. 2016, 15, 1–6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, C.-W.; Li, L.-H.; Su, C.-Y.; Li, S.-Y.; Yen, M.-Y. Changes in the six most common sequence types of Neisseria gonorrhoeae, including ST4378, identified by surveillance of antimicrobial resistance in northern Taiwan from 2006 to 2013. J. Microbiol. Immunol. Infect. 2016, 49, 708–716. [Google Scholar] [CrossRef] [Green Version]
- Unemo, M.; del Rio, C.; Shafer, W.M. Antimicrobial Resistance Expressed by Neisseria gonorrhoeae: A Major Global Public Health Problem in the 21st Century. Microbiol. Spectr. 2016, 4. [Google Scholar] [CrossRef] [Green Version]
- Chan, P.A.; Robinette, A.; Montgomery, M.; Almonte, A.; Cu-Uvin, S.; Lonks, J.R.; Chapin, K.C.; Kojic, E.M.; Hardy, E.J. Extragenital Infections Caused by Chlamydia trachomatis and Neisseria gonorrhoeae: A Review of the Literature. Infect. Dis. Obstet. Gynecol. 2016, 2016, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Anahtar, M.N.; Byrne, E.H.; Doherty, K.E.; Bowman, B.A.; Yamamoto, H.S.; Soumillon, M.; Padavattan, N.; Ismail, N.; Moodley, A.; Sabatini, M.E.; et al. Cervicovaginal Bacteria Are a Major Modulator of Host Inflammatory Responses in the Female Genital Tract. Immunity 2015, 42, 965–976. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mei, C.; Yang, W.; Wei, X.; Wu, K.; Huang, D. The Unique Microbiome and Innate Immunity During Pregnancy. Front. Immunol. 2019, 10, 2886. [Google Scholar] [CrossRef] [PubMed]
- Wira, C.R.; Grant-Tschudy, K.S.; Crane-Godreau, M.A. Epithelial Cells in the Female Reproductive Tract: A Central Role as Sentinels of Immune Protection. Am. J. Reprod. Immunol. 2005, 53, 65–76. [Google Scholar] [CrossRef]
- Wira, C.R.; Fahey, J.V.; Sentman, C.L.; Pioli, P.A.; Shen, L. Innate and adaptive immunity in female genital tract: Cellular responses and interactions. Immunol. Rev. 2005, 206, 306–335. [Google Scholar] [CrossRef] [PubMed]
- Shafer, W.M. Hrsg. Antimicrobial Peptides and Human Disease: With 4 Tables; Springer: Berlin/Heidelberg, Germany, 2006. [Google Scholar]
- Hickey, D.; Patel, M.; Fahey, J.; Wira, C. Innate and adaptive immunity at mucosal surfaces of the female reproductive tract: Stratification and integration of immune protection against the transmission of sexually transmitted infections. J. Reprod. Immunol. 2011, 88, 185–194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rotman, E.; Seifert, H.S. The Genetics of Neisseria Species. Annu. Rev. Genet. 2014, 48, 405–431. [Google Scholar] [CrossRef] [PubMed]
- Gibbs, C.P.; Meyer, T.F. Genome plasticity in Neisseria gonorrhoeae. FEMS Microbiol. Lett. 1996, 145, 173–179. [Google Scholar] [CrossRef]
- Masters, T.L.; Wachter, J.; Hill, S.A. Loop structures in the 5′ untranslated region and antisense RNA mediate pilE gene expression in Neisseria gonorrhoeae. Microbiology 2016, 162, 2005–2016. [Google Scholar] [CrossRef]
- Quillin, S.J.; Seifert, H.S. Neisseria gonorrhoeae host adaptation and pathogenesis. Nat. Rev. Genet. 2018, 16, 226–240. [Google Scholar] [CrossRef]
- Hill, S.A.; Samuels, D.S.; Carlson, J.H.; Wilson, J.; Hogan, D.; Lubke, L.; Belland, R.J. Integration host factor is a transcriptional cofactor of pilE in Neisseria gonorrhoeae. Mol. Microbiol. 1997, 23, 649–656. [Google Scholar] [CrossRef]
- McClure, R.; Sunkavalli, A.; Balzano, P.M.; Massari, P.; Cho, C.; Nauseef, W.M.; Apicella, M.A.; Genco, C.A. Global Network Analysis of Neisseria gonorrhoeae Identifies Coordination between Pathways, Processes, and Regulators Expressed during Human Infection. mSystems 2020, 5, e00729-19. [Google Scholar] [CrossRef] [Green Version]
- Cassat, J.E.; Skaar, E.P. Iron in Infection and Immunity. Cell Host Microbe 2013, 13, 509–519. [Google Scholar] [CrossRef] [Green Version]
- Palmer, L.D.; Skaar, E.P. Transition Metals and Virulence in Bacteria. Annu. Rev. Genet. 2016, 50, 67–91. [Google Scholar] [CrossRef] [Green Version]
- Hennigar, S.R.; McClung, J.P. Nutritional Immunity. Am. J. Lifestyle Med. 2016, 10, 170–173. [Google Scholar] [CrossRef] [PubMed]
- Srinivasan, S.; Liu, C.; Mitchell, C.M.; Fiedler, T.L.; Thomas, K.K.; Agnew, K.J.; Marrazzo, J.; Fredricks, D.N. Temporal Variability of Human Vaginal Bacteria and Relationship with Bacterial Vaginosis. PLoS ONE 2010, 5, e10197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roberts, S.A.; Brabin, L.; Diallo, S.; Gies, S.; Nelson, A.; Stewart, C.; Swinkels, D.W.; Geurts-Moespot, A.J.; Kazienga, A.; Ouedraogo, S.; et al. Mucosal lactoferrin response to genital tract infections is associated with iron and nutritional biomarkers in young Burkinabé women. Eur. J. Clin. Nutr. 2019, 73, 1464–1472. [Google Scholar] [CrossRef]
- Cornelissen, C.N. Subversion of nutritional immunity by the pathogenic Neisseriae. Pathog. Dis. 2017, 76, ftx112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weinberg, E.D. Iron availability and infection. Biochim. Biophys. Acta (BBA)-Gen. Subj. 2009, 1790, 600–605. [Google Scholar] [CrossRef] [Green Version]
- Andrews, S.C.; Robinson, A.K.; Rodríguez-Quiñones, F. Bacterial iron homeostasis. FEMS Microbiol. Rev. 2003, 27, 215–237. [Google Scholar] [CrossRef]
- Cornelis, P.; Wei, Q.; Andrews, S.C.; Vinckx, T. Iron homeostasis and management of oxidative stress response in bacteria. Metallomics 2011, 3, 540–549. [Google Scholar] [CrossRef]
- Yu, C.; McClure, R.; Nudel, K.; Daou, N.; Genco, C.A. Characterization of the Neisseria gonorrhoeae Iron and Fur Regulatory Network. J. Bacteriol. 2016, 198, 2180–2191. [Google Scholar] [CrossRef] [Green Version]
- Hollander, A.; Mercante, A.D.; Shafer, W.M.; Cornelissen, C.N. The Iron-Repressed, AraC-Like Regulator MpeR Activates Expression of fetA in Neisseria gonorrhoeae. Infect. Immun. 2011, 79, 4764–4776. [Google Scholar] [CrossRef] [Green Version]
- Nudel, K.; McClure, R.; Moreau, M.; Briars, E.; Abrams, A.J.; Tjaden, B.; Su, X.-H.; Trees, D.; Rice, P.A.; Massari, P.; et al. Transcriptome Analysis of Neisseria gonorrhoeae during Natural Infection Reveals Differential Expression of Antibiotic Resistance Determinants between Men and Women. mSphere 2018, 3, e00312-18. [Google Scholar] [CrossRef] [Green Version]
- Folster, J.P.; Shafer, W.M. Regulation of mtrF Expression in Neisseria gonorrhoeae and Its Role in High-Level Antimicrobial Resistance. J. Bacteriol. 2005, 187, 3713–3720. [Google Scholar] [CrossRef] [Green Version]
- Fillat, M.F. The FUR (ferric uptake regulator) superfamily: Diversity and versatility of key transcriptional regulators. Arch. Biochem. Biophys. 2014, 546, 41–52. [Google Scholar] [CrossRef] [PubMed]
- Bagg, A.; Neilands, J.B. Ferric uptake regulation protein acts as a repressor, employing iron(II) as a cofactor to bind the operator of an iron transport operon in Escherichia coli. Biochemistry 1987, 26, 5471–5477. [Google Scholar] [CrossRef]
- Sheikh, A.; Taylor, G.L. Crystal structure of the Vibrio cholerae ferric uptake regulator (Fur) reveals insights into metal co-ordination. Mol. Microbiol. 2009, 72, 1208–1220. [Google Scholar] [CrossRef]
- Yu, C.; Genco, C.A. Fur-Mediated Global Regulatory Circuits in Pathogenic Neisseria Species. J. Bacteriol. 2012, 194, 6372–6381. [Google Scholar] [CrossRef] [Green Version]
- Carpenter, B.M.; Gilbreath, J.J.; Pich, O.Q.; McKelvey, A.M.; Maynard, E.L.; Li, Z.-Z.; Merrell, D.S. Identification and Characterization of Novel Helicobacter pylori apo-Fur-Regulated Target Genes. J. Bacteriol. 2013, 195, 5526–5539. [Google Scholar] [CrossRef] [Green Version]
- Carpenter, B.M.; Whitmire, J.M.; Merrell, D.S. This Is Not Your Mother’s Repressor: The Complex Role of Fur in Pathogenesis. Infect. Immun. 2009, 77, 2590–2601. [Google Scholar] [CrossRef] [Green Version]
- Jackson, L.A.; Ducey, T.F.; Day, M.W.; Zaitshik, J.B.; Orvis, J.; Dyer, D.W. Transcriptional and Functional Analysis of the Neisseria gonorrhoeae Fur Regulon. J. Bacteriol. 2010, 192, 77–85. [Google Scholar] [CrossRef] [Green Version]
- Cornelissen, C.N.; Hollander, A. TonB-Dependent Transporters Expressed by Neisseria gonorrhoeae. Front. Microbiol. 2011, 2, 117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cornelissen, C.N.; Kelley, M.; Hobbs, M.M.; Anderson, J.E.; Cannon, J.G.; Cohen, M.S.; Sparling, P.F. The transferrin receptor expressed by gonococcal strain FA1090 is required for the experimental infection of human male volunteers. Mol. Microbiol. 1998, 27, 611–616. [Google Scholar] [CrossRef]
- Cornelissen, C.N.; Biswas, G.D.; Tsai, J.; Paruchuri, D.K.; Thompson, S.A.; Sparling, P.F. Gonococcal transferrin-binding protein 1 is required for transferrin utilization and is homologous to TonB-dependent outer membrane receptors. J. Bacteriol. 1992, 174, 5788–5797. [Google Scholar] [CrossRef] [Green Version]
- Ducey, T.F.; Carson, M.B.; Orvis, J.; Stintzi, A.P.; Dyer, D.W. Identification of the Iron-Responsive Genes of Neisseria gonorrhoeae by Microarray Analysis in Defined Medium. J. Bacteriol. 2005, 187, 4865–4874. [Google Scholar] [CrossRef] [Green Version]
- Ducey, T.F.; Jackson, L.; Orvis, J.; Dyer, D.W. Transcript analysis of nrrF, a Fur repressed sRNA of Neisseria gonorrhoeae. Microb. Pathog. 2009, 46, 166–170. [Google Scholar] [CrossRef] [Green Version]
- Mellin, J.R.; Goswami, S.; Grogan, S.; Tjaden, B.; Genco, C.A. A Novel Fur- and Iron-Regulated Small RNA, NrrF, Is Required for Indirect Fur-Mediated Regulation of the sdhA and sdhC Genes in Neisseria meningitidis. J. Bacteriol. 2007, 189, 3686–3694. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- The UniProt Consortium. UniProt: The universal protein knowledgebase in 2021. Nucleic Acids Res. 2021, 49, D480–D489. [Google Scholar] [CrossRef]
- Gallegos, M.T.; Schleif, R.; Bairoch, A.; Hofmann, K.; Ramos, J.L. Arac/XylS family of transcriptional regulators. Microbiol. Mol. Biol. Rev. 1997, 61, 393–410. [Google Scholar]
- Mercante, A.D.; Jackson, L.; Johnson, P.J.T.; Stringer, V.A.; Dyer, D.W.; Shafer, W.M. MpeR Regulates the mtr Efflux Locus in Neisseria gonorrhoeae and Modulates Antimicrobial Resistance by an Iron-Responsive Mechanism. Antimicrob. Agents Chemother. 2012, 56, 1491–1501. [Google Scholar] [CrossRef] [Green Version]
- Cortés-Avalos, D.; Martínez-Pérez, N.; Ortiz-Moncada, M.A.; Juárez-González, A.; Baños-Vargas, A.A.; Santos, P.E.-D.L.; Pérez-Rueda, E.; Ibarra, J.A. An update of the unceasingly growing and diverse AraC/XylS family of transcriptional activators. FEMS Microbiol. Rev. 2021, 45, fuab020. [Google Scholar] [CrossRef]
- Quillin, S.J.; Hockenberry, A.J.; Jewett, M.C.; Seifert, H.S. Neisseria gonorrhoeae Exposed to Sublethal Levels of Hydrogen Peroxide Mounts a Complex Transcriptional Response. mSystems 2018, 3, e00156-18. [Google Scholar] [CrossRef] [Green Version]
- Stohl, E.A.; Criss, A.K.; Seifert, H.S. The transcriptome response of Neisseria gonorrhoeae to hydrogen peroxide reveals genes with previously uncharacterized roles in oxidative damage protection. Mol. Microbiol. 2005, 58, 520–532. [Google Scholar] [CrossRef] [Green Version]
- Zughaier, S.M.; Kandler, J.L.; Shafer, W.M. Neisseria gonorrhoeae Modulates Iron-Limiting Innate Immune Defenses in Macrophages. PLoS ONE 2014, 9, e87688. [Google Scholar] [CrossRef]
- Becker, K.W.; Skaar, E.P. Metal limitation and toxicity at the interface between host and pathogen. FEMS Microbiol. Rev. 2014, 38, 1235–1249. [Google Scholar] [CrossRef] [Green Version]
- Wu, H.-J.; Seib, K.; Srikhanta, Y.; Kidd, S.; Edwards, J.L.; Maguire, T.L.; Grimmond, S.; Apicella, M.A.; McEwan, A.G.; Jennings, M.P. PerR controls Mn-dependent resistance to oxidative stress in Neisseria gonorrhoeae. Mol. Microbiol. 2006, 60, 401–416. [Google Scholar] [CrossRef]
- Jean, S.; Juneau, R.A.; Criss, A.K.; Cornelissen, C.N. Neisseria gonorrhoeae Evades Calprotectin-Mediated Nutritional Immunity and Survives Neutrophil Extracellular Traps by Production of TdfH. Infect. Immun. 2016, 84, 2982–2994. [Google Scholar] [CrossRef] [Green Version]
- Kidd, S.P.; Potter, A.J.; Apicella, M.A.; Jennings, M.P.; McEwan, A.G. NmlR of Neisseria gonorrhoeae: A novel redox responsive transcription factor from the MerR family: Neisseria MerR-like transcription factor. Mol. Microbiol. 2005, 57, 1676–1689. [Google Scholar] [CrossRef]
- Lim, K.H.L.; Jones, C.E.; Hoven, R.N.V.; Edwards, J.L.; Falsetta, M.L.; Apicella, M.A.; Jennings, M.P.; McEwan, A.G. Metal Binding Specificity of the MntABC Permease of Neisseria gonorrhoeae and Its Influence on Bacterial Growth and Interaction with Cervical Epithelial Cells. Infect. Immun. 2008, 76, 3569–3576. [Google Scholar] [CrossRef] [Green Version]
- Wu, H.-J.; Seib, K.L.; Srikhanta, Y.N.; Edwards, J.; Kidd, S.P.; Maguire, T.L.; Hamilton, A.; Pan, K.-T.; Hsiao, H.-H.; Yao, C.-W.; et al. Manganese regulation of virulence factors and oxidative stress resistance in Neisseria gonorrhoeae. J. Proteom. 2010, 73, 899–916. [Google Scholar] [CrossRef] [Green Version]
- Helmann, J.D.; Foster, A.W.; Osman, D.; Robinson, N.J. Specificity of Metal Sensing: Iron and Manganese Homeostasis in Bacillus subtilis. J. Biol. Chem. 2014, 289, 28112–28120. [Google Scholar] [CrossRef] [Green Version]
- Hood, M.I.; Skaar, E.P. Nutritional immunity: Transition metals at the pathogen–host interface. Nat. Rev. Genet. 2012, 10, 525–537. [Google Scholar] [CrossRef]
- Kehl-Fie, T.E.; Skaar, E.P. Nutritional immunity beyond iron: A role for manganese and zinc. Curr. Opin. Chem. Biol. 2010, 14, 218–224. [Google Scholar] [CrossRef] [Green Version]
- Seib, K.; Wu, H.-J.; Srikhanta, Y.; Edwards, J.L.; Falsetta, M.L.; Hamilton, A.J.; Maguire, T.L.; Grimmond, S.; Apicella, M.A.; McEwan, A.G.; et al. Characterization of the OxyR regulon of Neisseria gonorrhoeae. Mol. Microbiol. 2007, 63, 54–68. [Google Scholar] [CrossRef] [Green Version]
- Tseng, H.-J.; McEwan, A.G.; Apicella, M.A.; Jennings, M.P. OxyR Acts as a Repressor of Catalase Expression in Neisseria gonorrhoeae. Infect. Immun. 2003, 71, 550–556. [Google Scholar] [CrossRef] [Green Version]
- Christman, M.F.; Storz, G.; Ames, B.N. OxyR, a positive regulator of hydrogen peroxide-inducible genes in Escherichia coli and Salmonella typhimurium, is homologous to a family of bacterial regulatory proteins. Proc. Natl. Acad. Sci. USA 1989, 86, 3484–3488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schook, P.O.P.; Stohl, E.A.; Criss, A.K.; Seifert, H.S. The DNA-binding activity of the Neisseria gonorrhoeae LexA orthologue NG1427 is modulated by oxidation. Mol. Microbiol. 2010, 79, 846–860. [Google Scholar] [CrossRef] [Green Version]
- Newkirk, G.R. Pelvic inflammatory disease: A contemporary approach. Am. Fam. Physician 1996, 53, 1127–1135. [Google Scholar]
- Clark, V.L.; Knapp, J.S.; Thompson, S.; Klimpel, K.W. Presence of antibodies to the major anaerobically induced gonococcal outer membrane protein in sera from patients with gonococcal infections. Microb. Pathog. 1988, 5, 381–390. [Google Scholar] [CrossRef]
- Barth, K.R.; Isabella, V.M.; Clark, V.L. Biochemical and genomic analysis of the denitrification pathway within the genus Neisseria. Microbiology 2009, 155, 4093–4103. [Google Scholar] [CrossRef] [Green Version]
- Isabella, V.M.; Clark, V.L. Deep sequencing-based analysis of the anaerobic stimulon in Neisseria gonorrhoeae. BMC Genom. 2011, 12, 51. [Google Scholar] [CrossRef] [Green Version]
- Lissenden, S.; Mohan, S.; Overton, T.; Regan, T.; Crooke, H.; Cardinale, J.A.; Householder, T.C.; Adams, P.; O’Conner, C.D.; Clark, V.L.; et al. Identification of transcription activators that regulate gonococcal adaptation from aerobic to anaerobic or oxygen-limited growth. Mol. Microbiol. 2000, 37, 839–855. [Google Scholar] [CrossRef] [Green Version]
- Overton, T.; Whitehead, R.; Li, Y.; Snyder, L.; Saunders, N.; Smith, H.; Cole, J.A. Coordinated Regulation of the Neisseria gonorrhoeae-truncated Denitrification Pathway by the Nitric Oxide-sensitive Repressor, NsrR, and Nitrite-insensitive NarQ-NarP. J. Biol. Chem. 2006, 281, 33115–33126. [Google Scholar] [CrossRef] [Green Version]
- Whitehead, R.N.; Overton, T.W.; Snyder, L.A.S.; McGowan, S.J.; Smith, H.; Cole, J.A.; Saunders, N.J. The small FNR regulon of Neisseria gonorrhoeae: Comparison with the larger Escherichia coli FNR regulon and interaction with the NarQ-NarP regulon. BMC Genom. 2007, 8, 35. [Google Scholar] [CrossRef] [Green Version]
- Isabella, V.; Wright, L.F.; Barth, K.; Spence, J.M.; Grogan, S.; Genco, C.A.; Clark, V.L. cis- and trans-acting elements involved in regulation of norB (norZ), the gene encoding nitric oxide reductase in Neisseria gonorrhoeae. Microbiology 2008, 154, 226–239. [Google Scholar] [CrossRef] [Green Version]
- Isabella, V.M.; Jr, J.D.L.; Kennedy, E.M.; Clark, V.L. Functional analysis of NsrR, a nitric oxide-sensing Rrf2 repressor in Neisseria gonorrhoeae. Mol. Microbiol. 2008, 71, 227–239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van der Woude, M.W.; Bäumler, A.J. Phase and Antigenic Variation in Bacteria. Clin. Microbiol. Rev. 2004, 17, 581–611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bayliss, C.D.; Palmer, M.E. Evolution of simple sequence repeat-mediated phase variation in bacterial genomes. Ann. N. Y. Acad. Sci. 2012, 1267, 39–44. [Google Scholar] [CrossRef]
- Jordan, P.W.; Snyder, L.A.; Saunders, N.J. Strain-specific differences in Neisseria gonorrhoeae associated with the phase variable gene repertoire. BMC Microbiol. 2005, 5, 21. [Google Scholar] [CrossRef] [Green Version]
- Zelewska, M.A.; Pulijala, M.; Spencer-Smith, R.; Mahmood, H.-T.A.; Norman, B.; Churchward, C.; Calder, A.; Snyder, L.A.S. Phase variable DNA repeats in Neisseria gonorrhoeae influence transcription, translation, and protein sequence variation. Microb. Genom. 2016, 2, e000078. [Google Scholar] [CrossRef] [PubMed]
- Tan, A.; Atack, J.M.; Jennings, M.P.; Seib, K.L. The Capricious Nature of Bacterial Pathogens: Phasevarions and Vaccine Development. Front. Immunol. 2016, 7, 586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carson, S.; Stone, B.; Beucher, M.; Fu, J.; Sparling, P.F. Phase variation of the gonococcal siderophore receptor FetA. Mol. Microbiol. 2002, 36, 585–593. [Google Scholar] [CrossRef] [Green Version]
- Seib, K.L.; Srikhanta, Y.N.; Atack, J.M.; Jennings, M.P. Epigenetic Regulation of Virulence and Immunoevasion by Phase-Variable Restriction-Modification Systems in Bacterial Pathogens. Annu. Rev. Microbiol. 2020, 74, 655–671. [Google Scholar] [CrossRef]
- Srikhanta, Y.; Dowideit, S.J.; Edwards, J.L.; Falsetta, M.L.; Wu, H.-J.; Harrison, O.; Fox, K.L.; Seib, K.; Maguire, T.L.; Wang, A.H.-J.; et al. Phasevarions Mediate Random Switching of Gene Expression in Pathogenic Neisseria. PLOS Pathog. 2009, 5, e1000400. [Google Scholar] [CrossRef] [Green Version]
- Srikhanta, Y.N.; Fox, K.L.; Jennings, M.P. The phasevarion: Phase variation of type III DNA methyltransferases controls coordinated switching in multiple genes. Nat. Rev. Genet. 2010, 8, 196–206. [Google Scholar] [CrossRef]
- Faith, J.J.; Hayete, B.; Thaden, J.T.; Mogno, I.; Wierzbowski, J.; Cottarel, G.; Kasif, S.; Collins, J.J.; Gardner, T.S. Large-Scale Mapping and Validation of Escherichia coli Transcriptional Regulation from a Compendium of Expression Profiles. PLoS Biol. 2007, 5, e8. [Google Scholar] [CrossRef]
- Saint-André, V. Computational biology approaches for mapping transcriptional regulatory networks. Comput. Struct. Biotechnol. J. 2021, 19, 4884–4895. [Google Scholar] [CrossRef]
- Van Dam, S.; Võsa, U.; Van Der Graaf, A.; Franke, L.; De Magalhães, J.P. Gene co-expression analysis for functional classification and gene–disease predictions. Briefings Bioinform. 2018, 19, 575–592. [Google Scholar] [CrossRef]
- McDermott, J.E.; Taylor, R.C.; Yoon, H.; Heffron, F. Bottlenecks and Hubs in Inferred Networks Are Important for Virulence in Salmonella typhimurium. J. Comput. Biol. 2009, 16, 169–180. [Google Scholar] [CrossRef] [Green Version]
- Hou, J.; Ye, X.; Li, C.; Wang, Y. K-Module Algorithm: An Additional Step to Improve the Clustering Results of WGCNA Co-Expression Networks. Genes 2021, 12, 87. [Google Scholar] [CrossRef]
- Huynh-Thu, V.A.; Sanguinetti, G. Gene Regulatory Network Inference: An Introductory Survey. In Gene Regulatory Networks; Sanguinetti, G., Huynh-Thu, V.A., Eds.; Springer: New York, NY, USA, 2019; pp. 1–23. [Google Scholar]
- Gillis, J.; Pavlidis, P. “Guilt by Association” Is the Exception Rather Than the Rule in Gene Networks. PLoS Comput. Biol. 2012, 8, e1002444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lewis, F.M.T.; Bernstein, K.T.; Aral, S.O. Vaginal Microbiome and Its Relationship to Behavior, Sexual Health, and Sexually Transmitted Diseases. Obstet. Gynecol. 2017, 129, 643–654. [Google Scholar] [CrossRef]
- Huttenhower, C.; Gevers, D.; Knight, R.; Abubucker, S.; Badger, J.H.; Chinwalla, A.T.; Creasy, H.H.; Earl, A.M.; FitzGerald, M.G.; Fulton, R.S.; et al. Structure, function and diversity of the healthy human microbiome. Nature 2012, 486, 207–214. [Google Scholar] [CrossRef] [Green Version]
- Ravel, J.; Gajer, P.; Abdo, Z.; Schneider, G.M.; Koenig, S.S.K.; McCulle, S.L.; Karlebach, S.; Gorle, R.; Russell, J.; Tacket, C.O.; et al. Vaginal microbiome of reproductive-age women. Proc. Natl. Acad. Sci. USA 2011, 108 (Suppl. S1), 4680–4687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, N.-P.; Warnow, T.; Pop, M.; White, B. A perspective on 16S rRNA operational taxonomic unit clustering using sequence similarity. npj Biofilms Microbiomes 2016, 2, 16004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Finotello, F.; Mastrorilli, E.; Di Camillo, B. Measuring the diversity of the human microbiota with targeted next-generation sequencing. Briefings Bioinform. 2016, 19, 679–692. [Google Scholar] [CrossRef]
- Sycuro, L.K.; Fredricks, D.N. Microbiota of the Genitourinary Tract. In The Human Microbiota; Fredricks, D.N., Ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2013; pp. 167–210. [Google Scholar]
- Chen, C.; Song, X.; Chunwei, Z.; Zhong, H.; Dai, J.; Lan, Z.; Li, F.; Yu, X.; Feng, Q.; Wang, Z.; et al. The microbiota continuum along the female reproductive tract and its relation to uterine-related diseases. Nat. Commun. 2017, 8, 875. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitchell, C.M.; Haick, A.; Nkwopara, E.; Garcia, R.; Rendi, M.; Agnew, K.; Fredricks, D.; Eschenbach, D. Colonization of the upper genital tract by vaginal bacterial species in nonpregnant women. Am. J. Obstet. Gynecol. 2014, 212, 611.e1–611.e9. [Google Scholar] [CrossRef] [Green Version]
- Aldunate, M.; Srbinovski, D.; Hearps, A.C.; Latham, C.F.; Ramsland, P.A.; Gugasyan, R.; Cone, R.A.; Tachedjian, G. Antimicrobial and immune modulatory effects of lactic acid and short chain fatty acids produced by vaginal microbiota associated with eubiosis and bacterial vaginosis. Front. Physiol. 2015, 6, 164. [Google Scholar] [CrossRef] [PubMed]
- Plesniarski, A.; Siddik, A.B.; Su, R.-C. The Microbiome as a Key Regulator of Female Genital Tract Barrier Function. Front. Cell. Infect. Microbiol. 2021, 11, 790627. [Google Scholar] [CrossRef]
- Zhou, X.; Bent, S.J.; Schneider, M.G.; Davis, C.C.; Islam, M.R.; Forney, L.J. Characterization of vaginal microbial communities in adult healthy women using cultivation-independent methods. Microbiology 2004, 150, 2565–2573. [Google Scholar] [CrossRef] [Green Version]
- Gajer, P.; Brotman, R.M.; Bai, G.; Sakamoto, J.; Schütte, U.M.E.; Zhong, X.; Koenig, S.S.K.; Fu, L.; Ma, Z.; Zhou, X.; et al. Temporal Dynamics of the Human Vaginal Microbiota. Sci. Transl. Med. 2012, 4, 132ra52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peric, A.; Weiss, J.; Vulliemoz, N.; Baud, D.; Stojanov, M. Bacterial Colonization of the Female Upper Genital Tract. Int. J. Mol. Sci. 2019, 20, 3405. [Google Scholar] [CrossRef] [Green Version]
- Miles, S.M.; Hardy, B.L.; Merrell, D. Investigation of the microbiota of the reproductive tract in women undergoing a total hysterectomy and bilateral salpingo-oopherectomy. Fertil. Steril. 2017, 107, 813–820.e1. [Google Scholar] [CrossRef] [Green Version]
- Franasiak, J.M.; Werner, M.D.; Juneau, C.R.; Tao, X.; Landis, J.; Zhan, Y.; Treff, N.R.; Scott, R.T. Endometrial microbiome at the time of embryo transfer: Next-generation sequencing of the 16S ribosomal subunit. J. Assist. Reprod. Genet. 2016, 33, 129–136. [Google Scholar] [CrossRef]
- Moreno, I.; Codoñer, F.M.; Vilella, F.; Valbuena, D.; Martinez-Blanch, J.F.; Jimenez-Almazán, J.; Alonso, R.; Alamá, P.; Remohí, J.; Pellicer, A.; et al. Evidence that the endometrial microbiota has an effect on implantation success or failure. Am. J. Obstet. Gynecol. 2016, 215, 684–703. [Google Scholar] [CrossRef] [Green Version]
- Pelzer, E.S.; Willner, D.; Buttini, M.; Hafner, L.M.; Theodoropoulos, C.; Huygens, F. The fallopian tube microbiome: Implications for reproductive health. Oncotarget 2018, 9, 21541–21551. [Google Scholar] [CrossRef]
- Zhou, B.; Sun, C.; Huang, J.; Xia, M.; Guo, E.; Li, N.; Lu, H.; Shan, W.; Wu, Y.; Li, Y.; et al. The biodiversity Composition of Microbiome in Ovarian Carcinoma Patients. Sci. Rep. 2019, 9, 1691. [Google Scholar] [CrossRef]
- Zeng, J.; Yang, R.; He, W.; Zhong, X.; Liu, W.; Zhu, H.; Zhang, X.; Luo, Q. Modulation effect of vaginal mucosal microflora and susceptibility to Neisseria gonorrhoeae infections: A systematic review and meta-analysis. Arch. Gynecol. Obstet. 2019, 300, 261–267. [Google Scholar] [CrossRef]
- Kovachev, S. Defence factors of vaginal lactobacilli. Crit. Rev. Microbiol. 2017, 44, 31–39. [Google Scholar] [CrossRef]
- Witkin, S.S.; Linhares, I.M. Why do lactobacilli dominate the human vaginal microbiota? BJOG Int. J. Obstet. Gynaecol. 2017, 124, 606–611. [Google Scholar] [CrossRef] [Green Version]
- Foschi, C.; Salvo, M.; Cevenini, R.; Parolin, C.; Vitali, B.; Marangoni, A. Vaginal Lactobacilli Reduce Neisseria gonorrhoeae Viability through Multiple Strategies: An in Vitro Study. Front. Cell. Infect. Microbiol. 2017, 7, 502. [Google Scholar] [CrossRef]
- Spurbeck, R.R.; Arvidson, C.G. Inhibition of Neisseria gonorrhoeae Epithelial Cell Interactions by Vaginal Lactobacillus Species. Infect. Immun. 2008, 76, 3124–3130. [Google Scholar] [CrossRef] [Green Version]
- Spurbeck, R.R.; Arvidson, C.G. Lactobacillus jensenii Surface-Associated Proteins Inhibit Neisseria gonorrhoeae Adherence to Epithelial Cells. Infect. Immun. 2010, 78, 3103–3111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kainulainen, V.; Loimaranta, V.; Pekkala, A.; Edelman, S.; Antikainen, J.; Kylvaja, R.; Laaksonen, M.; Laakkonen, L.; Finne, J.; Korhonen, T.K. Glutamine Synthetase and Glucose-6-Phosphate Isomerase Are Adhesive Moonlighting Proteins of Lactobacillus crispatus Released by Epithelial Cathelicidin LL-37. J. Bacteriol. 2012, 194, 2509–2519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Płaczkiewicz, J.; Chmiel, P.; Malinowska, E.; Bącal, P.; Kwiatek, A. Lactobacillus crispatus and its enolase and glutamine synthetase influence interactions between Neisseria gonorrhoeae and human epithelial cells. J. Microbiol. 2020, 58, 405–414. [Google Scholar] [CrossRef]
- Cook, R.L.; Reid, G.; Pond, D.G.; Schmitt, C.A.; Sobel, J.D. Clue Cells in Bacterial Vaginosis: Immunofluorescent Identification of the Adherent Gram-Negative Bacteria as Gardnerella vaginalis. J. Infect. Dis. 1989, 160, 490–496. [Google Scholar] [CrossRef] [PubMed]
- Joseph, R.; Ser, H.-L.; Kuai, Y.-H.; Tan, L.; Arasoo, V.; Letchumanan, V.; Wang, L.; Pusparajah, P.; Goh, B.-H.; Ab Mutalib, N.-S.; et al. Finding a Balance in the Vaginal Microbiome: How Do We Treat and Prevent the Occurrence of Bacterial Vaginosis? Antibiotics 2021, 10, 719. [Google Scholar] [CrossRef]
- Redelinghuys, M.J.; Geldenhuys, J.; Jung, H.; Kock, M.M. Bacterial Vaginosis: Current Diagnostic Avenues and Future Opportunities. Front. Cell. Infect. Microbiol. 2020, 10, 354. [Google Scholar] [CrossRef]
- Pettit, R.K.; McAllister, S.C.; Hamer, T.A. Response of gonococcal clinical isolates to acidic conditions. J. Med. Microbiol. 1999, 48, 149–156. [Google Scholar] [CrossRef] [Green Version]
- Pettit, R.; Whelan, T.; Woo, K. Acid stress upregulated outer membrane proteins in clinical isolates of Neisseria gonorrhoeae, but not most commensal Neisseria. Can. J. Microbiol. 2001, 47, 871–876. [Google Scholar] [CrossRef]
- Rice, P.A.; Shafer, W.M.; Ram, S.; Jerse, A.E. Neisseria gonorrhoeae: Drug Resistance, Mouse Models, and Vaccine Development. Annu. Rev. Microbiol. 2017, 71, 665–686. [Google Scholar] [CrossRef] [Green Version]
- Gong, Z.; Tang, M.M.; Wu, X.; Phillips, N.; Galkowski, D.; Jarvis, G.; Fan, H. Arginine- and Polyamine-Induced Lactic Acid Resistance in Neisseria gonorrhoeae. PLoS ONE 2016, 11, e0147637. [Google Scholar] [CrossRef]
- Mehta, S.D.; Zhao, D.; Green, S.J.; Agingu, W.; Otieno, F.; Bhaumik, R.; Bhaumik, D.; Bailey, R.C. The Microbiome Composition of a Man’s Penis Predicts Incident Bacterial Vaginosis in His Female Sex Partner with High Accuracy. Front. Cell. Infect. Microbiol. 2020, 10, 433. [Google Scholar] [CrossRef] [PubMed]
- Borovkova, N.; Korrovits, P.; Ausmees, K.; Türk, S.; Jõers, K.; Punab, M.; Mändar, R. Influence of sexual intercourse on genital tract microbiota in infertile couples. Anaerobe 2011, 17, 414–418. [Google Scholar] [CrossRef] [PubMed]
- Mändar, R. Microbiota of male genital tract: Impact on the health of man and his partner. Pharmacol. Res. 2013, 69, 32–41. [Google Scholar] [CrossRef] [PubMed]
- Aragón, I.M.; Herrera-Imbroda, B.; Queipo-Ortuño, M.I.; Castillo, E.; Del Moral, J.S.-G.; Gómez-Millán, J.; Yucel, G.; Lara, M.F. The Urinary Tract Microbiome in Health and Disease. Eur. Urol. Focus 2018, 4, 128–138. [Google Scholar] [CrossRef]
- Kim, M.S.; Jung, S.I. The Urinary Tract Microbiome in Male Genitourinary Diseases: Focusing on Benign Prostate Hyperplasia and Lower Urinary Tract Symptoms. Int. Neurourol. J. 2021, 25, 3–11. [Google Scholar] [CrossRef]
- Nelson, D.E.; Van Der Pol, B.; Dong, Q.; Revanna, K.V.; Fan, B.; Easwaran, S.; Sodergren, E.; Weinstock, G.M.; Diao, L.; Fortenberry, J.D. Characteristic Male Urine Microbiomes Associate with Asymptomatic Sexually Transmitted Infection. PLoS ONE 2010, 5, e14116. [Google Scholar] [CrossRef]
- Nelson, D.E.; Dong, Q.; Van Der Pol, B.; Toh, E.; Fan, B.; Katz, B.P.; Mi, D.; Rong, R.; Weinstock, G.M.; Sodergren, E.; et al. Bacterial Communities of the Coronal Sulcus and Distal Urethra of Adolescent Males. PLoS ONE 2012, 7, e36298. [Google Scholar] [CrossRef] [Green Version]
- Dong, Q.; Nelson, D.E.; Toh, E.; Diao, L.; Gao, X.; Fortenberry, J.D.; Van Der Pol, B. The Microbial Communities in Male First Catch Urine Are Highly Similar to Those in Paired Urethral Swab Specimens. PLoS ONE 2011, 6, e19709. [Google Scholar] [CrossRef] [Green Version]
- Fouts, D.E.; Pieper, R.; Szpakowski, S.; Pohl, H.; Knoblach, S.; Suh, M.-J.; Huang, S.-T.; Ljungberg, I.; Sprague, B.M.; Lucas, S.K.; et al. Integrated next-generation sequencing of 16S rDNA and metaproteomics differentiate the healthy urine microbiome from asymptomatic bacteriuria in neuropathic bladder associated with spinal cord injury. J. Transl. Med. 2012, 10, 174. [Google Scholar] [CrossRef] [Green Version]
- Frølund, M.; Wikström, A.; Lidbrink, P.; Abu Al-Soud, W.; Larsen, N.; Harder, C.B.; Sørensen, S.J.; Jensen, J.S.; Ahrens, P. The bacterial microbiota in first-void urine from men with and without idiopathic urethritis. PLoS ONE 2018, 13, e0201380. [Google Scholar] [CrossRef] [Green Version]
- Rakhmatulina, M.R.; Boldyreva, M.N.; Lipova, E.V.; Chekmarev, A.S.; Galkina, I.S. Evaluation of the composition of the microbiota of the urethra in men with sexually transmitted infections. Urologiia 2019, 6, 31–37. [Google Scholar] [CrossRef]
- Saraf, V.S.; Sheikh, S.A.; Ahmad, A.; Gillevet, P.M.; Bokhari, H.; Javed, S. Vaginal microbiome: Normalcy vs dysbiosis. 2021, 203, 3793–3802. Arch. Microbiol. 2021, 203, 3793–3802. [Google Scholar] [CrossRef] [PubMed]
- Munckhof, E.H.A.V.D.; van Sitter, R.L.; Boers, K.E.; Lamont, R.F.; Witt, R.T.; le Cessie, S.; Knetsch, C.W.; van Doorn, L.-J.; Quint, W.G.V.; Molijn, A.; et al. Comparison of Amsel criteria, Nugent score, culture and two CE-IVD marked quantitative real-time PCRs with microbiota analysis for the diagnosis of bacterial vaginosis. Eur. J. Clin. Microbiol. 2019, 38, 959–966. [Google Scholar] [CrossRef] [PubMed]
- Nugent, R.P.; Krohn, M.A.; Hillier, S.L. Reliability of diagnosing bacterial vaginosis is improved by a standardized method of gram stain interpretation. J. Clin. Microbiol. 1991, 29, 297–301. [Google Scholar] [CrossRef] [Green Version]
- Gd, B.; Ca, H. Utility of Next-generation sequencing in Managing Bacterial Vaginosis: Examples from Clinical Practice. J. Women’s Health Care 2016, 5, 1–6. [Google Scholar] [CrossRef]
- Deng, Z.-L.; Gottschick, C.; Bhuju, S.; Masur, C.; Abels, C.; Wagner-Döbler, I. Metatranscriptome Analysis of the Vaginal Microbiota Reveals Potential Mechanisms for Protection against Metronidazole in Bacterial Vaginosis. mSphere 2018, 3, e00262-18. [Google Scholar] [CrossRef] [Green Version]
- Twin, J.; Bradshaw, C.S.; Garland, S.M.; Fairley, C.K.; Fethers, K.; Tabrizi, S.N. The Potential of Metatranscriptomics for Identifying Screening Targets for Bacterial Vaginosis. PLoS ONE 2013, 8, e76892. [Google Scholar] [CrossRef] [Green Version]
- Kumar, M.; Ji, B.; Zengler, K.; Nielsen, J. Modelling approaches for studying the microbiome. Nat. Microbiol. 2019, 4, 1253–1267. [Google Scholar] [CrossRef]
- Lachance, J.; Matteau, D.; Brodeur, J.; Lloyd, C.J.; Mih, N.; King, Z.A.; Knight, T.F.; Feist, A.M.; Monk, J.M.; Palsson, B.O.; et al. Genome-scale metabolic modeling reveals key features of a minimal gene set. Mol. Syst. Biol. 2021, 17, e10099. [Google Scholar] [CrossRef]
- Buchweitz, L.F.; Yurkovich, J.T.; Blessing, C.; Kohler, V.; Schwarzkopf, F.; King, Z.A.; Yang, L.; Jóhannsson, F.; Sigurjónsson, Ó.E.; Rolfsson, Ó.; et al. Visualizing metabolic network dynamics through time-series metabolomic data. BMC Bioinform. 2020, 21, 130. [Google Scholar] [CrossRef] [Green Version]
- Chowdhury, S.; Fong, S.S. Computational Modeling of the Human Microbiome. Microorganisms 2020, 8, 197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jean, S.; Huang, B.; Parikh, H.I.; Edwards, D.J.; Brooks, J.P.; Kumar, N.G.; Sheth, N.U.; Koparde, V.; Smirnova, E.; Huzurbazar, S.; et al. Multi-omic Microbiome Profiles in the Female Reproductive Tract in Early Pregnancy. Infect. Microbes Dis. 2019, 1, 49–60. [Google Scholar] [CrossRef]
- Fettweis, J.M.; Serrano, M.G.; Brooks, J.L.; Edwards, D.J.; Girerd, P.H.; Parikh, H.; Huang, B.; Arodz, T.J.; Edupuganti, L.; Glascock, A.L.; et al. The vaginal microbiome and preterm birth. Nat. Med. 2019, 25, 1012–1021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serrano, M.G.; Parikh, H.; Brooks, J.L.; Edwards, D.J.; Arodz, T.J.; Edupuganti, L.; Huang, B.; Girerd, P.H.; Bokhari, Y.A.; Bradley, S.P.; et al. Racioethnic diversity in the dynamics of the vaginal microbiome during pregnancy. Nat. Med. 2019, 25, 1001–1011. [Google Scholar] [CrossRef]
- Noecker, C.; Eng, A.; Srinivasan, S.; Theriot, C.M.; Young, V.B.; Jansson, J.K.; Fredricks, D.N.; Borenstein, E. Metabolic Model-Based Integration of Microbiome Taxonomic and Metabolomic Profiles Elucidates Mechanistic Links between Ecological and Metabolic Variation. mSystems 2016, 1, e00013-15. [Google Scholar] [CrossRef] [Green Version]
- Ceccarani, C.; Foschi, C.; Parolin, C.; D’Antuono, A.; Gaspari, V.; Consolandi, C.; Laghi, L.; Camboni, T.; Vitali, B.; Severgnini, M.; et al. Diversity of vaginal microbiome and metabolome during genital infections. Sci. Rep. 2019, 9, 14095. [Google Scholar] [CrossRef] [Green Version]
- Brotman, R.M.; Klebanoff, M.A.; Nansel, T.; Yu, K.F.; Andrews, W.W.; Zhang, J.; Schwebke, J.R. Bacterial Vaginosis Assessed by Gram Stain and Diminished Colonization Resistance to Incident Gonococcal, Chlamydial, and Trichomonal Genital Infection. J. Infect. Dis. 2010, 202, 1907–1915. [Google Scholar] [CrossRef]
- Brotman, R.M.; Bradford, L.L.; Conrad, M.; Gajer, P.; Ault, K.; Peralta, L.; Forney, L.J.; Carlton, J.M.; Abdo, Z.; Ravel, J. Association Between Trichomonas vaginalis and Vaginal Bacterial Community Composition Among Reproductive-Age Women. Sex Transm Dis 2012, 39, 807–812. [Google Scholar] [CrossRef] [Green Version]
- Chen, I.; Kelkar, Y.D.; Gu, Y.; Zhou, J.; Qiu, X.; Wu, H. High-dimensional linear state space models for dynamic microbial interaction networks. PLoS ONE 2017, 12, e0187822. [Google Scholar] [CrossRef] [Green Version]
- Lugo-Martinez, J.; Ruiz-Perez, D.; Narasimhan, G.; Bar-Joseph, Z. Dynamic interaction network inference from longitudinal microbiome data. Microbiome 2019, 7, 54. [Google Scholar] [CrossRef] [Green Version]
Figure 1.
Transcriptional regulators in N. gonorrhoeae respond to various stress stimuli. Nutritional immunity: Essential trace metals such as Fe3+, Zn2+, and Mn2+ are sequestered from pathogens by being stored in host proteins, such as lactoferricin (Fe3+), transferrin (Fe3+), and calprotectin (Zn2+, Mn2+). The transcriptional regulators Fur and PerR control the expression of genes involved in metal homeostasis, including surface-expressed metal transport proteins that scavenge metals for N. gonorrhoeae. ROS: the redox-responsive protein OxyR responds to the ROS and H2O2, and maintains redox homeostasis by regulating the expression of antioxidant genes. AMPs and antibiotics: efflux pumps that remove AMPs and antibiotics are negatively regulated by MtrR, which is itself negatively regulated by a second DNA-binding protein, MpeR, such that MpeR positively regulates efflux pumps indirectly. The figure was created with Biorender.com.
Figure 1.
Transcriptional regulators in N. gonorrhoeae respond to various stress stimuli. Nutritional immunity: Essential trace metals such as Fe3+, Zn2+, and Mn2+ are sequestered from pathogens by being stored in host proteins, such as lactoferricin (Fe3+), transferrin (Fe3+), and calprotectin (Zn2+, Mn2+). The transcriptional regulators Fur and PerR control the expression of genes involved in metal homeostasis, including surface-expressed metal transport proteins that scavenge metals for N. gonorrhoeae. ROS: the redox-responsive protein OxyR responds to the ROS and H2O2, and maintains redox homeostasis by regulating the expression of antioxidant genes. AMPs and antibiotics: efflux pumps that remove AMPs and antibiotics are negatively regulated by MtrR, which is itself negatively regulated by a second DNA-binding protein, MpeR, such that MpeR positively regulates efflux pumps indirectly. The figure was created with Biorender.com.

Figure 2.
Network analysis. To infer a gene co-expression network of a biological system transcriptomic data is first collected. Next, a network inference tool using correlation coefficient (e.g., Pearson), mutual information (e.g., Context Likelihood of Relatedness), random forest (e.g., GENIE3), or another method is used to calculate a co-expression value for each gene pair. Following this, only gene pairs that are highly co-expressed (either positively or negatively) are retained. Once this co-expression and filtering are done to all gene pairs a network of the most highly co-expressed genes can be inferred [96,100,101].
Figure 2.
Network analysis. To infer a gene co-expression network of a biological system transcriptomic data is first collected. Next, a network inference tool using correlation coefficient (e.g., Pearson), mutual information (e.g., Context Likelihood of Relatedness), random forest (e.g., GENIE3), or another method is used to calculate a co-expression value for each gene pair. Following this, only gene pairs that are highly co-expressed (either positively or negatively) are retained. Once this co-expression and filtering are done to all gene pairs a network of the most highly co-expressed genes can be inferred [96,100,101].

Figure 3.
Impact of a healthy vs dysbiotic microbiome on vaginal ecology. Left Panels: A healthy vaginal microbiome is less diverse and is dominated by Lactobacillus spp. The metabolites of Lactobacillus spp. including lactic acid, H2O2, and bacteriocins that create an environment that is not conducive for the growth of anaerobic bacteria or pathogens. Right Panels: Disruption to the core healthy microbiome, as in the case of BV, leads to an environment where Lactobacillus spp. is replaced by anaerobic facultative bacteria. This is associated with an increase in pH, SCFAs, and polyamines. Additionally, there is an induction of innate immune responses resulting in upregulation of cytokine and chemokine production, the influx of PMNs, and disrupted epithelial barrier increasing the susceptibility to STIs. DCs: dendritic cells; SCFAs: short-chain fatty acids; C & C: chemokines and cytokines; AMPs: antimicrobial peptides; LGT: lower genital tract; and PMNs: polymorphonuclear leukocytes. This figure was created with Biorender.com.
Figure 3.
Impact of a healthy vs dysbiotic microbiome on vaginal ecology. Left Panels: A healthy vaginal microbiome is less diverse and is dominated by Lactobacillus spp. The metabolites of Lactobacillus spp. including lactic acid, H2O2, and bacteriocins that create an environment that is not conducive for the growth of anaerobic bacteria or pathogens. Right Panels: Disruption to the core healthy microbiome, as in the case of BV, leads to an environment where Lactobacillus spp. is replaced by anaerobic facultative bacteria. This is associated with an increase in pH, SCFAs, and polyamines. Additionally, there is an induction of innate immune responses resulting in upregulation of cytokine and chemokine production, the influx of PMNs, and disrupted epithelial barrier increasing the susceptibility to STIs. DCs: dendritic cells; SCFAs: short-chain fatty acids; C & C: chemokines and cytokines; AMPs: antimicrobial peptides; LGT: lower genital tract; and PMNs: polymorphonuclear leukocytes. This figure was created with Biorender.com.

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Sunkavalli, A.; McClure, R.; Genco, C. Molecular Regulatory Mechanisms Drive Emergent Pathogenetic Properties of Neisseria gonorrhoeae. Microorganisms 2022, 10, 922. https://doi.org/10.3390/microorganisms10050922
AMA Style
Sunkavalli A, McClure R, Genco C. Molecular Regulatory Mechanisms Drive Emergent Pathogenetic Properties of Neisseria gonorrhoeae. Microorganisms. 2022; 10(5):922. https://doi.org/10.3390/microorganisms10050922
Chicago/Turabian StyleSunkavalli, Ashwini, Ryan McClure, and Caroline Genco. 2022. "Molecular Regulatory Mechanisms Drive Emergent Pathogenetic Properties of Neisseria gonorrhoeae" Microorganisms 10, no. 5: 922. https://doi.org/10.3390/microorganisms10050922
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.