
Soil Metabolomics Predict Microbial Taxa as Biomarkers of Moisture Status in Soils from a Tidal Wetland

 , , , , ,
, , , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Soil Collection and Incubation Conditions
2.1.1. Site
2.1.2. Soil Incubation
2.1.3. Soil Core Sampling and Analyses
2.2. DNA Extraction and 16S rRNA Sequence Analysis
2.3. Metabolome Extraction and GC–MS Analysis
3. Data Analyses
3.1. 16S rRNA Sequence Data Filtering
3.2. Metabolomics Data Univariate Analysis
3.3. Metabolite Data Processing for Random Forest Classification Approach
3.4. Predicting OTU Presence/Absence with Metabolomic Profiles
4. Results and Discussion
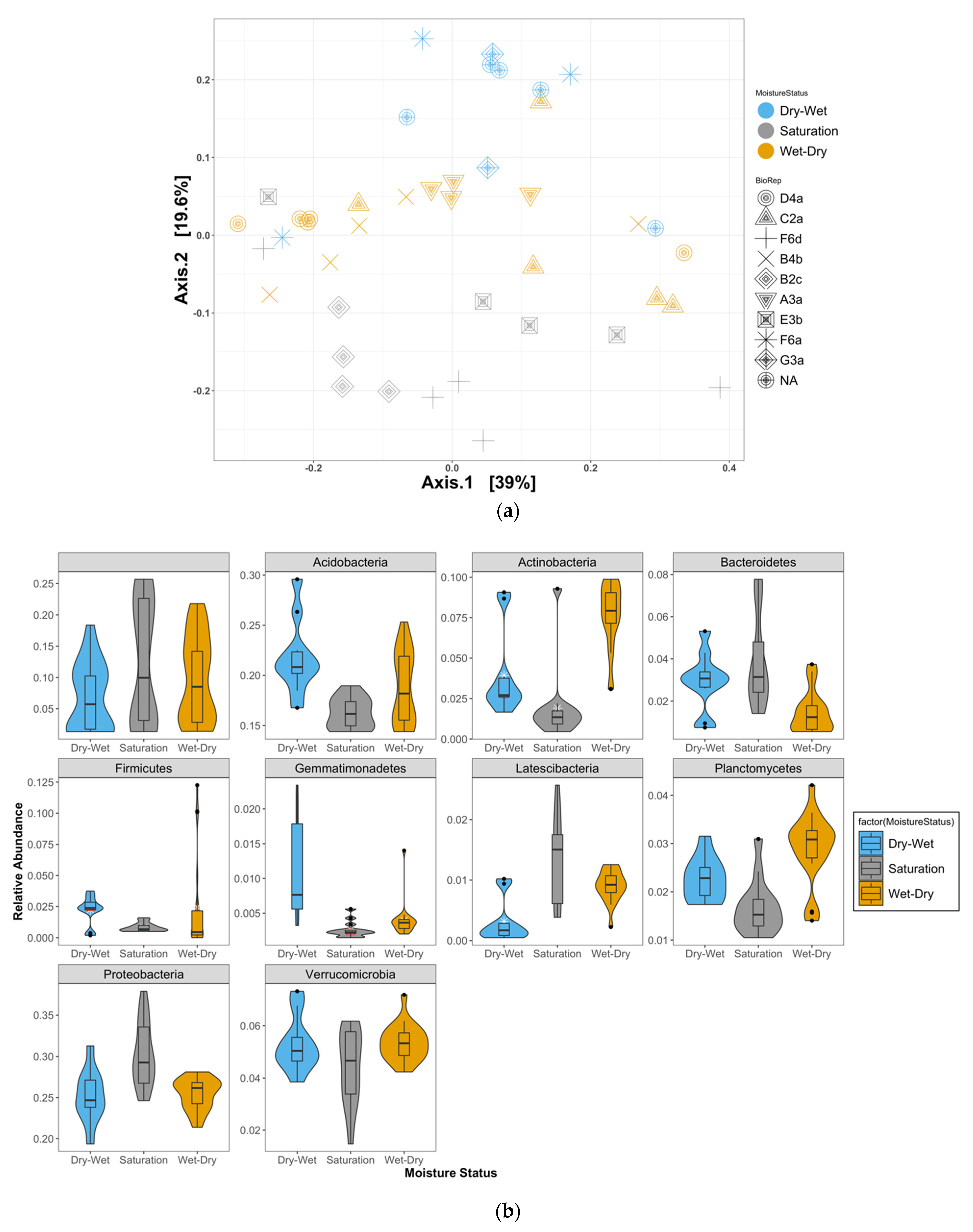
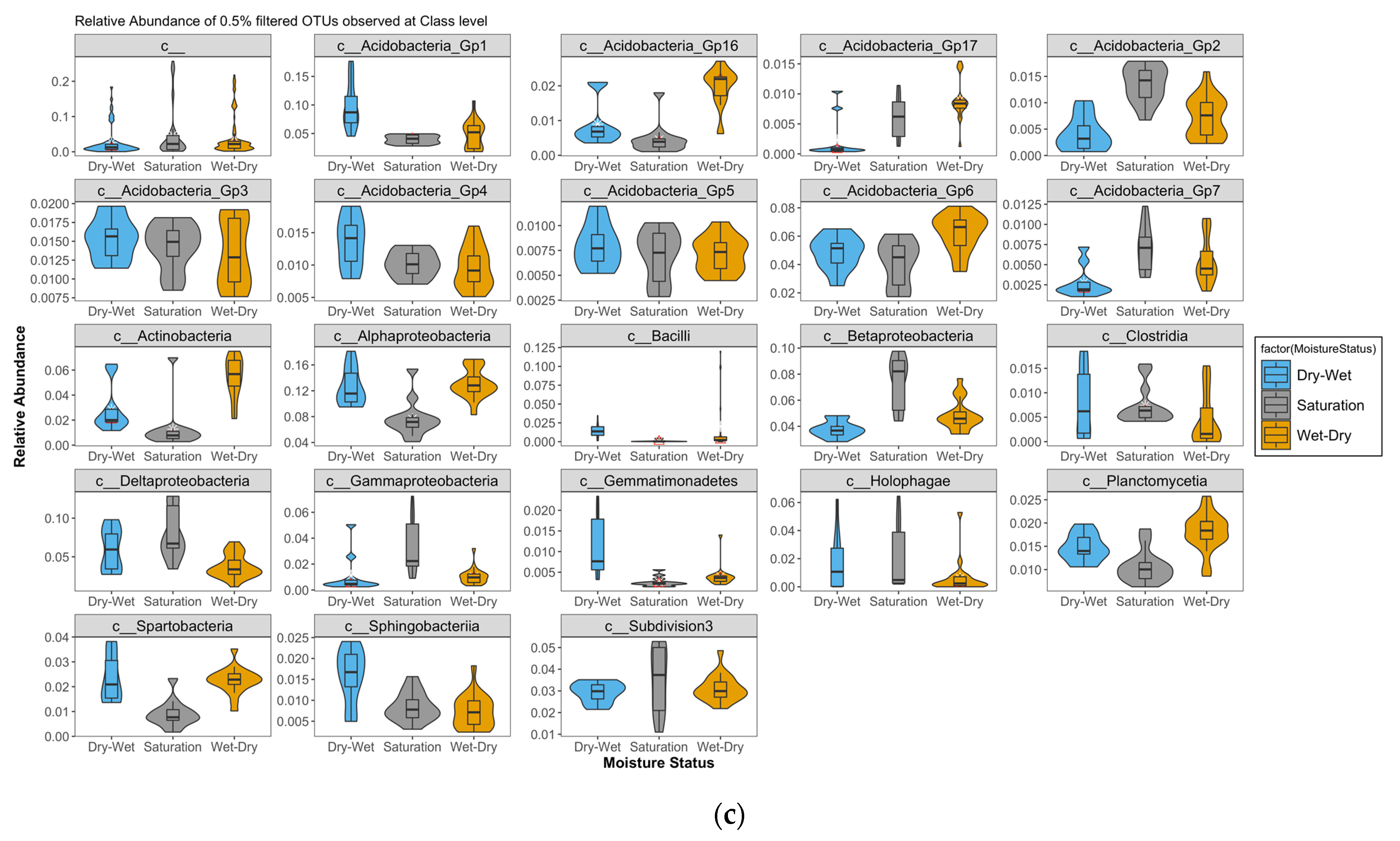
4.1. Shifts in Relative Abundances of Bacterial Taxa
4.2. Metabolite Shifts in Response to Hydrologic Shifts
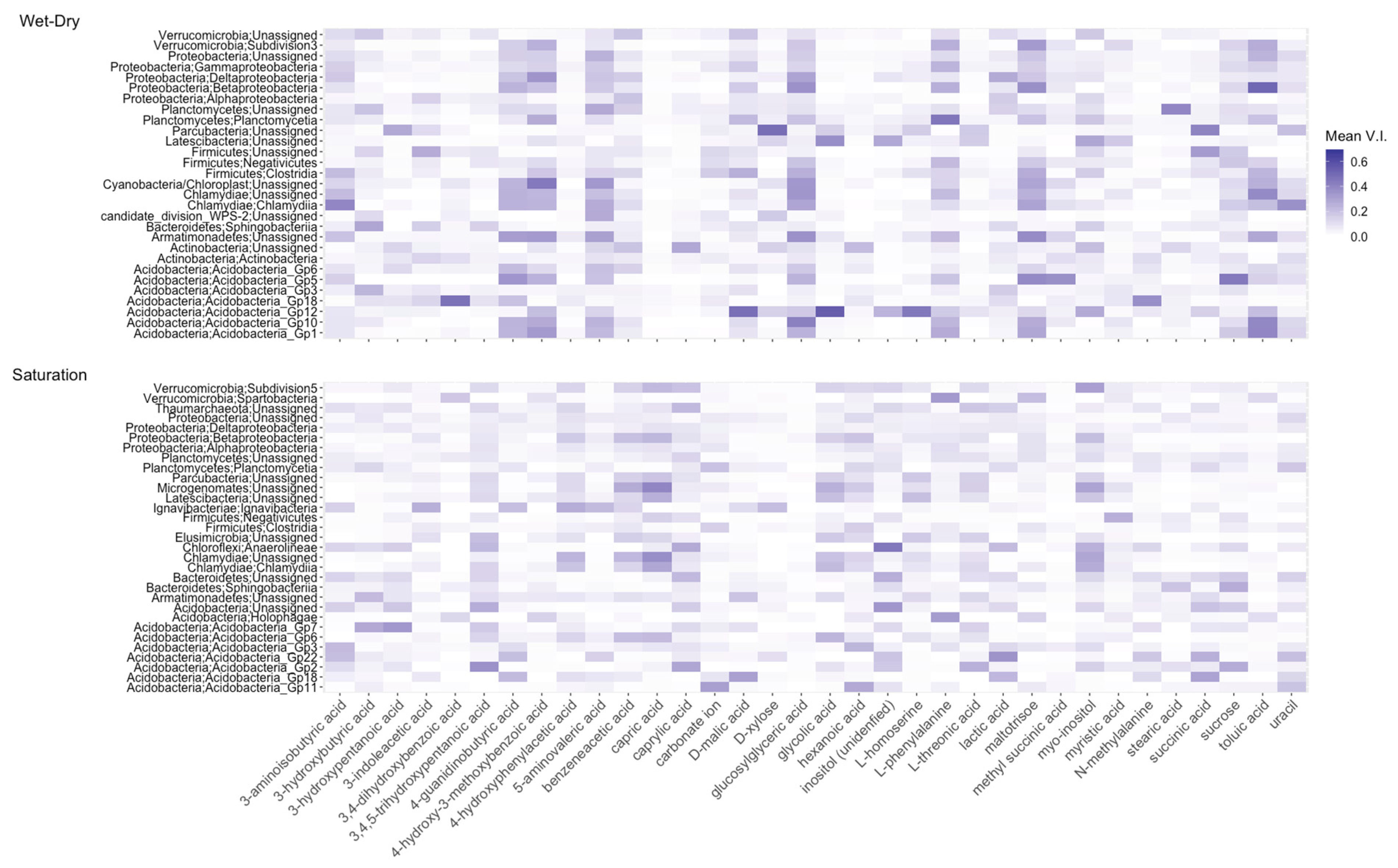
4.3. Metabolites Predict Taxa
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Barnard, R.L.; Osborne, C.A.; Firestone, M.K. Changing precipitation pattern alters soil microbial community response to wet-up under a Mediterranean-type climate. Isme J. 2015, 9, 946–957. [Google Scholar] [CrossRef] [PubMed]
- Kieft, T.; Soroker, E.; Firestone, M. Microbial biomass response to a rapid increase in water potential when dry soil is wetted. Soil Biol. Biochem. 1987, 19, 119–126. [Google Scholar] [CrossRef]
- RoyChowdhury, T.; Lee, J.-Y.; Bottos, E.M.; Brislawn, C.M.; White, R.A., III; Bramer, L.M.; Brown, J.N.; Zucker, J.D.; Kim, Y.-M.; Jumpponen, A.; et al. Metaphenomic response of a native prairie soil microbiome to moisture perturbations. mSystems 2019, 4, e00061-19. [Google Scholar]
- Xiang, S.; Doyle, A.; Holden, P.; Schimel, J. Drying and rewetting effects on C and N mineralization and microbial activity in surface and subsurface California grassland soils. Soil Biol. Biochem. 2008, 40, 2281–2289. [Google Scholar] [CrossRef]
- Evans, S.; Wallenstein, M. Soil microbial community response to drying and rewetting stress: Does historical precipitation regime matter? Biogeochemistry 2012, 109, 101–116. [Google Scholar] [CrossRef]
- Cong, J.; Yang, Y.; Liu, X.; Lu, H.; Liu, X.; Zhou, J.; Li, D.; Yin, H.; Ding, J.; Zhang, Y. Analyses of soil microbial community compositions and functional genes reveal potential consequences of natural forest succession. Sci. Rep. 2015, 5, 10007. [Google Scholar] [CrossRef]
- Swenson, T.; Karaoz, U.; Swenson, J.; Bowen, B.; Northen, T. Linking soil biology and chemistry in biological soil crust using isolate exometabolomics. Nat. Commun. 2018, 9, 19. [Google Scholar] [CrossRef]
- Widder, S.; Allen, R.J.; Pfeiffer, T.; Curtis, T.P.; Wiuf, C.; Sloan, W.T.; Cordero, O.X.; Brown, S.P.; Momeni, B.; Shou, W.Y.; et al. Challenges in microbial ecology: Building predictive understanding of community function and dynamics. Isme J. 2016, 10, 2557–2568. [Google Scholar] [CrossRef]
- Krause, S.; Le Roux, X.; Niklaus, P.A.; Van Bodegom, P.M.; Lennon, J.T.; Bertilsson, S.; Grossart, H.P.; Philippot, L.; Bodelier, P.L.E. Trait-based approaches for understanding microbial biodiversity and ecosystem functioning. Front. Microbiol. 2014, 5, 251. [Google Scholar] [CrossRef]
- Treseder, K.K.; Balser, T.C.; Bradford, M.A.; Brodie, E.L.; Dubinsky, E.A.; Eviner, V.T.; Hofmockel, K.S.; Lennon, J.T.; Levine, U.Y.; MacGregor, B.J.; et al. Integrating microbial ecology into ecosystem models: Challenges and priorities. Biogeochemistry 2012, 109, 7–18. [Google Scholar] [CrossRef]
- DeAngelis, K.; Silver, W.; Thompson, A.; Firestone, M. Microbial communities acclimate to recurring changes in soil redox potential status. Environ. Microbiol. 2010, 12, 3137–3149. [Google Scholar] [CrossRef] [PubMed]
- Weber, K.; Urrutia, M.; Churchill, P.; Kukkadapu, R.; Roden, E. Anaerobic redox cycling of iron by freshwater sediment microorganisms. Environ. Microbiol. 2006, 8, 100–113. [Google Scholar] [CrossRef] [PubMed]
- Pett-Ridge, J.; Silver, W.; Firestone, M. Redox fluctuations frame microbial community impacts on N-cycling rates in a humid tropical forest soil. Biogeochemistry 2006, 81, 95–110. [Google Scholar] [CrossRef]
- Pett-Ridge, J.; Firestone, M. Redox fluctuation structures microbial communities in a wet tropical soil. Appl. Environ. Microbiol. 2005, 71, 6998–7007. [Google Scholar] [CrossRef]
- Swenson, T.L.; Karaoz, U.; Swenson, J.M.; Bowen, B.P.; Northen, T.R. Linking soil biology and chemistry using bacterial isolate exometabolite profiles. bioRxiv 2017, 109330. [Google Scholar] [CrossRef]
- Swenson, T.L.; Jenkins, S.; Bowen, B.P.; Northen, T.R. Untargeted soil metabolomics methods for analysis of extractable organic matter. Soil Biol. Biochem. 2015, 80, 189–198. [Google Scholar] [CrossRef]
- Szymanski, J.; Jozefczuk, S.; Nikoloski, Z.; Selbig, J.; Nikiforova, V.; Catchpole, G.; Willmitzer, L. Stability of Metabolic Correlations under Changing Environmental Conditions in Escherichia coli—A Systems Approach. PLoS ONE 2009, 4, e7441. [Google Scholar] [CrossRef] [PubMed]
- Rosato, A.; Tenori, L.; Cascante, M.; Carulla, P.; dos Santos, V.; Saccenti, E. From correlation to causation: Analysis of metabolomics data using systems biology approaches. Metabolomics 2018, 14, 37. [Google Scholar] [CrossRef]
- van der Heijden, M.; Bardgett, R.; van Straalen, N. The unseen majority: Soil microbes as drivers of plant diversity and productivity in terrestrial ecosystems. Ecol. Lett. 2008, 11, 296–310. [Google Scholar] [CrossRef]
- Guijas, C.; Montenegro-Burke, J.; Warth, B.; Spilker, M.; Siuzdak, G. Metabolomics activity screening for identifying metabolites that modulate phenotype. Nat. Biotechnol. 2018, 36, 316–320. [Google Scholar] [CrossRef]
- RoyChowdhury, T.; Bramer, L.; Hoyt, D.; Kim, Y.; Metz, T.; Mccue, L.; Diefenderfer, H.; Jansson, J.; Bailey, V. Temporal dynamics of CO2 and CH4 loss potentials in response to rapid hydrological shifts in tidal freshwater wetland soils. Ecol. Eng. 2018, 114, 104–114. [Google Scholar] [CrossRef]
- Breiman, L. Random forests. Mach. Learn. 2001, 45, 5–32. [Google Scholar] [CrossRef]
- Delgado-Baquerizo, M.; Reith, F.; Dennis, P.; Hamonts, K.; Powell, J.; Young, A.; Singh, B.; Bissett, A. Ecological drivers of soil microbial diversity and soil biological networks in the Southern Hemisphere. Ecology 2018, 99, 583–596. [Google Scholar] [CrossRef] [PubMed]
- Kauffman, J.B.; Adame, M.F.; Arifanti, V.B.; Schile-Beers, L.M.; Bernardino, A.F.; Bhomia, R.K.; Donato, D.C.; Feller, I.C.; Ferreira, T.O.; Garcia, M.d.J.; et al. Total ecosystem carbon stocks of mangroves across broad global environmental and physical gradients. Ecol. Monogr. 2020, 90, e01405. [Google Scholar] [CrossRef]
- Thompson, L.R.; Sanders, J.; McDonald, D.; Amir, A.; Ladau, J.; KJ, L.; Prill, R.; Tripathi, A.; Gibbons, S.; Ackermann, G.; et al. The Earth Microbiome Project Consortium- A communal catalogue reveals Earth’s multiscale microbial diversity. Nature 2017, 551, 457–463. [Google Scholar] [CrossRef] [PubMed]
- Aronesty, E. Comparison of sequencing utility programs. Open Bioinform. J. 2013, 7, 1–8. [Google Scholar] [CrossRef]
- Bushnell, B. BBMap: A Fast, Accurate, Splice-Aware Aligner; Lawrence Berkeley National Laboratory (LBNL): Berkeley, CA, USA, 2014.
- Edgar, R.C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 2010, 26, 2460–2461. [Google Scholar] [CrossRef]
- Cole, J.R.; Wang, Q.; Fish, J.A.; Chai, B.L.; McGarrell, D.M.; Sun, Y.N.; Brown, C.T.; Porras-Alfaro, A.; Kuske, C.R.; Tiedje, J.M. Ribosomal Database Project: Data and tools for high throughput rRNA analysis. Nucleic Acids Res. 2014, 42, D633–D642. [Google Scholar] [CrossRef]
- Kim, Y.M.; Nowack, S.; Olsen, M.T.; Becraft, E.D.; Wood, J.M.; Thiel, V.; Klapper, I.; Kuhl, M.; Fredrickson, J.K.; Bryant, D.A.; et al. Diel metabolomics analysis of a hot spring chlorophototrophic microbial mat leads to new hypotheses of community member metabolisms. Front. Microbiol. 2015, 6, 209. [Google Scholar] [CrossRef]
- Hiller, K.; Hangebrauk, J.; Jager, C.; Spura, J.; Schreiber, K.; Schomburg, D. MetaboliteDetector: Comprehensive Analysis Tool for Targeted and Nontargeted GC/MS Based Metabolome Analysis. Anal. Chem. 2009, 81, 3429–3439. [Google Scholar] [CrossRef]
- Kind, T.; Wohlgemuth, G.; Lee, D.Y.; Lu, Y.; Palazoglu, M.; Shahbaz, S.; Fiehn, O. FiehnLib: Mass Spectral and Retention Index Libraries for Metabolomics Based on Quadrupole and Time-of-Flight Gas Chromatography/Mass Spectrometry. Anal. Chem. 2009, 81, 10038–10048. [Google Scholar] [CrossRef] [PubMed]
- Oksanen, J.; Blanchet, F.G.; Kindt, R. Vegan: Community Ecology Package; R Package Version 2.3-0; 2015; Available online: https://www.researchgate.net/publication/311493644_Vegan_commity_ecology_package_R_package_version_23-0 (accessed on 20 April 2022).
- McMurdie, P.J.; Holmes, S. phyloseq: An R Package for Reproducible Interactive Analysis and Graphics of Microbiome Census Data. PLoS ONE 2013, 8, e61217. [Google Scholar] [CrossRef] [PubMed]
- Bullard, J.H.; Purdom, E.; Hansen, K.D.; Dudoit, S. Evaluation of statistical methods for normalization and differential expression in mRNA-Seq experiments. BMC Bioinform. 2010, 11, 94. [Google Scholar] [CrossRef]
- Dalmasso, C.; Broet, P.; Moreau, T. A simple procedure for estimating the false discovery rate. Bioinformatics 2005, 21, 660–668. [Google Scholar] [CrossRef] [PubMed]
- Liaw, A.; Wiener, M. Classification and Regression by randomForest. R News 2002, 2, 18–22. [Google Scholar]
- Powers, D.M.W. Evaluation: From Precision, Recall and F-measure to ROC, Informedness, Markedness and Correlation. J. Mach. Learn. Technol. 2011, 2, 37–63. [Google Scholar]
- Basu, S.; Kumbier, K.; Brown, J.; Yu, B. Iterative random forests to discover predictive and stable high-order interactions. Proc. Natl. Acad. Sci. USA 2018, 115, 1943–1948. [Google Scholar] [CrossRef]
- Farag, I.; Youssef, N.; Elshahed, M. Global Distribution Patterns and Pangenomic Diversity of the Candidate Phylum “Latescibacteria” (WS3). Appl. Environ. Microbiol. 2017, 83, e00521-17. [Google Scholar] [CrossRef]
- DeBruyn, J.; Nixon, L.; Fawaz, M.; Johnson, A.; Radosevich, M. Global Biogeography and Quantitative Seasonal Dynamics of Gemmatimonadetes in Soil. Appl. Environ. Microbiol. 2011, 77, 6295–6300. [Google Scholar] [CrossRef]
- Herlemann, D.; Lundin, D.; Labrenz, M.; Jurgens, K.; Zheng, Z.; Aspeborg, H.; Andersson, A. Metagenomic De Novo Assembly of an Aquatic Representative of the Verrucomicrobial Class Spartobacteria. Mbio 2013, 4, e00569-12. [Google Scholar] [CrossRef]
- Chin, K.; Hahn, D.; Hengstmann, U.; Liesack, W.; Janssen, P. Characterization and identification of numerically abundant culturable bacteria from the anoxic bulk soil of rice paddy microcosms. Appl. Environ. Microbiol. 1999, 65, 5042–5049. [Google Scholar] [CrossRef] [PubMed]
- Janssen, P.; Schuhmann, A.; Morschel, E.; Rainey, F. Novel anaerobic ultramicrobacteria belonging to the Verrucomicrobiales lineage of bacterial descent isolated by dilution culture from anoxic rice paddy soil. Appl. Environ. Microbiol. 1997, 63, 1382–1388. [Google Scholar] [CrossRef] [PubMed]
- Pol, A.; Heijmans, K.; Harhangi, H.; Tedesco, D.; Jetten, M.; den Camp, H. Methanotrophy below pH1 by a new Verrucomicrobia species. Nature 2007, 450, 874–878. [Google Scholar] [CrossRef] [PubMed]
- Dunfield, P.; Yuryev, A.; Senin, P.; Smirnova, A.; Stott, M.; Hou, S.; Ly, B.; Saw, J.; Zhou, Z.; Ren, Y.; et al. Methane oxidation by an extremely acidophilic bacterium of the phylum Verrucomicrobia. Nature 2007, 450, 879–882. [Google Scholar] [CrossRef] [PubMed]
- Alexander, M. Introduction to Soil Microbiology; John Wiley: New York, NY, USA, 1977. [Google Scholar]
- Bicknell, B.; Owens, J. Utilization of methyl amines as nitrogen-sources by non-methylotrophs. J. Gen. Microbiol. 1980, 117, 89–96. [Google Scholar] [CrossRef]
- Anthony, C. Biochemistry of methylotrophic microorganisms. Sci. Prog. 1975, 62, 167–206. [Google Scholar]
- Lin, M.; Wagner, C. Purification and characterization of n-methylalanine dehydrogenase. J. Biol. Chem. 1975, 250, 3746–3751. [Google Scholar] [CrossRef]
- Warren, C. Do microbial osmolytes or extracellular depolymerisation products accumulate as soil dries? Soil Biol. Biochem. 2016, 98, 54–63. [Google Scholar] [CrossRef]
- Warren, C.R. Response of osmolytes in soil to drying and rewetting. Soil Biol. Biochem. 2014, 70, 22–32. [Google Scholar] [CrossRef]
- Tveit, A.; Urich, T.; Frenzel, P.; Svenning, M. Metabolic and trophic interactions modulate methane production by Arctic peat microbiota in response to warming. Proc. Natl. Acad. Sci. USA 2015, 112, E2507–E2516. [Google Scholar] [CrossRef]
- Angle, J.; Morin, T.; Solden, L.; Narrowe, A.; Smith, G.; Borton, M.; Rey-Sanchez, C.; Daly, R.; Mirfenderesgi, G.; Hoyt, D.; et al. Methanogenesis in oxygenated soils is a substantial fraction of wetland methane emissions. Nat. Commun. 2017, 8, 1567. [Google Scholar] [CrossRef] [PubMed]
- Nayak, D.; Metcalf, W. Methylamine-specific methyltransferase paralogs in Methanosarcina are functionally distinct despite frequent gene conversion. Isme J. 2019, 13, 2173–2182. [Google Scholar] [CrossRef] [PubMed]
- Narrowe, A.B.; Borton, M.A.; Hoyt, D.W.; Smith, G.J.; Daly, R.A.; Angle, J.C.; Eder, E.K.; Wong, A.R.; Wolfe, R.A.; Pappas, A.; et al. Uncovering the diversity and activity of methylotrophic methanogens in freshwater wetland soils. mSystems 2019, 4, e00320-19. [Google Scholar] [CrossRef] [PubMed]
- Healy, J.; Young, L.; Reinhard, M. Methanogenic decomposition of ferulic acid, a model lignin derivative. Appl. Environ. Microbiol. 1980, 39, 436–444. [Google Scholar] [CrossRef]
- Kielak, A.; Barreto, C.; Kowalchuk, G.; van Veen, J.; Kuramae, E. The Ecology of Acidobacteria: Moving beyond Genes and Genomes. Front. Microbiol. 2016, 7, 744. [Google Scholar] [CrossRef]
- Huber, K.; Wust, P.; Rohde, M.; Overmann, J.; Foesel, B. Aridibacter famidurans gen. nov., sp nov and Aridibacter kavangonensis sp nov., two novel members of subdivision 4 of the Acidobacteria isolated from semiarid savannah soil. Int. J. Syst. Evol. Microbiol. 2014, 64, 1866–1875. [Google Scholar] [CrossRef]
- Lee, S.; Ka, J.; Cho, J. Members of the phylum Acidobacteria are dominant and metabolically active in rhizosphere soil. Fems Microbiol. Lett. 2008, 285, 263–269. [Google Scholar] [CrossRef]
- Empadinhas, N.; da Costa, M. Osmoadaptation mechanisms in prokaryotes: Distribution of compatible solutes. Int. Microbiol. 2008, 11, 151–161. [Google Scholar] [CrossRef]
- Song, M.; Peng, W.; Zeng, F.; Du, H.; Peng, Q.; Xu, Q.; Chen, L.; Zhang, F. Spatial Patterns and Drivers of Microbial Taxa in a Karst Broadleaf Forest. Front. Microbiol. 2018, 9, 1691. [Google Scholar] [CrossRef]
- Schulz, S.; Conrad, R. Influence of temperature on pathways to methane production in the permanently cold profundal sediment of Lake Constance. Fems Microbiol. Ecol. 1996, 20, 1–14. [Google Scholar] [CrossRef]
- Ye, R.; Jin, Q.; Bohannan, B.; Keller, J.; Bridgham, S. Homoacetogenesis: A potentially underappreciated carbon pathway in peatlands. Soil Biol. Biochem. 2014, 68, 385–391. [Google Scholar] [CrossRef]
- Zalman, C.; Meade, N.; Chanton, J.; Kostka, J.; Bridgham, S.; Keller, J. Methylotrophic methanogenesis in Sphagnum-dominated peatland soils. Soil Biol. Biochem. 2018, 118, 156–160. [Google Scholar] [CrossRef]
- Liesack, W.; Bak, F.; Kreft, J.; Stackebrandt, E. Holophaga foetida gen-nov, sp-nov, a new, homoacetogenic bacterium degrading methoxylated aromatic-compounds. Arch. Microbiol. 1994, 162, 85–90. [Google Scholar] [CrossRef] [PubMed]
- Amador-Noguez, D.; Feng, X.; Fan, J.; Roquet, N.; Rabitz, H.; Rabinowitz, J. Systems-Level Metabolic Flux Profiling Elucidates a Complete, Bifurcated Tricarboxylic Acid Cycle in Clostridium acetobutylicum. J. Bacteriol. 2010, 192, 4452–4461. [Google Scholar] [CrossRef] [PubMed]
- George, H.; Johnson, J.; Moore, W.; Holdeman, L.; Chen, J. Acetone, isopropanol, and butanol production by Clostridium-beijerinckii (syn Clostridium-butylicum) and Clostridium-aurantibutyricum. Appl. Environ. Microbiol. 1983, 45, 1160–1163. [Google Scholar] [CrossRef]
- Stams, A. Metabolic interactions between anaerobic-bacteria in methanogenic environments. Antonie van Leeuwenhoek Int. J. Gen. Mol. Microbiol. 1994, 66, 271–294. [Google Scholar] [CrossRef]
- Conrad, R.; Rothfuss, F. Methane oxidation in the soil surface-layer of a flooded rice field and the effect of ammonium. Biol. Fertil. Soils 1991, 12, 28–32. [Google Scholar] [CrossRef]
- Frenzel, P.; Rothfuss, F.; Conrad, R. Oxygen profiles and methane turnover in a flooded rice microcosm. Biol. Fertil. Soils 1992, 14, 84–89. [Google Scholar] [CrossRef]
- Wang, J.; Krause, S.; Muyzer, G.; Meima-Franke, M.; Laanbroek, H.; Bodelier, P. Spatial patterns of iron- and methane-oxidizing bacterial communities in an irregularly flooded, riparian wetland. Front. Microbiol. 2012, 3, 64. [Google Scholar] [CrossRef]
- Peralta, A.; Ludmer, S.; Matthews, J.; Kent, A. Bacterial community response to changes in soil redox potential along a moisture gradient in restored wetlands. Ecol. Eng. 2014, 73, 246–253. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
RoyChowdhury, T.; Bramer, L.M.; Brown, J.; Kim, Y.-M.; Zink, E.; Metz, T.O.; McCue, L.A.; Diefenderfer, H.L.; Bailey, V. Soil Metabolomics Predict Microbial Taxa as Biomarkers of Moisture Status in Soils from a Tidal Wetland. Microorganisms 2022, 10, 1653. https://doi.org/10.3390/microorganisms10081653
RoyChowdhury T, Bramer LM, Brown J, Kim Y-M, Zink E, Metz TO, McCue LA, Diefenderfer HL, Bailey V. Soil Metabolomics Predict Microbial Taxa as Biomarkers of Moisture Status in Soils from a Tidal Wetland. Microorganisms. 2022; 10(8):1653. https://doi.org/10.3390/microorganisms10081653
Chicago/Turabian StyleRoyChowdhury, Taniya, Lisa M. Bramer, Joseph Brown, Young-Mo Kim, Erika Zink, Thomas O. Metz, Lee Ann McCue, Heida L. Diefenderfer, and Vanessa Bailey. 2022. "Soil Metabolomics Predict Microbial Taxa as Biomarkers of Moisture Status in Soils from a Tidal Wetland" Microorganisms 10, no. 8: 1653. https://doi.org/10.3390/microorganisms10081653